
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular thermogels from branched PCL-containing polyurethanes†
Qianyu Linb,
Jason Y. C. Lim a,
Kun Xuea,
Celestine P. T. Cheea and
Xian Jun Loh*a
a,
Kun Xuea,
Celestine P. T. Cheea and
Xian Jun Loh*a
aSoft Materials Department Institute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), Address: 2 Fusionopolis Way, Innovis, Singapore 138634. E-mail: lohxj@imre.a-star.edu.sg
bNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, 21 Lower Kent Ridge Rd, Singapore 119077
First published on 26th October 2020
Abstract
Thermogels are temperature-responsive hydrogels which are most commonly formed by supramolecular self-assembly of polymer amphiphiles comprising of both hydrophobic and hydrophilic segments. Although polyurethane thermogels have shown great promise as biomaterials, their synthesis by step-growth polymerisation of diols and diisocyanates can also result in formation of allophanate branches, which arise from the reaction between free isocyanate groups and urethane linkages along the polymer backbone. In this paper, we investigate the effects of different synthetic conditions on the degree of allophanate branching on polyurethane amphiphiles, and explore the influences of these branches on the polymers' critical micelle concentration (CMC), thermodynamics of micellization and subsequent thermogel properties. Our findings offer new insights into the relationship between polymer structure, micelle and gel properties. These results highlight the importance of taking polymer branching into account for understanding the hierarchical self-assembly of polymer amphiphiles and the resulting thermogel properties and behaviour.
Introduction
Thermogels are an important class of supramolecular polymeric hydrogel material that show sol–gel transitions as temperature increases, which is reversible upon cooling.1–4 This unique property makes thermogels highly appealing for a range of biomedical applications.5–10 For example, thermogels have been studied for sustained drug delivery,11–19 where a solution of the polymer and drug is first injected into the body and gelates in situ to form a localised depot for drug release when warmed up to body temperature.20–22 In addition to other exciting applications23 such as tissue engineering24–26 and wound healing,27–31 thermogels have very recently been demonstrated as vitreous endotamponades for treatment of vitreoretinal surgery.32 Thermogels are typically comprised of amphiphilic polymers possessing both hydrophilic and hydrophobic segments, and can take the forms of linear di-,33,34 tri-35–37 and alternating38–40 multi-block copolymers. These polymers can form thermogels by hierarchical self-assembly triggered by warming: dehydration of the hydrophobic polymer segments first brings about micelle formation due to hydrophobic interactions, which in turn then further self-assemble to form a non-covalently crosslinked network of micelles that entraps water to form stable gels.41–43 The exact mechanism of the thermogel formation from micelle self-assembly depends on the structure and composition of the amphiphilic block-copolymer backbone, with micelle aggregation, jamming and bridging42,44,45 all shown to be plausible.Compared to the vast majority of linear polymer amphiphiles, the supramolecular self-assembly of branched amphiphilic polymers to form thermogels remain poorly understood. Although molecular weight,46,47 choice of hydrophobic segment,48 and ratio of hydrophobic to hydrophilic segments49 are known to affect the rheological and mechanical properties of thermogels, the effects of polymer branching has been largely hitherto overlooked. In our previous studies,32 we have investigated random triblock polyurethanes as thermogelling polymers for biomedical applications. Other than acting as a linking group to join poly(ethylene glycol) (PEG), poly(propylene glycol) (PPG) and poly(caprolactone) (PCL) macromonomers together, the urethane linkages serve as hydrogen bond donors which further facilitates inter- and intrachain polymer interactions in aqueous solutions. However, polyurethane synthesis by step-growth polymerization reactions involving the polyaddition of diisocyanates and diols at elevated temperatures can lead to formation of allophanate linkages as well, which forms from the addition of a second isocyanate group to a pre-formed urethane moiety (Scheme 1).50,51 Compared to the predominant urethane linkages formed,52 the much smaller but significant numbers of allophanate linkages present create branch-points at random along the length of the polymer backbone. These will in turn affect the polymer's self-assembly properties to form micelles and thermogels. Indeed, studies have shown that grafting increasing numbers of hydrophilic branches onto the hydrophobic backbone of non-thermogelling comb-like amphiphilic copolymers can raise their cloud-point and reduce their ability to form micelles.53
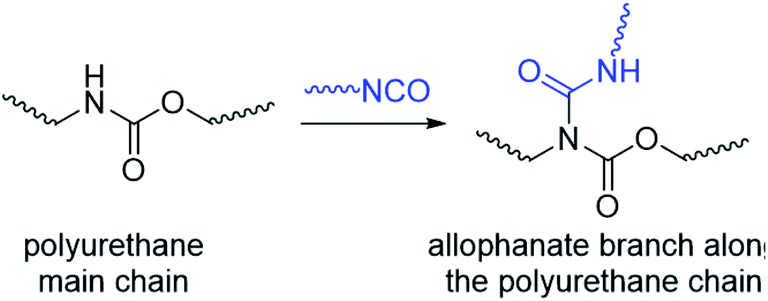 | ||
| Scheme 1 Formation of allophanate branch points along a polyurethane by reaction of isocyanates with pre-formed urethane groups. | ||
Therefore, in this manuscript, we explore how different degrees of branching in amphiphilic thermogelling polyurethanes influence their micellization and subsequent gelation behaviour for the first time. Firstly, by systematically varying the reaction concentration and duration for polymer synthesis, we generate a library of polyurethanes with different molecular weights and degrees of branching. By characterising the critical micelle concentrations (CMCs) and thermodynamic parameters of micellization, we show that these important parameters are dependent on both the molecular weights and branching density of the polymers. Unlike hyperbranched polymer architectures which can undergo extensive intermolecular chain entanglement,54,55 increases in the moderate densities of polymer branches on the polymers studied herein does not necessarily give rise to more mechanically-robust hydrogels. Our findings reinforce the necessity to take polymer branching, even at moderate extents, into account for the design and engineering of polyurethane thermogelling polymers for various applications, which are often-overlooked in materials design.
Results and discussion
Effects of reaction conditions on polymer structure
Allophanate-branched thermogelling polyurethanes are synthesized by the polyaddition of PEG, PPG and PCL macromonomer-diols in a 4.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.00:0.01 (wt/wt/wt) ratio with hexamethylene diisocyanate (HMDI) at 110 °C in anhydrous toluene, catalysed by dibutyltin dilaurate (DBTL), as shown in Scheme 2. As a small excess of isocyanates compared to alcohols are known to facilitate allophanate formation,56 a 3 mol% excess of HMDI over the diols were used. A range of monomer concentrations were investigated, as well as reaction durations ranging from 1 to 4 hours. 1H NMR characterization of the purified polyurethanes showed complete incorporation of all diol and diisocyanate reactants components (Fig. 1), indicating a high degree of polymer conversion in all cases. The polyurethanes were further characterized by gel permeation chromatography (GPC) and the degree of branching quantified spectroscopically and chemically (see Experimental section for more details). In addition, the size distribution and surface charge of the resulting micelles arising from the self-assembly of these amphiphilic polymers in water were determined by DLS and zeta potential measurements respectively. These results are summarized in Table 1.
:1.00:0.01 (wt/wt/wt) ratio with hexamethylene diisocyanate (HMDI) at 110 °C in anhydrous toluene, catalysed by dibutyltin dilaurate (DBTL), as shown in Scheme 2. As a small excess of isocyanates compared to alcohols are known to facilitate allophanate formation,56 a 3 mol% excess of HMDI over the diols were used. A range of monomer concentrations were investigated, as well as reaction durations ranging from 1 to 4 hours. 1H NMR characterization of the purified polyurethanes showed complete incorporation of all diol and diisocyanate reactants components (Fig. 1), indicating a high degree of polymer conversion in all cases. The polyurethanes were further characterized by gel permeation chromatography (GPC) and the degree of branching quantified spectroscopically and chemically (see Experimental section for more details). In addition, the size distribution and surface charge of the resulting micelles arising from the self-assembly of these amphiphilic polymers in water were determined by DLS and zeta potential measurements respectively. These results are summarized in Table 1.
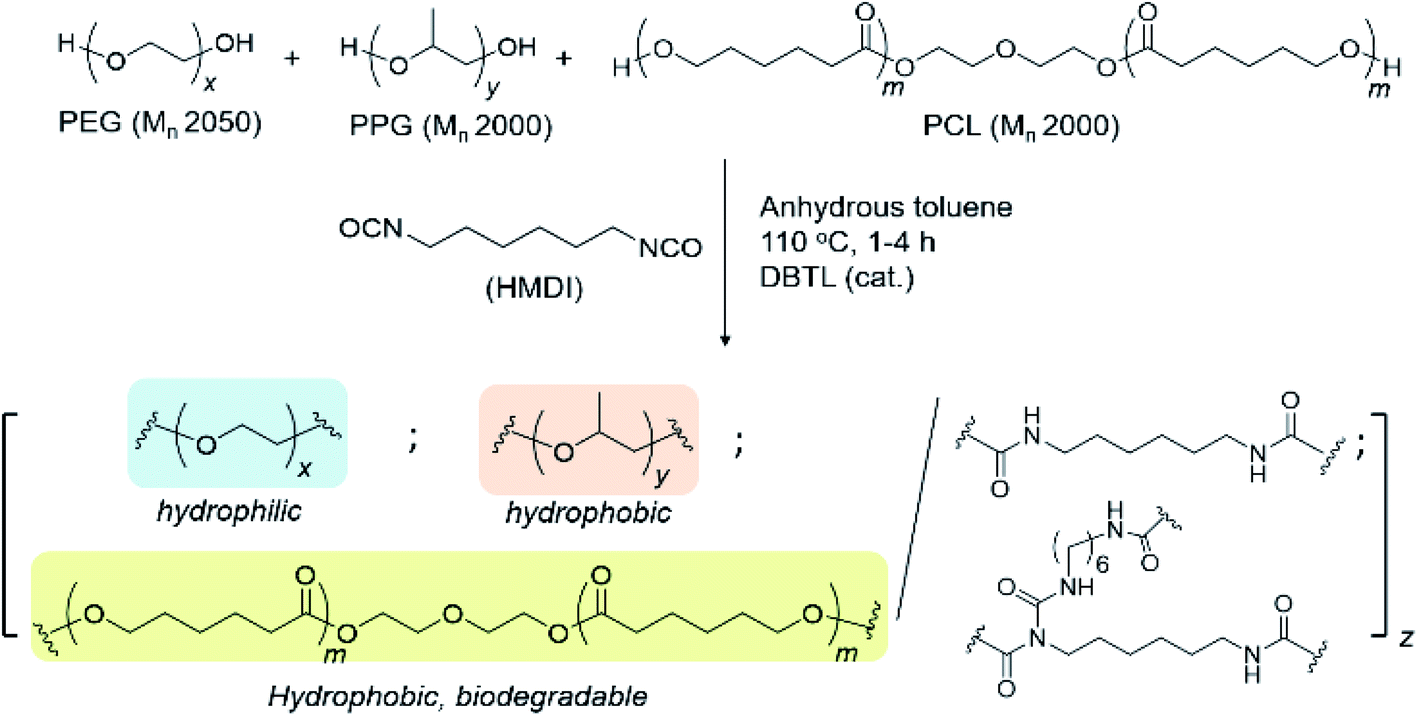 | ||
| Scheme 2 Synthesis of branched thermogelling polyurethanes. | ||
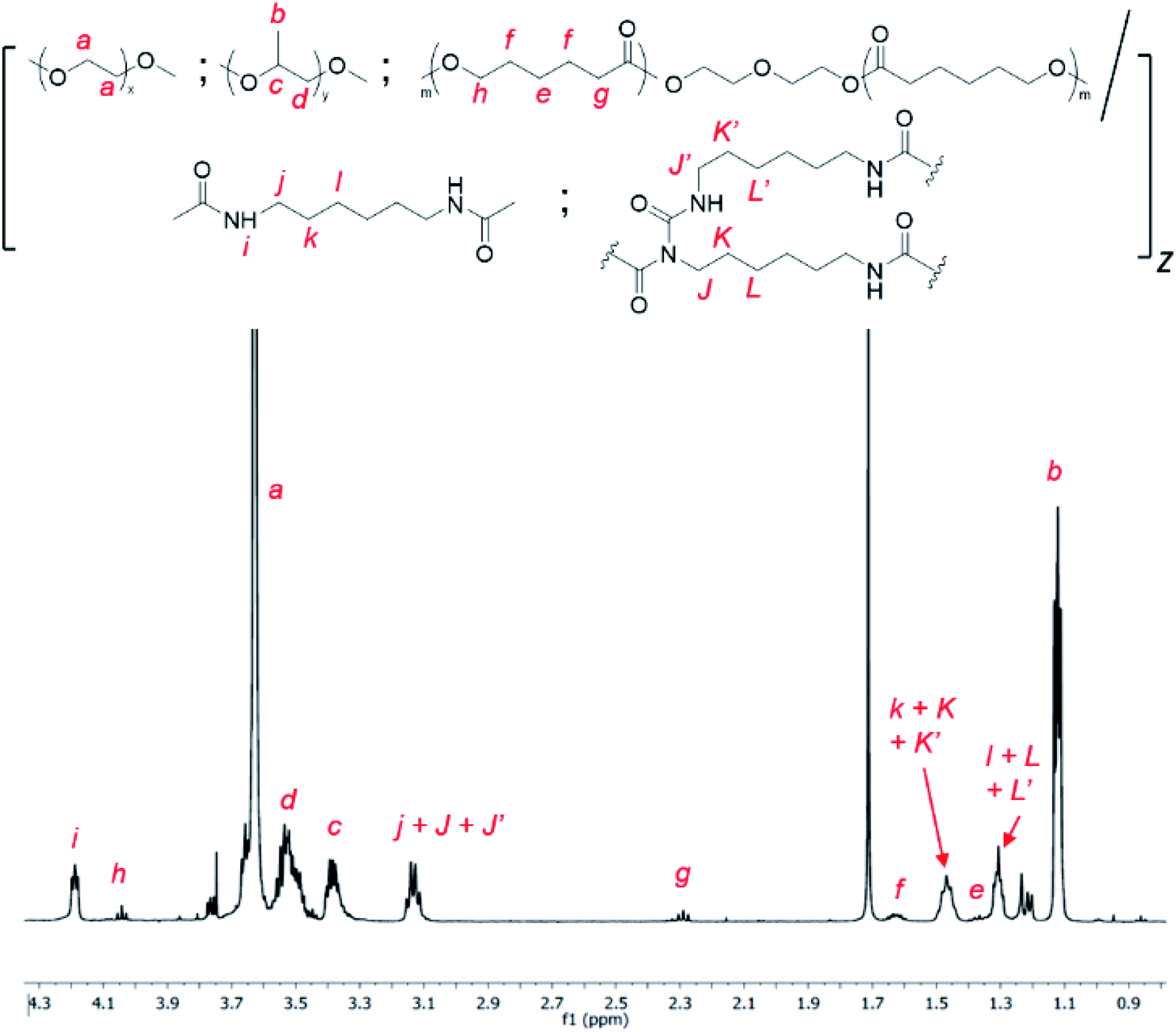 | ||
| Fig. 1 1H NMR spectrum of random triblock thermogelling polyurethane copolymer containing PEG:PPG and PCL in a 4.00:1.00:0.01 wt/wt/wt ratio (CDCl3). | ||
| S/N | Polymer name | Poly-diol concentration (g ml−1) | Synthesis duration (h) | Mna (kDa) | PDIa | E:P:C:H NMR molar ratiob |
Degree of branching (10−4 mol g−1) | Z-Avg size of micellesc (d nm) | Zeta potentialc (mV) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined using triple detection GPC in THF solvent.b Molar ratios of PEG (E), PPG (P), PCL (C), and HMDI (H) incorporated into each polymer, determined by integration of the 1H NMR resonances at 3.60–3.70 ppm, 1.10–1.15 ppm, 4.05 ppm and 3.10–3.15 ppm respectively.c Polymers were first dissolved at 4 °C overnight in deionised water at concentrations of 1.0 wt/v%, and they were warmed up to ambient temperature and equilibrated overnight before DLS experiments and zeta potential measurements were performed. | |||||||||
| 1 | 1P30 1h | 0.17 | 1 | 49.6 | 1.49 | 4.33:1.00:0.0545:5.60 |
4.6 | 60.2 ± 0.9 | −1.18 ± 0.25 |
| 2 | 1P30 2h | 0.17 | 2 | 62.2 | 1.55 | 4.34:1.00:0.0513:5.65 |
3.7 | 80.9 ± 0.8 | 0.038 ± 0.057 |
| 3 | 1P30 3h | 0.17 | 3 | 57.0 | 1.56 | 4.18:1.00:0.0490:5.52 |
4.7 | 81.6 ± 0.6 | −1.21 ± 0.30 |
| 4 | 1P35 1h | 0.14 | 1 | 37.7 | 1.71 | 4.48:1.00:0.0522:5.64 |
1.7 | 58.4 ± 0.5 | −1.70 ± 0.58 |
| 5 | 1P35 2h | 0.14 | 2 | 59.1 | 1.62 | 4.21:1.00:0.0458:5.62 |
2.6 | 72.6 ± 0.3 | −0.66 ± 0.34 |
| 6 | 1P35 3h | 0.14 | 3 | 60.0 | 1.50 | 4.19:1.00:0.0399:5.67 |
2.8 | 79.8 ± 4.0 | −0.44 ± 0.11 |
| 7 | 1P35 4h | 0.14 | 4 | 49.6 | 1.77 | 4.21:1.00:0.0410:5.66 |
3.9 | 79.8 ± 1.6 | −0.863 ± 0.11 |
| 8 | 1P40 1h | 0.13 | 1 | 31.2 | 1.94 | 4.33:1.00:0.0559:5.59 |
3.8 | 56.1 ± 0.7 | −1.57 ± 0.44 |
From Table 1, the degree of branching varies depending on the reaction conditions. At the highest reactant concentration investigated (0.17 g mol−1, entries 1–3), longer reaction times did not lead to significant changes in the extent of branching. In contrast, at the lower reactant concentration of 0.14 g mol−1, the degree of branching generally increased with reaction time, increasing from ≈2 × 10−4 mol g−1 of polymer after 1 hour (entry 4) to nearly ≈4 × 10−4 mol g−1 after 4 hours (entry 7). Furthermore, whilst only a small increase in molecular weight from ≈49 kDa to ≈62 kDa was observed at 0.17 g mol−1 upon increasing the reaction duration from 1 to 3 hours (entries 1 and 3), a much larger increase of ≈ 37 kDa to ≈ 60 kDa was observed for the lower concentration of 0.14 g mol−1 over the same duration. Generally, these differences are consistent with the greater rate of reactions between alcohols and isocyanates at the higher concentration57,58 to give a higher degree of polymerization. In the more concentrated reactions, this results in smaller quantities of unreacted isocyanate groups after an hour which are available for further urethane or allophanate formation. Indeed, the polyurethanes were able to self-assemble into micelles in dilute aqueous solutions (1 wt/v%), with micelle diameters ranging from 56–82 nm (Table 1). Generally, micelle sizes increased with larger molecular weights within each series. For the series comprising entries 1–3, where the polymers were synthesised at a concentration of 0.17 g ml−1, the micelle sizes increased from ≈60 nm to ≈80 nm as molecular weight increased from ≈49 kDa to ≈62 kDa. Similarly, for entries 4–7 where the polymers were synthesised at a concentration of 0.14 g ml−1, the micelle sizes increased from ≈58 nm to 80 nm as molecular weight increased from ≈37 kDa to ≈60 kDa. In addition, zeta potential measurements (Table 1) indicate that the micelles show a slight negative surface charge, possibly a result of a small degree of PCL hydrolysis occurring to generate ionised hydrophilic carboxylate groups.
Other than the molecular weight and branching density of the polyurethanes, the distribution of hydrophobic (PPG + PCL) and hydrophilic (PEG) blocks within the polymers can have profound implications on their micellization behaviour. Generally, secondary alcohols (such as those found on PPG) are known to react more slowly with isocyanates compared to primary alcohols (e.g. those in PEG) both in the absence59 and presence60,61 of Lewis acid catalysts. If PEG is preferentially reacted before PPG, this can result in the formation of a tapered diblock-copolymer structure, with a greater number of hydrophilic PEG blocks at one end of the polymer and more hydrophobic PPG blocks at the other. Thus, to ascertain the distribution of hydrophilic and hydrophobic blocks within our polyurethanes, we investigated the time-dependent evolution of block incorporation within the growing polymer chain. Polyurethane formation under identical reaction conditions were performed using a 2:1:3 molar ratio of PEG, PPG and HMDI,‡ catalysed by DBTL in anhydrous toluene at 110 °C. At regular reaction durations, a small aliquot of the reaction mixture was extracted and precipitated in hexane, and the oligomer/polymer was isolated by filtration. As the PPG macromonomer and HMDI reactant are miscible with hexane, any unreacted PPG and HMDI were removed from the mixture during filtration. Hence, any PPG and HMDI present in the isolated polymer would have been already integrated into the growing polymer chains. If PEG reacts much faster than PPG to form a tapered block-copolymer structure, there will be initially very low PPG incorporation compared to HMDI in the early stages of the reaction, which can be quantified by 1H NMR spectroscopy. As the reaction progresses, the molar ratio of PPG to HMDI blocks, will gradually increase with reaction duration to reach a theoretical maximum of 0.33 when complete monomer incorporation has been achieved. Conversely, the formation of a completely random block copolymer, without any preference for either PEG or PPG, will have a consistent theoretical PPG-to-HMDI molar ratio of 0.33 regardless of the extent and duration of the reaction.
The results of the experiment are shown in Fig. 2 and Table S1, ESI.† Even after short reaction durations at the start of polymer growth, a PPG-to-HMDI molar ratio of approximately 0.3 was obtained, which varied minimally throughout the course of the reaction. This indicates that even from the beginning, the reactions of isocyanates with PEG was not favored over PPG, and hence both components are very likely to be randomly distributed throughout the polymer structure, including any branches which may have formed. It is likely that at the elevated temperatures (110 °C) of the reaction, the rapid rates of nucleophilic addition catalysed by DBTL would have rendered any inherent differences in rate of reaction between primary and secondary alcohols too small to be significant. Hence, differences in subsequent micellization behavior and thermogel properties are likely to stem primarily from inherent differences in polymer molecular weight and branching density, rather than distribution of PEG and PPG segments along the polymer backbone.
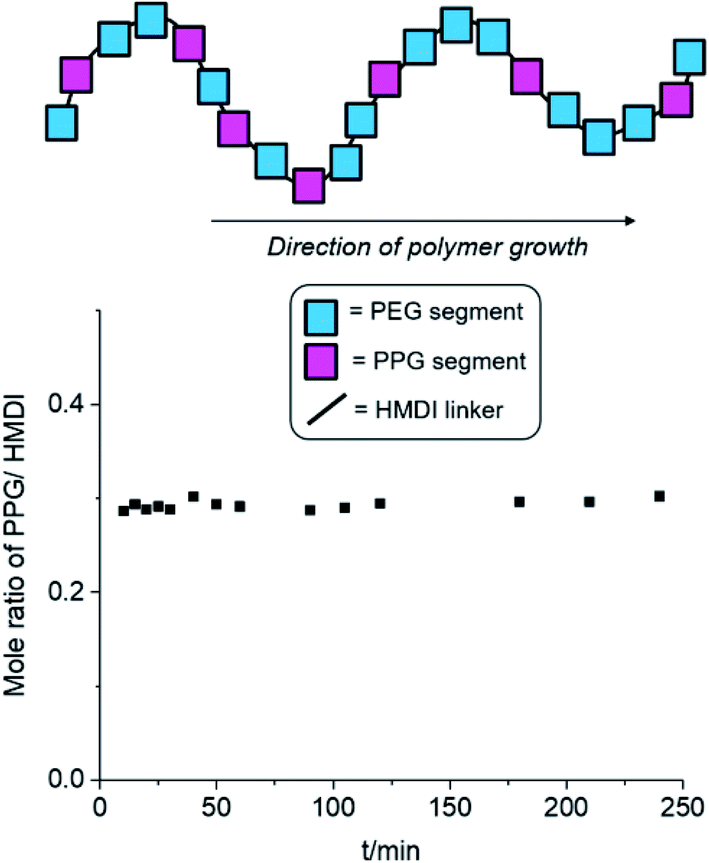 | ||
| Fig. 2 Changes in PPG-to-HMDI molar ratio of the growing polymer chain at different reaction durations. Synthesis of polyurethanes were performed using a 2:1:3 molar ratio of PEG, PPG and HMDI respectively at 110 °C in anhydrous toluene, catalysed by DBTL (see Table S1, ESI,† for further details). | ||
Effects of polymer branching on micellisation behaviour
The CMCs of the branched polyurethanes were determined using the dye solubilization method at 25 °C using the hydrophobic dye 1,6-diphenyl-1,3,5-hexatriene (DPH), as detailed in the Experimental section. The absorption coefficient of DPH at 378 nm increases in a hydrophobic environment (e.g. within the hydrophobic micelle core) compared to that in water.62 By varying the polymer concentration between 0.001 to 1.000 wt/v% at a fixed concentration of DPH, the CMC of the polymer was determined by monitoring the changes in DPH absorbance as a function of polymer concentration (Fig. 3 and Section S2, ESI†).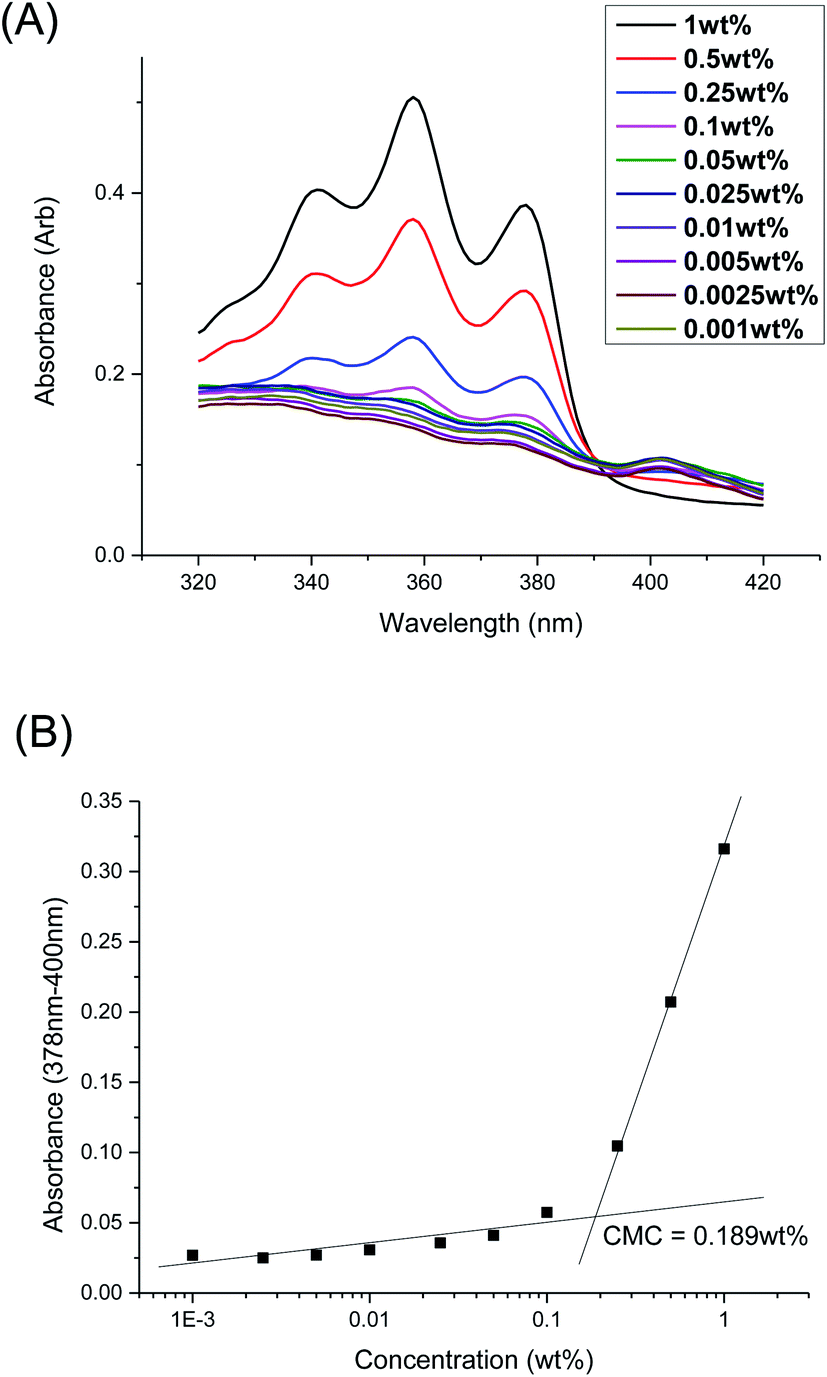 | ||
| Fig. 3 (A) UV-Vis absorbance spectra of DPH dye at different concentrations of polymer 1P30-1h at 25 °C; (B) difference in DPH absorbance at 378 and 400 nm as a function of polymer concentration of 1P30-1h at 25 °C. The CMC was determined by the intersection of the extrapolated linear best fit lines at low and high concentrations of polymer. | ||
Table 2 summarises the influences of polymer structure (molecular weight and branching density) on the properties of the micelles and resulting thermogels. Generally, polyurethanes of higher molecular weights result in lower CMCs, which is expected since the greater number of hydrophobic segments incorporated into the polymer structure will increase the overall hydrophobicity of the polymer.46,47 1P40-1h (entry 1) with the lowest molecular weight of 31.2 kDa is noted to have the one of the highest CMC value (0.173 wt%) amongst the samples while 1P35-3h (entry 6) with one of the highest molecular weight (60.0 kDa) has the lowest CMC value of 0.0747 wt%. However, polymers possessing high branching densities, such as 1P30-1h (Table 2, entry 2), 1P30-3h (Table 2, entry 4), and 1P30-2h (Table 2, entry 7) are notable exceptions to the trend as they have higher than expected CMC values. Despite having higher molecular weights as compared to their previous entries, they are noted to have higher CMCs and this may be attributed to their high branching densities (4.6 × 10−4 mol g−1 branches for 1P30-1h, 4.7 × 10−4 mol g−1 branches for 1P30-3h, and 3.7 × 10−4 mol g−1 branches for 1P30-2h). These results clearly demonstrate that polymer branching can exert important influences on the self-assembly of the amphiphilic polyurethane polymers into micelles in water. It is likely that the larger number of branches along the polymer backbone may sterically hinder the association of hydrophobic segments on adjacent polymer molecules, necessitating higher polymer concentrations for micelle formation.53,63
| S/N | Polymer name | Mn (kDa) | Degree of branch-ing (10−4 mol g−1) | CMC (wt%) | ΔH of micellsation (kJ mol−1) | ΔS of micellesation at 25 °C (kJ mol−1 K−1) | ΔG of micellesation at 25 °C (kJ mol−1) | Gelation temperature Tgela (°C) | Storage modulus of gel at 37 °Ca (Pa) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined by temperature-sweep rheology using 7 wt/v% solution of the polyurethanes in deionized water in all cases. | |||||||||
| 1 | 1P40 1h | 31.2 | 3.8 | 0.173 | 47.4 | 0.276 | −34.8 | 31.7 | 92.5 |
| 2 | 1P30 1h | 49.6 | 4.6 | 0.189 | 37.1 | 0.241 | −34.8 | 25.7 | 153 |
| 3 | 1P35 4h | 49.6 | 3.9 | 0.143 | 46.4 | 0.276 | −35.6 | 17.6 | 529 |
| 4 | 1P30 3h | 57.0 | 4.7 | 0.166 | 46.4 | 0.274 | −35.3 | 15.0 | 550 |
| 5 | 1P35 2h | 59.1 | 2.6 | 0.147 | 46.3 | 0.274 | −35.2 | 18.1 | 439 |
| 6 | 1P35 3h | 60.0 | 2.8 | 0.0747 | 27.3 | 0.217 | −37.5 | 17.4 | 544 |
| 7 | 1P30 2h | 62.2 | 3.7 | 0.147 | 54.7 | 0.303 | −35.5 | 20.0 | 335 |
The thermodynamics of micelle formation for each polyurethane were probed using Arrhenius plots, where the ln(XCMC) of the polymers were plotted as a function of T−1/K−1 (as shown for 1P30-1h in Fig. 4). The standard enthalpy (ΔH) and entropy of micellization (ΔS) can be extracted from the Arrhenius plot as detailed in the Experimental section, and the data are summarized in Table 2 (see Section S3, ESI† for the Arrhenius plots for the other polymers). In all cases, the negative values for the free energies of micellization (ΔG) indicates spontaneous formation of thermodynamically-stable micelles. Micelle formation is driven exclusively by entropy, as seen from the positive ΔS values, whilst the endothermic positive ΔH values indicates that the transfer of unimers from solution to micelles are enthalpically-disfavored. These observations are consistent with micelle formation being driven by desolvation of the hydrophobic polymer segments, where energy uptake during breakage of the unimer–water interactions is accompanied by a large increase in entropy accompanying the release of water molecules into the bulk solvent.62 Furthermore, enthalpy–entropy compensation was observed in all cases: more endothermic ΔH values are accompanied by a larger increase in ΔS. This can be understood as increased polymer interactions during micelle formation also drives a greater extent of desolvation of the polyurethane unimers.
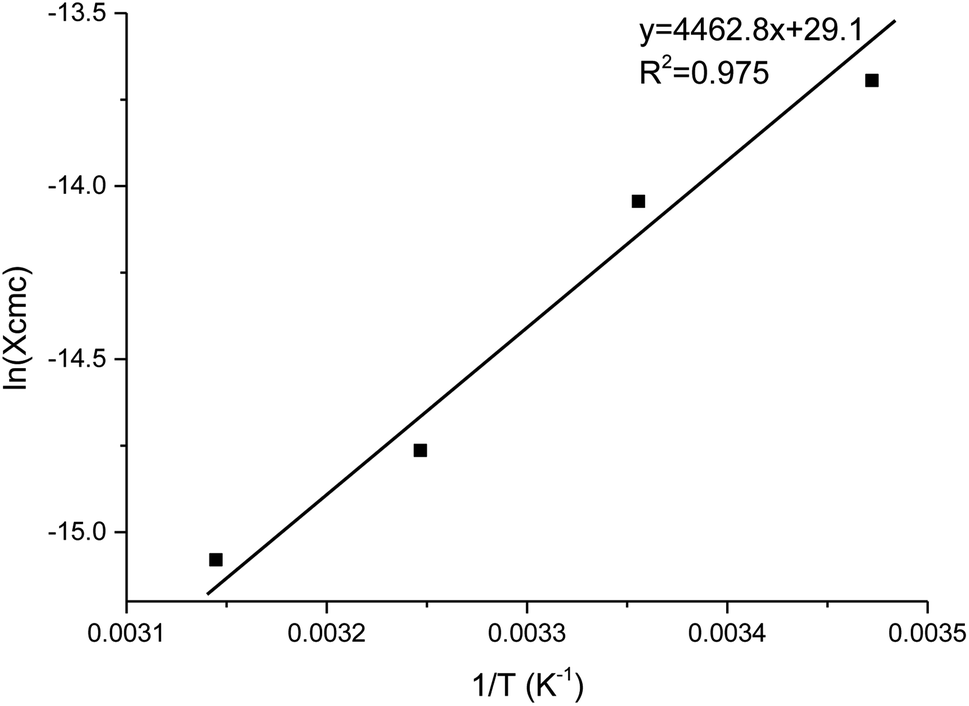 | ||
| Fig. 4 Arrhenius plot of ln(XCMC) versus T−1/K−1 for polyurethane 1P30-1h. The CMC (in mole fractions), or XCMC, was determined at 15, 25, 35 and 45 °C. A best fit line is drawn across and the standard enthalpy of micellisation (ΔH) is given by the product of the gas constant and the gradient. | ||
Like the CMC values, branching appears to affect the thermodynamics of micellization for polymers of similar molecular weights significantly. For samples of similar molecular weight, higher branching density leads to less endothermic enthalpy and also less positive entropy. This can be understood as higher branching density leads to better hydration of the polymer, thus less structured water is released from the polymer during micellisation, and this resulted in less heat being absorbed and less gain in entropy during micellisation. 1P30-1h (entry 2) and 1P35-4h (entry 3) have very similar molecular weights, but 1P30-1h (entry 2, 4.6 × 10−4 mol g−1) has higher branching density than 1P35-4h (entry 3, 3.9 × 10−4 g mol−1) and thus it is also observed that 1P30-1h (entry 2) has a less endothermic enthalpy and a less positive entropy gain, which is consistent with reasoning that higher branching density results in better polymer hydration. This is also supported by the CMC measurements; 1P30-1h (entry 2, 0.189 wt%) has a higher CMC value than 1P35-4h (entry 3, 0.143 wt%) which suggests greater hydrophilicity of 1P30-1h copolymer.
From Table 2, it is noted that 1P35-4h (entry 3) and 1P30-3h (entry 4) have very similar enthalpy of micellisation (∼+46.4 kJ mol−1) and very similar gain in entropy (∼0.27 kJ mol−1 K−1) but 1P30-3h (entry 4, 57.0 kDa) has a significantly higher molecular weight than 1P35-4h (entry 3, 49.6 kDa). It would be expected that 1P30-3h (entry 4) have a more endothermic enthalpy of micellisation and a larger gain in entropy due to its higher molecular weight and hence higher hydrophobicity. This is because more structured water is expected to be released from a more hydrophobic polymer and this would result in a higher amount of energy being absorbed during micellisation and also a greater gain in entropy. However, the results suggest that 1P30-3h's (entry 4) enthalpy of micellisation is less endothermic than expected and the gain in entropy is smaller than expected. This may be attributed to the higher branching density on 1P30-3h (entry 4, 4.7 × 10−4 mol g−1), which is likely to influence the overall degree of polymer hydration as well as the abilities of individual unimers to interact with each other during micellisation.
However, branching density is not the only factor that affects the thermodynamics of micellisation. For instance, factors such as the location of branches along the polymer backbone and their individual lengths are likely to also affect the self-assembly of unimers, and possibly the shapes and distributions of micelle sizes as well. From Table 2, 1P35-2h (entry 5) is a notable exception because although it has higher molecular weight (59.1 kDa) than 1P30-3h (entry 4, 57.0 kDa) and 1P35-4h (entry 3, 49.6 kDa); with the lowest branching density, all three samples are found to have similar enthalpies of micellisation (∼+46 kJ mol−1) and similar entropy gain (∼+0.27 kJ mol−1 K−1). It would be expected for 1P35-2h (entry 5) to have the most endothermic enthalpy of micellisation and greatest gain in entropy due to its largest molecular weight and lowest branching density. However, the observation that 1P35-2h has similar enthalpy and entropy of micellisation as 1P30-3h and 1P35-4h suggests the influence of the other aforementioned factors.
Effects of polymer branches on thermogel properties
Gelation is brought about by the aggregation and packing of the micelles into a three-dimensional matrix, and the micelle self-assembly mechanism depends heavily on the structure of the individual polymer constituents. We investigated how the molecular weight and branches of the polyurethanes affected the physical properties of the thermogels by performing rheological studies on all polyurethanes. Fig. 5 shows the rheological profile of a 7 wt/v% solution of the 1P30-1h polymer in deionised water. At low temperatures, the solution exists as a flowable mildly-viscous liquid-like state, reflected by the smaller values of the storage modulus (G′) compared with the loss modulus (G′′). As the temperature increases, the magnitude of G′ increases to a larger extent than G′′, indicating a rise in solution viscosity. Eventually, the curve of G′ intersects that of G′′ as the former's magnitude exceeds the latter, which shows that the solution now exists as a semi-solid state as a gel with a greater capacity to store deformation energy elastically. The temperature at which G′ intersects G′′ is known as the critical gelation temperature (Tgel). All polyurethane samples, regardless of degree of branching, showed temperature-responsive thermogelling properties, as confirmed by temperature-sweep rheology experiments (shown in Section S4, ESI†). The values of Tgel/°C and G′/Pa at the physiological temperature of 37 °C, which gives a measure of the stiffness of the gel, are summarized in Table 2.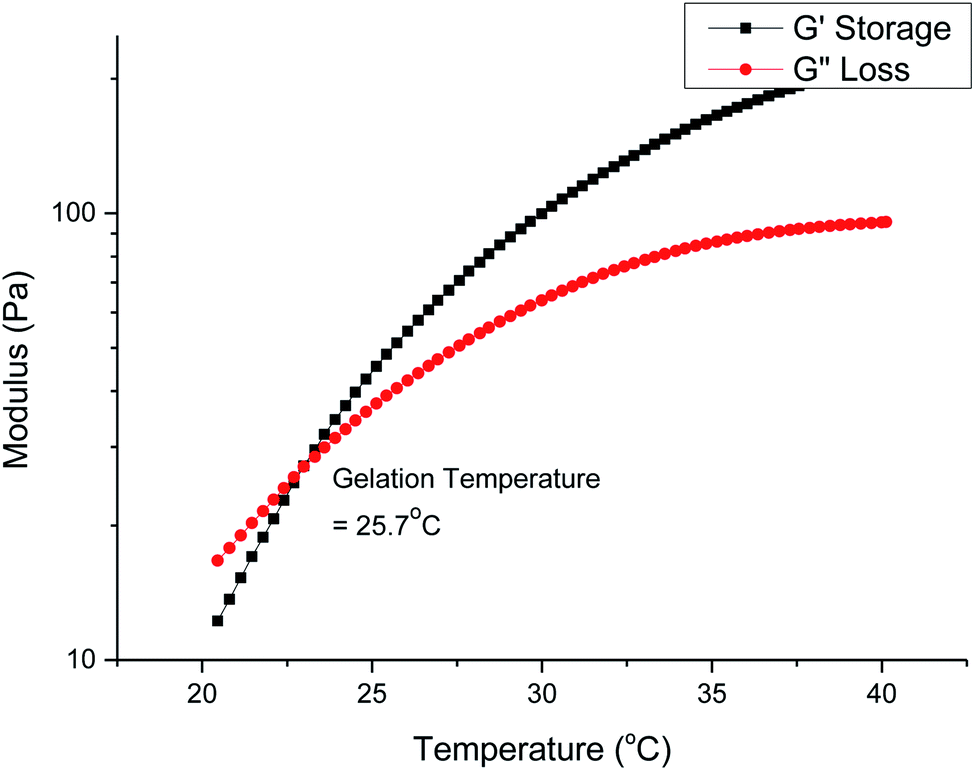 | ||
| Fig. 5 Temperature-sweep rheology for a 7 wt/v% solution of 1P30-1h in deionised water. Gelation occurs at the temperature when the storage modulus first becomes larger than the loss modulus and in this case it is 25.7 °C. | ||
From Table 2, higher polymer molecular weight is generally correlated with a lower Tgel (Fig. 6A) and stiffer gels (i.e. larger G′ values) and this is expected because higher molecular weight copolymers have higher propensity to form micelles and are also more likely to entangle with each other to form the three-dimensional supramolecular hydrogel matrix. This relationship generally holds true for polymers with similar branching densities but varying molecular weights. 1P30-1h (entry 2) has similar branching density with 1P30-3h (entry 4) of ∼4.6 × 10−4 mol g−1 but it has a lower molecular weight (49.6 kDa) than 1P30-3h (57.0 kDa). As expected, 1P30-3h has a ∼10 °C lower gelation temperature as compared to 1P30-1h and also a much higher storage modulus at 37 °C (550 Pa compared to 153 Pa), indicating that 1P30-3h gelates more readily and also forms a stiffer gel than 1P30-1h. A similar observation is noted between 1P40-1h (entry 1) and 1P35-4h (entry 3): both polymers have similar branching densities of ∼3.8 × 10−4 mol g−1 but the former has a lower molecular weight (31.2 kDa) than the latter (49.6 kDa). Unsurprisingly, 1P35-4h has a 14 °C lower gelation temperature as compared to 1P30-1h and it also forms a much stiffer gel than 1P30-1h (storage modulus at 37 °C: 529 Pa compared to 92.5 Pa). However, polymers 1P35-2h (entry 5) and 1P35-3h (entry 6) have very similar molecular weights (∼60 kDa) and branching densities (∼2.7 × 10−4 mol g−1), and as expected, they have very similar gelation temperatures (18.1 °C for 1P35 2h and 17.4 °C for 1P35-3h) and gel stiffness (439 Pa and 544 Pa respectively).
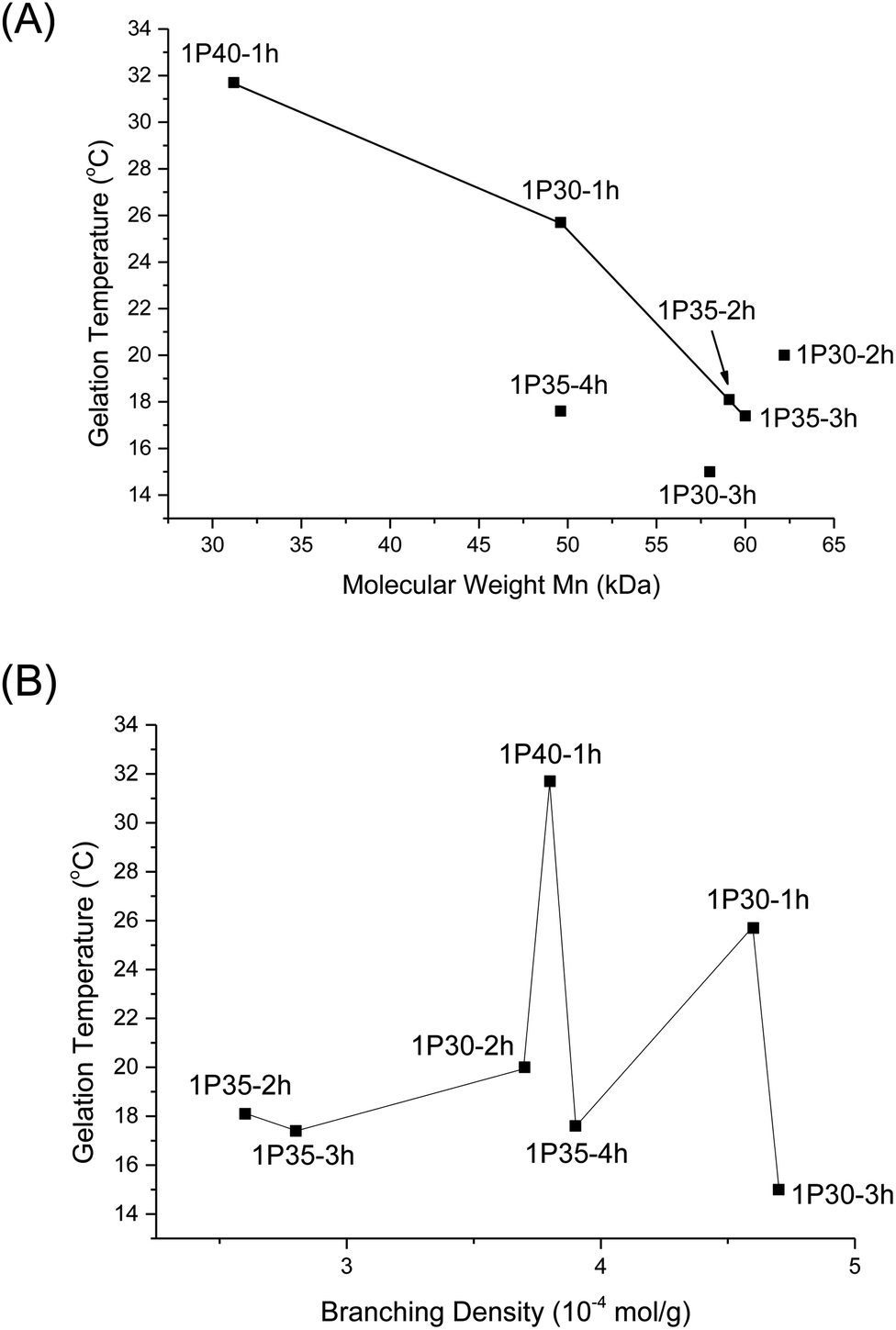 | ||
| Fig. 6 (A) Plot of Tgel of different polyurethanes as a function of polymer molecular weight (Mn); (B) plot of Tgel of different polyurethanes as a function of increasing degrees of branching. | ||
Nonetheless, 1P30-2h (Table 2, entry 7) is a notable exception that illustrates the complex relationships between molecular weight, branching, and gel property. 1P30-2h has similar branching density (∼3.8 × 10−4 mol g−1) as compared to 1P40-1h (entry 1) and 1P35-4h (entry 3) but 1P30-2h has the highest molecular weight (62.2 kDa) amongst the three. It would be expected that 1P30-2h has the lowest gelation temperature among the three and also forms the stiffest gel. However surprisingly, 1P30-2h has in fact a slightly higher gelation temperature (by ∼2 °C) and also lower stiffness as compared to 1P35-4h (529 Pa for 1P35-4h compared to 335 Pa for 1P30-2h). This may be due to other factors such as the location of branches along the polymer backbone, their individual lengths, as well as branching density. It is plausible that the branches on 1P30-2h (entry 7) has sterically hindered the formation of micelles and their subsequent aggregation, thereby resulting in a weaker gel.
On the other hand, no obvious correlation is seen between degree of branching and thermogel properties (Fig. 6B). This is understandable because the gel properties (gelation temperature and gel stiffness) is predominantly controlled by molecular weight though the branches may alter the overall gel properties. The effect of branch density on gel properties can be explored by comparing polymers of similar molecular weight but of different branching density. From Table 2, 1P30-1h (entry 2) and 1P35-4h (entry 3) have similar molecular weight of ∼49.6 kDa but 1P30-1h has higher branching density than 1P35-4h (4.6 × 10−4 mol g−1 compared to 3.9 × 10−4 mol g−1 branches), resulting in a significantly weaker gel with a higher gelation temperature (by ∼8 °C) and reduced stiffness (153 Pa compared to 529 Pa). This suggests that the higher branching density on 1P30-1h may have caused significantly higher steric hindrance and increased the difficulty of micelle formation and also gelation. This is supported by the fact that 1P30-1h also has a higher CMC value as compared to 1P35-4h (0.189 wt% compared to 0.143 wt%). The same relationship is observed between 1P35-3h (entry 6) and 1P30-2h (entry 7). Both polymers have similar molecular weights (∼60 kDa) but 1P30-2h has higher branching density (3.7 × 10−4 mol g−1) than 1P35-3h (2.8 × 10−4 mol g−1). 1P30-2h has slightly higher gelation temperature (∼2.5 °C) and forms a slightly weaker gel as compared to 1P35-3h (335 Pa compared to 544 Pa), with a higher CMC value as well (0.147 wt% compared to 0.0747 wt%).
Regardless of the amphiphilic nature of all known thermogelling polymers,64 a few distinct micellar aggregation mechanisms have been suggested for gel formation which are highly-dependent on the primary structure of the polymer. Amphiphilic tri-block polymers with hydrophilic-hydrophobic-hydrophilic structure, such as Pluronic F127, are likely to form micelles which pack together tightly and orderly to form the hydrogel matrix that entraps water (Fig. 7A).65 Alternatively, triblock hydrophobic–hydrophilic–hydrophobic polymers may achieve gelation by inserting their hydrophobic segments into separate adjacent micelles and connecting them together into a stable hydrogel matrix44,64,66,67 (Fig. 7B). For diblock copolymers with longer hydrophobic segments, ‘semi-bald’ micelles are likely to form as the hydrophilic segments are not long enough to completely envelope the hydrophobic micelle interior. These micelles can then aggregate via hydrophobic association to form a percolated matrix (Fig. 7C).45 Unlike these linear block-copolymer amphiphiles however, our branched multi-block polyurethanes contain a greater proportion of hydrophilic PEG groups over hydrophobic PPG and PCL groups randomly-distributed throughout the polymer structure, including along their branches. The complexity of the polymer structures studied herein result in unique micelle aggregation mechanism distinct from the aforementioned three, and may contain a combination of features from Pluronic-like and ‘micelle-bridging’ mechanisms. For instance, as shown in Fig. 8, it is possible for branches to fold back onto itself for the hydrophobic segments to interact intramolecularly with those on the main polymer chain. Under such circumstances, the polymer branches act to sterically hinder self-assembly of hydrophobic polymer segments for micelle formation and consequently gel formation. Conversely, some of these hydrophobic segments on branches protruding from the micelles may also interact intermolecularly with polymers on other similarly “hairy” micelles to form crosslinking bridges, or even penetrating the hydrophobic cores of adjacent micelles. Indeed, the contrasting effects of branching on reducing assembly at micelle level and potentially increasing micelle bridges at the gel level makes drawing overall correlations between degree of polymer branching and thermogels properties difficult. Nonetheless, the general trend of longer polymers forming stronger thermogels may be accounted for by the greater propensity to form micelles and the generally greater numbers of inter-micelle interactions statistically possible.
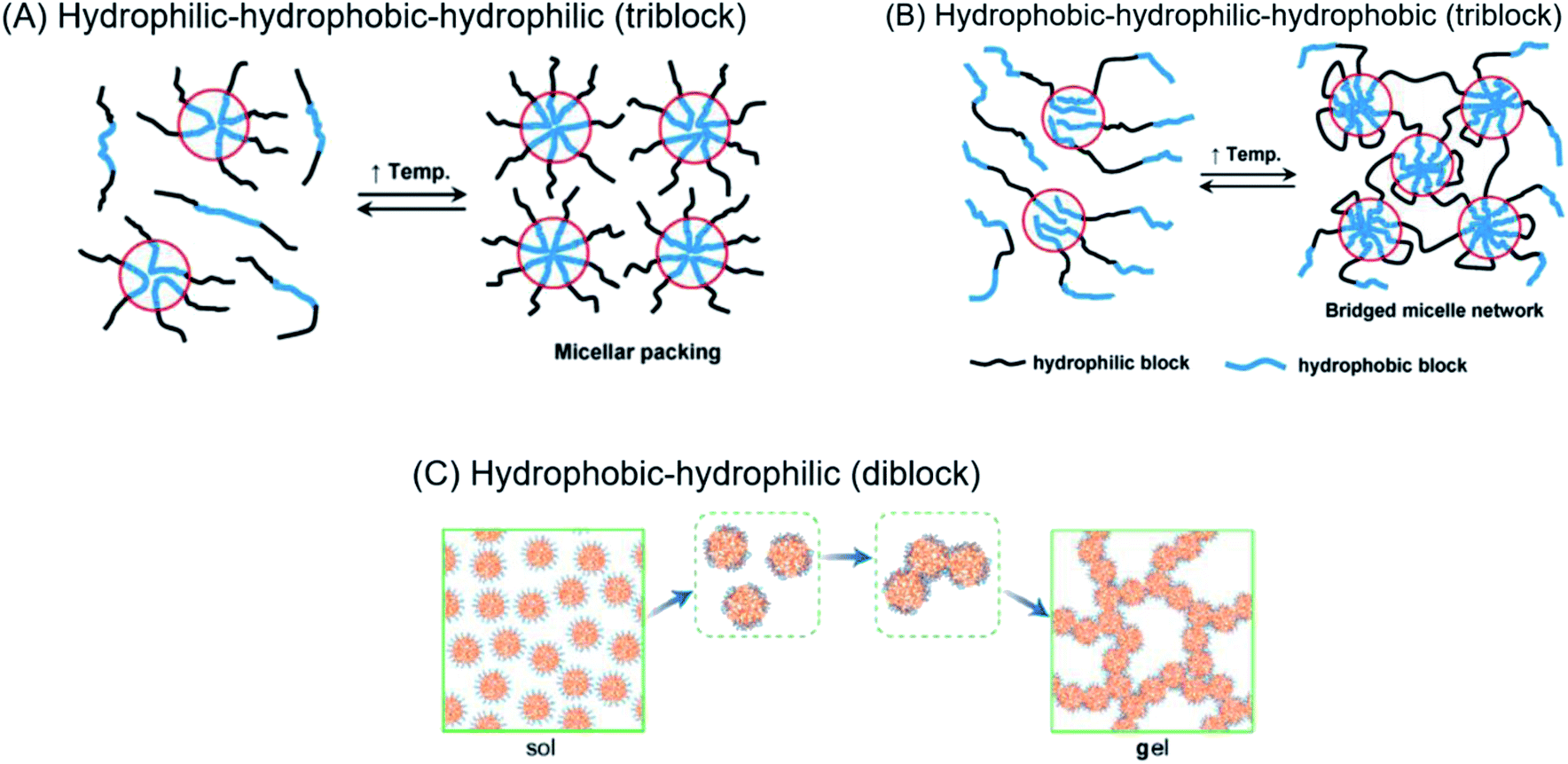 | ||
| Fig. 7 (A) Micelle formation and gelation via micelle packing (or jamming) for amphiphilic copolymers with hydrophilic–hydrophobic–hydrophilic structure. (B) Micelle formation and gelation via micelle bridging for amphiphilic copolymers with hydrophobic–hydrophilic–hydrophobic structure. Reprinted with permission from ref. 64 (C) micelle formation and gelation via aggregation of semi-bald micelles to form a percolated micelle network. Reprinted with permission from ref. 45. Copyright 2020 American Chemical Society. | ||
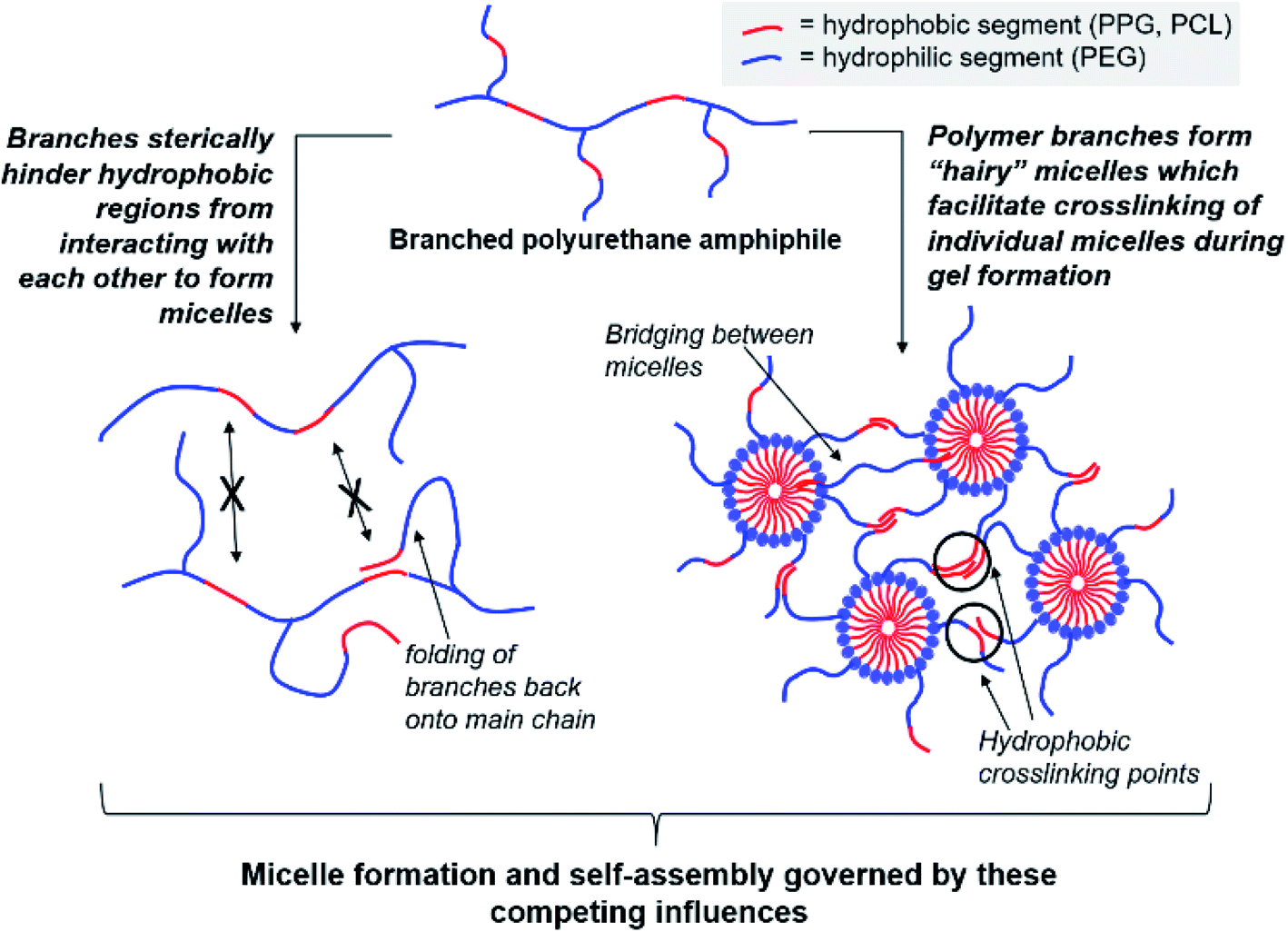 | ||
| Fig. 8 Competing effects of polymer amphiphile branching on micelle formation and self-assembly to form thermogels. | ||
Conclusion
In conclusion, branched poly(PEG/PPG/PCL urethane) polymeric amphiphiles can be produced when isocyanates react with urethane functional groups to form trifunctional allophanate linkages. The degree of branching and molecular weight of our polyurethanes may be tuned by varying the reaction concentrations and durations. At lower reaction concentrations, longer durations result in greater increases in molecular weight and degree of branching, compared to higher reaction conditions. The resulting multiblock polymers, which contained a random distribution of hydrophilic and hydrophobic segments along the polymer backbone and its branches, were capable of self-assembling into micelles and thermogels at elevated temperatures. Both molecular weights and degree of polymer branching exerts important influences on the CMCs, thermodynamics of micellisation and eventual thermogel properties. Longer polymers resulted in lower CMCs, gelation temperatures and stronger thermogels. However, a greater degree of polymer branching could reduce the CMC, which may be attributed to steric hindrance of the intermolecular polymer–polymer interactions necessary for micelle formation. However, the influences of polymer branching on the thermodynamics of micellisation and thermogel properties are less obvious, owing to the complex multilevel assembly and interactions of the polymers to first form micelles, then bulk thermogels. Further studies to understand the mechanism of gelation of our random multiblock copolymer micelles are underway.Experimental section
General considerations
Poly(ethylene glycol) of 2050 Da, poly(propylene glycol) of 2000 Da, poly(ε-caprolactone)-diol of 2000 Da, hexamethylene diisocyanate (HMDI) 99%, dibutyltin dilaurate 95% were purchased from Sigma Aldrich and used as received without further purification. Anhydrous toluene and anhydrous diethyl ether are sourced from TEDIA. Toluene was further dried and stored over pre-activated 4 Å molecular sieves.Gel permeation chromatography (GPC) was conducted on a Viscotek TDAmax which consists of three components – the GPCmax integrated solvent and sample delivery module, the TDA 302 Triple Detector Array, and the OmniSEC software. The TDA 302 array incorporates refractive index (RI) and light scattering detectors as well as a viscometer. 2 columns (2 × PLgel 10 μm mixed-B (500 to 10000000)) were applied in sequence for separation. THF was used as the eluent at 1.0 ml min−1 with column and detector temperature at 40 °C. Polystyrene standard (Mw 21960, dn/dc = 0.185, IV = 0.1471, PDI = 1.02) was used for the multi-detector calibration to calibrate the detector constants. Samples' concentration was input to determine their dn/dc and Mw. 1H nuclear magnetic resonance (NMR) spectra were recorded using a JEOL 500 MHz NMR spectrometer (Tokyo, Japan) at room temperature.
Synthesis of poly(PEG/PPG/PCL urethane)
4.00 g of PEG, 1.00 g of PPG, 0.050 g of PCL are weighed into a clean 250 ml round bottom flask. 20 ml of anhydrous toluene is added to dissolve the poly-diols at 60 °C. The polymers were dried by azeotropic distillation using a rotary evaporator twice, before the slurry was further dried under vacuum at 110 °C for 1 hour. Afterwards, the dried slurry was re-dissolved in anhydrous toluene (30 ml, 35 ml, or 40 ml to vary the starting concentrations and named as 1P30, 1P35, and 1P40 respectively) at 110 °C under a dry inert Argon atmosphere. To the stirred solution (stirring rate 300 RPM) was added DBTL (5 μl), followed by HMDI (0.41 ml, with NCO:OH mole ratio of 1.03:1.00), and the reaction was left to stir at 110 °C between 1 to 4 hours. Thereafter, the reaction was quenched with 5 ml of absolute ethanol, and the polymer was isolated by precipitation by slowly pouring the crude reaction mixture into vigorously-stirred anhydrous diethyl ether. The mixture was filtered to obtain the target polyurethanes as white strips, which were then dried in vacuo. Purification of the polymers were subsequently performed by re-dissolving them in CMOS-grade isopropyl alcohol (150 ml) at 60 °C before being transferred to a dialysis tubing (MWCO = 3.5 kDa) and dialysed against 2 l deionised water for three days. The dialysed solution was then frozen and lyophilised to obtain the purified polymer (typical yield = 4.5 g, ≈90%).
Characterization of degree of polymer branching§
A combination of spectroscopic and titrimetric techniques was used to determine the degree of polymer branching. As shown in Fig. 9, while branches originate from the main polymer backbone from allophanate linkages, individual branches can be terminated with either isocyanate (due to the slight excess of NCO compared to OH) or hydroxyl groups.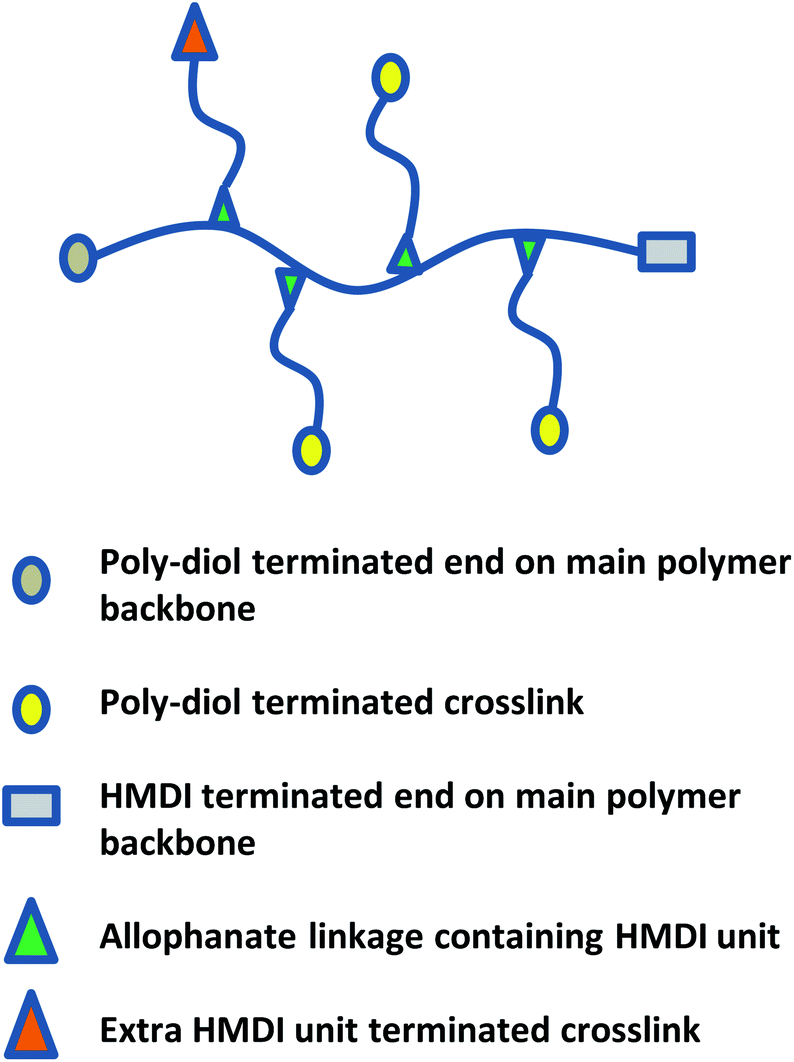 | ||
| Fig. 9 Schematic of poly(PEG/PPG/PCL urethane) polymer structure with crosslinks. | ||
1H NMR spectroscopy was used to quantify the branches terminated by NCO groups (Hc) per unit mass (g) of polymer. This was achieved by first calculating the molar composition of HMDI:PEG:PPG:PCL of the sample via NMR integration of the representative well-resolved resonance peaks at 3.10–3.15 ppm, 3.60–3.70 ppm, 1.10–1.15 ppm and 4.05 ppm respectively. If equimolar quantities of diol and HMDI molecules reacted to form the polymer, every polymer chain should be terminated with an NCO unit at one end and a OH group at the other. Accordingly, all the polymer branches will be terminated by OH groups as the NCO ends would have reacted with the urethanes to form allophanate linkages. Due to the slight excess of NCO units in the reaction, it thus follows that any excess HMDI detected from the NMR peak integrations will have originated from the termini of the branches. The quantity of crosslinks terminated by NCO units per gram of polymer can be calculated by using eqn (1): (derivation and experimental data are provided in Section S5, ESI†).
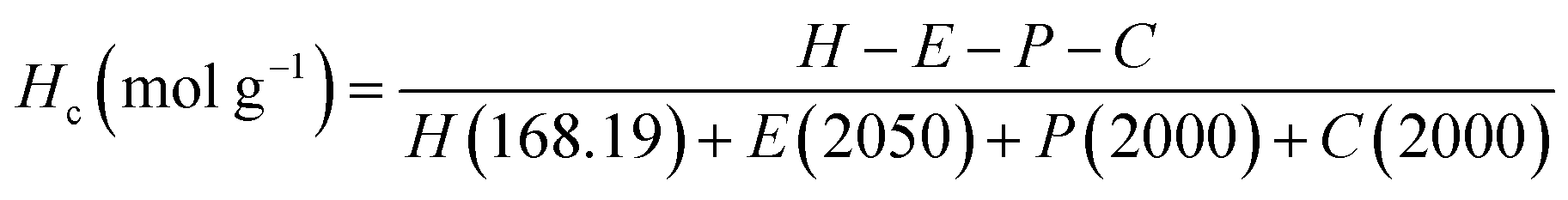 | (1) |
Hc: number of NCO-terminated branches in 1 g of sample.
H, E, P, C: molar composition ratios of HMDI, PEG, PPG, PCL in sample.
The numbers in parentheses are the molecular weight of each (macro)monomer.
The quantification of OH-terminated branches was achieved by titrimetry as previously reported,68,69 following the reaction shown in Scheme 3. Briefly, the reaction of acetic anhydride with an alcohol (ROH) produces an ester whilst liberating a free acetic acid molecule. After reaction of the polymer, the excess acetic anhydride is quenched with water to produce two molecules of acetic acid per molecule of acetic anhydride reacted. Compared to a blank control sample (without polymer), fewer acetic molecules are produced in the presence of alcohols (ROH), and the quantity of acetic acid produced can be determined by titration with aqueous sodium hydroxide in the presence of phenolphthalein as an indicator. The detailed experimental procedure for the titration is provided in Section S5, ESI.† The quantity of OH-terminated branches in 1 g of polymer can be calculated from eqn (2):
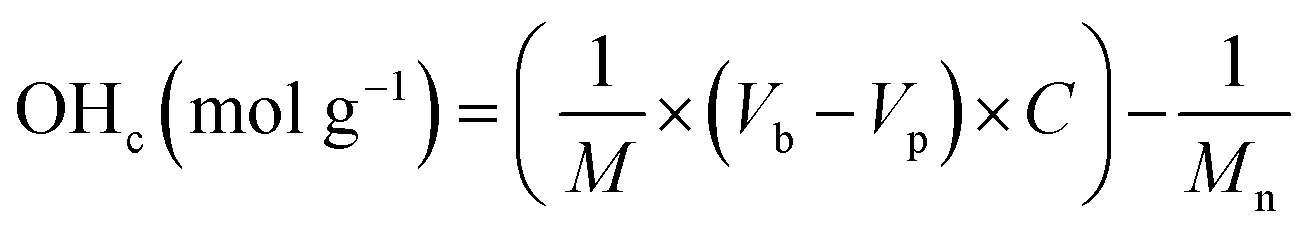 | (2) |
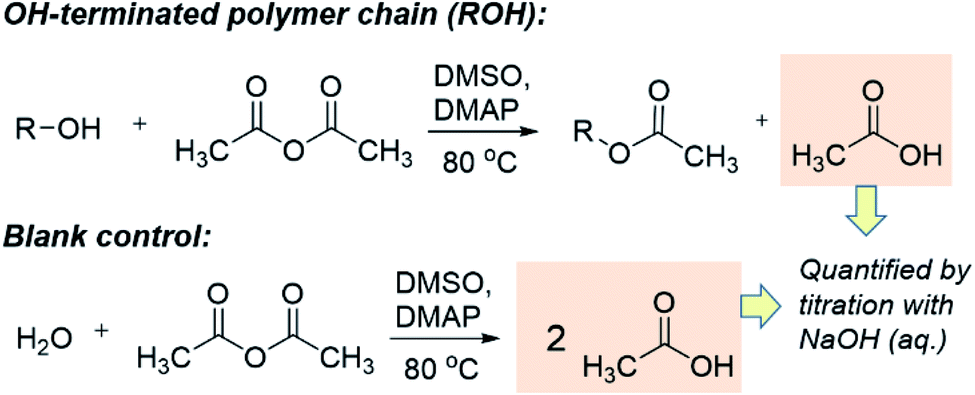 | ||
| Scheme 3 The working principle of the titration experiments with acetic anhydride to determine quantity of OH present. | ||
OHc: amount of OH-terminated branches in 1 g of polymer.
M: mass of sample polymer used for titrations.
Mn: number average molecular weight.
Vb: volume of NaOH (aq.) consumed for blank control reaction.
Vp: volume of NaOH (aq.) consumed for polymer sample. C: concentration of NaOH (aq.).
The total quantity of branches for each polymer = Hc + OHc.
Determination of critical micelle concentration
The critical micelle concentration of poly(PEG/PPG/PCL urethane) copolymers were determined by a dye solubilisation method.22,70 1,6-Diphenyl-1,3,5-hexatriene (DPH) was dissolved in methanol to form a solution. A total of 40 μl of 0.6 mM DPH solution was added to 4.0 ml of aqueous copolymer solution with concentrations ranging from 0.001 wt% to 1.0 wt% and equilibrated for 24 h at room temperature. Absorbance spectra were recorded using a Shimadzu UV-2501 PC UV-Vis spectrophotometer (Kyoto, Japan). Measurements were made in the range of 320–420 nm at 15 °C, 25 °C, 35 °C and 45 °C. The difference in absorbance at 378 nm and 400 nm were plotted against log(concentration). The CMC values of the copolymers were determined by the intersection of the extrapolation of linear fits of unimeric and micellar regimes.Determination of thermodynamic quantities of micellisation
Assuming a closed association of unimers into micelles,70 the thermodynamic parameters related to the micellisation process may be calculated based on the following equations. Free energy of micellisation, ΔG, may be calculated by eqn (3):|
ΔG = RTln(XCMC)
| (3) |
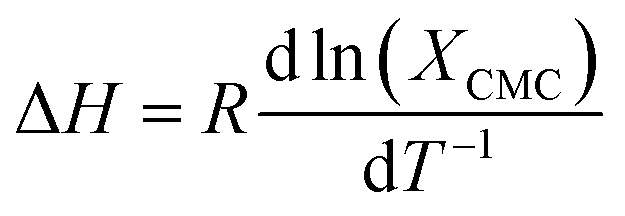
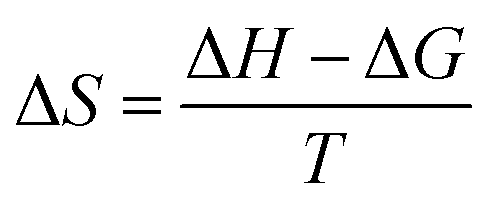
Rheological characterisation
Rheological measurements were performed using a TA Instruments Discovery DHR-3 hybrid rheometer (New Castle, DE, USA) fitted with 40 mm flat-plate geometry and a temperature-controlled Peltier base plate. Temperature sweep measurements were performed at 10–50 °C at a heating rate of 3 °C min, with strain fixed at 0.1% and frequency fixed at 1 rad s−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by an IAF-PP (HMBS Domain) grant H17/01/a0/013 (OrBID): OculaR BIomaterials and Device. The authors would like to acknowledge Zhao Wenguang (Institute of Chemical and Engineering Sciences, ICES) for assistance with triple detection GPC analysis of the polymer samples.References
- S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 CrossRef CAS.
- U. Rauwald, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Commun., 2011, 47, 6000–6002 RSC.
- L. Gan, G. R. Deen, X. Loh and Y. Gan, Polymer, 2001, 42, 65–69 CrossRef CAS.
- G. Barouti, S. S. Liow, Q. Dou, H. Ye, C. Orione, S. M. Guillaume and X. J. Loh, Chem.–Eur. J., 2016, 22, 10501–10512 CrossRef CAS.
- Z.-Y. He, K. Shi, Y.-Q. Wei and Z.-Y. Qian, Curr. Drug Metab., 2016, 17, 168–186 CrossRef.
- M. Patel, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomaterials, 2018, 159, 91–107 CrossRef CAS.
- H. S. Park, S. Y. Jung, H. Y. Kim, S. M. Chung, B. Jeong and H. S. Kim, European Archives of Oto-Rhino-Laryngology, 2016, 273, 3827–3834 CrossRef.
- K. Xue, X. Zhao, Z. Zhang, B. Qiu, Q. S. W. Tan, K. H. Ong, Z. Liu, B. H. Parikh, V. A. Barathi and W. Yu, Biomater. Sci., 2019, 7, 4603–4614 RSC.
- H. Shi, H. Chi, Z. Luo, L. Jiang, X. J. Loh, C. He and Z. Li, Front. Chem., 2019, 7, 683 CrossRef CAS.
- Z. Liu, S. S. Liow, S. L. Lai, A. Alli-Shaik, G. E. Holder, B. H. Parikh, S. Krishnakumar, Z. Li, M. J. Tan and J. Gunaratne, Nat. Biomed. Eng., 2019, 3, 598–610 CrossRef CAS.
- D. Cao, X. Zhang, M. Akabar, Y. Luo, H. Wu, X. Ke and T. Ci, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 181–191 CrossRef CAS.
- Y. Zhang, J. Zhang, W. Xu, G. Xiao, J. Ding and X. Chen, Acta Biomater., 2018, 77, 63–73 CrossRef CAS.
- Y.-L. Wu, H. Wang, Y.-K. Qiu and X. J. Loh, RSC Adv., 2016, 6, 44506–44513 RSC.
- V. M. Shah, D. X. Nguyen, D. A. Rao, R. G. Alany and A. W. Alani, Temp.-Responsive Polym., 2018, 313 CrossRef CAS.
- X. J. Loh, J. Appl. Polym. Sci., 2013, 127, 992–1000 CrossRef CAS.
- X. J. Loh, V. P. N. Nguyen, N. Kuo and J. Li, J. Mater. Chem., 2011, 21, 2246–2254 RSC.
- X. J. Loh, J. Gong, M. Sakuragi, T. Kitajima, M. Liu, J. Li and Y. Ito, Macromol. Biosci., 2009, 9, 1069–1079 CrossRef CAS.
- X. J. Loh, W. C. D. Cheong, J. Li and Y. Ito, Soft Matter, 2009, 5, 2937–2946 RSC.
- V. P. N. Nguyen, N. Kuo and X. J. Loh, Soft Matter, 2011, 7, 2150–2159 RSC.
- Y. L. Wu, H. Wang, Y. K. Qiu, S. S. Liow, Z. Li and X. J. Loh, Adv. Healthcare Mater., 2016, 5, 2679–2685 CrossRef CAS.
- X. J. Loh, W. Guerin and S. M. Guillaume, J. Mater. Chem., 2012, 22, 21249–21256 RSC.
- X. J. Loh, S. H. Goh and J. Li, J. Phys. Chem. B, 2009, 113, 11822–11830 CrossRef CAS.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mat. Today Adv., 2019, 3, 100021 CrossRef.
- S.-J. Wang, Z.-Z. Zhang, D. Jiang, Y.-S. Qi, H.-J. Wang, J.-Y. Zhang, J.-X. Ding and J.-K. Yu, Polymers, 2016, 8, 200 CrossRef.
- Y. Zhang, J. Zhang, F. Chang, W. Xu and J. Ding, Mater. Sci. Eng., C, 2018, 88, 79–87 CrossRef CAS.
- C. Celik, V. T. Mogal, J. H. P. Hui, X. J. Loh and W. S. Toh, in Hydrogels, Springer, Singapore, 2018, pp. 315–337 Search PubMed.
- W.-K. Xu, J.-Y. Tang, Z. Yuan, C.-Y. Cai, X.-B. Chen, S.-Q. Cui, P. Liu, L. Yu, K.-Y. Cai and J.-D. Ding, Chin. J. Polym. Sci., 2019, 37, 548–559 CrossRef CAS.
- M. A. A. Mohamed, V. Raeesi, P. V. Turner, A. Rebbapragada, K. Banks and W. C. Chan, Biomaterials, 2016, 97, 154–163 CrossRef.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS.
- M. Ma, Y. Zhong and X. Jiang, Carbohydr. Polym., 2020, 116096 CrossRef CAS.
- Z. Luo, K. Xue, X. Zhang, J. Y. C. Lim, X. Lai, D. J. Young, Z.-X. Zhang, Y.-L. Wu and X. J. Loh, Biomater. Sci., 2020, 8, 1364–1379 RSC.
- Z. Liu, S. S. Liow, S. L. Lai, A. Alli-Shaik, G. E. Holder, B. H. Parikh, S. Krishnakumar, Z. Li, M. J. Tan, J. Gunaratne, V. A. Barathi, W. Hunziker, R. Lakshminarayanan, C. W. T. Tan, C. K. Chee, P. Zhao, G. Lingam, X. J. Loh and X. Su, Nat. Biomed. Eng., 2019, 3, 598–610 CrossRef CAS.
- W.-S. Huang and I.-M. Chu, PloS One, 2019, 14 Search PubMed.
- H. J. Lee and B. Jeong, Small, 2019, 1903045 Search PubMed.
- X. Li, L. Chen, H. Lin, L. Cao, J. a. Cheng, J. Dong, L. Yu and J. Ding, Clin. Spine Surg., 2017, 30, E283–E290 CrossRef.
- H. Liu, Y. Cheng, J. Chen, F. Chang, J. Wang, J. Ding and X. Chen, Acta Biomater., 2018, 73, 103–111 CrossRef CAS.
- K. Shi, Y.-L. Wang, Y. Qu, J.-F. Liao, B.-Y. Chu, H.-P. Zhang, F. Luo and Z.-Y. Qian, Sci. Rep., 2016, 6, 19077 CrossRef CAS.
- B. Q. Y. Chan, H. Cheng, S. S. Liow, Q. Dou, Y.-L. Wu, X. J. Loh and Z. Li, Polymers, 2018, 10, 89 CrossRef.
- S. S. Liow, Q. Dou, D. Kai, Z. Li, S. Sugiarto, C. Y. Y. Yu, R. T. K. Kwok, X. Chen, Y. L. Wu and S. T. Ong, Small, 2017, 13, 1603404 CrossRef.
- C. Y. Wee, S. S. Liow, Z. Li, Y. L. Wu and X. J. Loh, Macromol. Chem. Phys., 2017, 218, 1700196 CrossRef.
- K. Xue, S. S. Liow, A. A. Karim, Z. Li and X. J. Loh, Chem. Rec., 2018, 18, 1517–1529 CrossRef CAS.
- P. Huang, H. Song, Y. Zhang, J. Liu, Z. Cheng, X.-J. Liang, W. Wang, D. Kong and J. Liu, Biomaterials, 2017, 145, 81–91 CrossRef CAS.
- O. Cally, D. J. Young and X. J. Loh, 2018.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2019, 52, 3697–3715 CrossRef CAS.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2018, 51, 6405–6420 CrossRef CAS.
- L. Chen, T. Ci, T. Li, L. Yu and J. Ding, Macromolecules, 2014, 47, 5895–5903 CrossRef CAS.
- L. Chen, T. Ci, L. Yu and J. Ding, Macromolecules, 2015, 48, 3662–3671 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Chang, Macromol. Rapid Commun., 2001, 22, 587–592 CrossRef.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J. P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- K. Dusek, M. Spirkova and I. Havlicek, Macromolecules, 1990, 23, 1774–1781 CrossRef CAS.
- R. P. Houghton and A. W. Mulvaney, J. Organomet. Chem., 1996, 518, 21–27 CrossRef CAS.
- M. Liu, J.-C. Leroux and M. A. Gauthier, Prog. Polym. Sci., 2015, 48, 111–121 CrossRef CAS.
- Z. Li and J. Li, J. Phys. Chem. B, 2013, 117, 14763–14774 CrossRef CAS.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS.
- K. Dušek, M. Špírková and M. Ilavský, Makromol. Chem., Macromol. Symp., 1991, 45, 87–95 CrossRef.
- E. G. Lovering and K. J. Laidler, Can. J. Chem., 1962, 40, 31–36 CrossRef CAS.
- A.-C. Draye, D. Tarasov and J.-J. Tondeur, React. Kinet. Catal. Lett., 1999, 66, 199–204 CrossRef CAS.
- S. Ephraim, A. Woodward and R. Mesrobian, J. Am. Chem. Soc., 1958, 80, 1326–1328 CrossRef CAS.
- A. Oberth and R. Bruenner, Ind. Eng. Chem. Fundam., 1969, 8, 383–388 CrossRef CAS.
- L. Rand, B. Thir, S. Reegen and K. Frisch, J. Appl. Polym. Sci., 1965, 9, 1787–1795 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- J. R. Finnegan, D. J. Lunn, O. E. Gould, Z. M. Hudson, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 13835–13844 CrossRef CAS.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Macromolecules, 2011, 44, 6629–6636 CrossRef CAS.
- T. Nicolai, F. Laflèche and A. Gibaud, Macromolecules, 2004, 37, 8066–8071 CrossRef CAS.
- P. Wang, W. Chu, X. Zhuo, Y. Zhang, J. Gou, T. Ren, H. He, T. Yin and X. Tang, J. Mater. Chem. B, 2017, 5, 1551–1565 RSC.
- R. Chen, J. Hao, C. Xiong and X. Deng, Adv. Eng. Mater., 2010, 12, B504–B510 CrossRef.
- S. Marks and R. Morrell, Analyst, 1931, 56, 428–429 RSC.
- M. A. Carey, S. L. Wellons and D. K. Elder, J. Cell. Plast., 1984, 20, 42–48 CrossRef CAS.
- X. J. Loh, Y. X. Tan, Z. Li, L. S. Teo, S. H. Goh and J. Li, Biomaterials, 2008, 29, 2164–2172 CrossRef CAS.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07426h |
| ‡ Due to the small quantity of PCL used in our polyurethanes, it was omitted from this experiment to simplify analysis. A 2:1 PEG/PPG molar ratio was chosen for ease of polymer extraction after various durations by precipitation in hexane. |
| § Determination of the degree of polymer branching by triple detection GPC analysis requires the use of a structurally-analogous linear polymer standard of comparable molecular weights as the polyurethanes discussed herein. As it is difficult to ensure lack of allophanate linkages by polyaddition, branching density was quantified via spectroscopic and titrimetric techniques. |
| This journal is © The Royal Society of Chemistry 2020 |