
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning-based prediction of toxicity of organic compounds towards fathead minnow†
Xingmei Chen,
Limin Dang *,
Hai Yang,
Xianwei Huang* and
Xinliang Yu‡
*
*,
Hai Yang,
Xianwei Huang* and
Xinliang Yu‡
*
Hunan Provincial Key Laboratory of Environmental Catalysis & Waste Regeneration, College of Materials and Chemical Engineering, Hunan Institute of Engineering, Xiangtan, Hunan 411104, China. E-mail: l3055155812@163.com; hxw1o3o@126.com; yxl@hnie.edu.cn; Fax: +86 731 58680125; Tel: +86 731 58680049
First published on 1st October 2020
Abstract
Predicting the acute toxicity of a large dataset of diverse chemicals against fathead minnows (Pimephales promelas) is challenging. In this paper, 963 organic compounds with acute toxicity towards fathead minnows were split into a training set (482 compounds) and a test set (481 compounds) with an approximate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Only six molecular descriptors were used to establish the quantitative structure–activity/toxicity relationship (QSAR/QSTR) model for 96 hour pLC50 through a support vector machine (SVM) along with genetic algorithm. The optimal SVM model (R2 = 0.756) was verified using both internal (leave-one-out cross-validation) and external validations. The validation results (qint2 = 0.699 and qext2 = 0.744) were satisfactory in predicting acute toxicity in fathead minnows compared with other models reported in the literature, although our SVM model has only six molecular descriptors and a large data set for the test set consisting of 481 compounds.
:1. Only six molecular descriptors were used to establish the quantitative structure–activity/toxicity relationship (QSAR/QSTR) model for 96 hour pLC50 through a support vector machine (SVM) along with genetic algorithm. The optimal SVM model (R2 = 0.756) was verified using both internal (leave-one-out cross-validation) and external validations. The validation results (qint2 = 0.699 and qext2 = 0.744) were satisfactory in predicting acute toxicity in fathead minnows compared with other models reported in the literature, although our SVM model has only six molecular descriptors and a large data set for the test set consisting of 481 compounds.
1. Introduction
With the development of science and technology, more and more chemicals are used in the world, which has caused great concern for their possible toxicity to aquatic organisms.1 Toxicity assessment of chemicals is necessary for all chemical industries, before releasing them into the market.2 Traditionally, the toxicities of chemicals are obtained from animal tests. However, these toxicological experiments are not only ethically problematic, but also expensive, labor-intensive and time-consuming.3,4 Fathead minnows (Pimephales promelas) are one of the most common fish in aquatic toxicity studies and 96 h LC50 denoting 96 hour 50% lethal concentration is used as a quantitative toxicity endpoint.Quantitative structure–activity/toxicity relationship (QSAR/QSTR) models are an important method to analysis toxic mechanisms and to predict the toxicity for organic chemicals,5–8 even for those that have not been synthesized. Many researchers have carried out QSAR studies for acute toxicity (LC50, logLC50 or −logLC50 (pLC50)) in fathead minnows.
Lozano et al. introduced 10 consensus linear models for the toxicity of 557 chemicals to fathead minnows using 4–17 descriptors.9 The coefficients of determination R2 are in the range of 0.62–0.73. Wang et al. developed a nonlinear model for the toxicity of 571 compounds to fathead minnows.10 Eight descriptors were used to develop the model that has coefficients of determination R2 of 0.826 for the training set and 0.802 for the test set. In et al. built linear and nonlinear QSAR models for the toxicity of 555 compounds to fathead minnows.11 Four QSARs models have coefficients of determination R2 as 0.553, 0.618, 0.632, and 0.605 on the test set, respectively. The consensus model consisting of three QSAR models shows good predictive capacity (R2 = 0.663) on the test set. Toropova et al. introduced MLR analysis of 568 acute toxicities in fathead minnows.12 The average correlation coefficients (R2) are 0.675, 0.824, and 0.787 for subtraining (n = 246–271, n: the number of compounds), calibration (n = 144–164), and test set (n = 148–158), respectively, which are acceptable.
Lyakurwa et al. established theoretical linear solvation energy relationship models for acute toxicity in fathead minnows with 3–5 quantum chemical descriptors.13 The four QSAR models based on 79–311 compounds have adjusted determination coefficient Radj2 ranging from 0.707 to 0.903. Cassotti et al. successfully constructed six-descriptor QSAR models for acute toxicity of 908 chemicals to fathead minnow with k nearest neighbor method.14 Correlation coefficients (R2) of the training set (726 molecules) and the test set (182 molecules) range from 0.62 to 0.73 and 0.61 to 0.77, respectively.
Drgan et al. built robust models for 566 toxicities to fathead minnows with a new in silico method, counter-propagation artificial neural network (ANN).4 Correlation coefficients (R) of the training set (340 compounds) and the test set (99–226 molecules) were in range of 0.93 and of 0.71–0.74, respectively. Wu et al. successfully developed a linear QSAR for a large dataset consisting of 963 organic compounds with acute toxicity towards fathead minnows.15 Eight molecular descriptors were used for the model that had good predictive capacity (R2 = 0.64) on the test set (192 compounds).
All these QSARs stated above were validated with small datasets (less than 250 organic compounds). Generally, the ratios of samples in training sets and test sets are 4:1–3:1, and the difficulty in developing successful QSAR models increases when more samples are included in the test set, especially for nonlinear QSARs. The aim of this paper is to develop a nonlinear QSAR model for acute toxicity of 482 chemicals to fathead minnows, which is to be validated with a large data set consisting of 481 organic chemicals, by applying support vector machine (SVM) together with genetic algorithm (GA).
2. Materials and methods
2.1 Datasets
Acute toxicity data of organic chemicals to fathead minnows were provided by the US Environmental Protection Agency, which later became important biochemical indicators and informative ecological parameters for regulatory ecotoxicology.12 The experiment pLC50 values were subjected to rigorous screening and comparison. These experiment values were removed when they were different by a factor of over 30 from the closest one in a set of at least three references. After that, arithmetic mean values were used for compounds with multiple experiment values. In addition, these compounds that lack well-defined structure or possess metallic elements were removed.15 In the end, 963 experimental toxicants tested against fathead minnow were obtained by Wu et al.,15 which are listed in Table S1 in ESI.† Experimental acute toxicities of chemicals were estimated with lethal concentration (mol L−1) causing death in 50% of test fathead minnows over a test duration of 96 hours (96 h LC50) and were converted into negative logarithmic scale, −logLC50 or pLC50. A compound with a larger pLC50 value has higher toxicity for fathead minnow. These toxicity data have been studied by Wu et al.15
2.2 Molecular descriptors
The molecular structures of 963 compounds in Table S1 in ESI† were generated with ChemDraw Ultra 8.0 in ChemOffice 2004, and optimized with semi-empirical AM1 method in MOPAC in Chem3D Ultra 8.0. 4885 molecular descriptors were calculated for each molecule with Dragon 6.0.16 After deleting those descriptors that equal a constant (or approximately constant) or whose pair-wise correlation coefficients are above 0.90, 1317 molecular descriptors were derived from Dragon soft. In addition, the octanol–water partition coefficient (CLOGP) was calculated with the CLogP Driver in Chem3D Ultra 8.0. Totally, 1318 descriptors were obtained for descriptor selection.2.3 Support vector machine
SVM algorithm is based on structural risk minimization principle and exhibits good prediction ability in classification and regression. The algorithm maps input data into a high-dimensional feature space, from which linear regression analysis is carried out.17,18 Support Vector Regression (SVR) algorithm approximates following regression model:
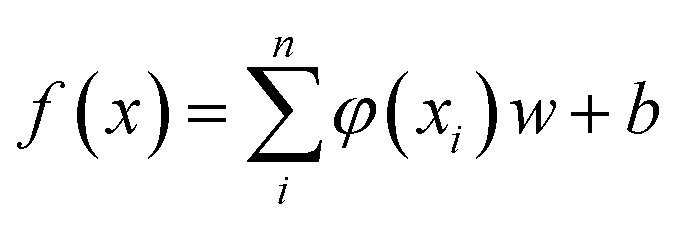 | (1) |
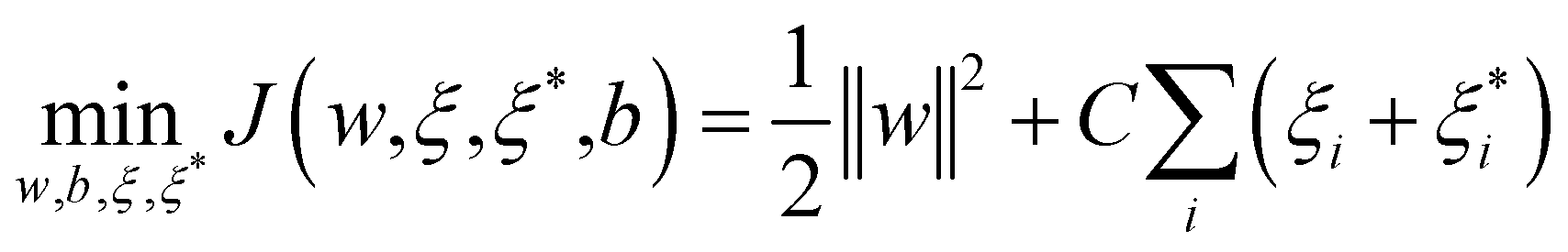 | (2) |
| yi − φT(xi)w − b ≤ ε + ξi | (3) |
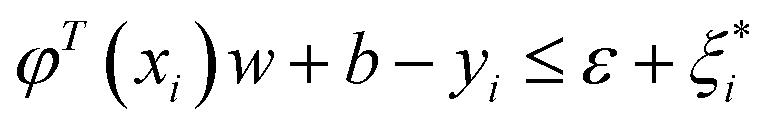 | (4) |
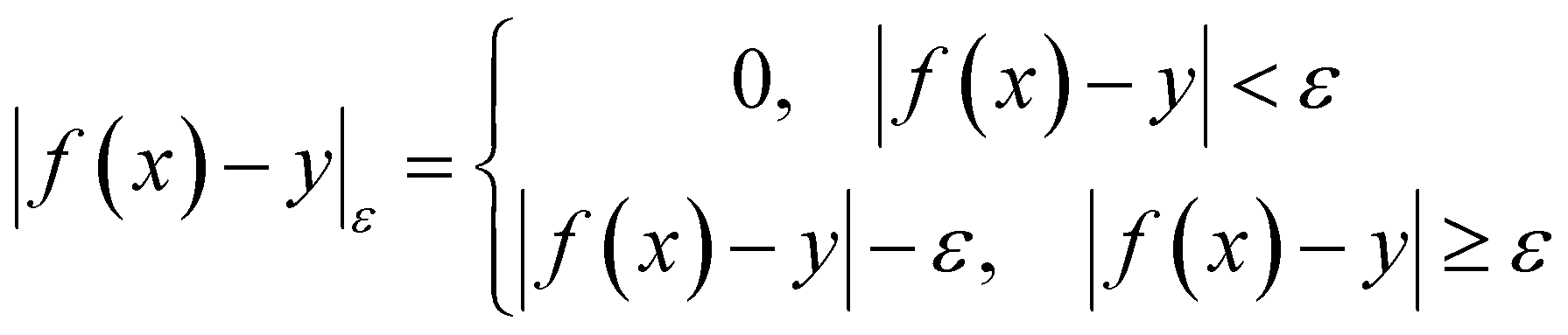 | (5) |
Thus, eqn (1) can be converted to eqn (6):
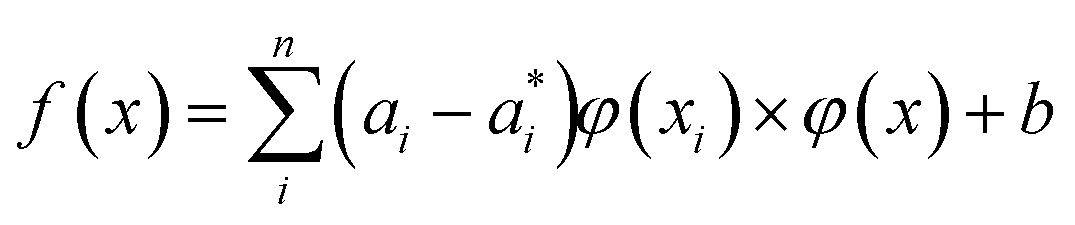 | (6) |
Here αi and 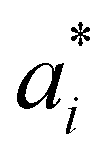 are Lagrange multipliers, which are introduced for solving the quadratic optimization problem. Introducing a kernel function k(x,y) into eqn (6) yields eqn (7):
are Lagrange multipliers, which are introduced for solving the quadratic optimization problem. Introducing a kernel function k(x,y) into eqn (6) yields eqn (7):
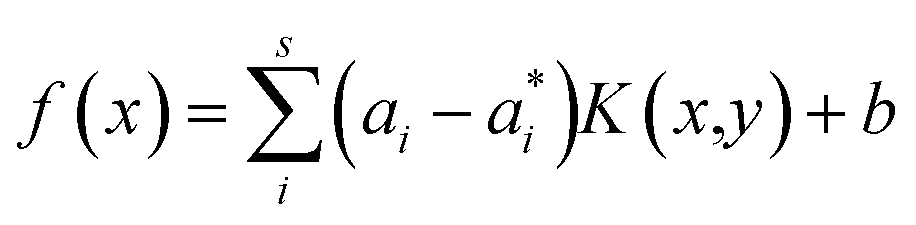 | (7) |
| K(Xi,Xj) = exp(−γ‖Xi − Xj‖2) | (8) |
3. Results and discussion
To select the optimal descriptor subset affecting pLC50 of organic chemicals towards fathead minnow, stepwise MLR analysis was performed to identify correlations between 963 pLC50 and 1618 molecular descriptors stated above, by applying IBM SPSS Statistical 19. Model summary obtained was listed in Table 1. The increment of determination coefficient ΔR2 > 0.01 was used as the criterion for introducing new variables. Therefore, the six molecular descriptors (CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA, and MOR10E) in Model 6 were used to develop regression eqn (9)–(12) with IBM SPSS Statistical 19. The definitions of molecular descriptors used in models were shown in Table S2† in Supplemental file.| Model | R | R square | Adjusted R square | Std. error of the estimate |
|---|---|---|---|---|
| a Predictors: (constant), CLOGP.b Predictors: (constant), CLOGP, SM6_B(P).c Predictors: (constant), CLOGP, SM6_B(P), NDB.d Predictors: (constant), CLOGP, SM6_B(P), NDB, nHM.e Predictors: (constant), CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA.f Predictors: (constant), CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA, MOR10E.g Predictors: (constant), CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA, MOR10E, B10[C–N].h Predictors: (constant), CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA, MOR10E, B10[C–N], MLOGP. | ||||
| 1 | 0.722a | 0.522 | 0.521 | 1.005398 |
| 2 | 0.773b | 0.598 | 0.597 | 0.922460 |
| 3 | 0.791c | 0.626 | 0.624 | 0.890379 |
| 4 | 0.806d | 0.649 | 0.648 | 0.862459 |
| 5 | 0.821e | 0.674 | 0.672 | 0.831622 |
| 6 | 0.830f | 0.689 | 0.687 | 0.812323 |
| 7 | 0.836g | 0.699 | 0.697 | 0.800239 |
| 8 | 0.840h | 0.706 | 0.703 | 0.791266 |
Moriguchi octanol–water partition coefficient (MLOGP) is calculated from Moriguchi logP model that is a regression equation based on 13 structural descriptors19 Ghose–Crippen–Viswanadhan octanol–water partition coefficient (ALOGP) is estimated with the AlogP model, a regression equation obtained with the hydrophobicity contribution of 115 atom types.19 The AlogP method is applicable for molecules possessing atoms of C, H, O, N, S, Se, P, B, Si, and halogens. Both MLOGP and ALOGP were calculated with Dragon 6.0. The octanol–water partition coefficient CLOGP can be calculated with the CLogP Driver in Chem3D Ultra 8.0 that adopts the fragment method. Fundamental fragments include isolating carbons, polar fragments, halogens, H-polar fragments, ions, double bonds, triple bonds, chain bonds, ring bonds, branch bonds, chain branch, group branch, electronic (or topological) interaction factors, intra-molecular hydrogen bonding, and so on. The CLogP method covers most neutral organic compounds. Furthermore, the CLOGP values are more accurate than the coefficients of MLOGP and ALOGP for molecules in the range of 20−45 atoms, especially for small molecules in the 1−20 atoms.20 CLOGP becomes valuable in many fields, including drug design and hazard assessment. When the descriptor CLOGP in model 6 (see Table S2† in ESI) was replaced with MLOGP (or ALOGP), the coefficient of determination (= 0.672 or 0.686) of models slightly deceases. Therefore, the octanol–water partition coefficient CLOGP was used to develop QSAR models.
The partition coefficient CLOGP measures the lipophilicity of a compound.21,22 While the lipophilicity describes the kinetics of uptake of chemicals from water and acts as the driving force in the whole interactions between toxic molecules and targets in fathead minnow.15,23,24 Thus, CLOGP has a positive correlation with toxicity pLC50, which can be reflected with eqn (9):
| pLC50 = 2.74 + 0.559 CLOGP, n = 963, R = 0.722, R2 = 0.522, Radj2 = 0.521, se = 1.005, F = 1.05×103 | (9) |
The spectral moment of order 6 from Burden matrix weighted by polarizability, SM6_B(P), belongs to 2D matrix-based descriptors. It is derived from an H-depleted molecular graph based on the Burden matrix. The diagonal elements are atomic carbon-scaled properties; the off-diagonal elements are related to pairs of bonded atoms and conventional bond orders; entries corresponding to terminal bonds are augmented by 0.1; and other elements are set to 0.001. SM6_B(P) measures the number of graph vertices and molecular polarizability.19 When SM6_B(P) is allowed to enter the model, eqn (9) becomes:
| pLC50 = −2.08 + 0.408 CLOGP + 0.656 SM6_B(P), n = 963, R = 0.773, R2 = 0.598, Radj2 = 0.597, se = 0.922, F = 713 | (10) |
The coefficient of determination R2 in eqn (10) increases obviously. As is shown in Fig. S2,† a larger SM6_B(P) value results in higher toxicity. A larger molecule in size usually has a high SM6_B(P) value than the small molecule, e.g. the diphenyl (3-phenyl-5-propan-2-ylphenyl) phosphate (no. 49 in Table S1†) and the methanol (no. 7).
Both NDB and nHM belong to constitutional indices. The former means the number of double bonds, and the later denotes the number of heavy atoms with principal quantum number L larger than 2. Perchloropentacyclodecane (no. 2 in Table S1†) possesses the maximum nHM value. Obviously, the two descriptors are related to molecular size. An increasing NDB (or nHM) causes an increase in the toxicity of the compounds.4 When they are also introduced, eqn (11) was obtained:
| pLC50 = −0.430 + 0.423 CLOGP + 0.394 SM6_B(P) + 0.255 NDB + 0.166 nHM, n = 963, R = 0.806, R2 = 0.649, Radj2 = 0.648, se = 0.862, F = 443 | (11) |
The correlation between experimental and predicted pEC10 with eqn (11) was depicted in Fig. S3,† which shows the sample points are relatively evenly and loosely distributed. Thus more descriptors should be introduced.
The edge adjacency index SPMAD_EA denotes spectral mean absolute deviation from edge adjacency matrix.25 The edge adjacency matrix is a square symmetric matrix of the dimension NBO × NBO and calculated with the H-depleted molecular graph that encodes the connectivity information between edges i and j of the graph. The entries of the matrix are equal to 1 if the bonds under consideration are adjacent. Otherwise, they equal zero. Similar to the descriptor CLOGP, SPMAD_EA describes different structural fragments (subgraphs) in the graph and is related to molecular two-dimensional shapes and polarization,15,26 although CLOGP measures the total number of fragments and SPMAD_EA represents their connectivity information.
Besides molecular two-dimensional shapes, molecular three-dimensional shapes also used to correlate with molecular toxicity pLC50. 3D-MoRSE means 3D-Molecule Representation of Structures based on Electron diffraction.27 The 3D-MoRSE descriptor MOR10E means signal 10/weighted by Sanderson electronegativity. It reflects the three-dimensional arrangement of the atoms in molecules.26,28 Thus MOR10E describing three dimensional shapes relates to molecular permeability in fathead minnow.
When SPMAD_EA and MOR10E that respectively reflect molecular two-dimensional and three-dimensional shapes were introduced to eqn (11), eqn (12) is obtained:
| pLC50 = −0.181 + 0.427 CLOGP + 0.793 SM6_B(P) + 0.283 NDB + 0.163 nHM − 2.62 SPMAD_EA + 0.387 MOR10E, n = 963, R = 0.830, R2 = 0.689, Radj2 = 0.687, se = 0.812, F = 354 | (12) |
When the descriptors B10[C–N] and MLOGP were entered, the statistical qualities of models 7 and 8 (see Table S2†) were not improved obviously. In addition, the coefficient of determination (R2 = 0.689) of eqn (12) is close to that (R2 = 0.698) of the eight-descriptor model for the same dataset.15 Thus the six descriptors in eqn (12) can be used as the optimal subset to develop QSAR models for toxicity pLC50.
Based on the six descriptors in eqn (12), the Kennard-Stone algorithm29 was adopted to divide the 963 experimental pLC50 data into a training set (482 compounds) and a test set (481 compounds) at the ratio close to 1:1. The data sets are listed in Table S1† in Supplemental file. A MLR model (eqn (13)) was obtained from the training set:
| pLC50 = −0.309 + 0.408 CLOGP + 0.725 SM6_B(P) + 0.305 NDB + 0.149 nHM − 2.25 SPMAD_EA + 0.390 MOR10E, n = 482, R = 0.825, R2 = 0.680, Radj2 = 0.676, se = 0.922, F = 168 | (13) |
The MLR model was validated with the test set. The coefficient of determination R2 for the test set is 0.675. The statistical characteristics of the six descriptors in eqn (13) are listed in Table 2. As can been seen from Table 2, these descriptors have sig.-values (or P-values) less than the default value of 0.05. Thus all the six descriptors make significant contribution to toxicity pLC50. Moreover, these descriptors have variance inflation factors (VIF) less than the default value of 10, which indicate that there is no obvious multicollinearity among descriptors; that is to say, each descriptor reflects different structure factors correlating with pLC50. The t-test is used to compare descriptor significance. A large absolute value of t-test indicates the corresponding descriptor more significant. According to the t-test in Table 2, the absolute values of t-test decrease in the sequence: CLOGP, NDB, SM6_B(P), SPMAD_EA, nHM and MOR10E, their significances to toxicity pLC50 decrease in the same sequence.
| Descriptor | Coefficients | Std. error | t-Test | Sig. | VIF |
|---|---|---|---|---|---|
| Constant | 0.309 | 0.510 | 0.605 | 0.546 | — |
| CLOGP | 0.408 | 0.025 | 16.1 | 0.00 | 1.89 |
| SM6_B(P) | 0.725 | 0.097 | 7.46 | 0.00 | 3.84 |
| NDB | 0.305 | 0.040 | 7.68 | 0.00 | 1.39 |
| nHM | 0.149 | 0.027 | 5.53 | 0.00 | 1.26 |
| SPMAD_EA | −2.25 | 0.363 | −6.21 | 0.00 | 2.54 |
| MOR10E | 0.390 | 0.075 | 5.22 | 0.00 | 1.15 |
The six molecular descriptors in eqn (13) were adopted to build SVM models for toxicity pLC50 of chemicals against fathead minnow. The LibSVM toolbox30 was used to train SVM models for 482 compounds in the training set, which was executed in the MATLAB R2014a software platform. The penalty factor C and the parameter γ of RBF nuclear function were optimized by genetic algorithm with the conditions of the searching ranges of C being [0, 2 × 103] and γ being [0, 1], the m (=5) in m-fold-cross-validation, the maximum generation (=200), the maximum population size (=20), and the ε (=0.01) in the ε -insensitive loss function.
The optimization results of parameters C being 320 and γ being 0.0127 were obtained. Leave-One-Out (LOO) cross-validation was carried out for the SVM model. An internal correlation coefficient qint2 of 0.699 was obtained, which is large than the default threshold of 0.5 and suggests the SVM model stable. Further, 481 compounds in the test set were used to validate the SVM model. The prediction pLC50 values are listed in Table S1† in ESI and sketched in Fig. 1, which shows the predicted pLC50 close to the experimental pLC50. The external validation correlation coefficient qext2 is 0.744, also greater than the default value of 0.5, which is satisfactory.
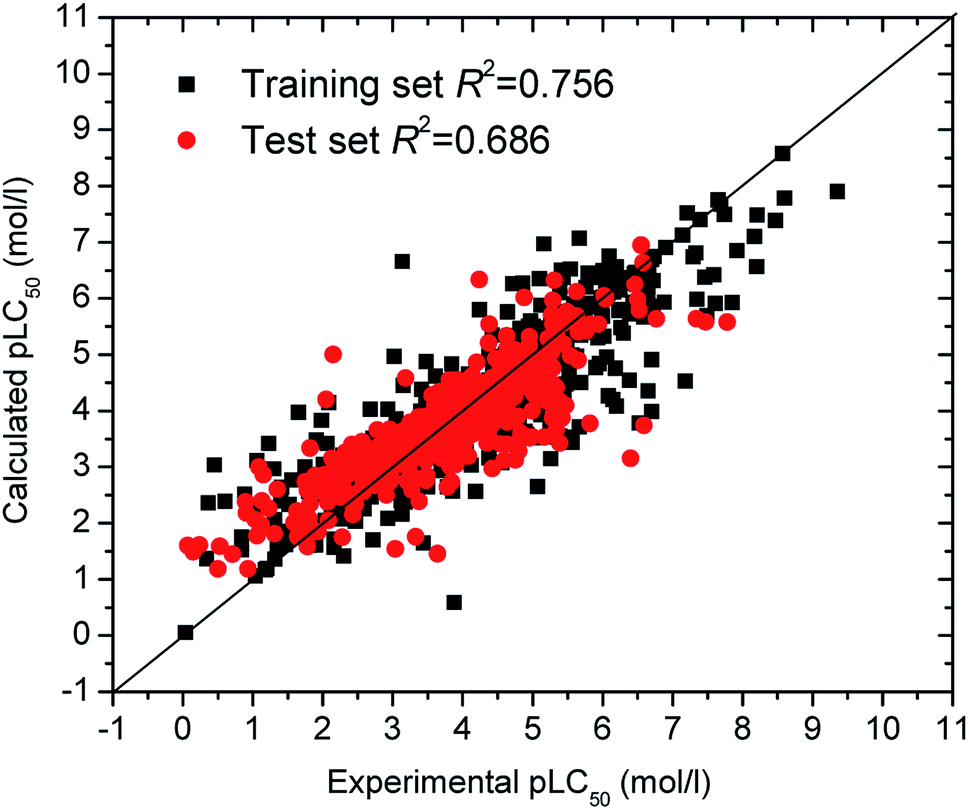 | ||
| Fig. 1 Plot of experimental versus predicted pLC50 with SVM model. | ||
Generally, it is difficult to develop QSAR models when more samples are included in the test set, especially for nonlinear QSARs. Moreover, successful QSARs should have fewer molecular descriptors, to reduce model complexity and multicollinearity. In this paper, the determination coefficients of the training set (482 compounds) and test set (481 compounds) are 0.756 and 0.686, respectively. Wu et al. investigated the same data set (963 compounds) in Table S1† with eight molecular descriptors.15 Their training set (771 compounds) and test set (192 compounds), respectively, have R2 of 0.70 and 0.64. Obviously, our SVM model has better prediction ability, although our SVM model possesses more samples in the test set and fewer molecular descriptors.
Table 3 shows the comparison of the current SVM model with previous relative works. The prediction results (n = 481 and R2 = 0.686) in our SVM model are still accurate and satisfactory, compared with other results: n = 201 and R2 = 0.60;9 n = 144 and R2 = 0.58;9 n = 110 and R2 = 0.553–0.663;11 n = 182 and R2 = 0.61–0.77;14 and n = 99–226 and R = 0.504–0.548.4 Therefore, the six descriptors, CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA and MOR10E, were successfully used to correlate with the acute toxicity pLC50, although many factors affect the toxicity of chemicals against fathead minnow.
| Algorithm | p | ntrain | Rtrain2 (or Radj2) | Ntest | Rtest2 (or qext2) | Reference |
|---|---|---|---|---|---|---|
| MLR | 3 | 556 | 0.65 | 219 | 0.51 | 9 |
| MLR | 3 | 556 | 0.65 | 169 | 0.41 | 9 |
| Consensus | — | 557 | 0.71 | 201 | 0.60 | 9 |
| Consensus | — | 557 | 0.71 | 144 | 0.58 | 9 |
| MLR + ANN | 5 | 445 | 0.712–0.776 | 110 | 0.553–0.632 | 11 |
| MLR | 3–5 | 63–247 | (0.707–0.903) | 16–62 | (0.660–0.858) | 13 |
| GA-kNN | 6 | 726 | 0.62–0.73 | 182 | 0.61–0.77 | 14 |
| MLR + ANN | 6 | 340 | 0.865 | 99–226 | 0.504–0.548 | 4 |
| GA-MLR | 8 | 771 | 0.70 | 192 | (0.641) | 15 |
| SVM | 6 | 482 | 0.756 | 481 | 0.686 | Current study |
The Williams plot (see Fig. 2) was adopted to reflect the relationships between leverages and standardized residuals from the SVM model. As can be seen from Fig. 2, the training and test sets respectively have six and three samples with absolutes values of standard residuals greater than 3, which suggest these compounds possessing chemical structures distinct from that in the training set. These outliers include oleic acid (no. 77 in Table S1 in ESI†), acrylaldehyde (no. 116), 4(1H)-pyridinone, 1-methyl-3-phenyl-5-[3-(trifluoromethyl)phenyl] (no. 137), N,N-dimethylhydrazine (no. 191), 1,3,5,7-tetraazaadamantane (no. 259), N,N-dimethyl-2,2-diphenyl-acetamide (no. 337), 1-methyl-4-(1-methylethenyl)-cyclohexene (no. 539), p-dihydroxybenzene (no. 953), 2-bromo-1-(2,5-dimethoxyphenyl)ethanone (no. 959). In addition, as is shown in Fig. 2, there are 12 samples whose leverages h greater than the warning leverage h* of 0.0436 (≈3 × (6 + 1)/482 = 3 × (p + 1)/n, where p and n are, respectively, the numbers of descriptors and compounds used in training set). But their absolutes values of standard residuals are lower than 3, which indicate that the SVM model built in this work possesses good prediction ability.2
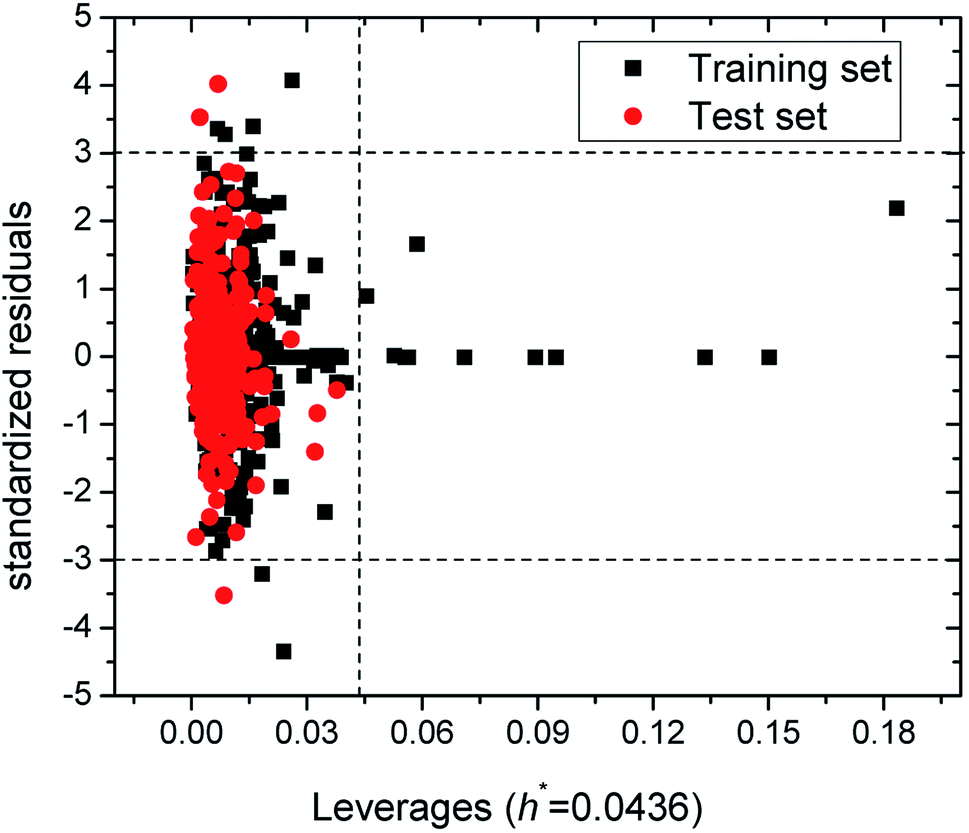 | ||
| Fig. 2 Williams plot with a warning leverage of 0.0436. | ||
4. Conclusions
Although many factors affect the acute toxicity of chemicals against fathead minnow, the six descriptors, CLOGP, SM6_B(P), NDB, nHM, SPMAD_EA and MOR10E, were successfully used to develop SVM model for 96 hour pLC50. After optimization with genetic algorithm, the optimal SVM model (C = 320 and γ = 0.0127) gives coefficients of determination R2 of 0.756 for the training set; and of 0.686 for the test set. Although our SVM model has fewer molecular descriptors and more samples in the test set, our SVM model has satisfactory prediction ability compared with other QSARs reported in the literature for the toxicity pLC50 of organic chemicals against fathead minnow.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Scientific Research Fund of Hunan Provincial Education Department (No. 16A047) and the Open Project Program of Hunan Provincial Key Laboratory of Environmental Catalysis & Waste Regeneration (No. 2018KF11 and 2020KFxx).References
- A. Stenzel, U. K. Goss and S. Endo, Determination of poly-parameter linear free energy relationship (pp-LFER) substance descriptors for established and alternative flame retardants, Environ. Sci. Technol., 2013, 47, 1399–1406 CrossRef CAS.
- X. Yu, Prediction of chemical toxicity to Tetrahymena pyriformis with four descriptor models, Ecotoxicol. Environ. Saf., 2020, 190, 110146 CrossRef.
- B. Peric, J. Sierra, E. Martí, R. Cruañas and M. A. Garau, Quantitative structure–activity relationship (QSAR) prediction of (eco)toxicity of short aliphatic protic ionic liquids, Ecotoxicol. Environ. Saf., 2015, 115, 257–262 CrossRef CAS.
- V. Drgan, Š. Župerl, M. Vračko, F. Como and M. Novič, Robust modeling of acute toxicity towards fathead minnow (Pimephales promelas) using counter-propagation artificial neural networks and genetic algorithm, SAR QSAR Environ. Res., 2016, 27, 501–519 CrossRef CAS.
- W. R. Brogan III and R. A. Relyea, Multiple mitigation mechanisms: effects of submerged plants on the toxicity of nine insecticides to aquatic animals, Environ. Pollut., 2017, 220, 688–695 CrossRef.
- C.-W. Cho and Y.-S. Yun, Application of general toxic effects of ionic liquids to predict toxicities of ionic liquids to Spodoptera frugiperda 9, Eisenia fetida, Caenorhabditis elegans, and Danio rerio, Environ. Pollut., 2019, 255, 113185 CrossRef CAS.
- S. K. Heo, U. Safder and C. K. Yoo, Deep learning driven QSAR model for environmental toxicology: effects of endocrine disrupting chemicals on human health, Environ. Pollut., 2019, 253, 29–38 CrossRef CAS.
- D. Wang, Q. Ning, J. Dong, B. W. Brooks and J. You, Predicting mixture toxicity and antibiotic resistance of fluoroquinolones and their photodegradation products in Escherichia coli, Environ. Pollut., 2020, 262, 114275 CrossRef CAS.
- S. Lozano, M.-P. Halm-Lemeille, A. Lepailleur, S. Rault and R. Bureau, Consensus QSAR related to global or MOA models: application to acute toxicity for fish, Mol. Inf., 2010, 29, 803–813 CrossRef CAS.
- Y. Wang, M. Zheng, J. Xiao, Y. Lu, F. Wang, J. Lu, X. Luo, W. Zhu, H. Jiang and K. Chen, Using support vector regression coupled with the genetic algorithm for predicting acute toxicity to the fathead minnow, SAR QSAR Environ. Res., 2010, 21, 559–570 CrossRef CAS.
- Y. In, S. K. Lee, P. J. Kim and K. T. No, Prediction of acute toxicity to fathead minnow by local model based qsar and global QSAR approaches, Bull. Korean Chem. Soc., 2012, 33, 613–619 CrossRef CAS.
- A. P. Toropova, A. A. Toropov, A. Lombardo, A. Roncaglioni, E. Benfenati and G. Gini, CORAL: QSAR Models for Acute Toxicity in Fathead Minnow (Pimephales promelas), J. Comput. Chem., 2012, 33, 1218–1223 CAS.
- F. Lyakurwa, X. Yang, X. Li, X. Qiao and J. Chen, Development and validation of theoretical linear solvation energy relationship models for toxicity prediction to fathead minnow (pimephales promelas), Chemosphere, 2014, 96, 188–194 CrossRef CAS.
- M. Cassotti, D. Ballabio, R. Todeschini and V. Consonni, A similarity-based QSAR model for predicting acute toxicity towards the fathead minnow (Pimephales promelas), SAR QSAR Environ. Res., 2015, 26, 217–243 CrossRef CAS.
- X. Wu, Q. Zhang and J. Hu, QSAR study of the acute toxicity to fathead minnow based on a large dataset, SAR QSAR Environ. Res., 2016, 27, 147–164 CrossRef CAS.
- Talete srl, DRAGON (Software for Molecular Descriptor Calculation), Version 6.0, 2012, http://www.talete.mi.it/ Search PubMed.
- Y. Yu, Extrapolation for Aeroengine Gas Path Faults with SVM Bases on Genetic Algorithm, J. Sound Vib., 2019, 53, 237–243 Search PubMed.
- X. Yu, L. Xu, Y. Zhu, S. Lu and L. Dang, Correlation between 13C NMR chemical shifts and complete sets of descriptors of natural coumarin derivatives, Chemom. Intell. Lab. Syst., 2019, 184, 167–174 CrossRef CAS.
- R. Todeschini and V. Consonni, Molecular Descriptors for Chemoinformatics, Wiley- VCH, Weinheim, 2009 Search PubMed.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods, J. Phys. Chem. A, 1998, 102, 3762–3772 CrossRef CAS.
- V. Aruoja, M. Moosus, A. Kahru, M. Sihtmäe and U. Maran, Measurement of baseline toxicity and QSAR analysis of 50 non-polar and 58 polar narcotic chemicals for the alga Pseudokirchneriella subcapitata, Chemosphere, 2014, 96, 23–32 CrossRef CAS.
- K. O. Kusk, A. M. Christensen and N. Nyholm, Algal growth inhibition test results of 425 organic chemical substances, Chemosphere, 2018, 204, 405–412 CrossRef CAS.
- H. J. M. Verhaar, C. J. Van Leeuwen and J. L. M. Hermens, Classifying environmental pollutants. 1: Structure–activity relationships for prediction of aquatic toxicity, Chemosphere, 1992, 25, 471–491 CrossRef CAS.
- G. Lucia, B. Francesco and R. Giacomo, Scrutinizing the interactions between bisphenol analogues and plasma proteins: insights from biomimetic liquid chromatography, molecular docking simulations and in silico predictions, Environ. Toxicol. Pharmacol., 2019, 68, 148–154 CrossRef.
- E. Estrada, Spectral Moments of the Edge-Adjacency Matrix of Molecular Graphs. 2. Molecules Containing Heteroatoms and QSAR Applications, J. Chem. Inf. Comput. Sci., 1997, 37, 320–328 CrossRef CAS.
- Y. Uesawa, H. Sakagami, H. Kagaya, M. Yamashita, K. Takao and Y. Sugita, Quantitative Structure-cytotoxicity Relationship of 3-Benzylidenechromanones, Anticancer Res., 2016, 36, 5803–5812 CrossRef CAS.
- X. Yu, R. Yu, L. Tang, Q. Guo, Y. Zhang, Y. Zhou, Q. Yang, X. He, X. Yang and K. Wang, Recognition of candidate aptamer sequences for human hepatocellular carcinoma in SELEX screening using structure–activity relationships, Chemom. Intell. Lab. Syst., 2014, 136, 10–14 CrossRef CAS.
- P. Ghaemian and A. Shayanfar, Quantitative Structure Activity Relationship (QSAR) of Methylated Polyphenol Derivatives as Permeability Glycoprotein (P-gp) Inhibitors: A Comparison of Different Training and Test Set Selection Methods, Lett. Drug Des. Discovery, 2017, 14, 999–1007 CAS.
- M. Daszykowski, S. Serneels, K. Kaczmarek, P. V. Espen, C. Croux and B. Walczak, TOMCAT: a MATLAB toolbox for multivariate calibration techniques, Chemom. Intell. Lab. Syst., 2007, 85, 269–277 CrossRef CAS.
- C. C. Chang and C. J. Lin, LIBSVM: a library for support vector machines, ACM Trans. Intell. Syst. Technol., 2011, 2, 27 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05906d |
| ‡ Present address: Donghu Road 18#, Xiangtan, Hunan 411104, China. |
| This journal is © The Royal Society of Chemistry 2020 |