
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resonance-assisted/impaired anion–π interaction: towards the design of novel anion receptors†
Juan Du a,
Changwei Wang*a,
Shiwei Yin*a,
Wenliang Wang*a and
Yirong Mo*b
a,
Changwei Wang*a,
Shiwei Yin*a,
Wenliang Wang*a and
Yirong Mo*b
aKey Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: upc.changweiwang@gmail.com; yin_sw@snnu.edu.cn; wlwang@gmail.com
bDepartment of Nanoscience Joint School of Nanoscience & Nanoengineering, University of North Carolina at Greensboro, Greensboro, NC 27401, USA. E-mail: y_mo3@uncg.edu
First published on 1st October 2020
Abstract
Substituents alter the electron density distribution in benzene in various ways, depending on their electron withdrawing and donating capabilities, as summarized by the empirical Hammett equation. The change of the π electron density distribution subsequently impacts the interaction of substituted benzenes or other cyclic conjugated rings with anions. Currently the design and synthesis of conjugated cyclic receptors capable of binding anions is an active field due to their applications in the sensing and removal of environmental contaminants and molecular recognition. By using the block-localized wavefunction (BLW) method, which is a variant of ab initio valence bond (VB) theory and can derive the reference resonance-free state self-consistently, we quantified the resonance-assisted (RA) or resonance-impaired (RI) phenomena in anion–π interactions from both structural and energetic perspectives. The frozen interaction, in which the electrostatic attraction is involved, has been shown to be the governing factor for the RA or RI interactions with anions. Energy analyses based on the empirical point charge (EPC) model indicated that the anion–π interactions can be simplified as the attraction between a negative point charge (anion) and a group of local dipoles, affected by the enriched or diminished π-cloud due to the resonance between the substituents and the conjugated ring. Hence, two strategies for the design of novel anion receptors can be envisioned. One is the enhancement of the magnitudes and/or numbers of local dipoles (polarized σ bonds), and the other is the reduction of π electron density in conjugated rings. For cases with the RI characteristics, “curved” aromatic molecules are preferred to be anion receptors. Indeed, extremely strong binding was found in complexes formed with fluorinated corannulene (F-CDD) and fluorinated [5]cycloparaphenylene (F-[5]CPP). Inspired by the RA phenomenon, complexes of p-, o- and m-benzoquinones with halides were revisited.
1. Introduction
Noncovalent interactions exist ubiquitously in all aggregates and materials, and provide us with directional “linkers” for the construction of chemical and biological substances.1–5 Unlike a chemical bond which involves sharing a pair of electrons and thus is strong, a noncovalent bond is usually a weak force of various natures such as electrostatic, van der Waals, polarization etc. The accumulation of many weak noncovalent interactions can form a strong binding force in such as proteins, nucleic acids and self-assembling materials. Based on the specific element providing the electrophilic cap for the contact, noncovalent interactions can be defined as hydrogen bonds, tetrel bonds, pnicogen bonds, chalcogen bonds, halogen bonds etc., in which the electrostatic attraction stems from the positively charged hydrogen or σ-hole,6–8 and the π face9–15 or lone pair region.16–20 Both experimental and computational studies of noncovalent interactions get broader over the time as new unconventional types and counterintuitive models, such as the resonance-assisted and resonance-impaired hydrogen bond (RAHB and RIHB),21–28 and anti-electrostatic hydrogen or halogen bonds29–31 have been identified and proposed. These new family members of noncovalent interactions, on one hand, potentially supply us with novel elementary “glues” for the architecture of complexes. On the other hand, they also call for updates of our understanding of noncovalent interactions. One of the particular instances is the anion–π interaction, which represents the attraction between the π face of a conjugated molecule and an anion.32–44 This novel interaction has sparked a surge of interests,32,33,38,40,45–50 and been remarkably utilized in anion recognition,38,40,46,48,50–55 supramolecular chemistry56–59 and catalysis.56–58,60–62 From the first sight, the anion–π interaction is counterintuitive as the interaction between the π-cloud of electrons and anion is expected to be electrostatically repulsive. Nevertheless, the key role of electrostatic attraction was implied in the pioneering experimental investigations decades ago,63–65 and was theoretically supported in the term of quadrupole inversion (opposite sign to benzene) due to the substitution of strong electron-withdrawing groups,66 which turns the negative electrostatic potential (ESP) on the π-face to be positive (π-hole).13–15,67,68 More interestingly, the anion–π interaction indisputably is an resonance-related phenomenon,33 since conjugated and often aromatic rings have been broadly adopted as anion receptors. However, the exact role of resonance has been barely explored largely due to the lack of proper references. From the view of the valence bond (VB) theory, the best reference is always the major Lewis structure of any anion receptor itself with all electrons localized between two bonding atoms or individual atoms. For instance, the resonance in benzene can be best elucidated with the reference of the Kekulé structure.Still, numerous computational investigations have been carried out to explore the nature of anion–π interactions within the molecular orbital (MO) theory or density functional theory (DFT).11,34,39,69 Politzer et al. found that the strength of anion–π interaction correlates well with the magnitude of the ESPs on the π-hole and its interacting anion.6,11 In other word, the anion–π interaction can be well interpreted with the electrostatic model. This is also supported by Wheeler et al., who observed the good correlation between the ESPs above the ring centroids and the predicted interaction energies.70 Moreover, evidences for the key role of the electrostatic attraction,71,72 as well as the significance of the polarization effect, have also been obtained quantitatively, using a variety of energy decomposition analysis (EDA) approaches.34,66,73–78 Differently, the strength of anion–π interaction in the I−⋯C6F6 complex was determined in a combined experimental and computational investigation by Anstöter et al., who found that the attraction is governed by the electron correlation. The latter accounts for about 41% of the interaction energy, with the rest from the frozen and polarization interactions.79 But this prominent role of the electron correlation could be compatible with the electrostatic explanation, because the destabilizing Pauli exchange repulsion is also included in the frozen term in their absolutely localized molecular orbital (ALMO) EDA analysis.80–82 In other words, if the Pauli repulsion is taken out of the frozen energy term, the remaining electrostatic stabilization would be comparably important as the electron correlation. Experimentally, novel anion–π associates were proposed by Kepler et al. in 2019, by using the p-benzoquinones as the halide receptors.83 Intriguingly, the bonding site in a p-benzoquinone is shifted away from the principal axis of the aromatic ring, which is deformed by the bonding with a considerable magnitude of covalency.83 Furthermore, the charge transfer nature was proved by means of the Natural Bond Orbital (NBO) analysis84,85 and Mulliken correlation.86–88 In another case, quinoid rings were utilized as iodide receptors in experiments in 2018, and the importance of charge transfer interactions was suggested by the color of crystals and DFT calculations.89 Thus, it is reasonable to conclude that all electrostatic attraction, polarization, charge transfer and dispersion (electron correlation) could be significant to specific anion–π contacts, making the anion–π interaction a diversified bonding family.
Apart from the nature of anion–π interactions, how the π electron cloud contributes to the interaction is another intriguing issue, as the understanding would allow researchers to modulate the π-cloud and consequently tune the anion–π interactions. The correlations between the bonding strength and the aromaticity criteria such as isodesmic stabilization energy and NICS90 of the anion receptor were found by Alkorta et al., thus highlighting the significance of π resonance.33 However, Kozuch suggested that the “π-hole” bond is a misleading term because it originates from the σ framework.14 For instance, fluorine, as one of the most wildly adopted electron-withdrawing substituents on the anion receptors,42 is a σ-electron acceptor but π-electron donor. As a consequence, an electrostatic attraction of C6F6 with anions comes from the accumulated σ-holes of F–C bonds in the σ framework, and is actually discounted by the enrichment of π-electrons (a RI phenomenon).14 Furthermore, persuasive evidence against the positive contribution of π electrons was presented in the complex formed by hexafluoroborazine and chloride (B3N3F6⋯Cl−), where the conjugated ring was twisted remarkably by the anion, indicating an unfavorable role of the resonance.14 The negative role of π-electrons in anion–π interactions was also supported by Wheeler and Houk, who reproduced the interaction energies for 83 anion–π complexes qualitatively, using a simple charge-dipole model, in which the π-polarization effect was turned off.70 Similar theoretical approach has also been successfully applied in the investigation of π/π stacking.91
It has been a textbook knowledge that a substituent can influence the π electron distribution and subsequently affect the reaction rates at different sites of the substituted benzene ring, as summarized by the empirical Hammett equation. Elucidating the impacts of conjugation between the cyclic receptor ring and substituents could clarify the different roles of σ and π electrons and provide us a more feasible pathway to modulate the strengths and bonding sites of anion–π interactions. In this regard, the popular RAHB,21–26 which refers to the strengthened interplay between H-bonding and the π resonance, is an advisable example for the current work. While the RAHB has been extensively investigated and utilized,92–112 recently the RIHB concept has also been proposed and confirmed.27,28 Notably, the enhanced electrostatic attraction due to the π-resonance was theoretically proved to be the main cause of RAHB by Mo and coworkers.98,110,113,114 Using the block-localized wavefunction (BLW) method which can disable the π resonance,115,116 they showed that the frozen term, in which the electrostatic interaction is included, becomes more stabilizing for exemplary cases, when the π-conjugation is “turned on”. Moreover, both RAHB and RIHB have been systemically examined, and the key difference lies in the flowing direction of π-electrons.117 Analogous to RAHB/RIHB, the contribution of resonance to the anion–π interactions is also a heuristic topic, yet less explored. The critical justification of the resonance effect requires a theoretical approach that can define electron-localized (resonance or Lewis) state as reference. This can be achieved by the VB theory, where a molecular wavefunction can be defines with a linear combination of resonance states and each resonance state can be defined with the Heitler–London–Slater–Pauling function.118–122 The BLW method is the simplest ab initio VB method that combines the computational efficiency of MO theory and the chemical intuition of VB theory, and can derive the optimal electron-localized state self-consistently. Besides, the binding energy can be decomposed into several physically meaningful terms based on BLW method (called BLW-EDA).123 We note that there are a range of EDA schemes, including the symmetry-adapted perturbation theory (SAPT),124 EDA-NOCV,125,126 and the Kitaura and Morokuma (KM) scheme127,128 that have been developed and extensively applied to the studies on the nature of chemical bonds.129 While the nature of π-hole and σ-hole interactions can be probed with the ALMO80–82 as mentioned in a recent review,12 this method is actually the same as the BLW method.
In this work, we intended to elucidate the impacts of π resonance on the strengths of anion–π interactions, by examining the changes of the bonding strengths after quenching the π-orbital mixing between conjugated rings and substituents. The contribution of resonance to each energy component was clarified by means of the BLW-EDA approach. Both the RA and RI phenomena and the dominating role of the electrostatic interaction were further confirmed using an empirical point charges (EPC) model, by replacing the anion or all atoms of the aromatic monomer with point charge(s). Key factors for the “π-hole” were illuminated by inspecting the evolution of ESP along the bonding direction. Finally, promising anion receptors were proposed based on our updated understanding of resonance in anion–π interactions.
2. Methodology and computational details
2.1 Block-localized wave function (BLW) method
MO theory is featured by Slater determinants composed of delocalized and orthogonal MOs, which lead to the high computational efficiency. In contrast, VB theory uses localized and nonorthogonal orbitals to construct HLSP functions and each HLSP function is a linear combination of 2n (n is the number of chemical bonds) Slater determinants. While the computational cost is much high in ab initio VB theory, resonance can be quantified with the energy difference between the ground state and the most stable VB structure. The essence of the BLW method lies in the partition of the whole molecule into several functional groups (fragments or blocks), and all orbitals are block-localized. In other words, orbitals are expanded only in one block. Orbitals belonging to the same block are constrained to be orthogonal (a MO characteristics), but orbitals belonging to different blocks are non-orthogonal (a VB characteristics). The final electron-localized state is defined with one Slater determinant composed of block-localized MOs that are self-consistently optimized. In this way, the BLW method is the simplest variant of VB theory, but it retains the high computational efficiency of MO theory.In this work, the diabatic state, in which the π electrons of the substituents on the ring are strictly localized, is expressed as
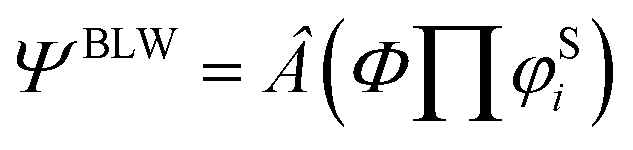 | (1) |
2.2 BLW energy decomposition (BLW-ED) analysis
In the BLW-ED analyses, both the anion and its receptor can be conveniently treated as two blocks respectively, and their binding energy (ΔEb) can be decomposed into five physically meaningful terms, including deformation (ΔEdef), frozen (ΔEF), polarization (ΔEpol), charge transfer (ΔECT), and dispersion correction (ΔEdisp) energies as| ΔEb = ΔEdef + ΔEF + ΔEpol + ΔECT + ΔEdisp = ΔEdef + ΔEint | (2) |
| ΔEdisp = EDFTdisp − EAdisp − EBdisp | (3) |
We note that the electron correlation itself is not an interaction term, as it has been largely included in the frozen term when DFT is adopted.
2.3 Empirical point charges (EPC) model
To inspect the role of electrostatic attraction explicitly, the EPC model was proposed to evaluate the interaction energy with an anion (halide in this work) replaced by a point charge (PC). Obviously, resonance within the receptor still contributes in this model. The interaction energy between the receptor and the PC can decomposed into electrostatic and polarization terms:| ΔEint = E(MPol + PC) − E(M0) − E(PC) = [E(M0 + PC) − E(M0) − E(PC)] + [E(MPol + PC) − E(M0 + PC)] = ΔEeles + ΔEpol | (4) |
The interaction energy can be alternatively computed with another scheme, by replacing each atom of the aromatic ring with a PC. In this case, the resonance is absent, as the PC values were derived from the diabatic wavefunction with the Natural Population Analysis (NPA).133 Only the electrostatic and polarization interactions between the anion, and a group of local-dipoles are left in this scheme, which thus is the simplest resonance free (RF) scheme.
3. Results and discussion
Two groups of complexes (Scheme 1) were studied in this work, including the extensively investigated prototypes, or substituted benzenes as receptors for halide ions, which form group 1, and systems with macrocyclic or new anion receptors which form group 2. The latter are constructed according to our improved understanding of RI and RA features. Numbers in Scheme 1 denote different anion receptors, and a, b or c stands for Cl−, Br− or I− respectively. Group 1 consists of complexes 1, 2 and 3, where hexafluorobenzene, 1,3,5-trinitrobenzene, and 1,3,5-tricyanobenzene were used as anion receptors where fluorine is the only π-electron-donating substituent. Group 2 is composed of complexes 4–9. The M062X functional134 was employed, as it has been proved to be a reliable method for non-covalent interactions.135–137 Computations were improved with the Grimme's dispersion correction.131,132 Pivotal observations were also confirmed using different functionals, and HF method as well. Full geometry optimizations were carried out, and all optimal structures were proved to be actual minima based on the analyses of harmonic frequencies for all complexes using the GAMESS (US) program.138 The cc-pVDZ basis set was adopted in the optimization and frequency calculations of group 2 species, but the cc-pVTZ basis set was used for all the rest calculations. Specifically, the small-core relativistic pseudopotential basis set cc-pVDZ-PP or cc-pVTZ-PP139,140 were employed for iodine. For systems in group 1, geometry optimizations and vibrational frequency calculations were also carried out using the spin component-scaled second-order Møller–Plesset perturbation theory (SCS-MP2),141–145 together with different DFTs for benchmarks, as shown in Table S1.† Sequences of binding strengths obtained with all DFTs remain unchanged, and consistent with the results at the SCS-MP2/aug-cc-pVTZ (-PP for iodine) level. Intriguingly, the overall attractions and the order of binding energies are also reproduced at the HF level, in spite of the underestimation of the binding strengths due to the missing of the electron correlations. Notably, the binding energy in complex 1c is −50.8 kJ mol−1 using M062X-D3, only 0.7 kJ mol−1 lower than the value observed by Anstöter et al. in their combined spectral and state-of-the-art computational investigation.79 It should be noted, however, that the analyses of group 1 were carried out at the geometries obtained from constrained optimizations, with the anion receptors constrained to planar, aiming at the rigorous separation of the π-subspace. This constraint is actually insignificant as the reduction in binding energies is less than 1 kJ mol−1 (Table S2†).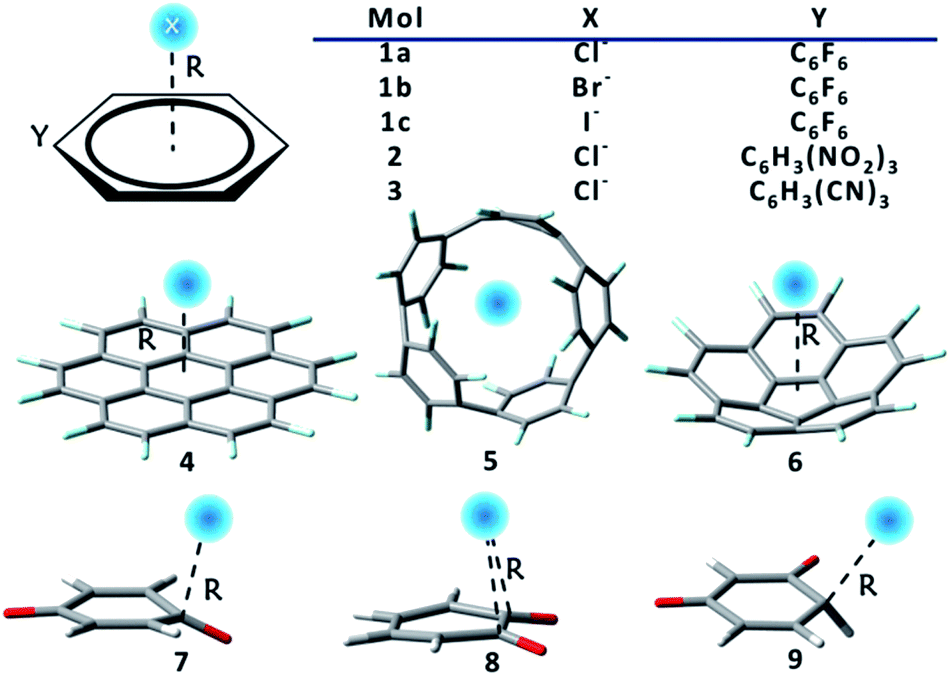 | ||
| Scheme 1 Complexes studied in this work. | ||
We first examined the impacts of π resonance between the substituent groups and the central benzene ring on the binding distances and energies with the halide anions in group 1. Table 1 compiles the bond distances (R) and energy components computed at optimal geometries of both the localized (BLW with the resonance from the substituent groups “turned off”) and delocalized (regular DFT with the resonance from the substituent groups “turned on”) states. According to the regular DFT results, frozen energy, in which the electrostatic interaction is included, is the key player, and polarization is the second important stabilizing factor, with both charge transfer and dispersion contribute slightly to the overall attraction. Bond strength decreases as the halide anion grows heavier (1a > 1b > 1c), mainly due to the reduced stabilizing frozen and polarization interactions, along with the elongated binding distances. Both the electrostatics and polarization decay fast with the stretching of the binding distance. Notably, both 1,3,5-trinitrobenzene (2), and 1,3,5-tricyanobenzene (3), with π-electron withdrawing substituents, attract chloride more intensely than hexafluorobenzene (1a), in which the fluorine, conversely, tends to enrich the π-cloud. The enhanced attraction in complexes 2 and 3 mainly stems from the frozen energy term, implying a possible RA characteristic in complexes 2 and 3, in contrast to the RI feature in complex 1a.
| Complex | R | ΔEdef | ΔEF | ΔEpol | ΔECT | ΔEdisp | ΔEint | ΔEb |
|---|---|---|---|---|---|---|---|---|
| DFT | ||||||||
| 1a | 3.107 | 0.8 | −36.2 | −23.3 | −3.5 | −0.1 | −63.2 | −62.3 |
| 1b | 3.301 | 0.6 | −33.3 | −18.7 | −4.2 | −0.1 | −56.4 | −55.8 |
| 1c | 3.523 | 0.5 | −29.8 | −15.4 | −5.5 | −0.1 | −50.8 | −50.3 |
| 2 | 2.604 | 1.5 | −67.4 | −31.3 | −5.7 | −0.4 | −104.8 | −103.3 |
| 3 | 2.503 | 0.2 | −54.9 | −30.9 | −2.6 | −0.4 | −88.8 | −88.6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| BLW | ||||||||
| 1a | 3.045 | 0.0 | −56.6 | −23.7 | −4.4 | −0.1 | −84.9 | −84.8 |
| 1b | 3.234 | 0.1 | −50.5 | −19.3 | −5.1 | −0.1 | −75.0 | −74.9 |
| 1c | 3.453 | 0.2 | −44.3 | −16.1 | −6.6 | −0.1 | −67.1 | −66.9 |
| 2 | 3.081 | 0.9 | −59.6 | −27.1 | −3.2 | −0.5 | −90.4 | −89.5 |
| 3 | 3.093 | 0.9 | −53.2 | −27.7 | −1.8 | −0.4 | −83.1 | −82.2 |
The most significant finding from Table 1 comes from the comparison of BLW and DFT states, which is the direct evidence for the RA and RI phenomena. With the deactivation of the π conjugation from the halogen atoms to the benzene rings, the binding energy of hexahalobenzenes (1a, 1b and 1c) with chloride increases by 33–36% with the shortening of the bonding distances by 0.06–0.07 Å. Energy decomposition analyses (Table 1) show that the energy changes are solely due to the increasing electrostatic attraction (from the frozen energy term), with polarization, charge transfer and dispersion energy components barely changed from the DFT to the BLW states. This is a strong evidence for the RI phenomenon where resonance weakens the binding to anions, and in accord with the understanding that halogen atoms are π electron donors, though they (particularly fluorine) can draw electrons via σ induction. In contrast, both nitro and cyano groups are π electron acceptors. With the deactivation of their conjugation with benzene rings, the binding with chloride in 2 and 3 notably weakens with the obvious elongation of the bonding distances. Thus, 2 and 3 are the examples for the RA phenomenon where resonance enhances the binding to anions. The study of group 1 reveals a role of thumb for modulating the anion–π interactions, i.e., increasing the π electron density in the conjugated ring would reduce the capability of binding anions, whereas removing the π electron density in the conjugated ring would enhance the π-hole and increase the capability of binding anions. The difference in flowing direction of π-electrons in 1–3 was also supported by the hyperconjugation evaluated with the NBO method (Table S3†).
As Table 1 shows that the RI or RA phenomena is solely reflected in the frozen energy term which consists of electrostatic, Pauli repulsion and DFT electron correlation, we further examined the characteristic of the frozen energy term using different functionals together with the HF method (Table 2). While it is obvious that different functionals result in different values for the frozen energy term since the electron correlation is considered at different magnitudes, it is interesting to note that the ΔΔEF term, which stands for the change of the frozen energy from the diabatic (BLW) to the adiabatic (DFT) state, is rather stable with different methods. Alternatively, we also evaluated the frozen energy term using a strictly localized model, in which not only the π electrons of all substituents are isolated, but the six π electrons are also strictly localized between two adjacent carbons on the 6-membered ring (e.g., the Kekulé structure). It turned out that the frozen energy is almost invariant by this further localization scheme (Table S4†), which means that the anion–π interaction is insensitive to the resonance within the aromatic ring.
| Complex | States | M062X | B3LYP | CAMB3LYP | ωB97X | HF |
|---|---|---|---|---|---|---|
| 1a | BLW | −56.6 | −36.0 | −43.5 | −52.6 | −31.2 |
| DFT | −36.2 | −18.1 | −24.3 | −31.4 | −14.7 | |
| ΔΔEF | 20.4 | 17.9 | 19.2 | 21.2 | 16.5 | |
| 1b | BLW | −50.5 | −29.4 | −36.6 | −46.4 | −24.9 |
| DFT | −33.3 | −14.4 | −20.4 | −28.8 | −11.5 | |
| ΔΔEF | 17.2 | 15.0 | 16.2 | 17.6 | 13.4 | |
| 1c | BLW | −44.3 | −22.7 | −29.7 | −39.5 | −18.4 |
| DFT | −29.8 | −10.7 | −16.6 | −24.8 | −7.6 | |
| ΔΔEF | 14.5 | 12.0 | 13.1 | 14.7 | 10.8 | |
| 2 | BLW | −59.6 | −35.0 | −43.7 | −55.2 | −40.4 |
| DFT | −67.4 | −43.6 | −51.3 | −62.1 | −45.7 | |
| ΔΔEF | −7.8 | −8.6 | −7.6 | −6.9 | −5.3 | |
| 3 | BLW | −53.2 | −32.2 | −39.1 | −50.9 | −27.9 |
| DFT | −54.9 | −34.1 | −41.1 | −52.7 | −29.4 | |
| ΔΔEF | −1.7 | −1.9 | −2.0 | −1.8 | −1.5 |
The changes of electrostatic interactions involved in the frozen energy term due to the resonance from the substituent groups to the aromatic benzene ring can be intuitively demonstrated using the variations in ESPs as shown in Fig. 1b, with the ESPs maps of the adiabatic (DFT) states as references in Fig. 1a. π-Holes in the aromatic rings were found in all substituted benzenes of group 1. Importantly, the most significant variation in ESP was observed in C6F6, which is turned to be less positive due to π electron movement from fluorine atoms to the central ring. Differently, the ESP of 1,3,5-trinitrobenzene becomes much more positive, and the π-hole in 1,3,5-tricyanobenzene is slightly enhanced by the π resonance as well.
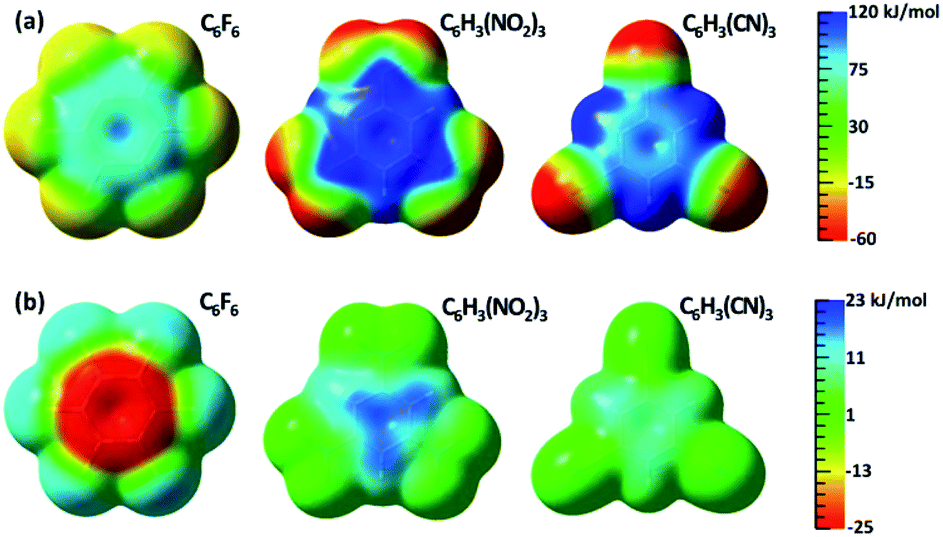 | ||
| Fig. 1 (a) ESP maps of free anion receptors calculated with DFT; (b) differences in ESP between the electron adiabatic (DFT) and diabatic (BLW) states mapped on an isodensity surface (0.001 eÅ−3). | ||
Fig. 2 shows the variations of ESP along the bonding direction in order to help understand and examine the π-hole concept. Maximum ESP values (positive) are observed in the centroid of each aromatic ring, in the order of C6F6 > C6H3(NO2)3 > C6H3(CN)3 (Fig. 2a). All ESPs decrease monotonically along the bonding direction and stay above zero for all anion receptors, forming the π-hole. Specifically, the C6F6 curve goes down the most steeply, because its π-cloud is enriched by fluorine atoms and results in an extra shielding on the centroid. For comparison, the evolution of the ESP of benzene was also displayed together in Fig. 2a. Similar decreasing trend was observed beginning at the centroid. Notably, ESP falls into the negative region at the distance of about 1 Å from the centroid, in sharp contrast to other substituted benzenes. This comes from the different electronegativities of hydrogen and halogens. For each C–X (X = F, Cl, I) σ bond, there are σ holes on the two ends of the bond. Thus, in the hexahalobenzene, the ring centroid is the merging point of six σ holes due to the six C–X σ bonds. But for C–H bonds in benzene, such σ holes are less obvious. At last, all ESPs tend to converge to zero in long range, making the whole curve of benzene non-monotone. Therefore, the key difference in ESP between benzene and anion receptors is ruled by the ring center, where the σ-holes merge. The different ESP distributions in the RA and RI phenomena can also be directly exhibited by the variations of ESP along the bonding direction due to the π resonance, as shown in Fig. 2b. The C6F6 curve with the RI characteristics lies below the horizontal axis, while curves of both C6H3(NO2)3 and C6H3(CN)3 with the RA characteristics stay above zero all the way. The variations of ESP decrease monotonously, and approach zero gradually for C6H3(NO2)3 and C6H3(CN)3. Differently, a minimum point was found on the C6F6 curve, implying an additional shielding of ESP on the centroid, possibly caused by the π-electrons donated by fluorine atoms.
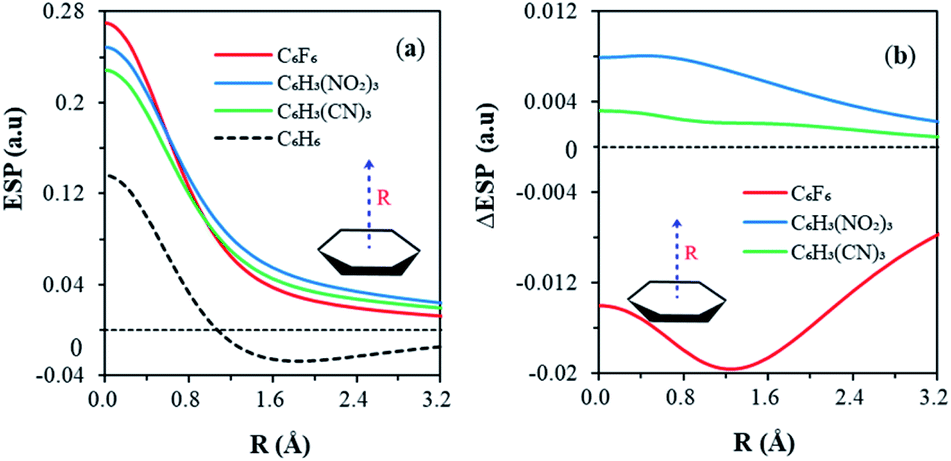 | ||
| Fig. 2 (a) Variations of ESP (in au) in the electron delocalized states of anion receptors 1a, 2, 3 and benzene, along the vertical distance (R in Å) above the centroid, and (b) variations of ESP due to resonance along the bonding direction. | ||
To further explore the role of π resonance in π–anion interactions, we first simplified the chloride anion with a point charge and used this empirical point charge (EPC) model to perform energy decomposition analyses in the cases where π resonance in receptors between substituents and the benzene ring is allowed (resonance contributing or RC scheme). Table 3 summarized the results. The comparison of Tables 1 and 3 show that the interaction energies can be well reproduced by simply replacing the anion with a point charge (RC scheme), with a relative error less than 7% at the M062X-D3 level. The electrostatic interactions are more stabilizing than the polarization interactions in the RC scheme, which is consistent with the BLW-EDA results in Table 1. In addition, the estimated electrostatic energy in Table 3 is very close to the frozen energy derived from the BLW-ED calculation in Table 1, with a difference less than 5.0 kJ mol−1. The similarity between the electrostatic energies in Table 3 and the frozen energies in Table 1 strongly suggests that the Pauli exchange repulsion and the electron correlation components in the frozen energy term are either both insignificant or roughly offset each other.
| Complex | RF scheme | RC scheme | ||||
|---|---|---|---|---|---|---|
| ΔEeles | ΔEpol | ΔEint | ΔEeles | ΔEpol | ΔEint | |
| 1a | −133.7 | −7.1 | −140.9 | −36.1 | −28.6 | −64.7 |
| 1b | −117.4 | −8.6 | −126.0 | −32.2 | −23.4 | −55.6 |
| 1c | −101.7 | −9.5 | −111.2 | −28.5 | −18.8 | −47.3 |
| 2 | −70.5 | −0.6 | −71.1 | −72.0 | −37.1 | −109.2 |
| 3 | 0.2 | −0.2 | 0.0 | −56.3 | −37.3 | −93.6 |
We continued to simplify each atom of the anion receptor with a PC whose value is gotten from the NPA calculations of the diabatic (BLW) state. The subsequent simplified EPC model is the resonance free (RF) scheme. Compared with the above RC scheme, interaction energies in the RF scheme increase in the complexes of hexahalobenzenes (RI cases), while decrease in complexes 2 and 3 (RA cases). In addition, the differences among interaction energies are largely governed by the electrostatics, which is consistent with the RA and RI features evidenced by the above ESP analyses. Interestingly, all energy components are close to zero in the RF scheme of complex 3, which shows the most significant RA phenomenon among all complexes. However, it should be emphasized that this very crude EPC model is just a simplified approach for understanding the role of electrostatic and polarization interactions, not an accurate computational method.
The above EPC model simplified the anion–π interaction as an attraction between a negative point charge (anion) and a group of local dipoles (C–F σ bonds for example), which is further influenced by resonance. Consequently, two strategies to strengthen the anion–π interaction can be envisioned. One is to increase the magnitudes and/or numbers of local dipoles,146 and the other is to either diminish the π electron delocalization from substituents to the ring or enhance the π delocalization from the ring to substituents. For the first strategy, perfluorocoronene147,148 (complex 4) is an appropriate candidate, since the number of C–F bonds is doubled compared with C6F6. Indeed, a considerable increment (68–77%) in binding energies of perfluorocoronene with halide anions can be found by comparing 4 in Table 4 with 1 in Table 1. Meanwhile, fluorinated [5]cycloparaphenylene (F-[5]CPP, complex 5), as one of the smallest nanohoop with the suitable size for halides, has also been tested. The binding energy of complex 5a reaches −205.5 kJ mol−1, or more than two times higher than the binding strength in 1a. Analogous to the complexes with C6F6, the overall attractions in all complexes constructed with perfluorocoronene and F-[5]CPP are dominated by the frozen energy term, and considerably strengthened by the polarization interaction, with slight contributions from both charge transfer and dispersion interactions. The electrostatics nature revealed by the BLW-EDA results is supported by the ESPs shown in Fig. S1 (see ESI†). In addition, a binding site outside the hoop of F-[5]CPP was tested (Fig. S2†) and proved to be less attractive than the centroid by the BLW-EDA results (Table S5†).
| ΔEdef | ΔEF | ΔEpol | ΔECT | ΔEdisp | ΔEint | ΔEb | |
|---|---|---|---|---|---|---|---|
| 4a | 1.5 | −48.6 | −48.2 | −8.6 | −1.0 | −106.3 | −104.9 |
| 4b | 1.4 | −48.5 | −39.5 | −8.6 | −1.0 | −97.6 | −96.2 |
| 4c | 1.2 | −49.2 | −31.8 | −8.3 | −1.0 | −90.3 | −89.1 |
| 5a | 5.7 | −119 | −85.9 | −5.7 | −0.7 | −211.2 | −205.5 |
| 5b | 10.7 | −92.5 | −77.9 | −16.8 | −0.6 | −187.8 | −177.2 |
| 5c | 31.7 | −65.6 | −66.8 | −23.9 | −0.5 | −156.8 | −125.2 |
| 6a | 4.1 | −78.3 | −48.3 | −4.0 | −0.5 | −131.1 | −127.0 |
| 6b | 3.4 | −74.2 | −40.8 | −5.9 | −0.5 | −121.4 | −118.0 |
| 6c | 2.1 | −69.3 | −33.8 | −8.1 | −0.4 | −111.7 | −109.6 |
| 7a | 15.4 | −4.4 | −36.3 | −46.2 | −0.2 | −87.0 | −71.6 |
| 7b | 13.2 | −8.7 | −26.5 | −37.8 | −0.2 | −73.3 | −60.0 |
| 7c | 12.1 | −9.6 | −20.5 | −34.0 | −0.2 | −64.3 | −52.2 |
| 8a | 10.6 | 3.6 | −36.2 | −54.2 | −0.2 | −86.9 | −76.3 |
| 8b | 9.8 | 0.5 | −26.8 | −47.4 | −0.2 | −73.8 | −64.0 |
| 8c | 9.5 | −0.4 | −20.9 | −44.3 | −0.1 | −65.8 | −56.3 |
| 9a | 124.9 | 394.4 | −380.2 | −451.1 | −0.2 | −437.1 | −312.2 |
| 9b | 110.8 | 346.0 | −288.3 | −442.7 | −0.2 | −385.2 | −274.4 |
| 9c | 99.7 | 293.5 | −195.0 | −447.1 | −0.2 | −348.8 | −249.1 |
The second strategy, in which the resonance from the substituent groups is reduced by curving the conjugated structure, was testified by exploring the anion–π complexes formed with fluorinated corannulene (F-CEE, complex 6). This was inspired by an excellent review by Haupt and Lentz, who summarized the experimental investigations of modified corannulene with electron-withdrawing substituents.149
Obviously, F-CEE could be one ideal protype of anion receptor because of three facts. Firstly, a positive ESP is set on both the concave and convex sides, mainly by the fluoro-substitutions (Fig. S1†). Secondly, the electron density is unequally distributed on two sides, because its bowl shape leads to a more positively charged concave side. At last, the delocalization of π electrons on fluorine can be impaired by the curvature, leading to an inhibition of the RI phenomenon. Table 4 shows that the anion–π bonding is doubly strengthened by replacing C6F6 (1) with F-CEE (6). Most importantly, the average contribution from each fluoro-substitution to the binding energy is the highest among all cases tested in this work, suggesting an extra stability gained by curving. The hypothetic flat structure of F-CEE (Fig. S2†), in which both the inhibition of RI phenomenon and the electrophilicity caused by curvity are absent, was also tried for comparison. Computations showed that and the binding of the flat F-CEE with anions is significantly reduced (about 40 kJ mol−1 shown in Table S5†). The binding on the convex side of 6 was also examined, and the binding energy is even lower than the coplanar structure (Table S5†). There could be more modified bucky bowls, nano hoops and even nano tubes as candidates for anion receptors for our future studies.
The RA phenomenon also reminds us to re-examine substituents with strong π-electron withdrawing capabilities. Notably, Kozuch theoretically proposed a genuine and extreme receptor, cyclohexanehexone.14 Rosokha et al. conducted experimental investigation of complexes formed with halogenated p-benzoquinones.83 Considering that benzoquinones can be good candidates for testing since the oxygen attracts both σ and π electrons strongly, here we studied p-, o- and m-benzoquinones (complexes 7–9 see Scheme 1). For complexes formed with p-benzoquinone (7a, 7b and 7c), enhanced binding can be observed compared with systems formed with C6F6, but the binding sites are shifted off the principal axis, forming the Meisenheimer structures.150 Surprisingly, the overall attraction turns out to be dominated by the charge transfer interaction, and strengthened by remarkable polarization interactions, indicating a characteristic of covalency. Both the charge transfer and the binding energies get higher from p-benzoquinone to o-benzoquinone (complexes 8a, 8b and 8c), with the frozen energy becoming repulsive obviously due to the increasing Pauli exchange repulsion. The latter is reflected by the shortened bonding distances in the complexes of o-benzoquinone with halides, where halides hang on the midpoints of substituted carbons. In contrast to both p- and o-benzoquinones, m-benzoquinone can form a covalent bond with a halide. As listed in Table 4, this is evidenced by the short C–X distances (for comparison, typical covalent bond lengths are 1.77, 1.94 and 2.14 Å for C–Cl, C–Br and C–I bonds) and the very high but offsetting frozen and charge transfer energy terms.
4. Conclusions
In this work, we explored how π resonance in anion receptors influences anion–π interactions, as it has been known that substituents to a conjugated cyclic ring alter its binding to anions. Based on the BLW method which can derive electron-localized diabatic states self-consistently, we demonstrated that the deactivation of the π resonance between substituents and the benzene ring can either enhance the binding to anions (a resonance-inhibited or RI phenomenon when substituents are π electron donors such as halogens), or inhibit the binding to anions (a resonance-assisted or RA phenomenon when substituents are π electron acceptors such as nitro and cyano). The subsequent energy decomposition analyses showed that the governing factor is the frozen energy term, in which the electrostatic interaction is involved, while all the other energy components are insensitive to resonance. This governing role of the frozen energy was also confirmed using different theoretical methods. The differences between the RA and RI characteristics are not only reflected by their different electrostatic interactions included in the frozen term, but also exhibited by the differences in ESP maps between the adiabatic and diabatic states visually: the π-hole is diminished by resonance in anion receptors with π electron-donating substituents, but strengthened in aromatic rings with π electron-withdrawing groups.The anion–π interaction was further simplified as an attraction between a negative point charge (anion) and a group of local dipoles, affected by the enriched/diminished π-cloud due to resonance, using an empirical point charge model. While in the resonance contributing or RC scheme, the interaction energies at DFT level can be well reproduced by replacing the anion with a point charge, the interaction energies turn to be more attractive in RI cases but less stabilizing in RA complexes in the resonance free or RF scheme, in which each atom of the receptor is replaced with a point charge.
Based on the BLW and EPC computations, we hypothesized that a better anion receptor can be designed with increased magnitudes and/or number of local dipoles and reduced π electron density in the conjugated ring. To this end, we tested perfluorocoronene with more local dipoles compared with the prototypical hexafluorobenzene, and demonstrated its remarkably enhanced binding to anions. Nevertheless, increasing the number of fluorine groups as substituents in perfluorocoronene also enhances the RI phenomenon at the same time, and this can be confirmed by the reduced binding energy per fluorine in perfluorocoronene compared with the value in hexafluorobenzene. A solution for this dilemma is to bend the cyclic receptor to impair the π conjugation from the substituents to the cyclic ring. Both fluorinated [5]cycloparaphenylene (F-[5]CPP) and fluorinated corannulene (F-CEE) are “curved” aromatic receptors, and computations show their remarkably enhanced binding capabilities for anion. For instance, the average binding energy in F-CEE contributed by each fluorine is increased by about 20%, compared with C6F6. Finally, p-, o- and m-benzoquinones were all revisited to confirm the RA phenomenon. Different from the rest cases studied in this work, the anion–π interactions with benzoquinones show considerable magnitude of covalency with binding sites shifting away from the ring centers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CW acknowledges the support from the Natural Science Foundation of China (No. 22073060), the Fundamental Research Funds for the Central Universities (No. GK201903043 and GK201901007), and the Talent Introduction Fund of Shaanxi Normal University. This work was performed in part at the Joint School of Nanoscience and Nanoengineering, a member of the Southeastern Nanotechnology Infrastructure Corridor (SENIC) and National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542174).Notes and references
- P. Hobza and K. Müller-Dethlefs, Non-covalent interactions: theory and experiment, Royal Society of Chemistry, 2010 Search PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- J. Y. Lee, B. H. Hong, W. Y. Kim, S. K. Min, Y. Kim, M. V. Jouravlev, R. Bose, K. S. Kim, I.-C. Hwang, L. J. Kaufman, C. W. Wong, P. Kim and K. S. Kim, Nature, 2009, 460, 498–501 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- J. S. Murray, M. C. Concha, P. Lane, P. Hobza and P. Politzer, J. Mol. Model., 2008, 14, 699–704 CrossRef CAS.
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2008, 14, 659–665 CrossRef CAS.
- Y. Mo, G. Subramanian, J. Gao and D. M. Ferguson, J. Am. Chem. Soc., 2002, 124, 4832–4837 CrossRef CAS.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef.
- S. Kozuch, Phys. Chem. Chem. Phys., 2016, 18, 30366–30369 RSC.
- V. Angarov and S. Kozuch, New J. Chem., 2018, 42, 1413–1422 RSC.
- I. Alkorta, J. Elguero and J. E. Del Bene, J. Phys. Chem. A, 2014, 118, 4222–4231 CrossRef CAS.
- I. Alkorta, J. Elguero and J. E. Del Bene, Chem. Phys. Lett., 2015, 641, 84–89 CrossRef CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2016, 17, 1608–1614 CrossRef.
- F. Blanco, I. Alkorta, I. Rozas, M. Solimannejad and J. Elguero, Phys. Chem. Chem. Phys., 2011, 13, 674–683 RSC.
- J. E. Del Bene, I. Alkorta, J. Elguero and G. Sánchez-Sanz, J. Phys. Chem. A, 2017, 121, 1362–1370 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917–4925 CrossRef CAS.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023–1028 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1994, 116, 909–915 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2004, 126, 3845–3855 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, A. Lyčka and G. Gilli, J. Am. Chem. Soc., 2002, 124, 13554–13567 CrossRef CAS.
- E. Masumian and A. Nowroozi, Theor. Chem. Acc., 2015, 134, 82 Search PubMed.
- J. M. Guevara-Vela, E. Romero-Montalvo, A. del Río-Lima, Á. Martín Pendás, M. Hernández-Rodríguez and T. Rocha Rinza, Chem. – Eur. J., 2017, 23, 16605–16611 CrossRef CAS.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481–487 CrossRef CAS.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274–6276 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593–8598 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250–1258 CrossRef CAS.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428, 10.1039/B800055G.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- D.-X. Wang, Q.-Y. Zheng, Q.-Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485–7488 CrossRef CAS.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem. – Eur. J., 2010, 16, 13053–13057 CrossRef CAS.
- Y. Chen, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2011, 47, 8112–8114 RSC.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Rev., 2015, 115, 8867–8895 CrossRef CAS.
- D. Quiñonero, P. M. Deyà, M. P. Carranza, A. M. Rodríguez, F. A. Jalón and B. R. Manzano, Dalton Trans., 2010, 39, 794–806 RSC.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2004, 43, 141 CrossRef.
- D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892–897 CrossRef CAS.
- Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650–4652 CrossRef CAS.
- M. Giese, M. Albrecht, T. Krappitz, M. Peters, V. Gossen, G. Raabe, A. Valkonen and K. Rissanen, Chem. Commun., 2012, 48, 9983–9985 RSC.
- L. Adriaenssens, G. Gil-Ramírez, A. Frontera, D. Quiñonero, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2014, 136, 3208–3218 CrossRef CAS.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202–7207 CrossRef CAS.
- O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506–508, 10.1039/B511570A.
- G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Angew. Chem., Int. Ed., 2008, 47, 4114–4118 CrossRef.
- O. B. Berryman, A. C. Sather, B. P. Hay, J. S. Meisner and D. W. Johnson, J. Am. Chem. Soc., 2008, 130, 10895–10897 CrossRef CAS.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS.
- P. Arranz-Mascarós, C. Bazzicalupi, A. Bianchi, C. Giorgi, M.-L. Godino-Salido, M.-D. Gutiérrez-Valero, R. Lopez-Garzón and M. Savastano, J. Am. Chem. Soc., 2013, 135, 102–105 CrossRef.
- Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940–9943 CrossRef CAS.
- Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101–2111 CrossRef CAS.
- Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS.
- Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785–11790 CrossRef CAS.
- Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582–11585 CrossRef CAS.
- L. Liu, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 7876–7879 CrossRef CAS.
- C. Wang, F. N. Miros, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2016, 55, 14422–14426 CrossRef CAS.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Phys. Chem., 1987, 91, 5294–5297 CrossRef CAS.
- H.-J. Schneider, F. Werner and T. Blatter, J. Phys. Org. Chem., 1993, 6, 590–594 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2003, 4, 1344–1348 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS.
- P. Politzer and J. S. Murray, J. Comput. Chem., 2018, 39, 464–471 CrossRef CAS.
- S. E. Wheeler and K. N. Houk, J. Phys. Chem. A, 2010, 114, 8658–8664 CrossRef CAS.
- Z. Aliakbar Tehrani, Z. Jamshidi and H. Farhangian, J. Mol. Model., 2013, 19, 4763–4772 CrossRef CAS.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Chem. Phys. Lett., 2012, 530, 145–150 CrossRef.
- D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 359, 486–492 CrossRef.
- C. Garau, D. Quiñonero, A. Frontera, A. Costa, P. Ballester and P. M. Deyà, Chem. Phys. Lett., 2003, 370, 7–13 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, New J. Chem., 2003, 27, 211–214 RSC.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Org. Lett., 2003, 5, 2227–2229 CrossRef CAS.
- D. Y. Kim, N. J. Singh and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1401–1407 CrossRef CAS.
- T. Brinck and A. N. Borrfors, J. Mol. Model., 2019, 25, 125 CrossRef.
- C. S. Anstöter, J. P. Rogers and J. R. R. Verlet, J. Am. Chem. Soc., 2019, 141, 6132–6135 CrossRef.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS.
- R. Z. Khaliullin, A. T. Bell and M. Head-Gordon, Chem. – Eur. J., 2009, 15, 851–855 CrossRef CAS.
- R. Z. Khaliullin, A. T. Bell and M. Head-Gordon, J. Chem. Phys., 2008, 128, 184112 CrossRef.
- S. Kepler, M. Zeller and S. V. Rosokha, J. Am. Chem. Soc., 2019, 141, 9338–9348 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Discovering Chemistry with Natural Bond Orbitals, Wiley, 2012 Search PubMed.
- A. I. Popov and R. F. Swensen, J. Am. Chem. Soc., 1955, 77, 3724–3726 CrossRef CAS.
- V. G. Shtyrlin, A. V. Zakharov, A. M. Kuznetsov, O. ga V. Kukushkina and P. P. Chernov, Eur. J. Inorg. Chem., 2002, 2002, 2947–2955 CrossRef.
- R. S. Mulliken and W. B. Person, Molecular complexes: a lecture and reprint volume, Wiley-Interscience, 1969 Search PubMed.
- K. Molčanov, G. Mali, J. Grdadolnik, J. Stare, V. Stilinović and B. Kojić-Prodić, Cryst. Growth Des., 2018, 18, 5182–5193 CrossRef.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- S. E. Wheeler and K. Houk, J. Am. Chem. Soc., 2008, 130, 10854–10855 CrossRef CAS.
- L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513–3560 CrossRef CAS.
- T. Steiner, Angew. Chem., 2002, 114, 50–80 CrossRef.
- P. Lenain, M. Mandado, R. A. Mosquera and P. Bultinck, J. Phys. Chem. A, 2008, 112, 10689–10696 CrossRef CAS.
- I. Alkorta, J. Elguero, O. Mó, M. Yáñez and J. E. D. Bene, Chem. Phys. Lett., 2005, 411, 411–415 CrossRef CAS.
- F. Fuster and S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 10078–10086 CrossRef CAS.
- C. Trujillo, G. Sánchez-Sanz, I. Alkorta, J. Elguero, O. Mó and M. Yáñez, J. Mol. Struct., 2013, 1048, 138–151 CrossRef CAS.
- J. F. Beck and Y. Mo, J. Comput. Chem., 2007, 28, 455–466 CrossRef CAS.
- J. M. Guevara-Vela, E. Romero-Montalvo, A. Costales, Á. M. Pendás and T. Rocha-Rinza, Phys. Chem. Chem. Phys., 2016, 18, 26383–26390 RSC.
- R. Kurczab, M. P. Mitoraj, A. Michalak and T. Ziegler, J. Phys. Chem. A, 2010, 114, 8581–8590 CrossRef CAS.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem. – Eur. J., 1999, 5, 3581–3594 CrossRef.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, J. Am. Chem. Soc., 2000, 122, 4117–4128 CrossRef.
- Y. Mo, J. Mol. Model., 2006, 12, 665–672 CrossRef CAS.
- R. I. Zubatyuk, Y. M. Volovenko, O. V. Shishkin, L. Gorb and J. Leszczynski, J. Org. Chem., 2007, 72, 725–735 CrossRef CAS.
- R. W. Góra, M. Maj and S. J. Grabowski, Phys. Chem. Chem. Phys., 2013, 15, 2514–2522 RSC.
- I. Alkorta, J. Elguero, O. Mó, M. Yáñez * and J. E. Del Bene, Mol. Phys., 2004, 102, 2563–2574 CrossRef CAS.
- P. Sanz, O. Mó, M. Yáñez and J. Elguero, ChemPhysChem, 2007, 8, 1950–1958 CrossRef CAS.
- P. Sanz, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. A, 2007, 111, 3585–3591 CrossRef CAS.
- P. Sanz, O. Mó, M. Yáñez and J. Elguero, Chem. – Eur. J., 2008, 14, 4225–4232 CrossRef CAS.
- X. Jiang, H. Zhang, W. Wu and Y. Mo, Chem. – Eur. J., 2017, 23, 16885–16891 CrossRef CAS.
- W. W. Xiaoyu JIANG and M. O. Yirong, Acta Phys.-Chim. Sin., 2018, 34, 278–285 Search PubMed.
- X. Lin, H. Zhang, X. Jiang, W. Wu and Y. Mo, J. Phys. Chem. A, 2017, 121, 8535–8541 CrossRef CAS.
- X. Lin, X. Jiang, W. Wu and Y. Mo, J. Org. Chem., 2018, 83, 13446–13453 CrossRef CAS.
- X. Lin, W. Wu and Y. Mo, J. Org. Chem., 2019, 84, 14805–14815 CrossRef CAS.
- Y. Mo and S. D. Peyerimhoff, J. Chem. Phys., 1998, 109, 1687–1697 CrossRef CAS.
- Y. Mo, L. Song and Y. Lin, J. Phys. Chem. A, 2007, 111, 8291–8301 CrossRef CAS.
- X. Lin, W. Wu and Y. Mo, Chemistry, 2018, 24, 1053–1056 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell university press Ithaca, New York, 1960 Search PubMed.
- D. Cooper, Valence bond theory, Elsevier, 2002 Search PubMed.
- S. S. Shaik and P. C. Hiberty, A chemist's guide to valence bond theory, John Wiley & Sons, 2007 Search PubMed.
- W. Wu, P. Su, S. Shaik and P. C. Hiberty, Chem. Rev., 2011, 111, 7557–7593 CrossRef CAS.
- G. A. Gallup, Valence Bond Methods: Theory and Applications, Cambridge University Press, Cambridge, 2002 Search PubMed.
- Y. Mo, P. Bao and J. Gao, Phys. Chem. Chem. Phys., 2011, 13, 6760–6775 RSC.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- K. Morokuma, Acc. Chem. Res., 1977, 10, 294–300 CrossRef CAS.
- W. Chen and M. S. Gordon, J. Phys. Chem., 1996, 100, 14316–14328 CrossRef CAS.
- J. Andrés, P. W. Ayers, R. A. Boto, R. Carbó-Dorca, H. Chermette, J. Cioslowski, J. Contreras-García, D. L. Cooper, G. Frenking, C. Gatti, F. Heidar-Zadeh, L. Joubert, Á. Martín Pendás, E. Matito, I. Mayer, A. J. Misquitta, Y. Mo, J. Pilmé, P. L. A. Popelier, M. Rahm, E. Ramos-Cordoba, P. Salvador, W. H. E. Schwarz, S. Shahbazian, B. Silvi, M. Solà, K. Szalewicz, V. Tognetti, F. Weinhold and É.-L. Zins, J. Comput. Chem., 2019, 40, 2248–2283 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, C. Mück-Lichtenfeld, G. Erker, G. Kehr, H. Wang, H. Beckers and H. Willner, Angew. Chem., Int. Ed., 2009, 48, 2592–2595 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS.
- K. S. Thanthiriwatte, E. G. Hohenstein, L. A. Burns and C. D. Sherrill, J. Chem. Theory Comput., 2011, 7, 88–96 CrossRef CAS.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef CAS.
- R. A. Bachorz, F. A. Bischoff, S. Höfener, W. Klopper, P. Ottiger, R. Leist, J. A. Frey and S. Leutwyler, Phys. Chem. Chem. Phys., 2008, 10, 2758–2766 RSC.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS.
- J. Rigby and E. I. Izgorodina, J. Chem. Theory Comput., 2014, 10, 3111–3122 CrossRef CAS.
- T. Takatani and C. David Sherrill, Phys. Chem. Chem. Phys., 2007, 9, 6106–6114 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, CrystEngComm, 2016, 18, 10–23 RSC.
- J. C. Sancho-García and A. J. Pérez-Jiménez, J. Chem. Phys., 2014, 141, 134708 CrossRef.
- S. Erkoç, F. Erkoç and L. Türker, J. Mol. Struct.: THEOCHEM, 2001, 538, 91–97 CrossRef.
- A. Haupt and D. Lentz, Chem. – Eur. J., 2019, 25, 3440–3454 CrossRef CAS.
- J. Meisenheimer, Justus Liebigs Ann. Chem., 1902, 323, 205–246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07877h |
| This journal is © The Royal Society of Chemistry 2020 |