
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCircular linkage of intramolecular multi-hydrogen bonding frameworks through nucleophilic substitutions of β-dicarbonyls onto cyanuric chloride and subsequent tautomerisation†
Ayano Awatani and
Masaaki Suzuki *
*
Graduate School of Natural Science and Technology, Shimane University, 1060 Nishikawatsu-cho, Matsue, Shimane 690-8504, Japan. E-mail: m-suzuki@riko.shimane-u.ac.jp
First published on 23rd October 2020
Abstract
Nucleophilic substitution reactions of cyanuric chloride with a series of β-dicarbonyls give triply β-dicarbonyl-embedded 1,3,5-triazines. Their subsequent but spontaneous tautomeric transformation leads to circularly linked, intramolecular, multi-hydrogen bonding networks. Their structural elucidation by X-ray crystallography showed elongated double bonds and shortened single bonds. This is likely due to a resonance hybrid formed via tautomerisation and simultaneous proton transfer.
Intramolecular hydrogen bonding contributes to various chemical phenomena, such as the stabilisation of molecules1 and/or three-dimensional arrangements of molecular frameworks.2 Normally, hydrogen bonding units are formed between heteroatomic functionalities, including alcohols, ketones, amines, imines, and halogens. When these are incorporated into a conjugated network, proton delivery alongside resonance, i.e. excited-state intramolecular proton transfer (ESIPT), can occur.3 Among numerous examples, an excited-state double-proton transfer (EDIPT) was reported by Chou et al.,4a,b where an ingenious arrangement of serial hydrogen bonding occurred in 1,8-dihydroxy-2-naphthaldehyde (DHNA, Chart 1a) that promoted a remarkably large Stokes shift. Adachi et al.4c also developed a triquinolonobenzene (TQB) system that could potentially exhibit a characteristic triple-ESIPT; however, TQB could not cause the third ESIPT (Chart 1b). This was likely due to high barriers to breaking the inherent stability of the Clar structure and amine-ketone skeletons of TQB.
 | ||
| Chart 1 Selected examples demonstrating intramolecular proton transfers: (a) 1,8-dihydroxy-2-naphthaldehyde (DHNA) and its excited-state double proton transfer (ESDPT), (b) triquinolonobenzene, and (c) tautomerism of 2,4,6-tris(β-monocarbonyl)-1,3,5-triazine (TMCT). | ||
Aromaticity is one of the best properties that stabilises molecules and is achieved via the delocalisation of π-electrons, as long as they satisfy Hückel's rule. Whenever possible, organic compounds will adopt aromatic skeletons, including non-classical Möbius systems, that are suitable for designated factors by undergoing protonation/deprotonation,5 macrocyclic distortion,6 or many other perturbations. There are several other mechanisms besides aromaticity that provide stability. One example is the amide–iminol tautomerism7 of cyanuric acid, whose original 2,4,6-triol form undergoes tautomerization to give isocyanuric acid, which adopts a 2,4,6-trione structure. The original structure is aromatic due to the 1,3,5-triazine core; however, an amide is more stable than an iminol. Therefore, both tautomers contribute to the cyanuric acid structure. These observations inspired us to create an organic molecular scaffold that could overcome aromatic stabilisation by other mechanisms.
An intramolecular hydrogen bonding framework would serve as a nice platform to realise our aforementioned idea, and we focused on the tautomerism of cyanuric acid. This process generated NH protons that could serve as hydrogen-bonding donors. When such a hydrogen-bonding donor is surrounded by acceptors, an intramolecular, multi-hydrogen bonding entity can be formed. A pioneering work that attempted to fabricate such a bonding entity focused on resonance-assisted hydrogen bonding (RAHB) systems.8 Liebscher and co-workers8b synthesised 2,4,6-tris(β-monocarbonyl)-1,3,5-triazine (TMCT) by the stepwise extension of the benzyl positions of 2,4,6-trialkyl-1,3,5-triazine and demonstrated the subsequent tautomerism into the corresponding 2,4,6-triylidene structures (Chart 1c). However, there were evident contributions from aromatic 1,3,5-triazine forms based on the broadened NH signals in their proton nuclear magnetic resonance (1H NMR) spectra. The design of our target molecule (B, Chart 2) began with cyanuric chloride (1) undergoing nucleophilic substitution with three β-dicarbonyls (i), which led to the desired triply conjugated 1,3,5-triazine derivative (A). Automatic tautomerization (ii) formed a circular array of multi-hydrogen bonding interactions amongst the molecular periphery to give B.9 As a result, the acceptors would be connected to the core by double bonds. Such a macrocyclic arrangement of multidentate RAHB arrays is the major difference between our compound and the reported TMCT systems, which possess independent monodentate RAHB sites.8b Compound B has been already reported, but the characterisation was limited by several, insufficient spectroscopic predictions (the NMR spectrum only showed its alkoxy protons).10 We predicted that proton transfer from the nitrogen atoms to the adjacent oxygen atoms (iii) would spontaneously occur without any stimuli, such as photoirradiation, owing to the decreased energy barrier that resulted from the directly conjugated hydrogen-bonding donor–acceptor assembly. A possible resonance form (C) was also depicted. The final goal of this project is to create highly delocalised, electronic platforms comparable to conventional carbon-rich materials.
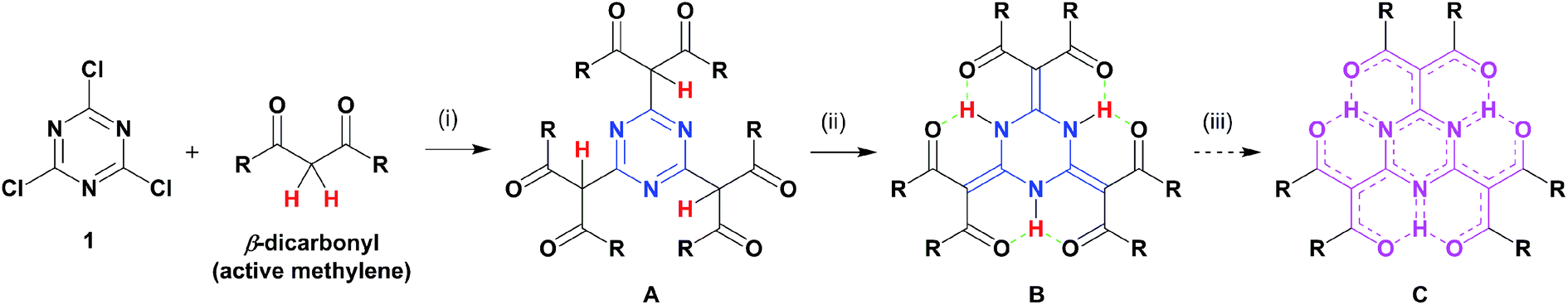 | ||
| Chart 2 Concept of this work: (i) nucleophilic substitution, (ii) tautomerism, and (iii) resonance. | ||
Several β-dicarbonyls (2) were tested for their performance in nucleophilic substitution reactions with 1.9,11 Diisopropylethylamine (DIEA) was chosen as the organic, non-nucleophilic base because its moderate boiling point would enable easy removal under reduced pressure (Scheme 1). DIEA and 2 were used in excess to avoid tedious chromatographic separation of the highly polar mono- and di-conjugates. The reaction employing Meldrum's acid (2a) gave a pale-yellow crystalline solid that was identified as 3a.
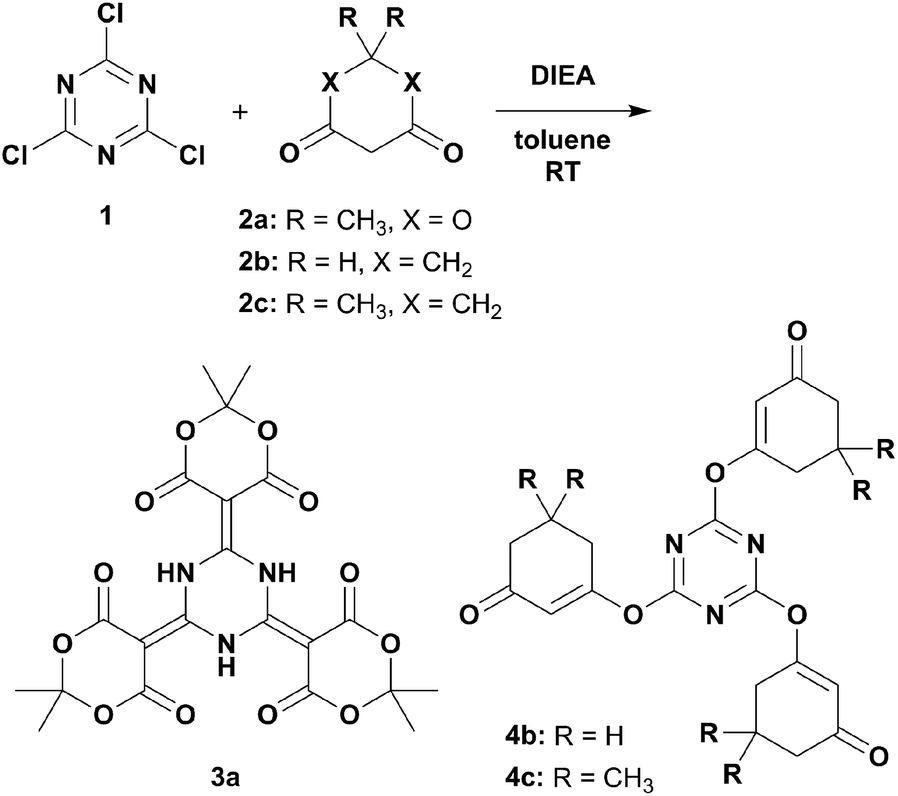 | ||
| Scheme 1 Nucleophilic substitution reactions of β-dicarbonyls onto cyanuric acid with diisopropylethylamine in toluene at room temperature. | ||
The 1H NMR spectrum of 3a (Fig. S1, ESI†) exhibited a pair of signals in the aliphatic region at 1.77 ppm and in a significantly downfield region at 15.13 ppm. These likely represented the methyl (Hb) and NH (Ha) protons, respectively, which reflects the highly symmetric structure of 3a. The deshielded NH signal suggests that the expected O⋯(N)H⋯O bidentate hydrogen bonding was occurring. This is supported by the signals of O⋯(N)H monodentate hydrogen bonding in not only TMCT systems, which exhibited their NH signals at 13.40–13.90 ppm,8a but also those in related mono-conjugates at ca.13.50 ppm.11d The 13C NMR spectrum of 3a (Fig. S2†) displays two aliphatic signals (C4 and C5) at 27.08 and 79.03 ppm, respectively, as well as two olefinic (C1 and C4) signals at 105.27 and 149.76 ppm, respectively. A carbonyl (C3) signal also appeared at 163.45 ppm. These signals further affirmed that the desired structure was successfully fabricated.
The final structures of 3a were elucidated by X-ray diffraction analysis (Fig. 1a and S21†), which revealed spectroscopic assignments that agreed with the expected molecular design. The N–O1 distances were 2.623(2)–2.680(3) Å, which indicated that certain hydrogen bonding interactions existed. The N–C1, C1–C2, C2–C3, and C3–O1 distances were 1.350(3)–1.369(4), 1.387(4)–1.392(3), 1.445(4)–1.464(5), and 1.209(3)–1.218(4) Å, respectively, which were estimated to be intermediate lengths of the alternating single and double C–C, C–N, and C–O bonds.12
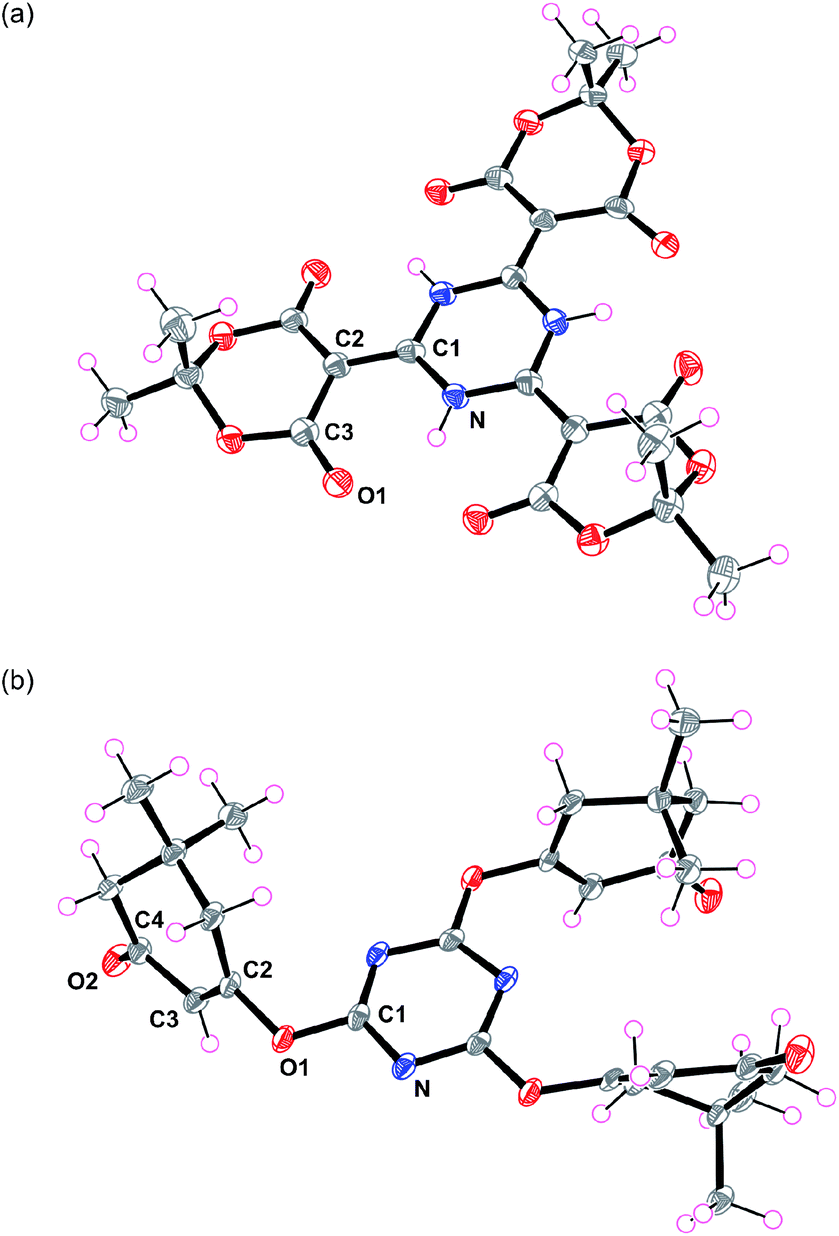 | ||
| Fig. 1 X-ray crystal structures of (a) 3a and (b) 4c. Thermal ellipsoids are set at 50% probability levels. | ||
On the other hand, when 1,3-cyclohexanedione (2b) was used as the β-dicarbonyl, the O-linked product 4b was obtained (Scheme 1). The 1H NMR spectrum of 4b (Fig. S9†) provided four signals in the ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1, where three signals (Hb, Hc, and Hd) were located in the aliphatic region and one signal (Ha) appeared in the olefinic region. The 13C NMR spectrum (Fig. S10†) showed seven signals: three aliphatic (C5, C6, and C7), two olefinic (C2, C3), one aromatic (C1), and one carbonyl (C4). Dimedone (5,5-dimethyl-1,3-cyclohexanedione, 2c) gave results similar to those of 2b. The crystal structure of 4c (Fig. 1b) revealed its authentic 1,3,5-triazine core with N–C1 distances of 1.322(2)–1.333(2) Å, as well as N–C1–N and C1–N–C1 bond angles of 127.5(2)–128.4(2) and 111.4(2)–112.2(2)°, respectively.13 Evident enol–enone moieties were also observed with the following bond distances: C1–O1, 1.342(2)–1.357(2); O1–C2, 1.410(3)–1.416(2); C2–C3, 1.309(3)–1.332(3); C3–C4, 1.463(3)–1.477(3); and C4–O2 1.214(3)–1.228(3) Å. No related condensation reactions occurred when acyclic β-dicarbonyls, such as acetylacetone (R = CH3), dibenzoylmethane (R = Ph), diethyl malonate (R = OEt), and hexafluoroacetylacetone (R = CF3), were used. These observations suggested that 2a may have several favourable factors contributing to its reaction with cyanuric acid: (1) a highly acidic active methylene,14 (2) reduced steric hindrance due to its cyclic structure, and (3) low contribution from the enol form. Interestingly, these reactions did not proceed in the presence of tetrahydrofuran.
:2:2:1, where three signals (Hb, Hc, and Hd) were located in the aliphatic region and one signal (Ha) appeared in the olefinic region. The 13C NMR spectrum (Fig. S10†) showed seven signals: three aliphatic (C5, C6, and C7), two olefinic (C2, C3), one aromatic (C1), and one carbonyl (C4). Dimedone (5,5-dimethyl-1,3-cyclohexanedione, 2c) gave results similar to those of 2b. The crystal structure of 4c (Fig. 1b) revealed its authentic 1,3,5-triazine core with N–C1 distances of 1.322(2)–1.333(2) Å, as well as N–C1–N and C1–N–C1 bond angles of 127.5(2)–128.4(2) and 111.4(2)–112.2(2)°, respectively.13 Evident enol–enone moieties were also observed with the following bond distances: C1–O1, 1.342(2)–1.357(2); O1–C2, 1.410(3)–1.416(2); C2–C3, 1.309(3)–1.332(3); C3–C4, 1.463(3)–1.477(3); and C4–O2 1.214(3)–1.228(3) Å. No related condensation reactions occurred when acyclic β-dicarbonyls, such as acetylacetone (R = CH3), dibenzoylmethane (R = Ph), diethyl malonate (R = OEt), and hexafluoroacetylacetone (R = CF3), were used. These observations suggested that 2a may have several favourable factors contributing to its reaction with cyanuric acid: (1) a highly acidic active methylene,14 (2) reduced steric hindrance due to its cyclic structure, and (3) low contribution from the enol form. Interestingly, these reactions did not proceed in the presence of tetrahydrofuran.
Next, we attempted to utilise sodium hydride as the base and found that only reactions in refluxing toluene provided good outcomes (Scheme 2). This procedure could address solid substrates that did not appear in a previous report.9 The suspensions gradually turned red, probably due to the sodium salts of the tri-conjugate intermediate. These precipitates containing excess sodium hydride were filtered out, purified by acidic aqueous treatment, and then recrystallised. This method could even be applied to weakly acidic and/or acyclic β-dicarbonyls that gave products 3b, 3c, and 3d (identical to a previously fabricated product9), whose NMR spectral trends were consistent with those of 3a. To our delight, the crystal structures of 3b and 3c showed slight yet significant differences from 3a regarding their bond lengths, which were caused by the electronic effects of substructures embedded at the carbonyl ends of 3b and 3c (Fig. S20 and S21†). These observations suggest that a potentially promising resonance hybrid involving proton transfers amongst the multi-hydrogen bonding entities could be fabricated and controlled by the attached functionalities (Chart S1a†).
 | ||
| Scheme 2 Nucleophilic substitution of β-dicarbonyls (2b–d) onto cyanuric acid with sodium hydride in toluene under reflux. | ||
UV-visible absorption spectra of 3a–d, 4b, and 4c are shown in Fig. 2. The absorption bands of 3a appeared at 319–333 nm, and 3d, belonging to the same ester family as 3a, also had an ill-defined absorption band at 317 nm. The wave shapes of 3b and 3c were similar to one another but were in sharp contrast to those of 3a and 3d, displaying twin maxima at 348–362 and 346–361 nm, respectively. Interestingly, they were accompanied by relatively intense bands at 236 and 238 nm, respectively, and similar absorptions were also observed in the UV-vis spectra of 4b and 4c. As a preliminary speculation, the absorption band in the high-energy region was assigned to the π–π* transition of an independent α,β-unsaturated carbonyl (enone) moiety,15 provided as a result of tautomerism and resonance (Chart S1b†). In this context, such differences between 3a/3d and 3b/3c may be ascribed to the peripheral attachments of 3b and 3c.
 | ||
| Fig. 2 UV-visible absorption spectra of the synthesised products in dichloromethane. | ||
In summary, we demonstrated synthetic protocols for reacting β-dicarbonyls with 1,3,5-triazine, where subsequent tautomerism gave intramolecular multi-hydrogen bonding structures. These AHB systems were fully characterised by spectroscopic analyses and X-ray crystallography. These scaffolds overcame aromatic stabilisation and potentially adopted resonance hybrid structures that involved proton transfers. This provided molecules with excellent delocalised electronic networks upon optimising their peripheral substructures. We expect that this work will be reinforced by future theoretical investigations and other instrumental examinations, where a novel, molecular motif replacing polyaromatic hydrocarbons could be discovered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant number 18K05082), and M. S. acknowledges the Electric Technology Research Foundation of Chugoku and the Tokuyama Science Foundation for financial support. We also thank Prof. Atsuhiro Osuka and Dr Takayuki Tanaka (Kyoto University) for their help with the X-ray measurements.Notes and references
- (a) B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601 CrossRef CAS; (b) S. Scheiner, Molecules, 2017, 22, 1521 CrossRef; (c) G. Sánchez, Molecules, 2019, 24, 2858 CrossRef; (d) G. Caron, J. Kihlberg and G. Ermondi, Med. Res. Rev., 2019, 39, 1707–1729 CrossRef CAS . and the references therein.
- (a) Y. Tanaka, J.-Y. Shin and A. Osuka, Eur. J. Org. Chem., 2008, 1341–1349 CrossRef CAS; (b) Y.-S. Xie, K. Yamaguchi, M. Toganoh, H. Uno, M. Suzuki, S. Mori, S. Saito, A. Osuka and H. Furuta, Angew. Chem., Int. Ed., 2009, 48, 5496–5499 CrossRef CAS; (c) T. Soya, W. Kim, D. Kim and A. Osuka, Chem.–Eur. J., 2015, 21, 8341–8346 CrossRef CAS; (d) C. Li, K. Zhang, M. Ishida, Q. Li, K. Shimomura, G. Baryshnikov, X. Li, M. Savage, X.-Y. Wu, S. Yang, H. Furuta and Y. Xie, Chem. Sci., 2020, 11, 2790–2795 RSC.
- (a) G. J. Woolfe, M. Melzig, S. Schneider and F. C. Dorr, Chem. Phys., 1983, 77, 213–221 CrossRef CAS; (b) S. Tobita, M. Yamamoto, N. Kurahayashi, R. Tsukagoshi, Y. Nakamura and H. Shizuka, J. Phys. Chem. A, 1998, 102, 5206–5214 CrossRef CAS; (c) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- (a) C.-Y. Peng, J.-Y. Shen, Y.-T. Chen, P.-J. Wu, W.-Y. Hung, W.-P. Hu and P.-T. Chou, J. Am. Chem. Soc., 2015, 137, 14349–14357 CrossRef CAS; (b) Y.-T. Chen, P.-J. Wu, C.-Y. Peng, J.-Y. Shen, C.-C. Tsai, W.-P. Hu and P.-T. Chou, Phys. Chem. Chem. Phys., 2017, 19, 28641–28646 RSC; (c) M. Mamada, K. Inada, T. Komino, W. J. Potscavage Jr, H. Nakanotani and C. Adachi, ACS Cent. Sci., 2017, 3, 769–777 CrossRef CAS.
- (a) W. Y. Cha, T. Soya, T. Tanaka, H. Mori, Y. Hong, S. Lee, K. H. Park, A. Osuka and D. Kim, Chem. Commun., 2016, 52, 6076–6078 RSC; (b) T. Soya, H. Mori, Y. Hong, Y. H. Koo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 3232–3236 CrossRef CAS; (c) M. Umetani, J. Kim, T. Tanaka, D. Kim and A. Osuka, Chem. Commun., 2019, 55, 10547–10550 RSC.
- (a) M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 CrossRef; (b) J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J.-Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568–13579 CrossRef CAS; (c) T. Soya, H. Mori and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 15882–15886 CrossRef CAS.
- D. P. Fairlie, T. C. Woon, W. A. Wickramasinghe and A. C. Willis, Inorg. Chem., 1994, 33, 6425–6428 CrossRef CAS.
- (a) H.-J. Schulz, J. Liebscher, P. Luger, M. Quian and J. Mulzer, J. Heterocycl. Chem., 1992, 29, 1125–1132 CrossRef CAS; (b) P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- Theoretical considerations of peripheral hydrogen bonding of aromatic rings were reported. (a) D. Rusinska-Roszak, Molecules, 2017, 22, 481 CrossRef; (b) I. V. Omelchenko, O. V. Shishkin, P. Dopieralski and Z. Latajka, J. Phys. Chem. A, 2019, 123, 2244–2251 CrossRef CAS.
- G. M. Vakhatova and L. N. Yakhontov, Chem. Heterocycl. Compd., 1980, 16, 433–436 CrossRef.
- (a) C. A. M. Afonso, N. M. T. Lourenço and A. d. A. Rosatella, Molecules, 2006, 11, 81 CrossRef CAS; (b) A. Chouai and E. E. Simanek, J. Org. Chem., 2008, 73, 2357–2366 CrossRef CAS; (c) K. Yamada, H. Fujita and M. Kunishima, Org. Lett., 2012, 14, 5026–5029 CrossRef CAS; (d) M. N. Kopylovich, K. T. Mahmudov, M. Haukkac and A. J. L. Pombeiro, New J. Chem., 2014, 38, 495–498 RSC; (e) N. Iranpoor and F. Panahi, Org. Lett., 2015, 17, 214–217 CrossRef CAS; (f) C. H. Lin, Y. C. Chou, M. W. Wang and R. J. Jeng, RSC Adv., 2016, 6, 17539–17545 RSC.
- Partially similar crystallographic considerations were stated. N. Kuhn, A. Al-Sheikh and M. Steimann, Z. Naturforsch., 2003, 58b, 381–384 Search PubMed.
- P. J. Wheatley, Acta Crystallogr., 1955, 8, 224–226 CrossRef CAS.
- (a) H. McNab, Chem. Soc. Rev., 1978, 7, 345–358 RSC; (b) S. Nakamura, H. Hirao and T. Ohwada, J. Org. Chem., 2004, 69, 4309–4316 CrossRef CAS.
- R. B. Woodward, J. Am. Chem. Soc., 1941, 63, 1123–1126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2014517–2014520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07677e |
| This journal is © The Royal Society of Chemistry 2020 |