
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured copper molybdates as promising bifunctional electrocatalysts for overall water splitting and CO2 reduction†
Atefeh Rahmania and
Hossein Farsi *ab
*ab
aDepartment of Chemistry, University of Birjand, Birjand, Iran. E-mail: hofarsi@birjand.ac.ir
bDeveloping Nanomaterials for Environmental Protection Research Lab, University of Birjand, Birjand, Iran
First published on 23rd October 2020
Abstract
Overall water splitting and CO2 reduction are two very important reactions from the environmental viewpoint. The former produces hydrogen as a clean fuel and the latter decreases the amount of CO2 emissions and thus reduces greenhouse effects. Here, we prepare two types of copper molybdate, CuMoO4 and Cu3Mo2O9, and electrochemically investigate them for water splitting and CO2 reduction. Our findings show that Cu3Mo2O9 is a better electrocatalyst for full water splitting compared to CuMoO4. It provides overpotentials, which are smaller than the overpotentials of CuMoO4 by around 0.14 V at a current density of 1 mA cm−2 and 0.10 V at −0.4 mA cm−2, for water oxidation and hydrogen evolution reactions, respectively. However, CuMoO4 adsorbs CO2 and the reduced intermediates/products more strongly than Cu3Mo2O9. Such different behaviors of these electrocatalysts can be attributed to their different unit cells.
1. Introduction
Fossil fuels are diminishing resources that contribute notably to environmental pollution, so developing sustainable energy sources suitable for our growing energy requirements is critical for the survival of humankind.1,2 An abiotic component of promising renewable energy infrastructure with no reliance on fossil fuels and no carbon dioxide emission is the splitting of water into hydrogen and oxygen.3,4 In photoelectrochemical (PEC) water splitting, solar energy is directly converted to fuel by light-absorbing semiconductors. As a new generation energy carrier, solar hydrogen will play an important role in our lives because it is storable, transportable, and convertible into efficient electricity in fuel cells, as well as being a viable and clean source of power due to its extraordinarily high energy density.5 During water splitting, the reductive half-reaction with a two-electron transfer mechanism is relatively easier than the oxidative half-reaction involving a four-electron transfer mechanism,6 which is the most energy-intensive step in the overall water splitting process. Therefore, the slow kinetics of the oxygen evolution reaction, OER, requires a substantial overpotential to generate measurable current densities.7 A large positive change in Gibbs free energy (ΔG0 = 237.13 kJ mol−1) causes the full water splitting reaction to be thermodynamically an uphill reaction.8 From this perspective, it is challenging to design highly effective photocatalysts while considering crucial parameters in the development of suitable semiconductors. These parameters include band gap width, optical absorption edge, stability against photocorrosion/lifetime, solar photons to current efficiency, cost-effectiveness, catalytic activity and surface structure.9–11 Furthermore, a high degree of crystallinity, i.e. a smaller amount of defects, is often required for water splitting because recombination between photogenerated carriers is particularly a serious problem in uphill reactions. Beginning with the discovery of Fujishima and Honda in the early 1970s, metal oxide or mixed-metal oxide semiconductors have been well studied as water splitting photoanodes due to their low cost and high stability in aqueous environments.12 Because the mixture of two metals in an oxide matrix can generate materials with unique chemical and physical properties, much consideration has been paid to the mixed-metal oxides, such as metal titanates, metal tungstates and metal molybdates.Moreover, the growing CO2 emission is an inevitable result of fossil fuels consumption because of the accessibility, diversity and high energy density of these fuels. However, the ever-increasing use of fossil fuels will cause dependence on them despite their limited reserves; additionally, global warming, drastic environmental changes and an intense threat to human survival are other issues associated with the use of fossil fuels. To resolve these issues, the best approach is to convert CO2 into carbon-containing fuels as a renewable energy resource.13,14 Recently, electrochemical reduction of accumulative CO2 has been considered as a feasible strategy for the conversion and utilization of this gas. This process was reported for the first time by Sir B. C. Brodie in 1873 and later in the 1990s by Hori et al.15,16 However, many efforts have been carried out to overcome usual challenges, such as poor efficiency because of kinetic barrier for stable molecules, weak selectivity resulting from reduction products competition, side reactions and hydrogen evolution reaction (HER).17,18 Therefore, probing new electrocatalysts that efficiently reduce CO2 into liquid fuels, especially in mild conditions, is extremely significant. For this purpose, copper (Cu) and Cu-based electrodes have been investigated as electrocatalyst candidates due to their intrinsic catalytic activity and variation of reduction products, which considerably depend on surface properties and morphology.19–30 Despite many experimental efforts and computational calculations, the mechanism of CO2 reduction on Cu electrodes is intricate and the exact reaction path is controversial.31,32
With regard to the above studies, it seems that Cu-based mixed oxides can be considered as bifunctional-electrocatalysts, which can electrochemically oxidize water and reduce CO2. For example, some of the widely studied copper-based photocatalysts for water splitting have been crystalline copper phosphide nanosheet,33 cuprous oxide composites,34–36 copper(II) tungstate37–39 and copper(II) borate.40 More recently, scientists have tried to find nanomaterials that exhibit remarkable properties to catalyze both hydrogen production and water oxidation reactions when the applied potentials fluctuate between oxidative and reductive conditions in the same reaction electrolyte.41 A copper-based catalyst composite film was found to be an interesting electrocatalyst for both HER and OER in the same electrolyte.42
To the best of our knowledge, there is no report on the application of copper molybdates for these properties. However, over the past several years, different preparations and structures of transition metal molybdates have been widely reported; these include doped,43,44 layered,45–47 nanosheet,48,49 powder,50 flower-like,51 nanoplate52 and micropompons53 structures. These structures are widely used in the areas of catalysis, magnetic application and energy storage. Furthermore, CuMoO4 is a thermochromic, trisochromic and piezochromic material, so it can be used in pressure and/or temperature sensors. Recently, extensive studies have been conducted on using transition metal molybdates in catalysis/photocatalysis,43,44,49,53–55 magnetic applications46,56–58 and lithium batteries;52 for example, preparation of hybrid Cu2O/CuMoO4 nanosheet electrodes for high-performance asymmetric supercapacitors,48 Cu3Mo2O9 nanoplates with excellent lithium storage performance52 and Cu3Mo2O9 micropompons with excellent performance in photocatalytic degradation of Congo red under visible light, photocurrent response and lithium storage.53 Two different compounds of copper molybdate exist with two different chemical formulae of CuMoO4 and Cu3Mo2O9. Both of them contain MoO4 tetrahedra, CuO6 octahedra and CuO5 pyramid units. In CuMoO4, six copper–oxygen polyhedra share edges to form a spiral-shaped chain fragment, interconnected by MoO4 tetrahedra,59 whereas sharing the edge oxygen atoms of Cu-based polyhedra makes a zigzag ribbon.60
The crystal structures of both compounds are shown in Fig. 1; these structures were reproduced by Mercury software61–63 from the Cambridge Crystallographic Data Center, using their CIF files. Recently, we reported that tungstate-derived copper has a good activity for electrochemical reduction of CO2,64 so it is expected here that we have molybdate-derived copper from two different sources, CuMoO4 and Cu3Mo2O9, with two different distribution of copper atoms and consequently different activities.
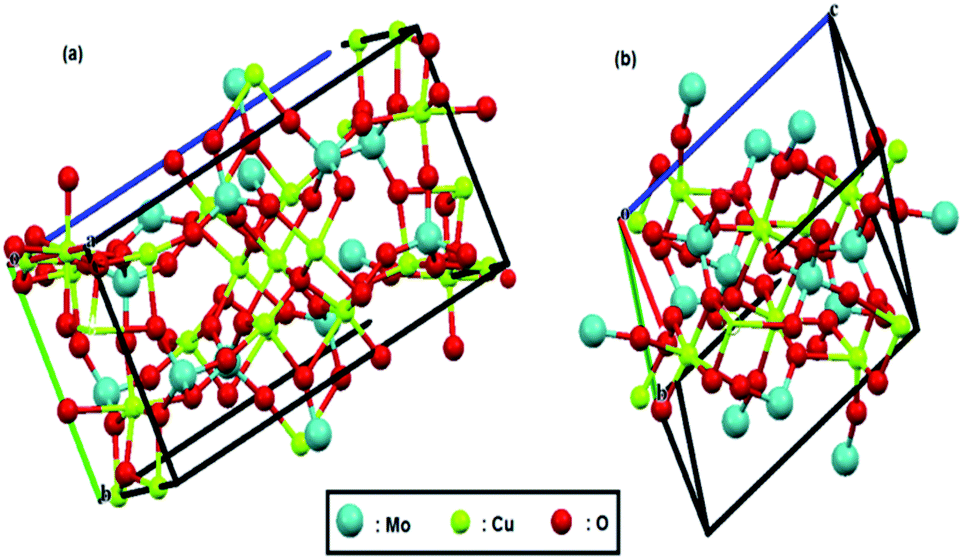 | ||
| Fig. 1 Crystal structure of (a) Cu3Mo2O9 and (b) CuMoO4. Red, green and pale blue balls represent oxygen, copper and molybdenum atoms, respectively. | ||
Due to these different crystal structures, their comparative study as bifunctional catalysts for electrochemical water splitting and CO2 reduction seems very interesting. In this study, we successfully prepare, characterize and electrochemically investigate both nano-CuMoO4 and nano-Cu3Mo2O9 structures.
2. Experimental
2.1. Materials preparation
The reactions were carried out under air at room pressure. High purity (>99%) AR grade copper nitrate Cu(NO3)2·3H2O, sodium molybdate Na2MoO4·2H2O, citric acid and ammonium hepta-molybdate, (NH4)6Mo7O24·4H2O, were obtained from commercial sources.For the synthesis of Cu3Mo2O9, 2.4195 g (0.01 mmol) Na2MoO4·2H2O was dissolved in 50 mL deionized water to form a transparent solution. Then, 2.4160 g (0.01 mmol) Cu(NO3)2·3H2O was dissolved in 10 mL deionized water to reach a dark blue solution, which was added to the first solution dropwise by a burette and stirred at room temperature for 30 min. After stirring, the beaker was subjected to 80 W cm−2 ultrasonic radiation under air for 30 min using an ultra sonicator operating at 40 kHz. The reaction was carried out in a glass balloon, which was heated in an oil bath at about 100 °C for 20 h. The balloon was connected to a reflux condenser system open to the atmosphere. Next, the resulted particles were filtered and washed several times with distilled water and ethanol and then dried in an oven at a temperature of 75 °C for 3 h. The obtained powders were treated under air at temperatures ranging from 25 °C to 400 °C for 2 h and were then remained at this temperature for 4 h. Finally, Cu3Mo2O9 was prepared as a brown-orange powder.
For the preparation of CuMoO4, 5 mL of copper nitrate solution (0.50 M) was added to molybdate solution, which was obtained by dissolving 0.4414 g ammonium heptamolybdate in 50 mL deionized water (pH = 4.7). The mixture was stirred about 15 min, and then a citric acid (CA) solution in a molar proportion of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CA:cation was added. By evaporating the homogeneous solution at 90 °C (low-temperature crystallization), a sky blue gel was gradually formed. The obtained gel was transferred to a highly porous Prussian blue matrix at 120 °C for 24 h; then, it was calcined over a wide range of temperatures and times as shown in Fig. 2 to optimize its surface area, crystallinity and photocatalytic activity.64 Finally, a green yellowish powder that was obtained at the end of the process was characterized.
:1 CA:cation was added. By evaporating the homogeneous solution at 90 °C (low-temperature crystallization), a sky blue gel was gradually formed. The obtained gel was transferred to a highly porous Prussian blue matrix at 120 °C for 24 h; then, it was calcined over a wide range of temperatures and times as shown in Fig. 2 to optimize its surface area, crystallinity and photocatalytic activity.64 Finally, a green yellowish powder that was obtained at the end of the process was characterized.
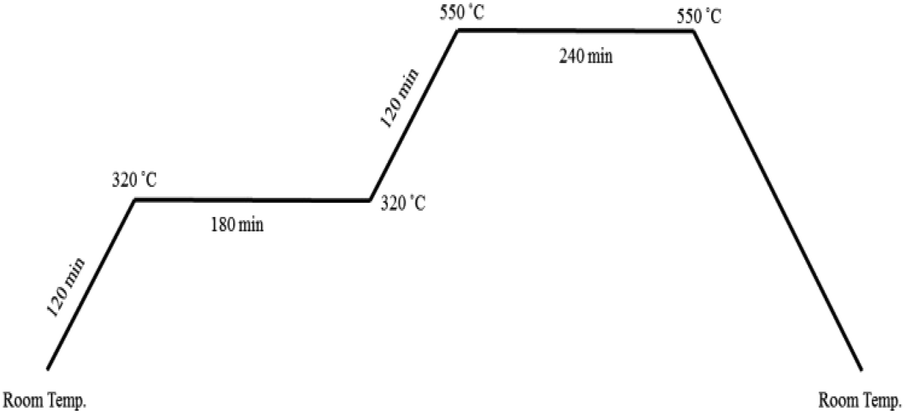 | ||
| Fig. 2 Temperature profile for calcination process. | ||
2.2. Electrode fabrication
For electrochemical studies, working electrodes were prepared by the electrophoretic method. Two 1 × 5 cm2 pieces of commercial stainless steel with a separation distance of 2 cm were inserted into a mixture, which was prepared by dispersing 0.0056 g of the prepared samples in 5 mL ethanol and five-minute exposure under 50 kHz ultrasonic waves. A potential of 20 V was applied between the two electrodes for 60 s. Next, the prepared electrodes were dried at 80 °C for 3 h and finally at 400 °C for 15 min.2.3. Material characterization
XRD pattern of CuMoO4 and Cu3Mo2O9 samples were recorded on an XPERT-PRO powder X-ray diffractometer. The CuKα (k = 1.5406 and 1.5443 Å) radiation was used, and diffractions were measured over 2θ range of 5–80°. Transmission electron microscopy (TEM) images were obtained with a CM120 electron microscope from Philips. The diffuse reflectance spectra were determined with a UV-Vis spectrophotometer Avantes (Avaspec-2048-TEC) with BaSO4 as the standard reference. The magnetic properties were measured using a vibrating sample magnetometer (VSM) system (Lake Shore7404).2.4. Electrochemical characterization
All electrochemical measurements on the surface of catalysts were conducted in a 0.1 M NaOH (pH = 13) solution by a standard three-electrode cell containing Ag/AgCl as the reference electrode, a platinum mesh as the auxiliary electrode and an electrophoretically deposited material electrode as the working electrode using a Solartron SI-1260 electrochemical interface. For Mott–Schottky analysis, a Solartron Phase Gain Analyzer SI1260 was used to determine electrochemical impedance spectra (EIS) by handling a frequency of 10000 Hz, an AC voltage amplitude of 10 mV in different scanning potential ranges for the deposited electrode vs. Ag/AgCl (between −0.35 and 0.2 V for CuMoO4, and between −0.45 and 0.4 V for Cu3Mo2O9) both in the dark and under illumination by a fluorescent lamp. A range of frequencies between 0.1 and 100000 Hz was used in EIS measurements with a 10 mV amplitude perturbation.
The electrocatalytic activities of the working electrodes were tested using Cyclic Voltammetry (CV) and Linear Sweep Voltammetry (LSV), at a scan rate of 10 mV s−1 in different potential ranges. All electrochemical tests were performed at ambient temperature and pressure. To reduce the additional ohmic resistance, bubbles that were generated on the electrode surface under OER and HER were dispersed by stirring the solution at 1000 rpm during the reaction. Moreover, to enhance the mass transport of CO2 during the electroreduction, CO2 was continuously bubbled.
3. Results and discussion
3.1. XRD analysis
As shown in Fig. 3a, the XRD patterns of the as-prepared CuMoO4 and Cu3Mo2O9 samples reveal their crystalline nature, which are consisted of a single-phase and orthorhombic Cu3Mo2O9 with space group Pna21 and anorthic CuMoO4 with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . As shown, the broadening of the diffraction peaks proves the high purity as well as the nanocrystalline nature of the as-prepared samples. The strong sharp diffraction peaks indicate that the samples are well crystallized. The peaks can be indexed to orthorhombic Cu3Mo2O9 (JCPDS 94-0728)60 and anorthic CuMoO4 (JCPDS 98-000-7372).59 The strong peaks at the defined 2θ values of 25.96, 27.02 and 25.24 can be indexed to (002), (140) and (131) planes of Cu3Mo2O9, and at 2θ values of 23.95, 26.53 and 27.16 to (012), (201) and (211) planes of CuMoO4 respectively. The (002) and (012) reflection peaks were used for the calculation of the crystallite size of Cu3Mo2O9 and CuMoO4, respectively. The average crystallite size calculated using the Debye–Scherrer equation from the FWHM (full-width half maxima) of the XRD peaks of Cu3Mo2O9 and CuMoO4 samples were found to be approximately 49 and 134 nm, respectively.
. As shown, the broadening of the diffraction peaks proves the high purity as well as the nanocrystalline nature of the as-prepared samples. The strong sharp diffraction peaks indicate that the samples are well crystallized. The peaks can be indexed to orthorhombic Cu3Mo2O9 (JCPDS 94-0728)60 and anorthic CuMoO4 (JCPDS 98-000-7372).59 The strong peaks at the defined 2θ values of 25.96, 27.02 and 25.24 can be indexed to (002), (140) and (131) planes of Cu3Mo2O9, and at 2θ values of 23.95, 26.53 and 27.16 to (012), (201) and (211) planes of CuMoO4 respectively. The (002) and (012) reflection peaks were used for the calculation of the crystallite size of Cu3Mo2O9 and CuMoO4, respectively. The average crystallite size calculated using the Debye–Scherrer equation from the FWHM (full-width half maxima) of the XRD peaks of Cu3Mo2O9 and CuMoO4 samples were found to be approximately 49 and 134 nm, respectively.
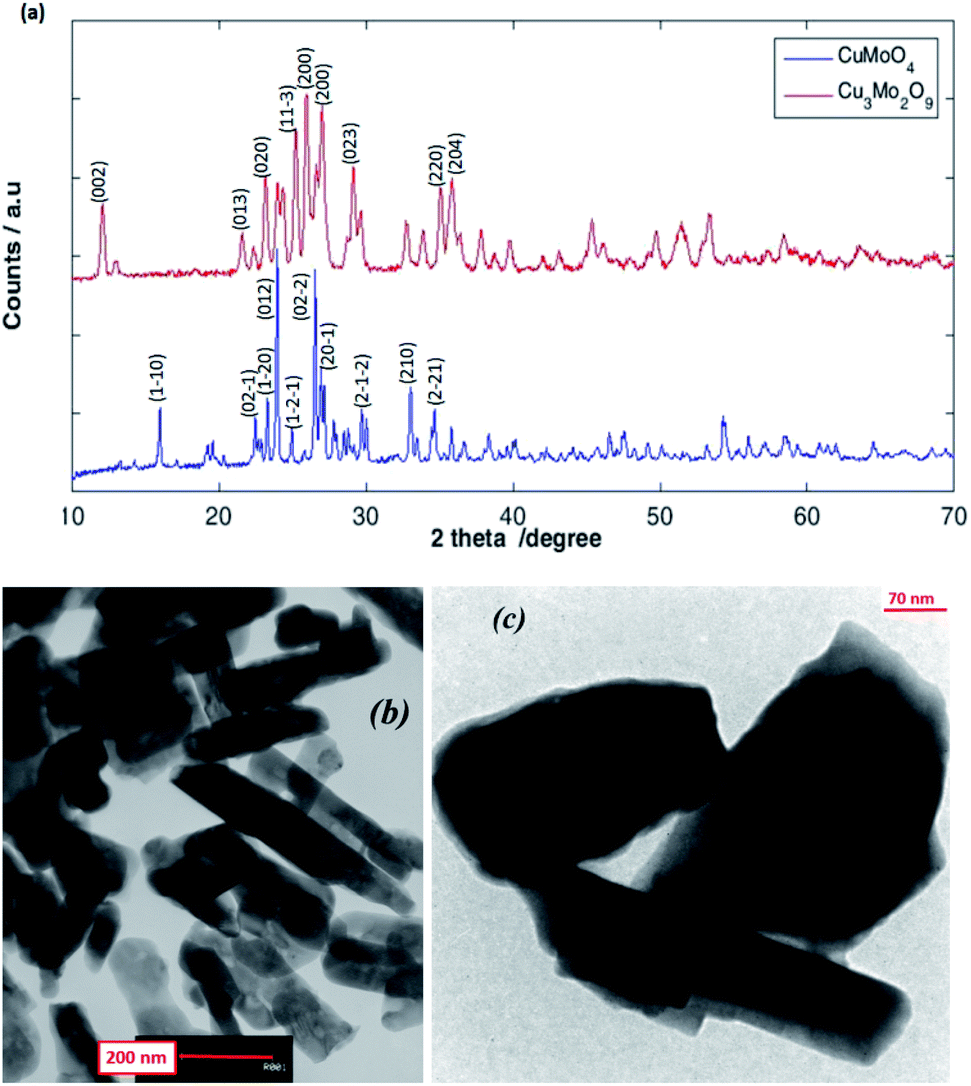 | ||
| Fig. 3 (a) XRD patterns, and TEM images for (b) Cu3Mo2O9 and (c) CuMoO4. | ||
3.2. TEM
TEM studies provide further insight into the nanostructure and topography of the treated samples. Fig. 3b and c shows the TEM images of Cu3Mo2O9 and CuMoO4 for the formation of rod-shaped orthorhombic and multiform shape particles of the samples; both monocrystalline particles have diameters that are in good agreement with the size obtained from powder XRD. The nanoparticles are a little agglomerated.3.3. Magnetic properties analysis
The relationships between the magnetization (M) and magnetic field (F) of Cu3Mo2O9 and CuMoO4 nanoparticles was determined by a vibrating sample magnetometer (VSM) system (Lake Shore 7404). Fig. 4a shows the M–F curve in the range from −18 to 18 kG with a saturated magnetization. A linear increment above 7 kG with a narrow hysteresis loop occurs between −2 and 2 kG; this pattern is the characteristic of both ferromagnetic and antiferromagnetic interactions for both samples.65,66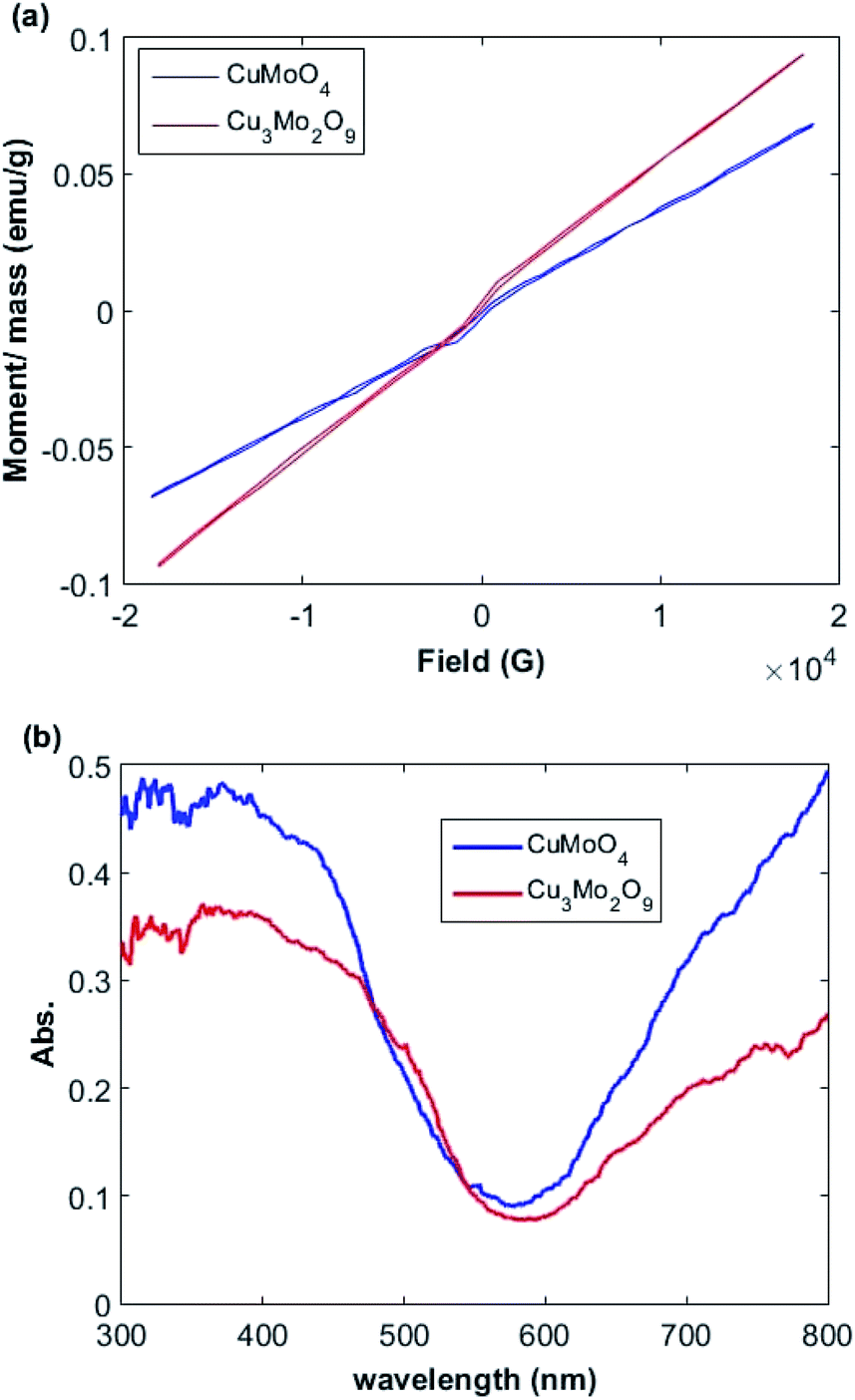 | ||
| Fig. 4 (a) Magnetization curves and (b) UV-Vis diffuse reflectance spectra of Cu3Mo2O9 and CuMoO4. | ||
3.4. Optical properties
The optical properties of the samples were measured by UV-Vis DRS spectrum. Fig. 4b demonstrates the DRS spectra of the prepared Cu3Mo2O9 and CuMoO4 samples. Two sequential absorption bands can be identified in these samples. One band is located above 600 nm, and the other one below 600 nm; these results are in good agreement with the color of the samples. The former can be mainly attributed to d–d transition of Cu2+ ions (3d9), whereas the latter may be due to the Cu(3d) ← O(2p) LMCT or Mo(4d) ← O(2p) LMCT and an Mo(4d) ← Cu(3d) MMCT, which is located at wavelengths higher than LMCT band.66,67The direct and indirect optical band gaps of nanoparticles were extracted according to Tauc's equation.68,69 According to Tauc's equation, the relation between the absorption coefficient (α) and the incident photon energy (hν) can be written as:
| (αhν)1/n = A(hν − Eg) | (1) |
The larger band gaps of both samples correspond to the direct electronic transition between the upper edge of O:2p valence band and the lower edge of Mo:4d conduction band. The calculated band gaps for these transitions are 2.4 and 2.6 eV for Cu3Mo2O9 and CuMoO4, respectively. The lower energy indirect transitions can be explained by assuming the existence of narrow band correlated-electron states in the band gap instead of a single wide band of uncorrelated-electron states that may correspond to Cu2+:3d9 band. We speculate that in addition to the contribution of Cu (3d) on the top of O (2p) valence band, the conduction band of both samples contain Cu (3d) characteristic, in agreement with our experimental observations. The Jahn–Teller distortion of Cu2+ gives rise to a d-orbital splitting in which the degeneracy of σ-antibonding orbitals is broken.38 The calculated Tauc's plot indirect band gaps are 2.1 and 2.3 eV for Cu3Mo2O9 and CuMoO4 samples, respectively, so their absorption edges are in the visible range of the spectrum. Ansari et al. reported that the electronic transitions of CuMoO4 are of indirect type and a band gap of 3.06 eV was obtained,71 whereas values of 2.38, 2.80 and 2.15 eVs were obtained by other researchers.72–74 Moreover, Dutta et al. found an indirect band gap of 2.32 eV.54
3.5. Mott–Schottky analysis
For Mott–Schottky analysis, one has to select a range of potentials in which Faraday's reaction does not occur. To determine this range, cyclic voltammetric technique was used. The characteristic of Faraday's processes is the evolution of peaks in cyclic voltammograms. For non-faradic or capacitive processes, no peak is observed and the voltammogram is almost rectangular. Cyclic voltammograms with a scan rate of 10 mV s−1 in potential ranges between 0.2 and −0.35 for CuMoO4 and between 0.25 and −0.45 V for Cu3Mo2O9 in 0.1 M NaOH were obtained. In this way, a range of potentials was determined over which the cyclic voltammograms showed no faradic current, and only a capacitive current was observed. In order to ensure that the observed behavior in the voltammograms was capacitive, the effect of different scan rates on the voltammograms was evaluated. If the observed behavior is capacitive, the anodic and cathodic currents should increase with increasing the scan rate. Therefore, the performance of samples in these potential regions can be considered as capacitive, as shown in Fig. S2.†When a semiconductor is exposed to an electrolyte, it reaches the thermodynamic equilibrium with the electrolyte by exchanging electrons through the interface and adjusting the Fermi level of the semiconductor to the Fermi level of the electrolyte, which results in the formation of a space charge layer. The Fermi level position of the electrolyte is constant because the number of available states in the electrolyte solution is typically more than the number of states in the semiconductor. With regard to electron transfer from/to a semiconductor to/from an electrolyte, an excess of immobilized charge in the semiconductor and an excess of opposite charge in the electrolyte are produced just behind the interface and in the space charge layer. This implies that the energy bands are bent,75 and the amount of bending depends on the difference between band edge energy in the semiconductor and at the interface; furthermore, the amount of bending corresponds to the potential drop in the space charge layer, US. When an external electrical source is applied, then US = Uapp − UFB, where UFB is the flat band potential, that is, a unique potential for which the potential drop between the bulk and surface is zero. Thus, by determining the flat band potential one can estimate the situations of conduction and valence band edges for n-type and p-type semiconductors, respectively.76 Mott–Schottky theory describes the properties of semiconductor electrolyte interface using space charge capacitance as follows77–79
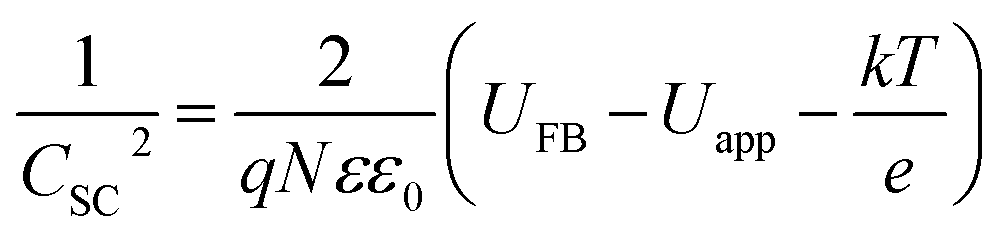 | (2) |
000 Hz in 0.1 M NaOH in the dark and under illumination.
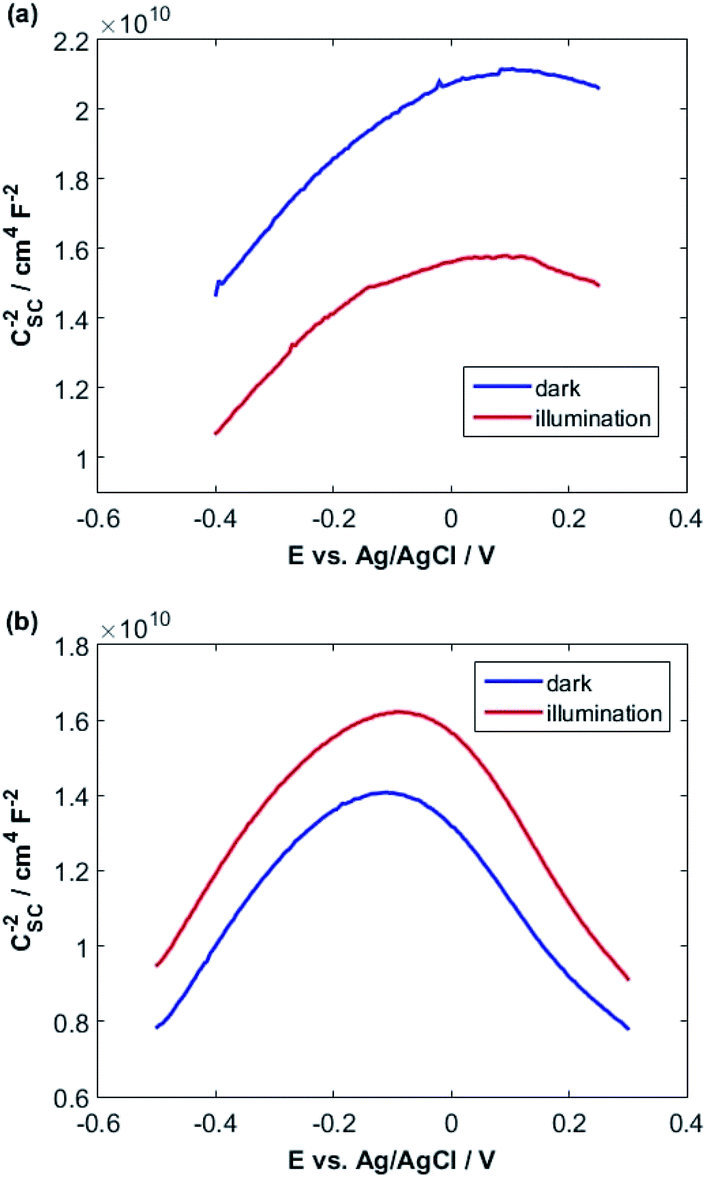 | ||
| Fig. 5 Mott–Schottky plots both in the dark and under illumination for (a) CuMoO4 and (b) Cu3Mo2O9. | ||
A fairly large frequency of 10000 Hz is chosen since fast processes such as charging of double layer and space charge layer capacitors respond at this frequency. Obviously, selecting and recognizing the linear region are difficult. Therefore, since at this frequency, the samples exhibit capacitive behavior and the capacity of the space charge layer is independent of potential, the Nyquist diagrams are in a range of potentials selected from the CVs. It is expected that these diagrams will coincide almost exactly with the region where only the space layer charging occurs.
For this purpose, Nyquist diagrams with 0.05 V potential interval and over a frequency range of 0.1 to 100000 Hz are plotted in the dark and under illumination in 0.1 M NaOH solution (Fig. S3 and S4†). As illustrated in these figures, Nyquist diagrams over the range −0.24 to −0.12 V for CuMoO4 and the range −0.30 to −0.18 V for Cu3Mo2O9 in the dark are fairly in good agreement. Therefore, these regions were used for the linear fitting of the Mott–Schottky equation, and the results are shown in Fig. S5 and S6.† A correlation coefficient close to 1 implies a good agreement between the experimental results and the results of the Mott–Schottky theory. The value of UFB can be determined from the intercept of the fitting line with the Mott–Schottky plot. In Fig. S5 and S6,† the slopes of Mott–Schottky plots for both electrodes are positive; this confirms that the electrodes are n-type semiconductors, and consequently, the lower conduction band edge practically merges with the quasi-Fermi level of electrons. The conduction band edge, EC, can be experimentally estimated using flat band potential (UFB).80 For this propose, the electrochemical potentials should be related to absolute energy, Eabs, and the effects of pH should be considered using the following equations at 25 °C:
| ERHE (V) = EAg/AgCl + 0.199 + 0.0592pH | (3) |
| Eabs (eV) = −4.5 − ERHE | (4) |
As can be seen, different values were obtained for the flat band potentials for both samples. The values of EC for both samples were determined using the UFB values reported in Table 1 and eqn (3) and (4).
| Sample | Condition | UFB/V | ND/cm−3 | WD/cm | L/cm | EC vs. NHE/V | EC (abs)/eV | EC (abs)/eV | Eg/eV |
|---|---|---|---|---|---|---|---|---|---|
| CuMoO4 | Dark | −1.50 | 1.25 × 1021 | 8.36 × 10−8 | 9.44 × 10−9 | −0.53 | −4.0 | −6.3 | 2.3 |
| Illumination | −1.29 | 1.38 × 1021 | 7.95 × 10−8 | 9.02 × 10−9 | −0.32 | −4.2 | −6.5 | 2.3 | |
| Cu3Mo2O9 | Dark | −1.18 | 1.42 × 1021 | 7.48 × 10−8 | 8.49 × 10−9 | −0.21 | −4.3 | −6.4 | 2.1 |
| Illumination | −1.36 | 1.47 × 1021 | 7.36 × 10−8 | 8.34 × 10−9 | −0.39 | −4.1 | −6.2 | 2.1 |
Then, with a method, which has already been described in the electrochemical studies of nanostructured nickel titanate81 and benefits from a combination of optical (DRS) and electrochemical (impedance) spectroscopies, we determined the valence band edges, EV, of both samples with subtracting the values of band gap energies from EC. All band edges for both samples are reported in Table 1 for in the dark and under illumination. According to these data, we outline the experimental electronic band structures of both samples as illustrated in Fig. 6. The O2−:2p and Cu2+:3d orbitals contribute to the valence band; additionally, O2−:2p and Mo6+:4d orbitals contribute to the conduction band for both compounds.82
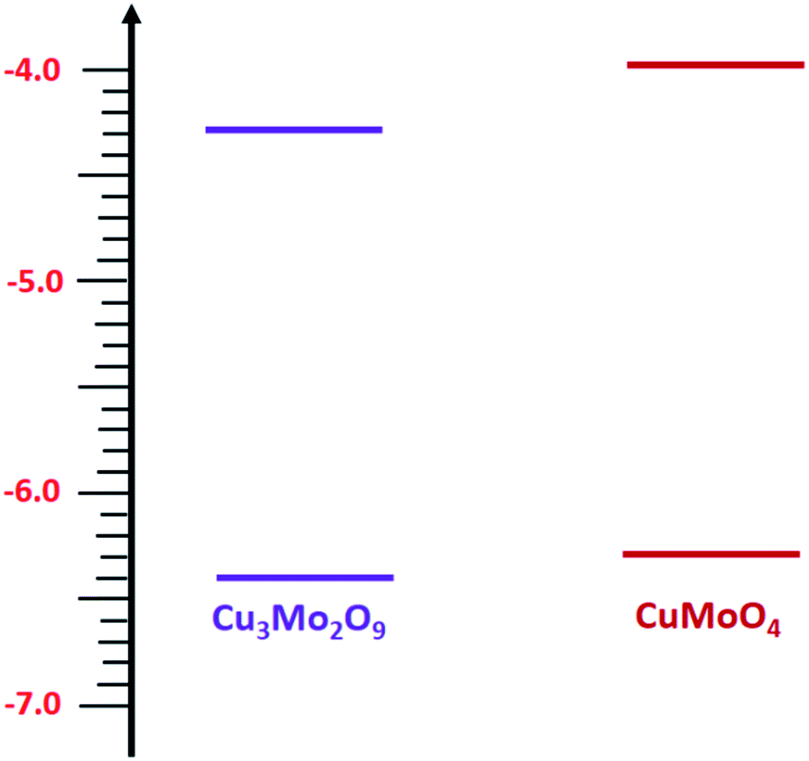 | ||
| Fig. 6 Energy states diagram of CuMoO4 and Cu3Mo2O9. | ||
The density of donor atoms can be calculated using the slope value of Mott–Schottky plots (Fig. S5 and S6†) and eqn (2). The calculated values are shown in Table 1. The density of donor atoms, especially copper atoms, in Cu3Mo2O9 is larger than CuMoO4. Moreover, using ND values, it is possible to estimate the diffusion length of minor charge carriers (here holes) using the following equations at 25 °C:
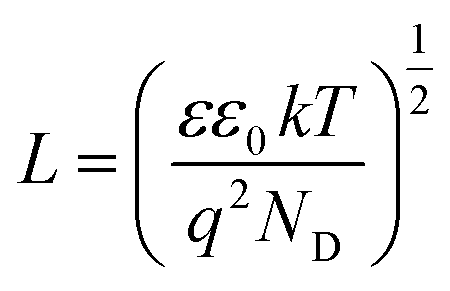 | (5) |
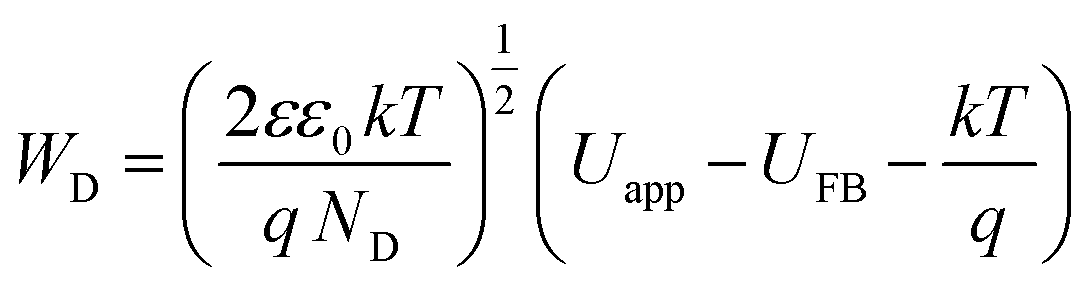 | (6) |
The obtained results are shown in Table 1 considering that the dielectric constants of Cu3Mo2O9 and CuMoO4 are equal to 7.2 (ref. 83) and 7.9,84 respectively.
One can calculate diffusion length of minority carrier, L, and the thickness of the depletion layer, WD, by using UFB, ND and eqn (2). The values of ND, L and WD (for Uapp − UFB = 1 V) were computed, and the results (accompanied with UFB) are listed in Table 1.
3.6. Electrochemical water splitting studies
Electrochemical water splitting includes oxygen evolution reaction, OER, on the anode surface and hydrogen evolution reaction, HER, on the cathode surface. Electrochemical water splitting is a pH-dependent reaction, so it has been widely investigated in both acidic and alkaline solutions. From the technological viewpoint, Schalenbach et al. showed that alkaline water electrolyzers are more efficient compared to acidic ones.85 Moreover, from the electrocatalyst design viewpoint, alkaline solutions allow us to use earth-abundant materials, especially for OER,86,87 whereas acidic solutions are limited to precious metals and metal oxides.88 However, the big disadvantage of alkaline solutions is the sluggish kinetics of OER in these media.89This is due to the four-electron-proton transfer mechanism in alkaline media, which is the rate-determining step at high overpotentials. Therefore, to compare CuMoO4 and Cu3Mo2O9 in electrochemical OERs, one should calculate their potentials and kinetic parameters such as exchange current density and Tafel slope. The overall reaction, which occurs on the surface of the electrocatalyst is:
| 4OH− + 4hVB+ →2H2O + O2 | (7) |
During this complex reaction, four electrons are transferred to the valence band of the semiconductor. As shown in Fig. 6, it is expected that the injection of the electrons of OH− to the valence band of Cu3Mo2O9 is more favorable compared to that of CuMoO4 because of its lower EV, and consequently, a higher OER activity than CuMoO4. Fig. 7a shows the comparative LSVs of both samples for OER measured in 0.1 M NaOH solution at 25 °C; they indicate close onset potentials. The dashed line shows the standard potential of OER. As Fig. 7a shows, OER overpotentials of 0.28 and 0.42 V at a current density of 1 mA cm−2 were obtained for nanostructured Cu3Mo2O9 and CuMoO4, respectively. These results are in agreement with the diagram of Fig. 6. In addition, these numerical values are in good agreement with the results of Gou et al., who reported an overpotential of 0.325 mV at 50 mA cm−2 in 1.0 M KOH for Cu3Mo2O9 nanosheet loaded on nickel foam.90
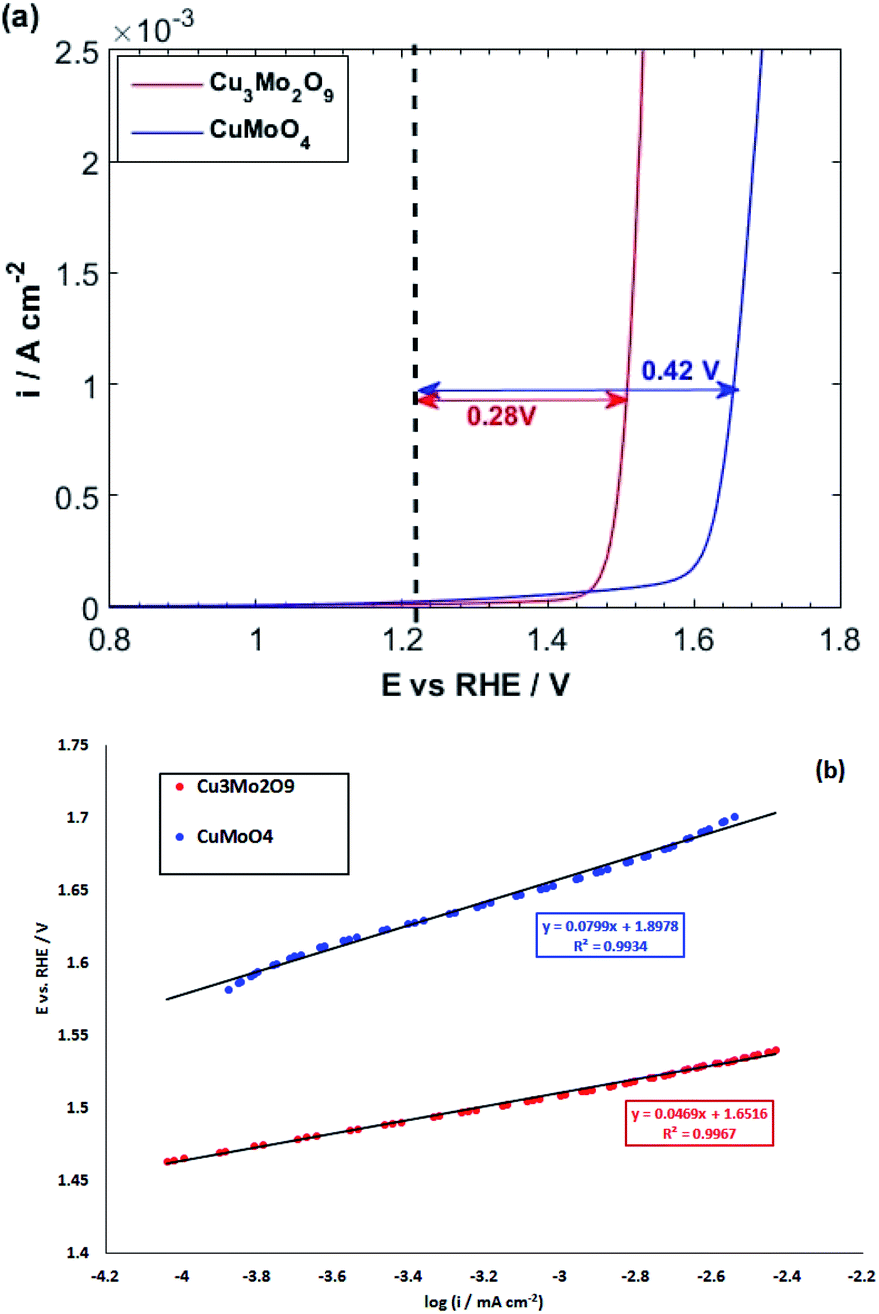 | ||
| Fig. 7 (a) LSVs and (b) linear parts of polarization curves of CuMoO4 and Cu3Mo2O9 for OER with a scan rate of 10 mV s−1 in 0.1 M NaOH. | ||
The kinetics of OER at electrode interfaces were reflected by their corresponding polarization curves. The linear curve fitting gives the Tafel slopes of 46.9 and 79.9 mV dec−1 for, respectively, CuMoO4 and Cu3Mo2O9, as illustrated in Fig. 7b. These slopes are less than that of Pt/C (99 mV dec−1), indicating a more rapid kinetics for OER on the surfaces of these electrocatalysts compared to Pt/C.91
The obtained Tafel slopes are close to those reported for some nickelates and cobaltates by Bockris and Otagawa,92 who investigated OER on the surface of different perovskites and developed a common mechanism for OER by emphasizing on electrochemical adsorption of OH− ions followed by desorption of OH and formation of H2O2 as an intermediate that is decomposed into O2. Such a mechanism has also been suggested for RuO2 (ref. 93) and TiO2.94 Scheme 1 was inspired by their work.
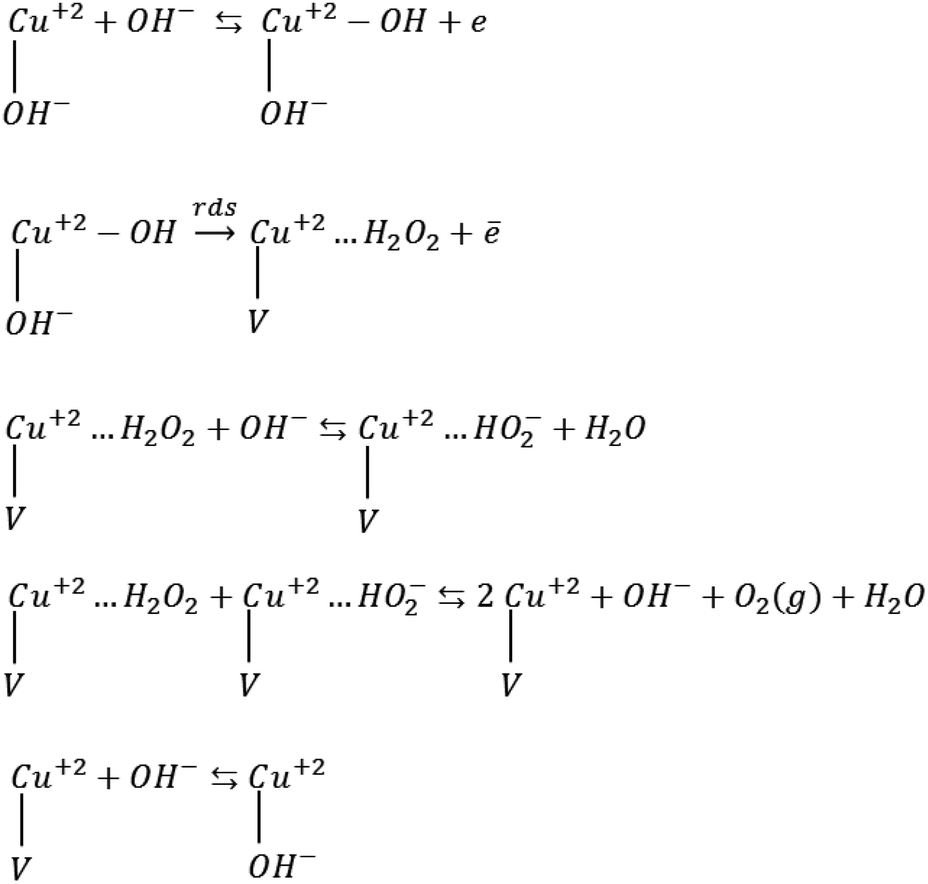 | ||
| Scheme 1 OER mechanism by emphasizing on the electrochemical desorption of OH− as the rate-determining step (rds). V denotes an OH− vacancy and dots show physisorbed species. | ||
Mott–Schottky analysis showed that Cu2+ ions are the localized charges in the space charge layer, thus they should be involved in interfacial phenomena. Accordingly, for the adapted mechanism, we supposed that the adsorption of OH− ions occurs on the copper ions. The first step in Scheme 1 is the formation of the adsorbed OH radicals, which can occur via discharging of both hydroxyl ions and H2O molecules. However, in alkaline solution, hydroxyl ions reach the surface of electrodes faster than water molecules because of their diffusion coefficient.95,96 The second step, which is rds, includes the electrochemical desorption of OH radicals, the formation of physically adsorbed hydrogen peroxide and OH− vacancy on the surface of these compounds. The next two steps are fast steps and show desorption and decomposition of hydrogen peroxide and the formation of O2 molecules. The final fast step is surface diffusion of OH− into the oxygen vacancy sites. The exchange current densities (i0) (measured by extrapolation of the Tafel plot) were calculated, and the values of 6.8 × 10−10 and 3.1 × 10−9 mA cm−2 were obtained for CuMoO4 and Cu3Mo2O9, respectively. Generally, the exchange current density is expected to be proportional to the surface density of catalytically active sites; here, Cu2+ ions correspond to the below mechanism.
After investigating the OER activity, the electrocatalytic performances of the samples were comparatively studied in terms of HER in 0.1 M NaOH solution at 25 °C. For this purpose, the LSVs of both samples were taken by starting from positive potentials to negative potentials up to the occurrence of HER, which is identified by a sharp increase in cathodic current. The resulting LSVs are depicted in Fig. 8a. It is observed that some cathodic peaks exist before hydrogen evolution. Liu et al. showed that the redox reaction of Mo in copper molybdate does not occur during electrochemical measurement, so the redox behavior of Mo has no contribution to the measured capacitance.97 Moreover, our previous study on the comparative electrochemical behaviors of CuWO4 and CuO showed that these peaks can be attributed to Cu(II)/Cu(I) and Cu(I)/Cu(0) redox reactions.64 Cyclic voltammograms (Fig. S7†) show more detail on these redox reactions for both samples. These cyclic voltammograms clearly show that the reduction of copper ions to copper atoms occurs before HER. Therefore, HER occurs on the surface of molybdate-derived copper atoms. In fact, HER takes place in acidic media more facile than in alkaline solution. However, using acidic electrolyte has some disadvantages, such as the corrosion of the electrolyzer.98
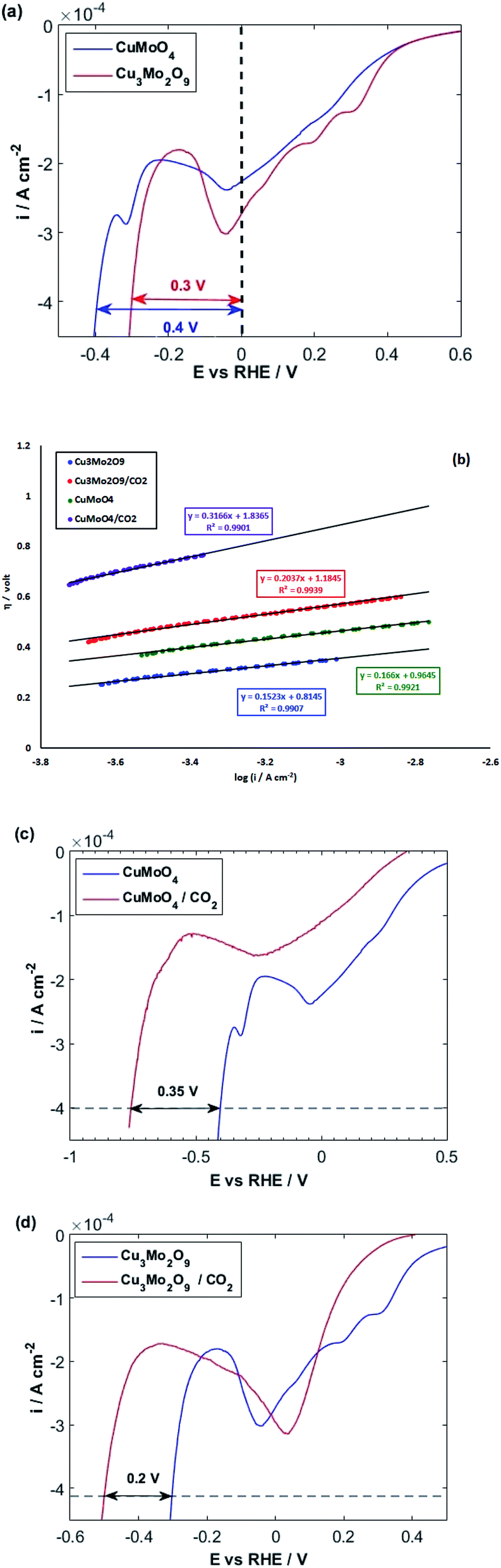 | ||
| Fig. 8 (a) LSVs and (b) linear parts of polarization curves of CuMoO4 and Cu3Mo2O9 for OER with a scan rate of 10 mV s−1 in 0.1 M NaOH and (c and d) polarization curves of CuMoO4 and Cu3Mo2O9 in the presence/absence of CO2. | ||
Moreover, the reaction needs more energy to produce proton by O–H bond cleavage of water molecules in alkaline solution.99 The elementary steps for HER in alkaline solution accompanied by their Tafel slopes are:
Volmer step (120 mV dec−1):
| H2O + M + e → M–H + OH− | (8) |
Heyrovsky step (40 mV dec−1):
| H2O + M–H + e → M + H2 + OH− | (9) |
Tafel step (30 mV dec−1):
| M–H + M–H → 2M + H2 | (10) |
Based on these steps, two mechanisms of Volmer–Heyrovsky and Volmer-Tafel are possible. The Volmer step, which is the water dissociation step, is common in both mechanisms. The energy required for this step can be provided by the adsorption of water molecules on the surface of the electrocatalyst.100 The stronger the water molecules adsorption the more the energy provided for the Volmer step. In addition, adsorption of OH− ions on the active sites has a poisoning effect and increases the overpotential by lowering the activity because of limiting the number of sites in alkaline solution. The hydrogen binding energy is another important factor that governs the HER activity of the catalysts. In fact, a volcano-type correlation between HER exchange current densities and H-binding energy values was demonstrated, which was supported by both DFT and experimental studies.101,102 Furthermore, the crystallographic planes of the catalysts103 and the existence of an active component for water dissociation104 can affect the kinetics of the reaction.
As illustrated in Fig. 8a, the HER overpotential on the surface of Cu3Mo2O9 has a value of around 0.1 V, which is smaller than that of CuMoO4 at a current density of 0.4 mA cm−2. Because the adsorbed molecules (here, water) and the active sites (here, copper atoms) are the same in nature, the predominant factor should be the number of active sites. It is clear that the surface density of copper atoms on the surface of Cu3Mo2O9 is larger than that of CuMoO4, so more surface sites are available for HER on the surface of Cu3Mo2O9; this lowers the overpotential. Moreover, although a precise look indicates copper active sites, copper atoms have different arrangements and different sublayers. Reduction of copper ions to copper occurs on the surface layer of copper molybdates, and the oxygen species, which exist in the sublayers, facilitate water molecules adsorption, causing HER promotion.105,106 Our previous work on the comparative studies of CuWO4 and CuO showed that the sublayer atoms have a considerable effect on lowering HER overpotentials.64 The effects of the nature of sublayer atoms and their configurations on reducing the HER overpotential on the surface of copper were already reported by Zhang et al.; they investigated full water splitting on sulfur-doped copper oxide in comparison with metallic copper.107
Fig. 8b shows a Tafel slope close to 120 mV dec−1 for both samples highlighting that the Volmer step is the rate-determining step in HER mechanism for both samples. The Tafel slopes of 166 and 152 mV dec−1 were obtained, and accordingly, exchange current densities of 1.5 × 10−6 and 4.5 × 10−6 A cm−2 were calculated for CuMoO4 and Cu3Mo2O9, respectively. It means that the rate of HER on the surface of Cu3Mo2O9 is three times faster compared to CuMoO4, which can be expected because of the more accessible active sites of this compound. The number of surface sites was also used for identifying the smaller Tafel slope for HER on the surface of MoS2.108,109 Additionally, these results are in agreement with our previous findings that compared CuWO4 and CuO.64 We showed that exchange current density for HER on the surface of CuO is much larger than that of CuWO4. Similarly, as shown in Fig. 1, the structural difference between CuMoO4 and Cu3Mo2O9 is that the latter has one CuO unit for each CuMoO4 pair, i.e., Cu3Mo2O9 ≡ 2CuMoO4 + CuO, and because of the existing CuO unit, Cu3Mo2O9 has a larger exchange current density.
At the final step of our electrochemical studies, we investigated the ability of both samples for CO2 reduction. Therefore, we purged the 0.1 M NaOH solution with CO2 for at least 30 min. The pH of the solution decreased from 13 to around 7 because of the following equilibrium:
| OH−(aq) + CO2(aq) ⇆ HCO3−(aq) | (11) |
| OH−(aq) + HCO3−(aq) ⇆ CO32−(aq) | (12) |
At ambient temperature and natural pH, the major species inside the solution is HCO3−.110 Fig. 8c and d illustrate that HER takes place in more negative potentials when CO2 is bubbled into the solution, and consequently, CO2 and its related species HCO3− and CO32− are present in the solution, i.e., the HER overpotential increases for both samples.
For investigating this process in more detail, we compared the cyclic voltammograms of both compounds with and without CO2 bubbling. The results are presented in Fig. S8.†
Both cathodic and anodic peaks of the samples in CO2 saturated 0.1 M NaOH solution show a significant decrease in height. This indicates that the adsorption of CO2 and maybe its reduction intermediates occur on the same sites where HER occurs. Furthermore, the adsorption of CO2 has a poisoning effect on HER and inhibits it.
In fact, there is a competition between H2O and CO2 molecules to be reduced on the surface of copper atoms.111,112 However, according to Fig. 8c and d, when CO2 is blown into the NaOH solution, the increasing of HER overpotential for CuMoO4 is more than that of Cu3Mo2O9 by around 0.15 V.
It means that the dissolved CO2 species in the solution or the adsorbed CO2 reduction intermediates occupy more HER active sites on the surface of CuMoO4 compared to Cu3Mo2O9. This implies the fact that the density of active sites (here, copper atoms) on the surface of Cu3Mo2O9 is higher compared to CuMoO4. Moreover, the atoms and their configuration in the sublayer have a considerable effect on the adsorption of CO2.
The Tafel slope analysis for HER in the presence of CO2 shows the same value of exchange current density of 1.5 × 10−6 A cm−2, for both samples, whereas Cu3Mo2O9 has a larger surface density of active sites for HER. Actually, it is possible that when HER starts on Cu3Mo2O9, some sites have still been occupied by CO2 reduction species. According to our previous report,64 which showed that, in the presence of CO2-species in the solution, HER occurs more readily than CuWO4. Therefore, it is possible here that HER occurs on the surface of copper atoms, which have come from CuO unit of Cu3Mo2O9, while CO2-species involved in the reaction on the surface of copper atoms have originated from CuMoO4 units of Cu3Mo2O9. This means that the adsorption of CO2-species is strong on the surface of CuMoO4.
In fact, the existence of oxygen in sublayers causes the strong adsorption of CO2, which will produce carbon-rich CO2-reduction products such as alcohols, carboxylic acids and hydrocarbons instead of CO. This conclusion can be supported by other reports. For instance, it has been reported that oxide-derived copper produces methanol,113–115 ethylene and ethanol with increased current efficiency.116 Moreover, the existence of tin oxide layer over tin effectively produces formate117,118 and formic acid.119 Tayyebi et al. used DFT calculation for electrochemical reduction of CO2 for producing methanol, methane and formic acid over different transition metal oxides, and showed that there is a volcano-shaped behavior through the scaling relations of adsorbed intermediates.120
4. Conclusion
In this study, two types of copper molybdate, CuMoO4 and Cu3Mo2O9, were synthesized successfully in nanoscale. Our results showed that the difference between their electrochemical behaviors strongly depends on their crystal structure. In fact, the existence and addition of CuO unit in the unit cell of Cu3Mo2O9 caused a modified behavior corresponding to a combination of CuO and CuMoO4.The value of the Tafel slope for Cu3Mo2O9 (as OER) was much lower than that for CuMoO4. Therefore, Cu3Mo2O9 shows a better catalytic performance for both HER and OER in 0.1 M alkaline solution. The literature review indicated that Cu3Mo2O9 could be a bifunctional water splitting catalyst under alkaline conditions, and show better activity at low cell voltage than CuMoO4. The superior OER and HER activities for Cu3Mo2O9 can be attributed to the more active sites arising from oxygen vacancies or the unique topology of Cu3Mo2O9 crystals that maximizes the number of exposed active sites. However, our investigation showed that CO2-species are adsorbed on the surface of CuMoO4 more strongly than on the surface of Cu3Mo2O9, so they may produce more stable and carbon-rich products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the financial support from University of Birjand.References
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 1–8 CrossRef.
- X. Xu, Y. Ge, M. Wang, Z. Zhang, P. Dong, R. Baines, M. Ye and J. Shen, ACS Appl. Mater. Interfaces, 2016, 8, 18036–18042 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 18, 2655–2661 CrossRef.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- S. N. Lou, J. Scott, A. Iwase, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2016, 4, 6964–6971 RSC.
- J. Bisquert and S. Giménez, Photoelectrochemical Solar Fuel Production, Springer, Switzerland, 2016 Search PubMed.
- K. Maeda, Phys. Chem. Chem. Phys., 2013, 15, 10537–10548 RSC.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- M. Kitano and M. Hara, J. Math. Chem., 2010, 20, 627–641 RSC.
- E. Nurlaela, S. Ould-Chikh, M. Harb, S. del Gobbo, M. Aouine, E. Puzenat, P. Sautet, K. Domen, J.-M. Basset and K. Takanabe, Chem. Mater., 2014, 26, 4812–4825 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS.
- B. C. Brodie, Proceedings of the Royal Society of London, London, 1873–1874, vol. 22, p. 245 Search PubMed.
- Y. Hori, O. Koga, A. Aramata and M. Enyo, Bull. Chem. Soc. Jpn., 1992, 65, 3008–3010 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- W. Zhu, R. Michalsky, O. N. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Kato, M. Azuma and T. Sakata, J. Electrochem. Soc., 1991, 138, 3382–3389 CrossRef CAS.
- G. Kyriacou and A. Anagnostopoulos, J. Electroanal. Chem., 1992, 322, 233–246 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Katoh, T. Sakata and M. Azuma, J. Electroanal. Chem., 1991, 305, 319–328 CrossRef CAS.
- M. Schwartz, R. L. Cook, V. M. Kehoe, R. C. Macduff, J. Patel and A. F. Sammells, J. Electrochem. Soc., 1993, 140, 614–618 CrossRef CAS.
- J. F. Xie, Y. X. Huang, W. W. Li, X. N. Song, L. Xiong and H. Q. Yu, Electrochim. Acta, 2014, 139, 137–144 CrossRef CAS.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- X. Guo, Y. Zhang, C. Deng, X. Li, Y. Xue, Y.-M. Yan and K. Sun, Chem. Commun., 2015, 51, 1345–1348 RSC.
- R. S. Kumar, S. S. Kumar and M. A. Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 CrossRef.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, 17–19 CrossRef.
- W. C. Li and M. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef.
- Y. Lan, C. Gai, P. J. A. Kenis and J. Lu, ChemElectroChem, 2014, 1, 1577–1582 CrossRef CAS.
- Y. Lan, S. Ma, J. Lu and P. J. A. Kenis, Int. J. Electrochem. Sci., 2014, 9, 7300–7308 Search PubMed.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- K. J. P. Schouten, Z. S. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- A. Han, H. Zhang, R. Yuan, H. Ji and P. Du, ACS Appl. Mater. Interfaces, 2017, 9, 2240–2248 CrossRef CAS.
- Z. Lou, Y. Li, L. Zhu, W. Xie, W. Niu, H. Song, Z. Ye and S. Zhang, J. Mater. Chem. A, 2017, 5, 2732–2738 RSC.
- Y. Kwon, A. Soon, H. Han and H. Lee, J. Mater. Chem. A, 2015, 3, 156–162 RSC.
- C. Li, J. Wang, Z. Jiang and P. Hu, New J. Chem., 2015, 39, 4562–4567 RSC.
- D. Hu, P. Diao, D. Xu, M. Xia, Y. Gu, Q. Wu, C. Li and S. Yang, Nanoscale, 2016, 8, 5892–5901 RSC.
- K. J. Pyper, J. E. Yourey and B. M. Bartlett, J. Phys. Chem. C, 2013, 117, 24726–24732 CrossRef CAS.
- J. E. Yourey and B. M. Bartlett, J. Mater. Chem., 2011, 21, 7651–7660 RSC.
- J. Liu, S. Wen, X. Zou, F. Zuo, G. J. O. Beran and P. Feng, J. Mater. Chem. A, 2013, 1, 1553–1556 RSC.
- S. Cobo, J. Heidkamp, P. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS.
- X. Liu, H. Zheng, Z. Sun, A. Han and P. Du, ACS Catal., 2015, 5, 1530–1538 CrossRef CAS.
- T. K. Ghorai, D. Dhak, S. Dalai and P. Pramanik, J. Mol. Catal. A: Chem., 2007, 273, 224–229 CrossRef CAS.
- K. S. Makarevich, N. V. Lebukhova, P. G. Chigrin and N. F. Karpovich, Inorg. Mater., 2010, 46, 1359–1364 CrossRef CAS.
- B. Wang, K. Zhou, B. Wang, Z. Gui and Y. Hu, Ind. Eng. Chem. Res., 2014, 53, 12355–12362 CrossRef CAS.
- S. H. Qiblawi and R. L. LaDuca, Inorg. Chim. Acta, 2014, 413, 115–120 CrossRef CAS.
- S. Mitchell, A. Gómez-Avilés, C. Gardner and W. Jones, J. Solid State Chem., 2010, 183, 198–207 CrossRef CAS.
- D. Du, R. Lan, W. Xu, R. Beanland, H. Wang and S. Tao, J. Mater. Chem. A, 2016, 4, 17749–17756 RSC.
- L. Zhang, T. Li, Y. Deng, Y. Zhang, S. Hu and W. Sun, J. Iran. Chem. Soc., 2014, 11, 407–414 CrossRef CAS.
- M. Benchikhi, R. E. Ouatib, S. Guillemet-Fritsch, J.-Y. Chane-Ching, L. Er-Rakho and B. Durand, Ceram. Int., 2014, 40, 5371–5377 CrossRef CAS.
- Z. Shahri, M. Salavati-Niasari, N. Mir and G. Kianpour, J. Cryst. Growth, 2014, 386, 80–87 CrossRef CAS.
- J. Xia, L. X. Song, W. Liu, Y. Teng, L. Zhao, Q. S. Wang and M. M. Ruan, Dalton Trans., 2015, 44, 13450–13454 RSC.
- J. Xia, L. X. Song, W. Liu, Y. Teng, Q. S. Wang, L. Zhao and M. M. Ruan, RSC Adv., 2015, 5, 12015–12024 RSC.
- D. P. Dutta, A. Rathore, A. Ballal and A. K. Tyagi, RSC Adv., 2015, 5, 94866–94878 RSC.
- W. G. Chu, H. F. Wang, Y. J. Guo, L. N. Zhang, Z. H. Han, Q. Q. Li and S. S. Fan, Inorg. Chem., 2009, 48, 1243–1249 CrossRef CAS.
- T. Ito, H. Takagi and T. Asano, Chem. Mater., 2009, 21, 3376–3379 CrossRef CAS.
- S. Hara and H. Sato, J. Phys. Soc. Jpn., 2013, 82, 054801–054805 CrossRef.
- C. Canevali, F. Morazzoni, R. Scotti, D. Cauzzi, P. Moggi and G. Predieri, J. Mater. Chem., 1999, 9, 507–513 RSC.
- S. C. Abrahams, J. L. Bernstein and P. B. Jamieson, J. Chem. Phys., 1968, 48, 2619–2629 CrossRef CAS.
- B. L. Kihlborg, R. Norrestam and B. Olivecrona, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2066–2070 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- H. Farsi, S. Moghiminia, M. Raygan, E. Dana, S. Hosseini, M. Behforooz, T. Zubkov, I. V. Lightcap and Z. Li, J. Phys. Chem. C, 2019, 123, 25941–25948 CrossRef CAS.
- S. Vilminot, G. Andre and M. Kurmoo, Inorg. Chem., 2009, 48, 2687–2692 CrossRef CAS.
- T. Ito, H. Takagi and T. Asano, Chem. Mater., 2009, 21, 3376–3379 CrossRef CAS.
- X. Fu, J. Ji, W. Tang, W. Liu and S. Chen, Mater. Chem. Phys., 2013, 141, 719–726 CrossRef CAS.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef CAS.
- E. A. Davis and N. F. Mott, Philos. Mag. A, 1970, 22, 903–922 CrossRef CAS.
- T. Ansari, A. Pandit, M. Prasad and R. Singh, Indian J. Phys., A, 1898, 63, 784–792 Search PubMed.
- G. Steiner, R. Salzer and W. Reichelt, Fresenius. J. Anal. Chem., 2001, 370, 731–734 CrossRef CAS.
- M. Sadeghi, J. Mater. Sci.: Mater. Electron., 2016, 27, 5796–5801 CrossRef CAS.
- W. Zhang, J. Yin, F. Min, L. Jia, D. Zhang, Q. Zhang and J. Xie, J. Mol. Struct., 2017, 1127, 777–783 CrossRef CAS.
- T. Soga, Nanostructured materials for solar energy conversion, Elsevier, Amesterdam, The Netherlands, 2006 Search PubMed.
- M. Radecka, M. Rekas, A. Trenczek-Zajac and K. Zakrzewska, J. Power Sources, 2008, 181, 46–55 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, J. Bisquert, P. Bogdanoff and A. Zaban, J. Electrochem. Soc., 2003, 150, E293–E298 CrossRef CAS.
- A. Pitarch, J. Bisquert and G. Garcia-Belmonte, J. Non-Cryst. Solids, 2003, 324, 196–200 CrossRef CAS.
- M. C. K. Sellers and E. G. Seebauer, Thin Solid Films, 2011, 519, 2103–2110 CrossRef CAS.
- S. R. Morrison, Electrochemistry at Semiconductor and Oxidized Metal Electrodes, Plenum Press, New York, NY, USA, 1980 Search PubMed.
- S. Moghiminia, H. Farsi and H. Raissi, Electrochim. Acta, 2014, 132, 512–523 CrossRef CAS.
- J. C. Hill, Y. Ping, G. A. Galli and K. Choi, Energy Environ. Sci., 2013, 6, 2440–2446 RSC.
- N. Joseph, J. Varghese, T. Siponkoski, M. Teirikangas, M. T. Sebastian and H. Jantunen, ACS Sustainable Chem. Eng., 2016, 4, 5632–5639 CrossRef CAS.
- W. Wen, C. Li, Y. Sun, Y. Tang and L. Fang, J. Electron. Mater., 2017, 47, 1003–1008 CrossRef.
- M. Schalenbach, G. Tjarks, M. Carmo, W. Lueke, M. Mueller and D. Stolten, J. Electrochem. Soc., 2016, 163, F3197–F3208 CrossRef CAS.
- H. Zhang, H. Jiang, Y. Hu, Y. Li, Q. Xu, S. Petr and C. Li, J. Mater. Chem. A, 2019, 7, 7548–7552 RSC.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855–7865 CrossRef CAS.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 10960–10964 CrossRef CAS.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- Y. Gou, L. Yang, Z. Liu, A. M. Asiri, J. Hu and X. Sun, Inorg. Chem., 2018, 57, 1220–1225 CrossRef CAS.
- X. Yue, S. Huang, J. Cai, Y. Jin and P. K. Shen, J. Mater. Chem. A, 2017, 5, 7784–7790 RSC.
- J. O. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS.
- L. D. Burke, O. J. Murphy, J. F. O'Neill and S. Venkatesan, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1659–1671 RSC.
- P. Clechet, C. Martelet, J. R. Martin and R. Olier, Electrochim. Acta, 1979, 24, 457–461 CrossRef CAS.
- D. A. Denton, J. A. Harrison and R. I. Knowles, Electrochim. Acta, 1981, 26, 1197–1251 CrossRef CAS.
- B. E. Conway, Ionic Hydration in Chemistry and Biophysics, 1981, Elsevier, New York, p. 572 Search PubMed.
- X. Liu, Q. Gao, Y. Zhang, F. Li and Y. Zhang, Mater. Technol., 2016, 31, 653–657 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed. Engl., 2012, 51, 12495 CrossRef CAS.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS.
- N. M. Markovića, S. T. Sarraf, H. A. Gasteigert and P. N. Ross, J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., 2012, 124, 12663–12666 CrossRef.
- P. F. B. D. Martins, P. P. Lopes, E. A. Ticianelli, V. R. Stamenkovic, N. M. Markovic and D. Strmcnik, Electrochem. Commun., 2019, 100, 30–33 CrossRef CAS.
- S. Yamamoto, K. Andersson, H. Bluhm, G. Ketteler, D. E. Starr, T. Schiros, H. Ogasawara, L. G. M. Pettersson, M. Salmeron and A. Nilsson, J. Phys. Chem. C, 2007, 111, 7848–7850 CrossRef CAS.
- X. Zhang, X. Cui, Y. Sun, K. Qi, Z. Jin, S. Wei, W. Li, L. Zhang and W. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 745–752 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS.
- J. Benson, M. Li, S. Wang, P. Wang and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2015, 7, 14113–14122 CrossRef CAS.
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, J. Phys. Chem. C, 2015, 119, 55–61 CrossRef CAS.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS.
- Y. J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef CAS.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS.
- S. Ohya, S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Catal. Today, 2009, 148, 329–334 CrossRef CAS.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2714–2821 CrossRef.
- J. Wu, F. G. Risalvato, S. Ma and X. D. Zhou, J. Mater. Chem. A, 2014, 2, 1647–1651 RSC.
- R. Zhang, W. Lv and L. Lei, Appl. Surf. Sci., 2015, 356, 24–29 CrossRef CAS.
- C. W. Lee, N. H. Cho, K. D. Yang and T. Nam, ChemElectroChem, 2017, 4, 2130–2136 CrossRef CAS.
- E. Tayyebi, J. Hussain, Y. Abghui and E. Skulason, J. Phys. Chem. C, 2018, 122, 10078–10087 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07783f |
| This journal is © The Royal Society of Chemistry 2020 |