
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and bioevaluation of novel 6-substituted aminoindazole derivatives as anticancer agents†
Ngo Xuan Hoanga,
Van-Hai Hoangb,
Thi-Thu-Trang Luuc,
Hung N. Luu de,
Thien Ngof,
Duong Van Hieua,
Nguyen Huu Longa,
Le Viet Anha,
Son Tung Ngogh,
Yen Thi Kim Nguyeni,
Byung Woo Hani,
Thanh Xuan Nguyenj,
Dinh Thi Thanh Haia,
Tran Thi Thu Hienk and
Phuong-Thao Tran*a
de,
Thien Ngof,
Duong Van Hieua,
Nguyen Huu Longa,
Le Viet Anha,
Son Tung Ngogh,
Yen Thi Kim Nguyeni,
Byung Woo Hani,
Thanh Xuan Nguyenj,
Dinh Thi Thanh Haia,
Tran Thi Thu Hienk and
Phuong-Thao Tran*a
aHanoi University of Pharmacy, 13-15 Le Thanh Tong, Hanoi 100000, Vietnam. E-mail: thaotp119@gmail.com
bLaboratory of Medicinal Chemistry, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea
cCollege of Pharmacy, Natural Products Research Institute, Seoul National University, Seoul 08826, Republic of Korea
dDivision of Cancer Control and Population Sciences, UPMC Hillman Cancer Center, University of Pittsburgh, Pittsburgh, PA 15232, USA
eDepartment of Epidemiology, Graduate School of Public Health, University of Pittsburgh, PA 15261, USA
fFaculty of Pharmacy, Thai Binh University of Medicine and Pharmacy, Thai Binh City 410000, Vietnam
gLaboratory of Theoretical and Computational Biophysics, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
hFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City 700000, Vietnam
iCollege of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea
jDepartment of Surgical Oncology, Viet-Duc University Hospital, Hanoi 100000, Vietnam
kVietnam University of Traditional Medicine, 2 Tran Phu, Ha Dong, Hanoi 100000, Vietnam
First published on 22nd December 2020
Abstract
In the present study, a series of 6-substituted aminoindazole derivatives were designed, synthesized, and evaluated for bio-activities. The compounds were initially designed as indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors based on the structural feature of five IDO1 inhibitors, which are currently on clinical trials, and the important anticancer activity of the indazole scaffold. One of them, compound N-(4-fluorobenzyl)-1,3-dimethyl-1H-indazol-6-amine (36), exhibited a potent anti-proliferative activity with an IC50 value of 0.4 ± 0.3 μM in human colorectal cancer cells (HCT116). This compound also remarkably suppressed the IDO1 protein expression. In the cell-cycle studies, the suppressive activity of compound 36 in HCT116 cells was related to the G2/M cell cycle arrest. Altogether, the current findings demonstrate that compound 36 would be promising for further development as a potential anticancer agent.
Introduction
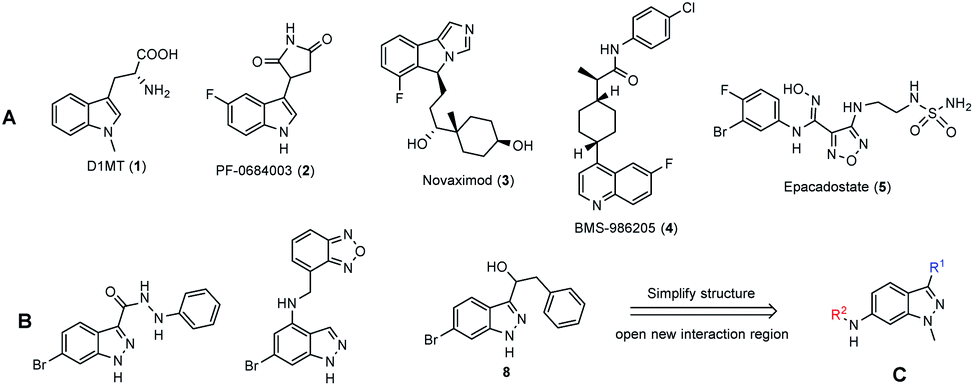
Immunotherapy is a type of cancer treatment that exposes cancer cells and/or enhances the immune system to fight cancer cells along with chemotherapy, radiation and surgery. Although these approaches elicit advanced benefits cancer patients, drug resistance has become one of the big challenges in cancer treatment.1–6 The resistance mechanisms are still under debate, but most scientists support the immunosuppressive tumor microenvironment concept.7–10Indoleamine 2,3-dioxygenase 1 (IDO1) is one of the heme-containing enzymes involved in the immune system suppression process.11 It catalyzes the oxidative ring-opening of tryptophan: the first and rate-limiting step of the kynurenine pathway.12 IDO1 suppresses the immune system via the kynurenine pathway by two mechanisms: (1) tryptophan depletion and (2) toxicity of the metabolites in the kynurenine pathway. Tryptophan depletion inhibits T-cell proliferation and induces cell cycle arrest and the apoptosis of T lymphocytes, while tryptophan metabolites promote the activity of regulatory T cells, a types of T cell inhibits the maturation and cytotoxicity of a T cell.12–14 IDO1 is an overexpression in different types of cancer cells and tumors, such as prostate, colorectum, pancreas, cervix, stomach, ovary and lung, and helps these cancers escape the immune system.15,16 Prior studies have reported that high IDO1 expression could be related to inadequate outcome of chemotherapy, radiotherapy17,18 and other immunotherapies.10
During the last decades, thousands of compounds have been identified as IDO1 inhibitors for cancer immunotherapy, but none of these was approved and brought to the market. Indoximod (D1MT, 1), PF-0684003 (2), novaximod (3), BMS-986205 (4), and epacadostat (5) (Fig. 1A) are five compounds currently in clinical trials.19 They have been generally combined with cytotoxic chemicals and immune checkpoint agents on clinical trials.19,20 The combined therapies improved the efficacy on the tumors of immune checkpoint and cytotoxic agents in both preclinical-21,22 and clinical studies.23 The benefits come from the ability to restore immunosurveillance and blunt tumor neovascularization via re-programming inflammatory processes.24 However, the combination of two drugs is able to cause drawbacks, such as drug–drug interactions, different pharmacokinetics and inconveniences in pharmaceutics and treatment.25 Therefore, a new “multitarget” concept was enunciated for complex diseases such as cancer.25,26 Based on this new concept, a few X/IDO1 dual inhibitors were reported, such as HDAC/IDO1 dual inhibitors27 and DNA/IDO1 dual targeting agents.28
 | ||
| Fig. 1 (A) Clinical IDO1 inhibitors; (B) some IDO1 inhibitors bearing the indazole structure; (C) novel designed indazole derivatives. | ||
Indazole is an important scaffold in drug discovery, which is present in different anticancer agents. Most of them are receptor tyrosine kinase- (EGFR, FGFR, VEGFR) or serine/threonine kinase (aurora kinase, cyclin-dependent kinase) inhibitors.29–31 The diversity comes from the structure, in which two nitrogen atoms can tautomerize and can be introduced to different groups. In addition, indazole, a bioisostere of the indole ring in tryptophan, was used as the skeletal structure for design new IDO1 inhibitors.32–35 Indazoles bearing 3-hydrazide groups (6)32 were first studied in 2016 with IC50 values near the nanomolar range. Next, some 4-amino (7)33,34 and 3-(hydroxymethyl)35 (8) indazoles were also reported as IDO1 inhibitors at the sub-nanomolar range (Fig. 1B). To date, all indazole derivatives bear a huge group at position 4 and a small group (normally a halogen) at position 6.
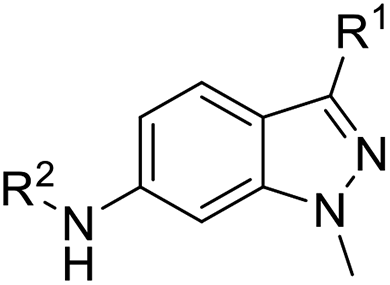
In this study, we chose indazole as the core structure for the rational design of new IDO1- and cytotoxic agents. We simplified and considered the derivatization abilities of the indazole structure, and then synthesized a library of 6-substituted aminoindazoles (Fig. 1C). Next, we evaluated their toxicities on five cancer cell lines and investigated the IDO1 inhibitory activities in vitro. We further performed docking studies to calculate the binding energy and analyzed the specific binding interactions between the selected inhibitors and the IDO1 active site. Finally, we examined the potential mechanisms of the most active compound on the cell cycle arrest in human colorectal cancer cells.
Results and discussion
Chemistry
The synthesis of 6-substituted-indazole derivatives was performed, as illustrated in Scheme 1. Briefly, 6-nitro indazole derivatives 9, 10 were methylated by iodomethane using potassium carbonate in DMF to get two isomers: the major products, N1-alkylindazoles 12, 13 and the minor products N2-alkylindazoles 14, 15. The structures of two isomers were confirmed by 2D-NMR before carrying out a reduction reaction under hydrogen gas to afford corresponding 6-amino indazole derivatives 11, 16–19. Next, the amines were reductively aminated with a ketone or an aldehyde in the presence of a mild and selective reductive agent, namely sodium cyanoborohydride, to afford secondary amines 24–51. Moreover, acetamide derivatives 20–23 were prepared from the corresponding amine and acetic anhydride under basic conditions.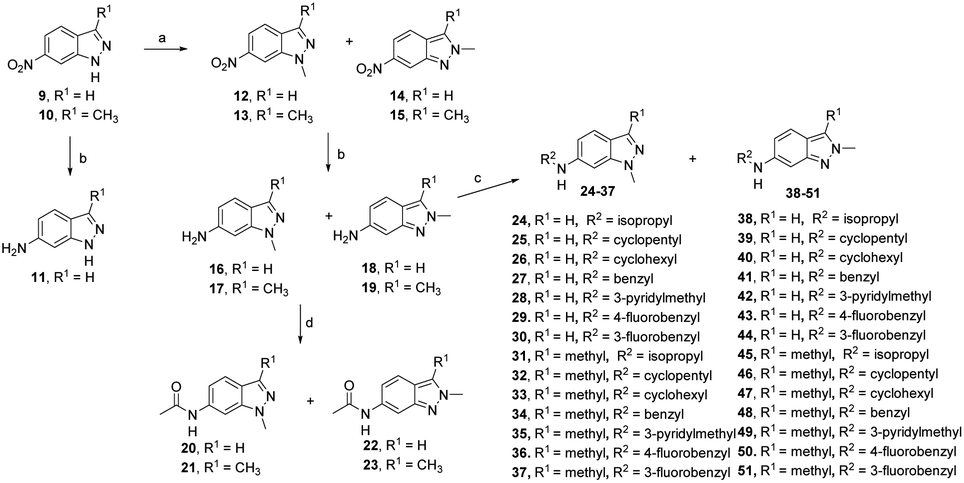 | ||
| Scheme 1 Reagents and conditions: (a) CH3I (2.0 equiv.), excess K2CO3, DMF, 60 °C, 4 h, 95–98%; (b) H2, Pd/C (10 mol%), r.t., 4 h, 85–96%; (c) ketone or aldehyde (1.0 equiv.), NaBH3CN (5.0 equiv.), AcOH, MeOH, 40 °C, 4 h, 73–96%; (d) excess Ac2O, TEA, DMC, r.t., 2 h, 89–92%. | ||
Anti-proliferative activity
The as-synthesized indazole derivatives were evaluated for the in vitro anti-proliferative activity in five different cancer cell lines including HCT116, A549, SK-HEP-1, SNU-638, and MDA-MB-231 using the sulforhodamine B method. Etoposide was used as the positive control. The results are summarized in Tables 1 and 2.
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | R1 = H (IC50a (μM)) | R1 = CH3 (IC50a (μM)) | ||||||||||
| Cpd | A549 | SK-HEP-1 | SNU-638 | MDA-MB-231 | HCT116 | Cpd | A549 | SK-HEP-1 | SNU-638 | MDA-MB-231 | HCT116 | |
| a IC50: the concentration (μM) of compounds that produces a 50% reduction cell growth. All the data were calculated from the experiments conducted in triplicate and repeated at least three times. Data represent mean ± SD. | ||||||||||||
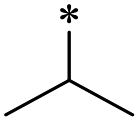 |
24 | >100 | >100 | >100 | >100 | >100 | 31 | >100 | >100 | >100 | >100 | >100 |
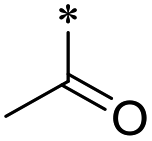 |
20 | >100 | >100 | >100 | >100 | 26.7 ± 15.7 | 21 | >100 | >100 | >100 | >100 | >100 |
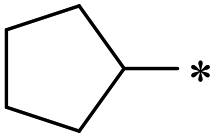 |
25 | >100 | >100 | 64.8 ± 28.4 | >100 | >100 | 32 | >100 | 36.8 ± 11.2 | 3.8 ± 3.9 | >100 | >100 |
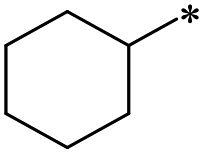 |
26 | >100 | >100 | >100 | >100 | >100 | 33 | 27.0 ± 18.7 | 39.6 ± 22.4 | 13.7 ± 9.1 | >100 | 14.5 ± 16.3 |
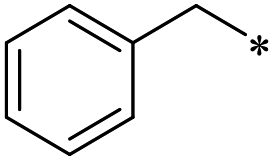 |
27 | >100 | >100 | >100 | >100 | >100 | 34 | 5.1 ± 23.3 | 28.1 ± 20.0 | 1.9 ± 2.0 | >100 | 7.5 ± 5.0 |
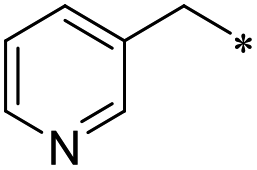 |
28 | 19.8 ± 6.1 | >100 | >100 | >100 | 47.2 ± 12.9 | 35 | 30.0 ± 18.5 | 33.9 ± 22.6 | >100 | >100 | 2.7 ± 2.3 |
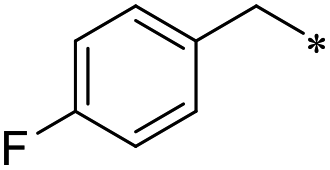 |
29 | 0.7 ± 0.1 | >100 | 1.2 ± 1.2 | >100 | 34.9 ± 21.3 | 36 | 6.8 ± 2.4 | 50.1 ± 23.4 | 1.8 ± 2.2 | 47.5 ± 8.4 | 0.4 ± 0.3 |
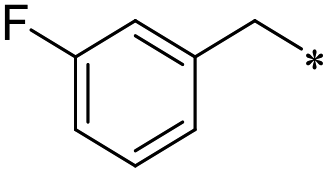 |
30 | 19.9 ± 10.6 | >100 | 1.3 ± 1.0 | >100 | >100 | 37 | >100 | 42.2 ± 15.0 | 2.5 ± 2.6 | 81.7 ± 25.2 | 2.2 ± 0.9 |
| Etoposide | 0.27 ± 0.1 | 0.30 ± 0.1 | 0.61 ± 0.2 | 7.35 ± 2.3 | 1.27 ± 0.8 | |||||||
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | R1 = H (IC50a (μM)) | R1 = CH3 (IC50a (μM)) | ||||||||||
| Cpd | A549 | SK-HEP-1 | SNU-638 | MDA-MB-231 | HCT116 | Cpd | A549 | SK-HEP-1 | SNU-638 | MDA-MB-231 | HCT116 | |
| a IC50: the concentration (μM) of compounds that produces a 50% reduction cell growth. All the data were calculated from the experiments conducted in triplicate and repeated at least three times. Data represent mean ± SD. | ||||||||||||
 |
38 | 38.9 ± 13.2 | >100 | 30.4 ± 27.0 | 68.3 ± 27.3 | 33.4 ± 11.7 | 45 | >100 | >100 | >100 | >100 | >100 |
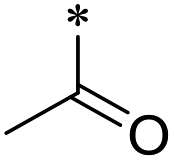 |
22 | 58.7 ± 23.9 | >100 | >100 | 20.6 ± 1.7 | 2.5 ± 1.6 | 23 | >100 | >100 | >100 | >100 | >100 |
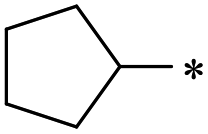 |
39 | 1.8 ± 1.4 | >100 | 2.8 ± 1.3 | 1.7 ± 1.1 | >100 | 46 | >100 | >100 | >100 | >100 | >100 |
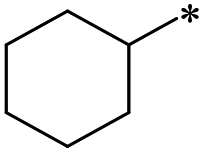 |
40 | 37.4 ± 10.2 | >100 | >100 | >100 | >100 | 47 | 37.0 ± 22.7 | 32.7 ± 11.2 | 33.0 ± 17.5 | >100 | 26.1 ± 13.8 |
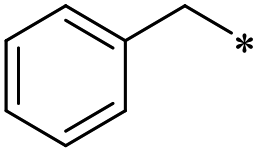 |
41 | >100 | >100 | >100 | >100 | 49.7 ± 3.6 | 48 | >100 | >100 | >100 | >100 | >100 |
 |
42 | >100 | >100 | >100 | >100 | >100 | 49 | >100 | >100 | >100 | 19.4 ± 17.0 | 19.1 ± 13.5 |
 |
43 | >100 | >100 | 63.3 ± 13.8 | >100 | >100 | 50 | 19.4 ± 20.6 | 37.3 ± 6.3 | 10.8 ± 11.2 | 8.0 ± 1.7 | 13.8 ± 6.5 |
 |
44 | >100 | >100 | >100 | >100 | >100 | 51 | >100 | >100 | >100 | 61.3 ± 32.3 | 12.7 ± 8.5 |
| Etoposide | 0.27 ± 0.1 | 0.30 ± 0.1 | 0.61 ± 0.2 | 7.35 ± 2.3 | 1.27 ± 0.8 | |||||||
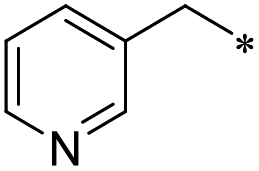
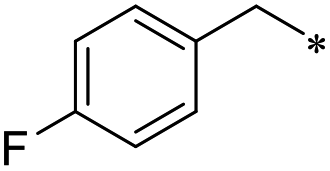
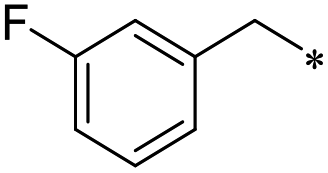
First, we examined the structure–activity relationship (SAR) of the as-synthesized compounds on colon cancer cell (HCT116) proliferation, in which required IDO activation increases the expression of β-catenin and its targeting genes including cyclin D and Axin2 for development.36 As demonstrated in Tables 1 and 2, the introduction of a methyl group at the C-3 position of the indazole ring 33–37 caused a potential toxicity against HCT116. Among them, the substitution of an aryl group in indazoles 34–37 showed a better cytotoxicity than the cyclohexyl-substituted compound 33. A replacement of benzyl by 3-pyridylmethyl 35 or fluorobenzyl 36, 37 increased the toxic potency from 2.7 to 18.8-fold as compared to 34. Of note, compound 36 had a great anti-proliferative activity (IC50 = 0.4 ± 0.3 μM), which indicates the importance of the fluoro group. Changing the position of the fluoro group from para to meta caused a 5.5 times activity drop. In contrast, introducing alkyl groups to 31, 32 resulted in non-toxic compounds. Similarly, the toxicity of acetamide derivative 21 was abolished. Removing the methyl group from the C-3 position of the indazole ring caused a reduction in the toxicity against HCT116, except for the acetylated compound 20 (IC50 = 26.7 ± 15.7 μM). Besides, relocating the methyl group from N1 to N2 decreased the anti-proliferative activity of most of the compounds (Table 2), excluding isopropyl- and acetyl compounds. In particular, compound 22 showed a potent activity with an IC50 value of 2.5 ± 1.6 μM. The most effective groups, 3-pyridylmethyl 49, 4-fluorobenzyl 50 and 3-fluorobenzyl 51, demonstrated less cytotoxicity of 5.8 to 34.5-fold than the corresponding N1-methyl isomers. Overall, the absence of the methyl group at the 3-position in the indazole ring favored a small alkyl- or an acetyl-substituted group in displaying the anti-proliferative activity. In contrast, the substitution of an aryl group preferred 3-methylindazole compounds in presenting cytotoxic activities.
Next, the SAR of all as-synthesized compounds against the other four cell lines including A549, SK-HEP-1, SNU-638, and MDA-MB-231 (Tables 1 and 2) was examined. Most of them showed a mild suppressive effect on SK-HEP-1 and MDA-MB-231 (IC50 > 20 μM) excluding compounds 39, 49, 50. We observed that compound 39 had a great suppressive potency on MDA-MB-231, A549, and SNU-638 cells with IC50 = 1.7 ± 1.1, 2.8 ± 1.3, and 1.8 ± 1.4 μM, respectively.
In addition, the N-aromatic substitution of 6-aminoindazole derivatives 29, 30, 34, 36, 37 exhibited considerable cytotoxicity towards A549 and SNU-638 cancer cell lines with IC50 values ranging from 0.7 to 10 μM. The cytotoxicity changed dramatically among substituted groups. Moreover, there was no correlation between the cytotoxicity and structure observed. In addition, the IC50 values of compounds 22, 34–37 on the normal cell MRC5 were higher than the IC50 of them on HCT116 cancer cell line. Specially, compound 36 demonstrated 29.6-fold selectivity against HCT116 than MRC5 (Table S1 – ESI file†).
IDO1 inhibition assay
In order to examine the effects on IDO1 expression, which can be well correlated to human colon cancer cell growth,37,38 we selected the compounds with the most anti-proliferative potency against HCT116 for the analysis of the IDO1-suppressive activity (Fig. 2). We found that at the concentration of 10 μM, the expression of IDO1 in HCT16 cells seemed unaffected by compound 35 or 37 (Fig. 2); moreover, it was suppressed by a treatment with compound 22 or 36 (Fig. 2). Furthermore, the effects of compound 36 on IDO1 expression were observed in a concentration-dependent manner (Fig. 3 and S2 – ESI file†).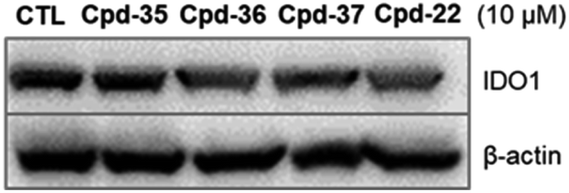 | ||
| Fig. 2 Effect of 6-substituted aminoindazole derivatives on the IDO1 expression in human colorectal cancer cells. HCT116 were treated with 10 μM of indicative compounds for 24 h. The immunoblotting analysis was performed to determine IDO1 and β-actin expression. | ||
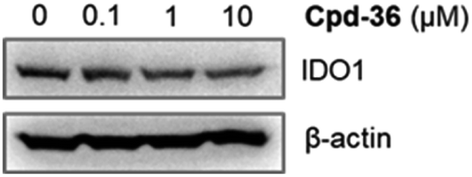 | ||
| Fig. 3 HCT116 cells were treated with compound 36 at various concentrations. The immunoblotting analysis was performed to determine IDO1 and β-actin protein levels. | ||
Docking simulation analysis of IDO1 and its inhibitors
To gain insight into the interaction between inhibitors and IDO1, we performed molecular docking simulation studies on the active site region of IDO1. To validate the docking program Autodock Vina, we performed a control docking experiment with the 5PK compound in the IDO1 complex structure (PDB ID: 5EK3). 5PK was predicted to dock into the active site of IDO1 as in the actual complex structure with a binding energy of −9.5 kcal mol−1 (described in Fig. S1 of the ESI file†). We found that both compounds 35 and 36 bound to IDO1 in a comparable way to that of the 5PK–IDO1 complex structure (Fig. 4A). In addition, the binding energy of compound 35 (−8.1 kcal mol−1) was higher than that of compound 36 (−8.9 kcal mol−1), suggesting a higher binding affinity towards IDO1 of compound 36 than that of compound 35. In the docking model, compounds 35 and 36 were recognized and/or stabilized by well-known active site residues Tyr126, Phe163, Phe164, Ser167, Phe226, Phe227, Arg231, Ser263, and His346,39 which play a similar role in the 5PK–IDO1 complex structure (Fig. 4B). The docking models showed that the N2 indazole group of compound 36 formed a hydrogen bond with the hydroxy group of Ser167, and the flurophenyl/pyridinyl ring formed a π–π interaction with Phe226 (Fig. 4C). In addition, the 6-NH group in the compounds also formed a hydrogen bond with the 7-propionate of the heme ion (Fig. 4C).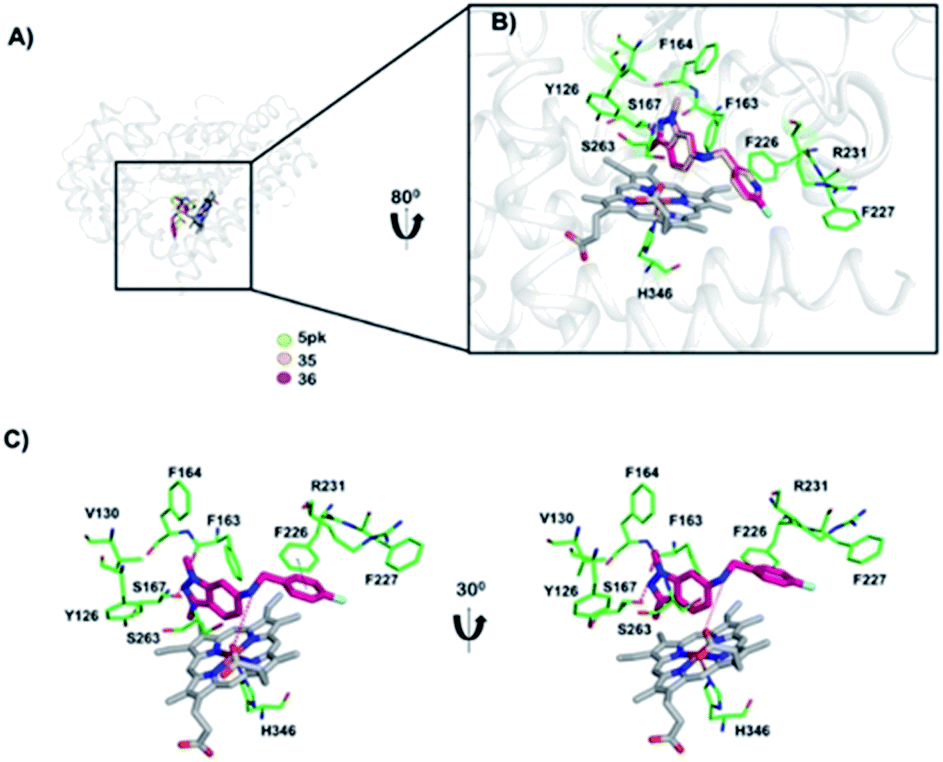 | ||
| Fig. 4 Docking model of IDO1 with 5PK and compounds. (A) The docking models of 5PK, compound 35, and compound 36 are shown in yellow green, light yellow, and pink models, respectively. (B) A close-up view of docked compounds in the active site cavity of IDO1. (C) The interaction mode of IDO1 with compound 36. The IDO1 residue recognizing ligands are shown in the green stick model. The heme ion is shown in a grey stick. Hydrogen bonds and π–π interactions are shown in red and black dots, respectively. | ||
Study mechanism of compound 36 on human colorectal cancer cells
Compound 36 demonstrated a concentration-dependent suppressive effect on the activity of the HCT116 viability (Fig. 5). On other cell lines, despite a modest effect on the IDO expression, compound 36 still demonstrated a remarkable inhibition of cell viability. This phenomenon suggested that other mechanisms may also be responsible for the anti-proliferative effect of compound 36. Therefore, to further explore the possible mechanism of action of the anti-proliferative activity of compound 36, the effect of compound 36 on the modulation of the cell cycle was determined.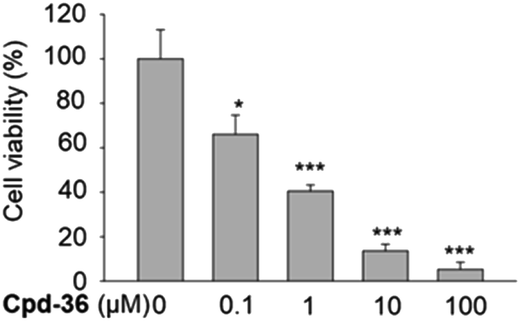 | ||
| Fig. 5 Anti-proliferative effect of compound 36 on HCT116 cells. HCT116 cells were treated with compound 36 at various concentrations for 72 h, and cell proliferation was determined by the SRB assay. The data were presented as mean ± SD. The experiment was performed in triplicate (n = 3). | ||
A cell cycle analysis indicated that a treatment with 10 μM of compound 36 stimulated the G2/M cell cycle arrest in HCT116 cells within 24 h of post-treatment (Fig. 6A). Furthermore, a concentration-dependent concentration simulative effect on the G2/M cell cycle arrest was observed at 48 h after the treatment (Fig. 6B). We further analyzed the expression of the G2/M cell cycle regulatory proteins by immunoblot. We found that the expression of p-CDC25C (Thr48), CDC25C, Chk1, and p-cyclin B1 were down-regulated (Fig. 6C). These findings suggested that the anti-proliferative activity of compound 36 was associated, at least partially, with the G2/M phase cell cycle arrest through regulating cell cycle modulators in human colorectal cancer cells.
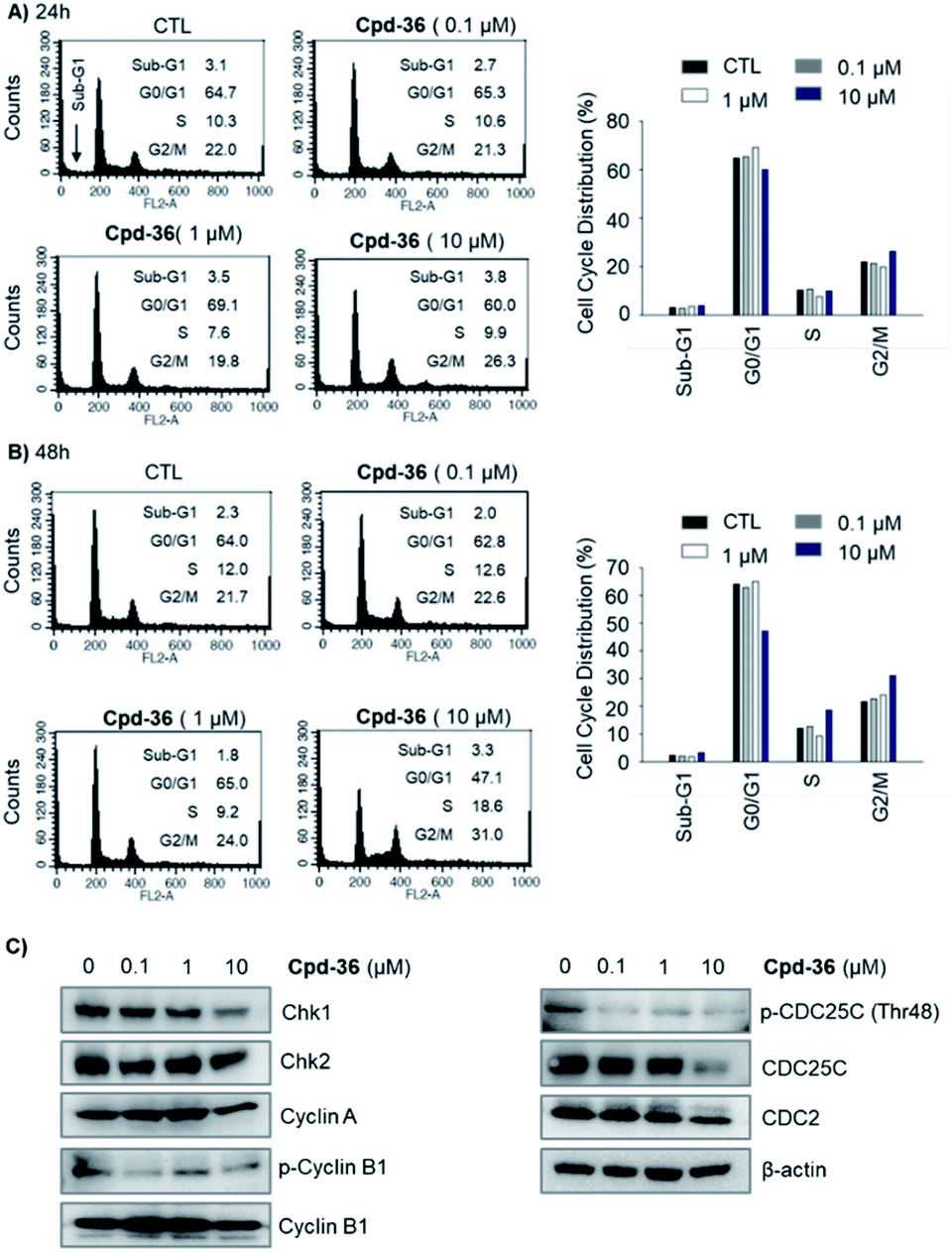 | ||
| Fig. 6 Effect of compound 36 on the cell cycle distribution in human colorectal cancer cells. (A and B) HCT116 cells were treated with compound 36 for 24 h or 48 h. The cell cycle distribution was analyzed via flow cytometry. (C) Effects of 36 on the expression of cell cycle-related proteins in HCT116 cells. HCT116 cells were treated with 36 for 48 h. Subsequently, the protein expression levels of cell cycle-related proteins were analyzed by the immunoblotting assay. | ||
Experimental section
Chemistry
All chemical reagents were commercially available and used directly without any purification. Thin layer chromatography, which was performed using Whatman® 250 μm Silica Gel GF Uniplates and visualized under UV light at 254 nm, was used to check the progress of reactions and preliminary evaluation of the homogeneity of the compounds. In all cases, the compounds achieved a purity of 97% or above as determined by HPLC. Melting points were measured using a Gallenkamp Melting Point Apparatus (LabMerchant, London, United Kingdom) and were uncorrected. The purification of compounds was carried out using crystallization methods and/or open silica gel column flash chromatography employing Merck silica gel 60 (240 to 400 mesh) as the stationary phase. Nuclear magnetic resonance spectra (1H NMR, 13C NMR) were recorded on a JEOL JNM-LA 300 at 300 MHz and Bruker Analytik, DE/AVANCE Digital 500 at 500 and 125 MHz spectrometers. Tetramethylsilane was used as the internal standard. Chemical shifts were reported in parts per million (ppm) downfielded from tetramethylsilane. Mass spectra with different ionization modes including electron ionization (EI) and electrospray ionization (ESI) were recorded using PE Biosystems API2000 (PerkinElmer, Palo Alto, CA, USA) and Mariner® (Azco Biotech, Inc. Oceanside, CA, USA) or LC-MSD-Trap-SL (Agilent, CA, USA) mass spectrometers. All reagents and solvents were purchased from Aldrich or Fluka Chemical Corp. (Milwaukee, WI, USA) or Merck unless noted otherwise. Solvents were used directly as purchased unless otherwise indicated.Cell culture
Human colorectal cancer cells (HCT116), gastric cancer cells (SNU638), breast cancer cells (MDA-MB-231), liver cancer cells (SK-HEP-1) were purchased from American Type Culture Collection (Manassas, VA, USA). HCT116, SNU638, MDA-MB-231, SK-HEP-1 were cultured, as described previously.40Sulforhodamine B assay
The effect of the indicated compounds on cell proliferation was calculated as described previously.40 In brief, HCT116 cells (2 × 104 cells per mL) were seeded in 96-well plates with different compounds. After incubation, the cells were fixed with a 50% trichloroacetic acid (TCA) solution for 1 h, and cellular proteins were stained with 0.4% sulforhodamine B (SRB) in 1% acetic acid. The stained cells were dissolved in 10 mM Tris buffer (pH 10.0). The effect of the investigated compound on cell proliferation was calculated as a percentage relative to a solvent-treated control, and the IC50 values were evaluated via nonlinear regression analysis (percent survival versus concentration). Etoposide was used as the positive control.Flow cytometry for cell cycle analysis
HCT116 cells (2 × 105 cells per mL) were plated in a 36 mm culture dish and incubated for 24 h. A fresh medium containing the indicated concentration of compound 36 was added to culture dishes. Following a 24 h or 48 h incubation, the cells were harvested (via trypsinization and centrifugation), rinsed twice with pre-cooled phosphate buffered saline (PBS), and prepared for the cell cycle analysis as described previously.40Western blot analysis
HCT116 cells (2 × 105 cells per mL) were treated with various concentrations of 36 for the indicated times. Western blot analysis was carried out as described previously.41 The antibody for IDO1 (ab55305) was purchased from Abcam (Abcam, Cambridge, UK). Antibodies for CDC25C, phosphor-CDC25C (Thr48), Chk1, Chk2, and phosphor-cyclin B1 (Ser147) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies for cyclin A (H-432), CDC2, cyclin B1 (H-433), and β-actin (C4) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).Statistical analysis
All experiments were repeated at least three times. The data are shown as the means ± standard deviation (SD) for the indicated number of independently performed experiments. The student's t-test was used for the determination of statistical significance, and IC50 values were determined by nonlinear regression using the Sigma Plot (Systat Software, Inc., San Jose, CA, USA). The difference was considered statistically significant when p (*) < 0.05, (**) < 0.001, (***) < 0.0001.Molecular simulation docking
The structure of IDO1 was acquired from the protein data bank (PDB ID: 5EK3), and the structure of compound 35 and compound 36 were drawn by the ChemDraw program (version Pro 16.0, Cambridge soft, Cambridge, MA). The structure of the control compound 5PK was extracted from the complex structure of 5PK–IDO1 (PDB ID: 5EK3). The compounds were docked into the active site region of IDO1 using AutoDock Vina (version 1.1.2, La Jolla, CA, USA)42 with a grid map defined in 20 × 20 × 20 Å3 dimensions to contain the proposed active sites and hem, which are essential for the enzyme activity.43 The conformations of the ligands and receptors were fixed. The program was run in score mode with an exhaustiveness value of 20.Conclusions
In summary, four series of 6-substituted aminoindazoles were designed based on an anticancer privileged structure, indazole, targeting to inhibit the IDO1 activity, and the designed compounds were synthesized through 3 steps. Of them, the acetyl, pyridinylmethyl, and fluorobenzyl substituted compounds (22, 35–37) showed potential cytotoxicity towards colorectal cancer cells HCT116. A study on the IDO1 activity indicated that compound 36 had a moderate inhibition activity of IDO1 towards the HCT116 cell line. The molecular modeling analysis showed that compound 36 fitted well to the IDO1 active site and created hydrogen bonds with Ser167 and 7-propionate in the heme structure. Further studies indicated that compound 36 arrested the G2/M cell cycle of HTC116 through regulating the cell cycle modulators. Due to the good cytotoxicity on colorectal cancer cells, and having a small structure, we believe that compound 36 might be useful for the design of new anticancer agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 108.05-2020.01. We would like to thank Professor Sang Kook Lee and Dr Duc-Hiep Bach (College of Pharmacy, Natural Products Research Institute, Seoul National University, Seoul 08826, Republic of Korea) for the biological part.References
- D. H. Bach, D. Kim, S. Y. Bae, W. K. Kim, J. Y. Hong, H. J. Lee, N. Rajasekaran, S. Kwon, Y. Fan, T. T. Luu, Y. K. Shin, J. Lee and S. K. Lee, Mol. Ther.--Nucleic Acids, 2018, 11, 455–467 CrossRef CAS.
- D. H. Bach, T. T. Luu, D. Kim, Y. J. An, S. Park, H. J. Park and S. K. Lee, Mol. Ther.--Nucleic Acids, 2018, 12, 817–828 CrossRef CAS.
- D. Kim, D. H. Bach, Y. H. Fan, T. T. Luu, J. Y. Hong, H. J. Park and S. K. Lee, Cell Death Dis., 2019, 10, 361 CrossRef.
- T. T. Luu, D. H. Bach, D. Kim, R. Hu, H. J. Park and S. K. Lee, Anticancer Res., 2020, 40, 1855–1866 CrossRef CAS.
- P. T. Huong, L. T. Nguyen, X. B. Nguyen, S. K. Lee and D. H. Bach, Cancers, 2019, 11, 240 CrossRef CAS.
- D. H. Bach, J. Y. Hong, H. J. Park and S. K. Lee, Int. J. Cancer, 2017, 141, 220–230 CrossRef CAS.
- N. P. Restifo, M. J. Smyth and A. Snyder, Nat. Rev. Cancer, 2016, 16, 121 CrossRef CAS.
- J. Tang, L. Pearce, J. O'Donnell-Tormey and V. M. Hubbard-Lucey, Nat. Rev. Drug Discovery, 2018, 17, 783–784 CrossRef CAS.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS.
- A. Botticelli, B. Cerbelli, L. Lionetto, I. Zizzari, M. Salati, A. Pisano, M. Federica, M. Simmaco, M. Nuti and P. Marchetti, J. Transl. Med., 2018, 16, 1–6 CrossRef.
- D. H. Munn and A. L. Mellor, Trends Immunol., 2013, 34, 137–143 CrossRef CAS.
- S. Löb, A. Königsrainer, H.-G. Rammensee, G. Opelz and P. Terness, Nat. Rev. Cancer, 2009, 9, 445 CrossRef.
- M. Platten, N. von Knebel Doeberitz, I. Oezen, W. Wick and K. Ochs, Front. Immunol., 2014, 5, 673 Search PubMed.
- Y. W. Moon, J. Hajjar, P. Hwu and A. Naing, J. Immunother. Cancer, 2015, 3, 51 CrossRef.
- J. Godin-Ethier, L.-A. Hanafi, C. A. Piccirillo and R. Lapointe, Clin. Cancer Res., 2011, 17, 6985–6991 CrossRef CAS.
- C. Uyttenhove, L. Pilotte, I. Théate, V. Stroobant, D. Colau, N. Parmentier, T. Boon and B. J. Van den Eynde, Nat. Med., 2003, 9, 1269–1274 CrossRef CAS.
- B. C. Creelan, S. Antonia, G. Bepler, T. J. Garrett, G. R. Simon and H. H. Soliman, Oncoimmunology, 2013, 2, e23428 CrossRef.
- H. Liu, Z. Shen, Z. Wang, X. Wang, H. Zhang, J. Qin, X. Qin, J. Xu and Y. Sun, Sci. Rep., 2016, 6, 21319 CrossRef CAS.
- T. Weng, X. Qiu, J. Wang, Z. Li and J. Bian, Eur. J. Med. Chem., 2018, 143, 656–669 CrossRef CAS.
- J. E. Cheong and L. Sun, Trends Pharmacol. Sci., 2018, 39, 307–325 CrossRef CAS.
- A. J. Muller, J. B. DuHadaway, P. S. Donover, E. Sutanto-Ward and G. C. Prendergast, Nat. Med., 2005, 11, 312 CrossRef CAS.
- M. Li, A. R. Bolduc, M. N. Hoda, D. N. Gamble, S.-B. Dolisca, A. K. Bolduc, K. Hoang, C. Ashley, D. McCall and A. M. Rojiani, J. Immunother. Cancer, 2014, 2, 21 CrossRef.
- M. Liu, X. Wang, L. Wang, X. Ma, Z. Gong, S. Zhang and Y. Li, J. Hematol. Oncol., 2018, 11, 100 CrossRef.
- G. C. Prendergast, A. Mondal, S. Dey, L. D. Laury-Kleintop and A. J. Muller, Trends Cancer, 2018, 4, 38–58 CrossRef CAS.
- M. L. Bolognesi, Curr. Med. Chem., 2013, 20, 1639–1645 CrossRef CAS.
- R. R. Ramsay, M. R. Popovic-Nikolic, K. Nikolic, E. Uliassi and M. L. Bolognesi, Clin. Transl. Med., 2018, 7, 3 Search PubMed.
- K. Fang, G. Dong, Y. Li, S. He, Y. Wu, S. Wu, W. Wang and C. Sheng, ACS Med. Chem. Lett., 2018, 9, 312–317 CrossRef CAS.
- K. Fang, S. Wu, G. Dong, Y. Wu, S. Chen, J. Liu, W. Wang and C. Sheng, Org. Biomol. Chem., 2017, 15, 9992–9995 RSC.
- J. Dong, Q. Zhang, Z. Wang, G. Huang and S. Li, ChemMedChem, 2018, 13, 1490–1507 CrossRef CAS.
- Y. Wan, S. He, W. Li and Z. Tang, Anti-Cancer Agents Med. Chem., 2018, 18, 1228–1234 CrossRef CAS.
- D.-H. Bach, D. Kim and S. K. Lee, in Natural Products for Cancer Chemoprevention: Single Compounds and Combinations, ed. J. M. Pezzuto and O. Vang, Springer International Publishing, Cham, 2020, pp. 469–488 Search PubMed.
- N. Pradhan, S. Paul, S. J. Deka, A. Roy, V. Trivedi and D. Manna, ChemistrySelect, 2017, 2, 5511–5517 CrossRef CAS.
- S. Qian, T. He, W. Wang, Y. He, M. Zhang, L. Yang, G. Li and Z. Wang, Bioorg. Med. Chem., 2016, 24, 6194–6205 CrossRef CAS.
- L. Yang, Y. Chen, J. He, E. M. Njoya, J. Chen, S. Liu, C. Xie, W. Huang, F. Wang and Z. Wang, Bioorg. Med. Chem., 2019, 27, 1087–1098 CrossRef CAS.
- H. Tsujino, T. Uno, T. Yamashita, M. Katsuda, K. Takada, T. Saiki, S. Maeda, A. Takagi, S. Masuda and Y. Kawano, Bioorg. Med. Chem. Lett., 2019, 29, 126607 CrossRef CAS.
- A. I. Thaker, M. S. Rao, K. S. Bishnupuri, T. A. Kerr, L. Foster, J. M. Marinshaw, R. D. Newberry, W. F. Stenson and M. A. Ciorba, Gastroenterology, 2013, 145, 416–425 CrossRef CAS.
- M. Ogawa, M. Watanabe, T. Hasegawa, K. Ichihara, K. Yoshida and K. Yanaga, Mol. Clin. Oncol., 2017, 6, 701–704 CrossRef CAS.
- J. Godin-Ethier, L. A. Hanafi, C. A. Piccirillo and R. Lapointe, Clin. Cancer Res., 2011, 17, 6985–6991 CrossRef CAS.
- J. E. Cheong, A. Ekkati and L. Sun, Expert Opin. Ther. Pat., 2018, 28, 317–330 CrossRef CAS.
- D. H. Bach, S. H. Kim, J. Y. Hong, H. J. Park, D. C. Oh and S. K. Lee, Mar. Drugs, 2015, 13, 6962–6976 CrossRef CAS.
- D.-H. Bach, J.-Y. Liu, W. K. Kim, J.-Y. Hong, S. H. Park, D. Kim, S.-N. Qin, T.-T.-T. Luu, H. J. Park, Y.-N. Xu and S. K. Lee, Bioorg. Med. Chem., 2017, 25, 3396–3405 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- Y.-H. Peng, S.-H. Ueng, C.-T. Tseng, M.-S. Hung, J.-S. Song, J.-S. Wu, F.-Y. Liao, Y.-S. Fan, M.-H. Wu, W.-C. Hsiao, C.-C. Hsueh, S.-Y. Lin, C.-Y. Cheng, C.-H. Tu, L.-C. Lee, M.-F. Cheng, K.-S. Shia, C. Shih and S.-Y. Wu, J. Med. Chem., 2016, 59, 282–293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental chemistry, biology and molecular simulation docking. Additional data of 1H NMR, 13C NMR & MS spectra of the compounds. See DOI: 10.1039/d0ra09112j |
| This journal is © The Royal Society of Chemistry 2020 |