
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of new heteroaryl nitrones with spin trap properties†
G. Barriga-González *a,
C. Aliagabc,
E. Chamorrod,
C. Olea-Azare,
E. Norambuenaa,
W. Porcalf,
M. Gonzálezf and
H. Cerecettof
*a,
C. Aliagabc,
E. Chamorrod,
C. Olea-Azare,
E. Norambuenaa,
W. Porcalf,
M. Gonzálezf and
H. Cerecettof
aDepartamento de Química, Facultad de Ciencias Básicas, Universidad Metropolitana de Ciencias de la Educación, Av. José Pedro Alessandri 774, Ñuñoa, Santiago, Chile. E-mail: german.barriga@umce.cl
bFacultad de Química y Biología, Universidad de Santiago de Chile, Av. Bernardo O'Higgins 3363, Santiago, Chile
cCentro para el Desarrollo de la Nanociencia y la Nanotecnología, CEDENNA, Chile
dDepartamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andrés Bello, Avenida República 275, 8370146 Santiago, Chile
eDepartamento de Química Inorgánica y Analítica, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Santiago, Chile
fGrupo de Química Orgánica Medicinal, Laboratorio de Química Orgánica, Facultad de Ciencias/Facultad de Química, Universidad de la República, Montevideo, Uruguay
First published on 4th November 2020
Abstract
A new series of heteroaryl nitrones were synthesized and evaluated as free radical traps due to the results showed in our previous report. The physicochemical characterization of these new nitrones by electron spin resonance (ESR) demonstrated their high capability to trap and stabilize different atom centered free radicals generated by the Fenton reaction. Additionally, we intensely studied them in terms of their physicochemical properties. Kinetic studies, including the use of a method based on competition and the hydroxyl adduct decay, gave the corresponding rate constants and half-lives at the physiological pH of these newly synthesized nitrones. New nitrones derived from quinoxaline 1,4-dioxide heterocycles were more suitable than DMPO to trap hydroxyl free radicals with a half-life longer than two hours. We explain some of the results using computational chemistry through density functional theory (DFT).
Introduction
Due to natural metabolic activity in the human body, some of the essential by-products found are reactive oxygen and nitrogen species,1,2 which mediate various physiological oxidative processes. The ability to reliably identify and quantify such short-lived oxidant species is critical to determine their role in human disease. In the past three decades, spin trapping, in conjunction with electron spin resonance spectrometry (ESR), has proven to be an essential experimental technique to accomplish this goal. In the last years, the spin-trapping technique was used to deeply study the behavior and role played by oxygen and nitrogen centered free radicals.3,4 The most widely used spin trap has been the non-cyclic nitrones α-phenyl-N-t-butylnitrone and α-4-pyridyl-1-oxide-N-t-butylnitrone (PBN and POBN, respectively, Fig. 1A) and the cyclic one 5,5-dimethylpyrroline-N-oxide (DMPO, Fig. 1B).5–8 In general terms, these available spin traps present many disadvantages such as low water solubility (in the case of PBN), sensitivity to nucleophilic attack or other degradation reactions, and low stability of the generated spin adduct. Some new spin traps with better trapping abilities like BMPO, EMPO, and DEPMPO (Fig. 1B)9,10 have been developed. However, despite the efforts, there are still limitations in their employment. In the past years, we found significant advances in the development of new free radical spin traps. Some new free radical spin traps, such as isoindole-based nitrones, have shown they can trap the nitric oxide radical (TMINO11–13 and 3-TF-TMINO14). | ||
| Fig. 1 Structures of most used nitrones as spin trap agents today. (A) PBN and POBN, (B) DMPO, BMPO, EMPO, and DEPMPO, and (C) furoxanyl nitrone (FxBN) described by us. | ||
We have described a new series of heteroaryl nitrones in previous work, bearing furoxanyl and thiadiazolyl moieties with the capability to trap and stabilize oxygen-, carbon-, sulfur-, and nitrogen-centered free radicals.15 The spin trapping properties of the nitrones depends in great part on the connectivity and the nature of substituents on the nitrone group. As part of our ongoing investigations in the development of new nitrone-free radical trapping, we set out to design and synthesizer new heteroarylnitrones starting from suitable precursors available in our heterocyclic library. Notably, the 4-furoxanyl nitrone FxBN (Fig. 1C) showed good solubility in aqueous solution, and FxBN adducts formed with hydroxyl and superoxide radicals exhibited distinct and characteristic ESR spectral patterns.
Herein, we study and assay the spin trapping capabilities of three new heteroaryl nitrones (nitrone 1, nitrone 2, and nitrone 3, Fig. 2) against oxygen-, carbon-, sulfur-, and nitrogen-centered free radicals, i.e., hydroxyl-, superoxide-, 1-hydroxyethyl-, methyl-, sulfur trioxide anion-, and azidyl-free radicals. Thus, kinetic constants and decay rates were obtained for the corresponding hydroxyl adduct at pH 7.4 in aqueous solution. Additionally, we performed assays for the trap of nitric oxide-free radicals and the behavior of these heteroaryl nitrones in biological models.
 | ||
| Fig. 2 Structures of the heteroaryl nitrones described in the present work. | ||
Experimental
Materials
The newly developed heteroaryl nitrones studied as spin traps are summarized in Fig. 2. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO, Fig. 1), dimethylsulfoxide, anhydrous sodium sulfite, monobasic potassium phosphate, sodium hydroxide, N-methyl-D-glucamine (MGD), diethylenetriaminepentaacetic acid, xanthine, and xanthine oxidase were purchased from Sigma-Aldrich. Sodium azide, hydrogen peroxide aqueous solution (30%), anhydrous ethanol, dibasic sodium phosphate heptahydrate, methanol, carbon disulfide, and sodium nitrite were purchased from Merck. Ferrous ammonium sulfate hexahydrate was purchased from Mallinckrodt Baker.Synthesis
Before using, all solvents were distilled, and experimental conditions for analytical TLC and column chromatography purification was used according to previous work. The electrothermal 9100 apparatus was used to determining melting points, and they are uncorrected. Microanalyses were performed on a Fisons EA 1108 CHNS-O instrument and were within ±0.4% of the calculated compositions. Bruker DPX-400 spectrometer was used to record NMR spectra. The assignment of chemical shifts is based on standard NMR experiments (1H, 13C, 1H-COSY, HSQC, HMBC, and NOE). The chemical shift values are expressed in ppm relative to tetramethylsilane as an internal standard. Shimadzu DI-2010 was used to determine the mass spectra.α(Z)-(3-Methylfuroxan-4-yl)-N-cyclohexylnitrone (1). Pale yellow solid (60%); mp 118.0–120.0 °C. Found: C, 52.9; H, 6.5; N, 18.6. C10H15N3O3 requires C, 53.3; H, 6.7; N, 18.7. 1H-NMR (CDCl3) δ: 1.21 (m, 1H, CH), 1.40 (m, 2H, CH2), 1.73 (m, 1H, CH), 1.95 (m, 4H, CH2), 2.15 (m, 2H, CH2), 2.41 (s, 3H, CH3), 4.01 (m, 1H, CH–N), 7.57 (s, 1H, CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 13C-NMR (CDCl3) δ: 10.3 (
N). 13C-NMR (CDCl3) δ: 10.3 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 24.9 (cyclohexyl-H2), 25.1 (cyclohexyl-H2), 31.2 (cyclohexyl-H2), 76.1 (cyclohexyl-H), 112.5 (furoxanyl-N→O), 119.8 (nitrone-HN→O), 150.6 (furoxanyl-N). ESI-MS, m/z: 248 (M+˙ + Na), 226 (M+˙ + H), 144, 100.
H3), 24.9 (cyclohexyl-H2), 25.1 (cyclohexyl-H2), 31.2 (cyclohexyl-H2), 76.1 (cyclohexyl-H), 112.5 (furoxanyl-N→O), 119.8 (nitrone-HN→O), 150.6 (furoxanyl-N). ESI-MS, m/z: 248 (M+˙ + Na), 226 (M+˙ + H), 144, 100.
α(Z)-(1,4-Dioxide-2,3-dimethylquinoxalin-6-yl)-N-t-butylnitrone (2). Yellow solid (35%); mp 209.4–212.3 °C. Found: C, 62.0; H, 6.7; N, 14.3. C15H19N3O3 requires C, 62.3; H, 6.6; N, 14.5. 1H-NMR (CDCl3) δ: 1.67 (s, 9H, (CH3)3C), 2.75 (s, 6H, CH3), 7.85 (s, 1H, quinoxalinyl-C5H), 8.62 (d, 1H, J = 8.8 Hz, quinoxalinyl-C7H), 9.03 (d, 1H, J = 8.8 Hz, quinoxalinyl-C8H), 9.13 (s, 1H, CH
N). 13C-NMR (CDCl3) δ: 14.7 (H3-quinoxalinyl), 28.4 ((H3)3C), 72.3 ((CH3)3), 119.7 (nitrone-HN→O), 120.4 (quinoxalinyl-N→O), 128.2 (quinoxalinyl 7-C), 130.5 (quinoxalinyl 6-C), 130.6 (quinoxalinyl 5-C), 133.9 (quinoxalinyl 8-C), 136.3 (quinoxalinyl quaternary-C), 141.6 (quinoxalinyl quaternary-C). EI-MS m/z (%): 289 (M+˙, 83), 273 (21), 257 (4), 233 (100), 216 (86), 199 (33).
α(Z)-(1,4-Dioxidequinoxalin-2-yl)-N-t-butylnitrone (3). Yellow solid (95%); mp 191.8–193.5 °C. Found: C, 59.6; H, 5.9; N, 15.8. C10H15N3O3 requires C, 59.8; H, 5.8; N, 16.1. 1H-NMR (CDCl3) δ: 1.67 (s, 9H, (CH3)3C), 7.86 (m, 2H, quinoxalinyl-C6H and quinoxalinyl-C7H), 8.61 (m, 2H, quinoxalinyl-C5H and quinoxalinyl-C8H), 8.70 (s, 1H, quinoxalinyl-C3H), 10.30 (s, 1H, CH
N). 13C-NMR (CDCl3) δ: 28.3 ((H3)3C), 74.4 ((CH3)3), 119.8 (nitrone-HN→O), 120.3 (quinoxalinyl-N→O), 120.6 (quinoxalinyl-N→O), 129.9 (quinoxalinyl 6-C and quinoxalinyl 7-C), 131.6 (quinoxalinyl 8-C), 132.2 (quinoxalinyl 5-C), 135.8 (quinoxalinyl quaternary-C), 137.6 (quinoxalinyl quaternary-C). EI-MS m/z (%): 261 (M+˙, 23), 245 (13), 205 (19), 189 (37), 172 (35), 129 (26), 105 (24), 57 (100).
Free radical generation and ESR measurements
Radicals were generated by employing a Fenton type reaction. Aqueous solutions of Fe2+ (1.0 mM of ferrous ammonium sulfate hexahydrate) and hydrogen peroxide (1%) in phosphate buffer (pH 7.4) was loaded into the ESR cell. Thus, for 1-hydroxyethyl, methyl, sulfur trioxide anion, and azidyl radicals, we employed as generation source the following reactants: anhydrous ethanol (200 mM), anhydrous dimethylsulfoxide (200 mM), anhydrous sodium sulfite (200 mM), and anhydrous sodium azide (200 mM), respectively. POBN (50 mM) and DMPO (50 mM) were used as a control to check the generation of 1-hydroxyethyl, hydroxyl, and superoxide free radical, respectively.16The xanthine/xanthine oxidase couple was used for superoxide anion radical, and the detection procedure was modified from literature17 with the following conditions: the assayed nitrone (50 mM) was incubated with xanthine (0.4 mM) and xanthine oxidase (0.06 units per mL) in phosphate-buffered saline (PBS, 100 mM, pH 7.4, containing 1.0 mM DTPA). In order to verify the proper generation of superoxide anion free radical, DMPO was used as spin trap control.
The following procedure was used for nitric oxide radical generation: MDG (50 mM),18 Fe2+ (10 mM), NaNO2 (100 mM), and the corresponding nitrone (50 mM). In order to verify the proper generation of nitric oxide free radical, MGD–Fe2+ complex was used as spin trap control.
Bruker ECS 106 ESR (X-band) spectrometer equipped with a rectangular cavity was used to record the ESR spectra at room temperature. The spectrometer conditions were microwave frequency, 9.80 GHz; modulation frequency, 50 kHz; microwave power, 20 mW; modulation amplitude, 0.9 G; time constant, 81.92 ms; the number of scans, 12.
ESR simulation
The NIEHS WinSim software was used for the ESR spectra simulation.19 The simulations of the spectra give us a systematic study based on the dependencies of certain spectral features with some magnetic parameters and accurate parameter extraction from obtained experimental data.Rate constant and half-life estimation for ˙nitrone–OH adduct
The measure of the decay constant for the nitrone–hydroxyl adduct used the same procedure described above for the generation of hydroxyl radical via Fenton reaction. Following the decrease of an appropriate ESR line was used to monitoring the decay of the spin adduct.20,21 Pseudo-first-order decay of the spin adduct was measured by continuously recording the ESR signal intensity. Although the decay of a spin adduct should follow second-order kinetics, under the experimental conditions used in the present work, it can be treated as pseudo-first-order due to the spin trap concentration used. This was in a significant excess compared to the radical concentration and making us consider the concentration of radical trapping constant throughout the experiment. The only variable that changes through the duration of the experiment was the concentration of the generated free radical species. The decay rate was also calculated from the double integral of the ESR spectra. Therefore, it may differ from the rate calculated from the concentration. The relative spin trapping rate for hydroxyl radical was determined using a competitive-trapping method.21 In this method, two spin traps are mixed (DMPO and nitrones studied), and the ESR signal intensities are compared using the value of the double integral of the recorded spectra. The reaction scheme for spin trapping in the competition method for the hydroxyl radical with both spin traps compounds is:


First, we assumed first-order rates for the formation of the two spin adducts, and the intensities found can be related as follows:

Spin trapping in biological systems studies
The spin trap capability of the new nitrones was assessed in mouse mammary adenocarcinoma TA3 cell line exposed to 5-nitroindazole, a free radical producer agent. ESR spectra were obtained using the following conditions: studied nitrones (50 mM), mouse mammary adenocarcinoma TA3 cells (4.0 mg mL−1), and 5-nitroindazole (25 mM), in phosphate buffer, pH 7.4. The mixture was transferred to a 100 μL capillary. A Bruker ECS 106 spectrometer (rectangular cavity and 50 kHz field modulation) was used to record ESR spectra at the X band (9.80 GHz). After 12 scans, all the spectra were registered on the same scale.23Computational calculations
In order to explain results obtained in the trapping of the other radicals using these new heteroaryl nitrones, we used Density Functional Theory (DFT)24,25 calculations using Jaguar software.26 We used the B3LYP/6-31++G** level for optimization of the geometries and to obtain the spin density isosurfaces. Solvent effects were investigated using the polarizable continuum model (PCM).27–29Results and discussion
The previous results with the furoxanyl nitrone FxBN (Fig. 1C) conduct us to use it as a basic template to produce new analogs with modified physicochemical properties. In this sense, in order to study the stability of the generated spin adduct, we designed and synthesized nitrone 1 (Fig. 2), where FxBN t-butyl moiety was substituted by a cyclohexyl group. On the other hand, due to FxBN possesses a cyclic nitrone group in the furoxanyl ring, we aimed to analyze the effect of other nitrone containing cyclic N-oxides. To do this, we selected the well-known quinoxaline 1,4-dioxide heterocycle, which involves two cyclic-nitrone moieties, and additionally, we selected two different positions for the non-cyclic extra nitrone (nitrone 2, and nitrone 3, Fig. 2). The different non-cyclic nitrone positions and substituents on the quinoxaline nucleus could modify the physicochemical properties of these compounds.The new nitrones were synthesized through the condensation between heteroaromatic aldehydes and N-t-butylhydroxylamine or N-cyclohexylhydroxylamine with moderate to excellent yields (Scheme 1).30
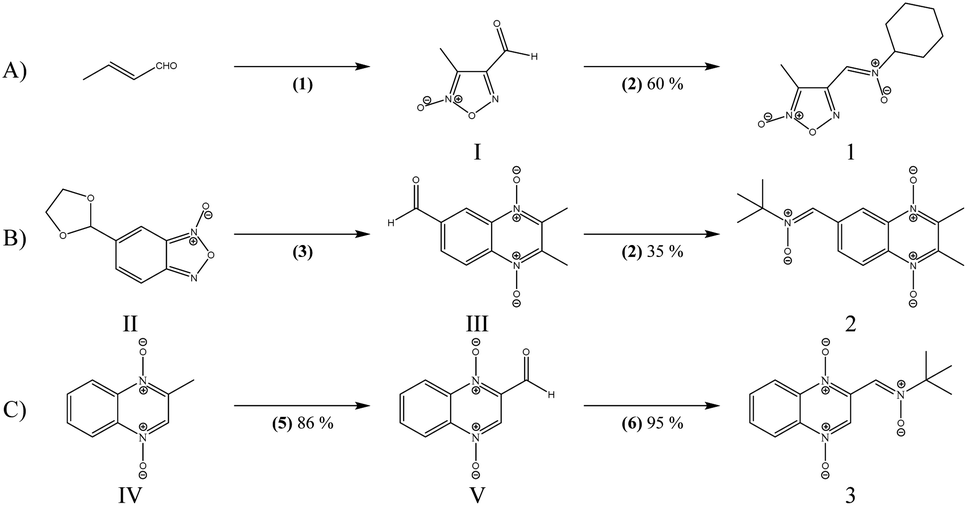 | ||
| Scheme 1 Synthesis of heteroarylnitrones 1–3. Experimental conditions are: (A) (1) NaNO2/AcOH, (2) C6H11–NHOH·HCl/NaHCO3/EtOH/60 °C. (B) (3) (i) butanone/NH3 (g), (ii) HCl (c), (4) (CH3)3–NHOH·AcOH/NaHCO3/EtOH/60 °C. (C) (5) SeO2/dioxane/H2O/reflux, (6) (CH3)3C–NHOH·HAc/NaHCO3/EtOH/60 °C. | ||
The formylfuroxan I was obtained from crotonaldehyde through reaction with NaNO2 in AcOH by intramolecular cyclization (Scheme 1A).30 We employed the synthetic route showed in Scheme 1B to obtain the desired formylquinoxaline 1,4-dioxide III. Beirut reaction of bezofuroxan II with butanone in the presence of NH3 (g) gave the compound III in good yield.31 Using the Beirut reaction, the 2-methylquinoxaline 1,4-di-N-oxide intermediate IV (Scheme 1C) was synthesized, from benzofuroxan reacting with acetone in the presence of pyrrolidine.32 Then we proceeded to the oxidation of the methyl group in position 2 of the quinoxaline ring, using SeO2 as the oxidant agent in order to obtain the carboxaldehyde derivative V. Finally, the desired heteroaryl nitrones, 1–3, were obtained for the reaction between the corresponding aldehydes I, III, V and N-substituted hydroxylamine in the presence of NaHCO3 (Scheme 1).
Spin trapping assays
Firstly, the spin trapping properties of these new nitrones were investigated against different centered free radicals (O, C, N, and S atoms, respectively). The radicals generated were hydroxyl, 1-hydroxyethyl, azidyl, and sulfur trioxide anion. All radicals mentioned above were generated through Fenton's reagents, using aqueous solutions of Fe2+ and hydrogen peroxide (1%), ethanol, sodium azide, or sodium sulfite, respectively.Each of the nitrones tested gave an ESR-detectable spin adduct with other radicals (Fig. 3A and B).
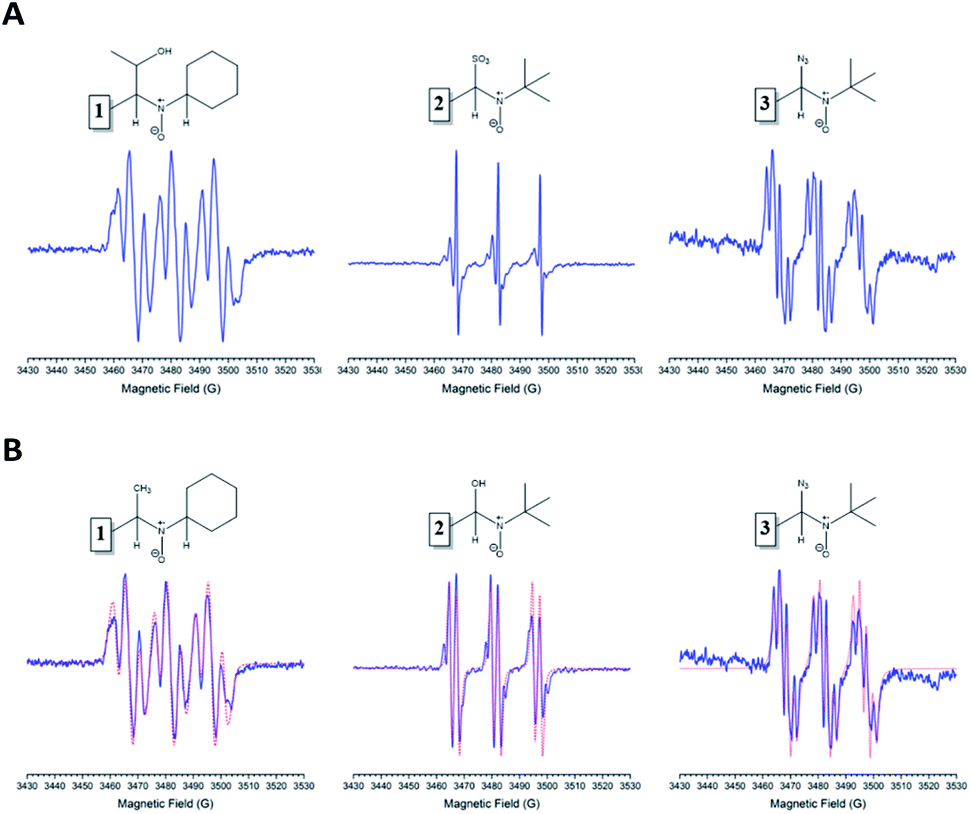 | ||
| Fig. 3 (A) The left spectrum corresponds to the trap of 1-hydroxyethyl radical, nitrone 1 was used as a spin trap. The center spectrum showed the hyperfine splitting pattern for the trap of sulfur trioxide anion radical when nitrone 2 was used. It is possible to observe some minor artifacts in the spectra. The right spectrum is nitrone 3 trapping azidyl radical. (B) Experimental spectra are shown in blue, and the corresponding simulations are remarked with red color. From left to right, simulations of the trap of methyl, hydroxyl, and azidyl radical with nitrones 1, 2, and 3, respectively. | ||
The obtained spin adducts spectrum was composed as follows: for hydroxyl and 1-hydroxyethyl radicals, the pattern obtained was composed of six-line (triplet of doublet) due to coupling of the unpaired electron with nitrogen and hydrogen at α-carbon, except for nitrone 1, which the obtained pattern was composed by nine lines (triplet of triplet, Fig. 3A) probably due to the extra hydrogen located in the cyclohexyl moiety. In order to explain this different behavior, we proceeded to perform the corresponding computational calculations using DFT level, explaining the hyperfine pattern found. From the analysis of the spectrum, it was obtained that a second H atom with a smaller hyperfine coupling constant would explain the observed hyperfine pattern. The spin density reveals that this is delocalized to a part of the cyclohexyl group. Fig. 4 shows the atoms contributing to the hyperfine pattern found, confirming that this substituent group influences the spectrum pattern.
 | ||
| Fig. 4 Spin density isosurface for ˙nitrone 1–OH spin adduct using DFT calculus. The arrows indicate the atoms in which the unpaired electron is delocalized and forms the hyperfine pattern are observed experimentally. | ||
The ESR spectrum when methyl radical was trapped was composed of a triplet of doublet due to coupling with a nitrogen atom (triplet) and β-hydrogen atom (doublet) at the α-carbon atom, except for nitrone 1 (Fig. 3B). The ESR spectrum for nitrone 1 trapping methyl radical was composed again by nine lines. The spin adducts with the azidyl radical exhibit twelve-line (Fig. 3A and B) attributed to a triplet of triplet of doublets. This pattern is explained due to two different nitrogen atoms, one atom from the nitrone group and the other atom from the azidyl radical. To complete the pattern explanation, we considered the contribution of the β-hydrogen atom to this splitting. For nitrone 1, the intensity of the signal is weak, and the spectrum is not well resolved. Finally, the spin trapping properties of these new nitrones were investigated against sulfur-centered free radical produced by sulfur trioxide anion free radical. The spin adducts from these nitrones showed symmetrical patterns in all the cases. The ESR spectrum for nitrone 1 was composed of nine lines (triplet of triplet). For nitrones 2 and 3, the hyperfine pattern is similar, and it can be observed in the value of the hyperfine splitting constant (Fig. 3A). Fig. 3B showed the ESR simulations for all data correlated well with experimental ESR spectra.
Table 1 resumes the hyperfine constants coupling for the different generated spin adducts.
| ˙CH3 |
|
˙N3 | ˙OH | ˙SO3− | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aN | aH | aH | aN | aH | aH | aN | aN | aH | aN | aH | aH | aN | aH | aH | |
| a No signal. | |||||||||||||||
 |
14.7 | 5.0 | 4.1 | 14.7 | 5.1 | 4.0 | 15.0 | 5.5 | 3.9 | 14.8 | 5.1 | 3.9 | 14.6 | 5.2 | 4.2 |
| g value | 2.00925 | 2.00919 | 2.01435 | 2.01119 | 2.01135 | ||||||||||
 |
15.1 | 2.8 | nsa | 15.5 | 3.1 | ns | 14.3 | 2.1 | 1.6 | 15.1 | 2.8 | ns | 15.0 | 1.9 | ns |
| g value | 2.00953 | 2.00702 | 2.00841 | 2.00879 | 2.00885 | ||||||||||
 |
14.4 | 2.2 | ns | 14.4 | 2.1 | ns | 14.4 | 2.3 | 2.5 | 14.4 | 2.2 | ns | 14.5 | 2.0 | ns |
| g value | 2.00829 | 2.00863 | 2.00840 | 2.00846 | 2.00834 | ||||||||||

In the case of nitrones 2 and 3, the nitrogen hyperfine constants between the trapping of the hydroxyl, methyl, 1-hydroxyethyl, and sulfite radical have the same values. For the hydrogen atom, these hyperfine constants showed a slight difference. It can be appreciated that nitrone 1 has very different hyperfine coupling constants compared with nitrones 2 and 3. Among the other radicals trapped, the hyperfine splitting constant showed similar values for nitrogen atom and the same behavior for the two hydrogen atoms (except for azidyl radical).
Determination of spin adduct stability and the decay rate constant for ˙nitrone–OH spin adducts
Regardless of this, all nitrones under study showed a behavior as spin traps. To get insight into the physicochemical properties of ˙nitrone–OH spin adducts, stability, and kinetic studies in PBS (pH = 7.4) were performed. The stability of spin adducts was studied through the monitoring of the disappearance of the ESR signal intensity of ˙nitrone–OH spin adducts (see example in Fig. 5a).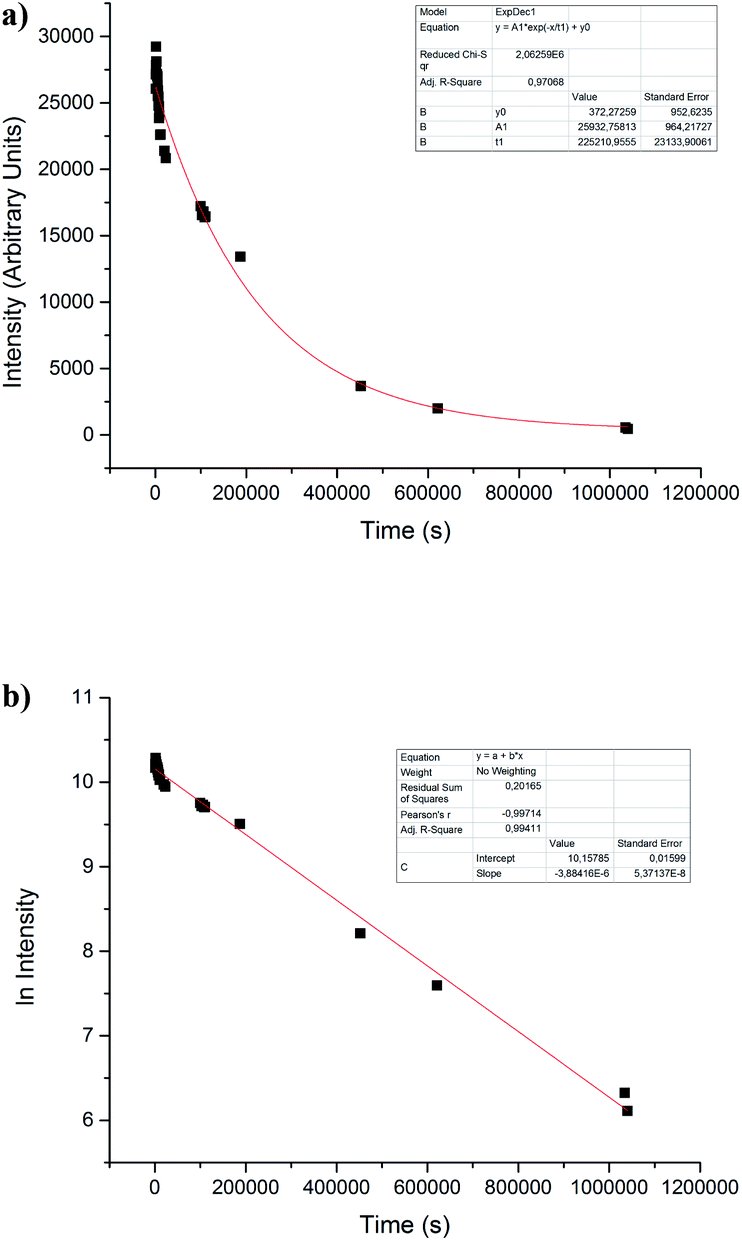 | ||
| Fig. 5 (a) The time course of the ESR signal intensity for ˙nitrone 2–OH adduct, in PBS (pH = 7.4). The decay agrees with a first-order reaction. (b) The time course of the ESR signal intensity is expressed as the natural logarithm of intensity for ˙nitrone 2–OH adduct, confirming a process of first-order decay. | ||
It could be attributed to a solvolysis process as the mechanism of decay.7,33,34 The half-life of ˙nitrone–OH spin adduct was determined, assuming that the decay is of first-order. Both nitrones 2 and 3 showed excellent stability with a t1/2 more than 100-fold higher the value for ˙DMPO–OH adduct3 (Table 2).
| Spin adduct | Half-life (s) |
|---|---|
| ˙DMPO–OH | 3300 |
| ˙PBN–OH | ∼38 |
| ˙FxBN–OH | 7567 |
| ˙Nitrone 1–OH | 3398 |
| ˙Nitrone 2–OH | 8923 |
| ˙Nitrone 3–OH | 7270 |
We also included the values for the previously reported FxBN nitrone15
Until this stage of experiments, we discussed some aspects concerning the structural characteristics of these new nitrones. First, unlike that, it was observed in our previous work furoxanyl substituent did not exert an excellent stabilizing effect in the trapped radicals as it happens with the nitrone 1 (compare t1/2 for ˙FxBN–OH and ˙nitrone 1–OH in Table 2). However, the use of a cyclohexyl substituent gives us the possibility to obtain a hyperfine pattern. Although it was similar in all cases, it was more sensitive to changes in the hyperfine coupling constants compared with t-butyl substituent used in the nitrones 2 and 3. Instead, the usage of a quinoxalinyl substituent conferred an essential aid in stabilizing the other radicals trapped (compare t1/2 for ˙FxBN–OH and ˙nitrone 2–OH in Table 2). Probably this substituent imparts a planar structure that helps the electronic delocalization and the consequent stabilization of the corresponding spin adduct.
Competitive studies for ˙OH-trapping
We determined the ratio of constants through a competitive study, (k2/k1) of hydroxyl radical for the studied nitrones, the previously reported nitrone FxBN and DMPO (Table 3), being for DMPO k1 = 3.4 × 109 dm3 mol−1 s−1.35| Nitrone | kST (dm3 mol−1 s−1) | k2/k1 |
|---|---|---|
| DMPO | 3.4 × 109 | 1.00 |
| FxBN | 1.22 × 1010 | 3.59 |
| Nitrone 1 | 1.17 × 109 | 0.34 |
| Nitrone 2 | 1.70 × 1010 | 5.00 |
| Nitrone 3 | 1.61 × 1010 | 4.74 |
It was possible to observe that nitrone 2 and 3 exhibited the fastest rate in trapping hydroxyl radical when was compared with DMPO and the previously reported FxBN (see example in Fig. 7a and results in Table 3). However, when the same kind of competitive experiment was performed with nitrone 1, a slower trapping process compared to DMPO occurred (Fig. 6b and Table 3).
 | ||
| Fig. 6 ESR spectra obtained after the generation of hydroxyl radical using Fenton reaction, ˙DMPO–OH spin adduct, was marked with red dots. (a) Nitrone 3 competitive spectrum obtained versus DMPO trapping hydroxyl radical. (b) Nitrone 1 competitive spectrum obtained versus DMPO for the same radical mentioned above. | ||
It could be correlated with the different structural moieties with the different reactivity and stability between these nitrones36 claiming the relevance of the quinoxaline 1,4-dioxide substructure on nitrone 2 and 3 in the spin adduct stabilization.
Trapping of nitric oxide and superoxide anion radicals
Nitric oxide (NO) is a signaling and regulatory molecule in many physiological and pathological processes.37 Several attempts have been performed to detect NO in aqueous solutions and biological systems by using spin trapping methodology.38 However, the traditional nitrone spin traps such as PBN and DMPO appeared to be unsuitable as they did not form stable and characteristic spin adducts with NO.38 These limitations suggest the need to develop new spin trapping agents, which can specifically trap NO generated under physiological conditions. In this context, the ability of the newly synthesized nitrones to trap the nitric oxide radical was studied. Thus, we performed the same test with the FxBN nitrone, in which it was clear that it can trap the ˙NO2 radical (Fig. 7). | ||
| Fig. 7 Hyperfine patterns for the trapping of nitrogen dioxide radical (˙NO2). (a) Nitrone 2 spectrum of ˙NO2 radical trapping process, in blue experimental results and in red the corresponding simulation. (b) The spectrum for FxBN trapping ˙NO2 radical process, in blue experimental results and in red the corresponding simulation. | ||
The analysis of the intensities of the lines by double integration allows us to point out that the nitrone 2 is better compared to FxBN.
The spectra obtained were composed of six lines (triplet of doublet) for nitrones 2 and 3. FxBN nitrone exhibited the same spectrum as described above. Because the obtained pattern was not than expected, computational calculations were performed in order to determine the trapped species. Structures were optimized for nitrones 2 and 3, simulating the trapping of NO and ˙NO2 free radicals. If the nitrones trap NO free radical (Fig. 8 for nitrone 2), according to the calculus, it should be observed a hyperfine pattern composed by two nitrogen atoms, one from the trapped radical and the other from the nitrone group, with no signal being observed for the β-position hydrogen (Table 4).
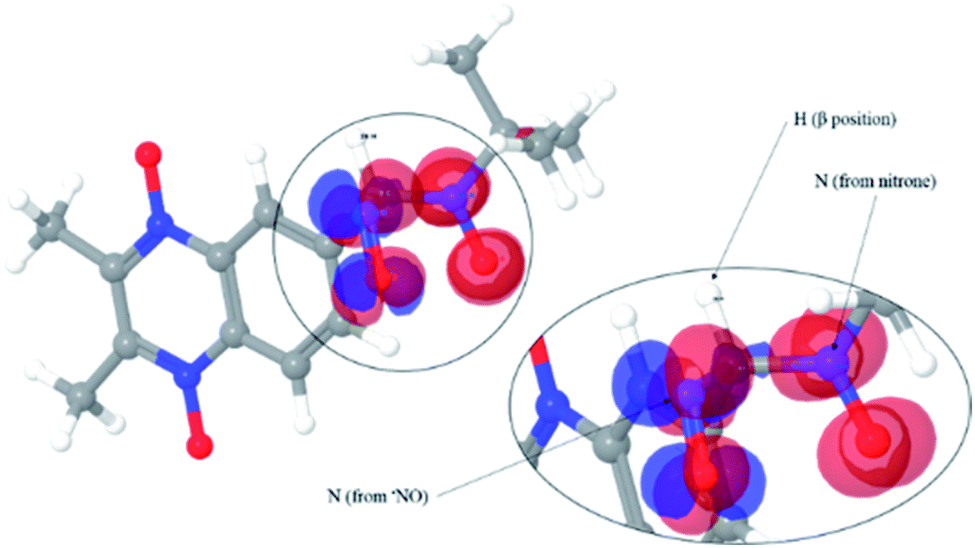 | ||
| Fig. 8 Spin density isosurface for ˙nitrone 2–NO spin adduct using DFT. | ||
| ˙NO2 | ||
|---|---|---|
| aN | aH | |
| Nitrone 1 | ns | ns |
| Nitrone 2 | 15.0 | 2.6 |
| Nitrone 3 | 15.0 | 2.0 |
| FxBN | 15.0 | 2.3 |
The isosurface showed that the spin density is mainly located on each of the mentioned nitrogen atoms.
When we performed the same analysis using ˙NO2 as trapped free radical, it was observed that the spin density was primarily located between the nitrogen and hydrogen in the β-position of the nitrone group (Fig. 9).
 | ||
| Fig. 9 Spin density isosurface for ˙nitrone 2–NO2 spin adduct using DFT calculus. In this model, we could corroborate that the nitrones were not capable of trapping nitric oxide radical, and the experimental spectra corresponded to the trap of nitrogen dioxide radical. | ||
This result showed that the observed spectra would correspond to a triplet of doublet as observed in experimental spectra (Fig. 7) with some minor influences from the nitrogen atom from the nitrogen dioxide radical. Therefore, under the conditions in which was generated the NO radical, these nitrones are not capable of trapping the NO free radical in contrast to that occurs when using the complex MGD–Fe2+ as spin traps.
For the generation of superoxide anion radical was used as the system xanthine/xanthine oxidase. The results showed that the nitrone 1 was not able to trap this radical. Instead, the nitrones 2 and 3 showed similar hyperfine patterns for this radical trapping. Fig. 10 shows the experimental spectra of the hyperfine patterns displayed by nitrones 2 and 3. Hyperfine coupling constants for nitrones 2 and 3 are summarized in Table 5.
 | ||
| Fig. 10 ESR spectra for the ˙nitrone–OOH adducts were obtained with nitrone 2 (left) and nitrone 3 (right). | ||
| ˙O2− | |||
|---|---|---|---|
| aN | aH | aH | |
| Nitrone 1 | ns | ns | ns |
| Nitrone 2 | 15.3 | 2.9 | ns |
| Nitrone 3 | 15.3 | 2.0 | ns |
| FxBN | 15.6 | 1.9 | 1.2 |
In order to corroborate the obtained spectra, we made the computational calculations for the ˙nitrone–OOH spin adduct for nitrone 2 and 3. In Fig. 11, it is observed that the spin densities were located mainly between the nitrogen atom and the hydrogen atom located in β-position from the nitrone group.
 | ||
| Fig. 11 Spin density isosurface for ˙nitrone 2–OOH spin adducts using DFT calculus. The experimental spectra correspond to a delocalization of the spin density over the atoms indicated with arrows. | ||
In this sense, the theoretical results confirm that the experimental spectra correspond to the trapping of the superoxide anion radical.
Spin trapping in biological systems studies
As it was described in our previous work,15 the new nitrones were tested in biological models to corroborate their trapping capacity. We used mouse mammary adenocarcinoma TA3 cells and a nitroindazole (NI, 5-nitroindazole) derivative. The main characteristic of this nitro derivative is its capability to be a substrate of nitroreductases and undergoing a reduction in the presence of oxygen by one or two-electron transfer mechanism.39 The mono electronic reductions catalyzed by nitroreductases in the presence of molecular oxygen generate superoxide anion radical, and the nitro compounds are regenerated to their initial state. This redox cycle can cause oxidative stress due to the production of superoxide anion radical. Finally, the superoxide anion radical decays to hydroxyl radical, which may trap by using spin traps. Our results showed that nitrone 1 was not able to trap any free radicals in this model. Instead, nitrones 2 and 3 showed sharp spectra (Fig. 12). | ||
| Fig. 12 (Left) The ESR-spectrum obtained in the absence of NI. Center: the ESR-spectrum obtained when nitrone 2 traps exclusively hydroxyl free radical produced by NI in the presence of mouse mammary adenocarcinoma TA3 cells. (Right) The ESR-spectrum obtained when FxBN was used in the same biological system. Both spectra were presented in the same conditions. | ||
The analysis of the hyperfine coupling constant points out that the radical trapped corresponds to hydroxyl generated by the NI. Additionally, as Fig. 12 shows, nitrone 2 was more sensitive to hydroxyl radical trapping in this biological model than FxBN.
Conclusions
New heteroaryl nitrones (1, 2, and 3) were designed, synthesized and analyzed for their ability to act as spin-trapping agents. These heteroaryl nitrones exhibit a high capacity to trap and stabilize O-, C-, S-, and N-centered radicals. Nitrone 1 showed different ESR-spectra, and the hyperfine coupling constant allows us to distinguish the different radicals trapped easily, but with lower half-life compared to nitrone 2 and 3. On the other hand, nitrone 2 and 3 showed better spin traps capabilities due to the high stability of the spin adduct formed. In some cases, it was not so easy to distinguish the radical trapped using nitrones 2 and 3 (specifically hydroxyl and methyl radicals), but the g value helped us to differentiate the trapped species. We found that these new nitrones were capable of trapping superoxide anion radical generated using xanthine/xanthine oxidase couple. Thus, we performed the trap of nitric oxide radical, in which the results obtained showed that these new nitrones are not capable of trapping this radical, but they can trap nitrogen dioxide radicals. In aqueous solution, we found that nitrones 2 and 3 can form a spin adduct with hydroxyl radical. The half-life for these spins adducts shows a value higher than two hours. In the competition assays, the results indicated that these new nitrones (2 and 3) are more sensitive than DMPO to trap hydroxyl free radicals. Computational calculations supported the experimental data and allowed us to explain the different phenomena observed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Germán Barriga-González thanks financial support by FONDECYT grant no. 3120241 and UMCE APIX 2019 (second semester). Germán Barriga González also thanks to Patricio Canales Volpone from Unidad Segundo Idioma at UMCE for correcting the manuscript. Carolina Aliaga thanks CEDENNA AFB180001 project. Eduardo Chamorro thanks financial support by FONDECYT no. 1100277 and UNAB NUCLEO DI-219-12/N. Eduardo Chamorro acknowledges the continuous support provided by Fondo Nacional de Ciencia y Tecnología (FONDECYT-Chile) through Project No. 1181582. We also thank Cecilia Chavarria and Marcos Nieves for their collaboration in the synthetic procedures.Notes and references
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS.
- J. M. Gutteridge and B. Halliwell, Ann. N. Y. Acad. Sci., 2000, 899, 136–147 CrossRef CAS.
- F. A. Villamena and J. L. Zweier, Antioxid. Redox Signaling, 2004, 6, 619–629 CrossRef CAS.
- C. Olea-Azar, C. Rigol, F. Mendizabal and R. Briones, Mini-Rev. Med. Chem., 2006, 6, 211–220 CrossRef CAS.
- E. G. Janzen and J. I. P. Liu, J. Magn. Reson., 1969, 9, 510–512 Search PubMed.
- G. S. Timmins, K. J. Liu, E. J. Bechara, Y. Kotake and H. M. Swartz, Free Radical Biol. Med., 1999, 27, 329–333 CrossRef CAS.
- Y. Kotake and E. G. Janzen, J. Am. Chem. Soc., 1991, 113, 9503–9506 CrossRef CAS.
- R. A. Floyd, K. Hensley, M. J. Forster, J. A. Kelleher-Andersson and P. L. Wood, Mech. Ageing Dev., 2002, 123, 1021–1031 CrossRef CAS.
- G. Olive, A. Mercier, F. Le Moigne, A. Rockenbauer and P. Tordo, Free Radicals Biol. Med., 2000, 28, 403–408 CrossRef CAS.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 CrossRef CAS.
- S. E. Bottle and A. S. Micallef, Org. Biomol. Chem., 2003, 1, 2581–2584 RSC.
- S. E. Bottle, G. R. Hanson and A. S. Micallef, Org. Biomol. Chem., 2003, 1, 2585–2589 RSC.
- B. Hatano, H. Sato, T. Ito and T. Ogata, Synlett, 2007, 2007, 2130–2132 CrossRef.
- B. Hatano, K. Miyoshi, H. Sato, T. Ito, T. Ogata and T. Kijima, Tetrahedron Lett., 2010, 51, 5399–5401 CrossRef CAS.
- G. Barriga, C. Olea-Azar, E. Norambuena, A. Castro, W. Porcal, A. Gerpe, M. González and H. Cerecetto, Bioorg. Med. Chem., 2010, 18, 795–802 CrossRef CAS.
- K. Sakurai, D. A. Stoyanovsky, Y. Fujimoto and A. I. Cederbaum, Free Radical Biol. Med., 2000, 28, 273–280 CrossRef CAS.
- K. Stolze, N. Udilova and H. Nohl, Free Radicals Biol. Med., 2000, 29, 1005–1014 CrossRef CAS.
- L. A. Shinobu, S. G. Jones and M. M. Jones, Acta Pharmacol. Toxicol., 1984, 54, 189–194 CrossRef CAS.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef CAS.
- M. Kamibayashi, S. Oowada, H. Kameda, T. Okada, O. Inanami, S. Ohta, T. Ozawa, K. Makino and Y. Kotake, Free Radical Res., 2006, 40, 1166–1172 CrossRef CAS.
- Y. Sueishi, C. Yoshioka, C. Olea-Azar, L. A. Reinke and Y. Kotake, Bull. Chem. Soc. Jpn., 2002, 75, 2043–2047 CrossRef CAS.
- E. Finkelstein, G. M. Rosen and E. J. Rauckman, J. Am. Chem. Soc., 1980, 102, 4994–4999 CrossRef CAS.
- C. Olea-Azar, C. Rigol, L. Opazo, A. Morello, J. D. Maya, Y. Repetto, G. Aguirre, H. Cerecetto, R. Di Maio, M. González and W. Porca, J. Chil. Chem. Soc., 2003, 48, 77–79 CAS.
- Density functional methods in chemistry, ed. J. K. Labanowski and J. W. Andzelm, Springer-Verlag New York, Inc., 1991 Search PubMed.
- J.-L. Calais, Int. J. Quantum Chem., 1993, 47, 101 CrossRef.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- W. Porcal, P. Hernandez, M. Gonzalez, A. Ferreira, C. Olea-Azar, H. Cerecetto and A. Castro, J. Med. Chem., 2008, 51, 6150–6159 CrossRef CAS.
- H. Cerecetto, E. Dias, R. Di Maio, M. Gonzalez, S. Pacce, P. Saenz, G. Seoane, L. Suescun, A. Mombru, G. Fernandez, M. Lema and J. Villalba, J. Agric. Food Chem., 2000, 48, 2995–3002 CrossRef CAS.
- C. H. Issidorides and M. J. Haddadin, Novel process for the synthesis of quinoxaline and benzimidazole-N-oxides, US Pat. US4866175A, 1989.
- P. R. Marriott, M. J. Perkins and D. Griller, Can. J. Chem., 1980, 58, 803–807 CrossRef CAS.
- A. J. Carmichael, K. Makino and P. Riesz, Radiat. Res., 1984, 100, 222–234 CrossRef CAS.
- R. Sridhar, P. Beaumont and E. Powers, J. Radioanal. Nucl. Chem., 1986, 101, 227–237 CrossRef CAS.
- S. Goldstein and P. Lestage, Curr. Med. Chem., 2000, 7, 1255–1267 CrossRef CAS.
- S. Pou, L. Keaton, W. Surichamorn, P. Frigillana and G. M. Rosen, Biochim. Biophys. Acta, 1994, 1201, 118–124 CrossRef CAS.
- K. J. Reszka, P. Bilski and C. F. Chignell, Nitric Oxide, 2004, 10, 53–59 CrossRef CAS.
- D. W. Bryant, D. R. McCalla, M. Leeksma and P. Laneuville, Can. J. Microbiol., 1981, 27, 81–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07720h |
| This journal is © The Royal Society of Chemistry 2020 |