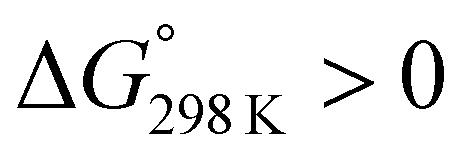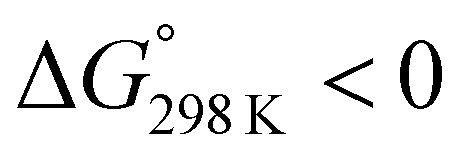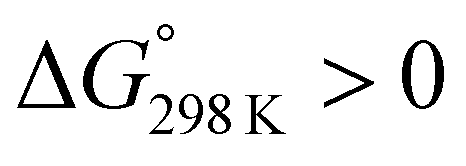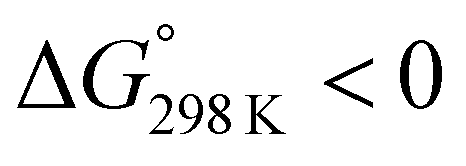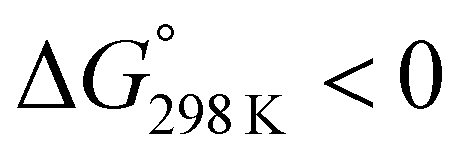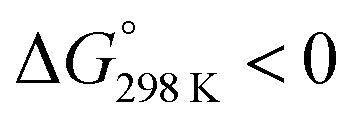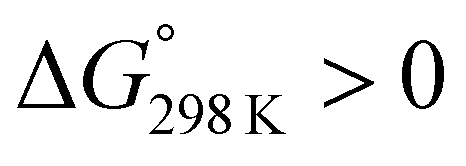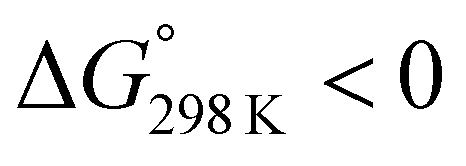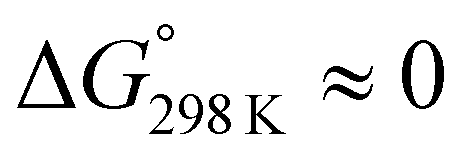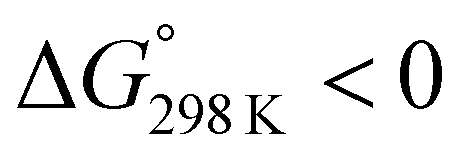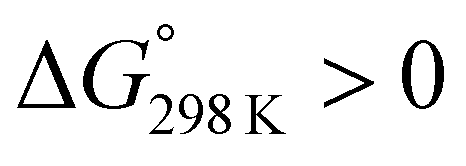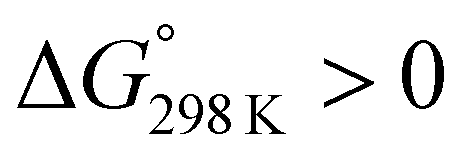DOI:
10.1039/D0RA07396B
(Paper)
RSC Adv., 2020,
10, 33317-33326
NMR-based quantitative studies of the conformational equilibrium between their square and folded forms of ascidiacyclamide and its analogues†
Received
6th August 2020
, Accepted 3rd September 2020
First published on 9th September 2020
Abstract
Ascidiacyclamide [cyclo(-Ile1,5-oxazoline2,6-D-Val3,7-thiazole4,8-)] (1) is a cytotoxic cyclic peptide from the ascidian, or sea squirt. Through structural analyses using asymmetric analogues [Xxx1: Ala (2), Val (3), Leu (4), Phe (5), cyclohexylalanine (6) and phenylglycine (7)], we previously showed 1 to exist in a conformational equilibrium between square and folded forms. In the present study, five new asymmetric analogues [Xxx1: 2-aminobutyric acid (8), 2-aminopentyric acid (9), tert-butylalanine (10), cyclohexylglycine (11) and tert-leucine (12)] were synthesized, and their structures were analyzed with X-ray diffraction and CD spectral measurements. Variable temperature 1H NMR measurements were performed to determine their equilibrium constants and their thermodynamic parameters. The use of two reference peptides made these quantitative studies possible. T3ASC, which contains three thiazole rings as a result of replacing oxazoline2 with thiazole, and dASC, in which the two oxazoline rings were deleted, were respectively used as square and folded reference peptides. The estimated parameters enabled more detailed discussion of the relationship between the bulkiness of substituents and the conformational free energies (ΔG°) of the peptides as well as the relationship between structure and cytotoxicity. The ΔG° values for peptides 1, 2, 3, 8, 9 and 11 decreased with decreases in the bulkiness of their substituents. We suggest that spontaneous folding is promoted as the bulkiness of substituents decreases. Peptides 7 and 12, which have large positive ΔG° values independently of temperature, did not exhibit spontaneous folding at any temperature; that is, their conformations were very stable in the square form. Peptides 4, 5, 6 and 10 had negative ΔG° values, despite their bulky substituents. Peptides with a positive ΔG° value showed cytotoxicity, and peptides with a negative ΔG° value showed reduced or no cytotoxicity. However, peptides 5 and 6 showed cytotoxicity equal to or stronger than 1. Those ten peptides except for 5 and 6 showed a clear structure–cytotoxicity relationship based on ΔG° values.
Introduction
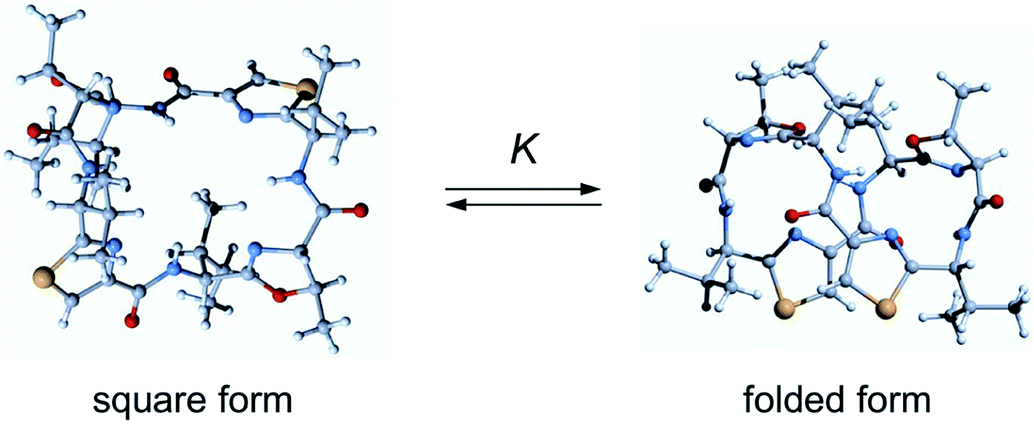
Ascidiacyclamide (1) is a cytotoxic cyclic peptide isolated from the marine invertebrate ascidian.1 Peptide 1 has a C2-symmetric sequence cyclo(-Ile1-Oxz2-D-Val3-Thz4-Ile5-Oxz6-D-Val7-Thz8-) containing the unusual amino acids (4S, 5R)-5-methyl-oxazoline-4-carboxylic acid (Oxz) and thiazole-4-carboxylic acid (Thz) (Fig. 1). Previous structural analyses revealed that 1 assumes two major conformations: a “square form” in which the Oxz and Thz rings are located at the four corners of the open peptide backbone, and a “folded form” in which the peptide backbone is folded at around the two Oxz rings and the Thz rings face each other.2–4 In solution, peptide 1 is in an equilibrium between the square and folded conformations (Fig. 2).5 We have also introduced asymmetric modifications in which the Ile1 residue was replaced with other amino acids [Xxx1: Ala (2), Val (3), Leu (4), Phe (5), cyclohexylalanine (Cha) (6) and phenylglycine (Phg) (7)]6–9 (Fig. 1). These modifications showed that the repulsive steric interactions between the side chains of the Xxx1 and Ile5 residues, which are close to one another within the square form, contribute to the conformational equilibrium between the square and folded conformers, and are related to their cytotoxicity.6–10
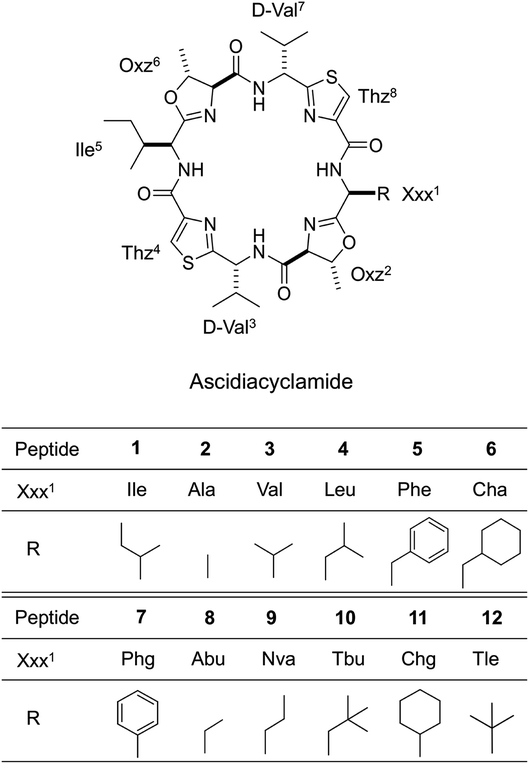 |
| | Fig. 1 Chemical structures of 1 and the side chains (R) of the Xxx1 residues in the asymmetric analogues (2–12). Peptides 1–7 are the previously synthesized.2–9 Peptides 8–12 are new asymmetric analogues. | |
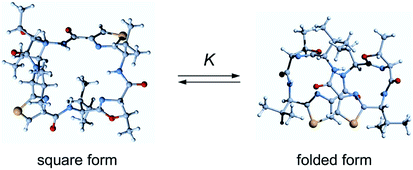 |
| | Fig. 2 Conformational equilibrium of ascidiacyclamides between the square and folded forms. | |
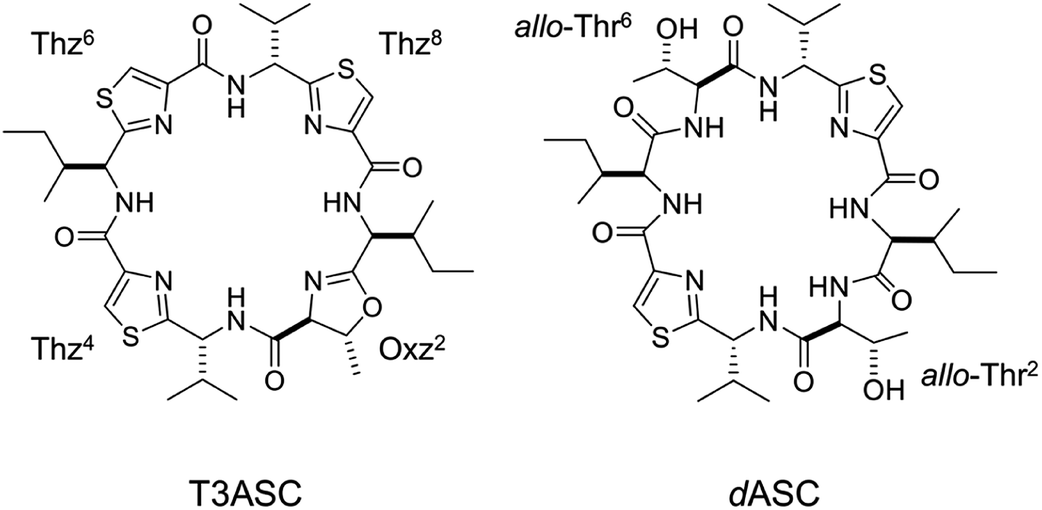
In 1955, the A values of substituent as the free energy difference between the axial and equatorial isomers of monosubstituted cyclohexanes were defined by Winstein and Holness.11 The equatorial isomer becomes predominant as the corresponding size of substituent grows, and the A values increase concomitantly. This parameter is a very useful resource to assess the steric size of variety of substituents. Many other conformational analyses using cyclohexane and its analogues have been carried out in an effort to understand steric repulsion within a molecule.12–17 We were interested in assessing the conformational equilibrium quantitatively to better understand the conformational behavior of ascidiacyclamides. In order to determine the equilibrium constants experimentally, it is necessary to observe an independent signal from each conformer. However, the signals from a small peptide like ascidiacyclamides equilibrate rapidly between the two conformers on the NMR time scale. It is often possible to slow down the equilibration of the signals by measuring at low temperatures, but in the case of ascidiacyclamides, the signals corresponding to the two conformers were still not separated when the temperature was lowered to −60 °C in acetonitrile-d3 solution. To overcome this limitation with small peptides, NMR-based quantitative studies of β-sheet populations have been carried out using reference peptides for the folded and unfolded states.18,19 For instance, to determine the β-sheet populations of flexible linear peptides, disulfide-linked cyclic peptides (or backbone cyclization) and residue truncated peptides were used as controls to provide reference chemical shifts for the fully folded and random coil states, respectively. We therefore searched for square and folded reference peptides to apply this strategy to ascidiacyclamides. The results showed that the T3ASC and dASC analogues were potential models for the fully square and folded states, respectively (Fig. 3). T3ASC, cyclo(-Ile1-Thz2-D-Val3-Thz4-Ile5-Oxz6-D-Val7-Thz8-) contains three Thz rings, as the Oxz2 ring was replaced with a Thz2 ring. The crystal structure of T3ASC exhibited a square form very similar to that of 1 (The RMS deviation = 0.351 Å) (Fig. S43†), and most T3ASC molecules reportedly retain the square form even in solution.20 On the other hand, dASC, cyclo(-Ile-alloThr-D-Val-Thz-)2, which lacks the Oxz rings of 1, assumes a folded form in both solid and solution states (Fig. S44†).21 Asymmetric modifications were also made in dASC, but unlike 1, all dASC analogues were folded, regardless of the bulkiness of the side chain of the replaced amino acid. The folded conformation of dASC was stabilized by four intramolecular hydrogen bonds, making it unable to transform to the square form from the folded form, and it exhibited no cytotoxicity.21,22
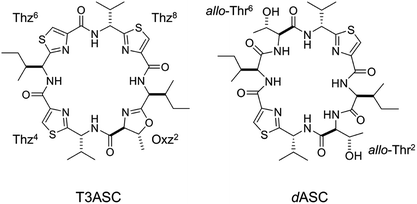 |
| | Fig. 3 Chemical structures of T3ASC and dASC. | |
In the present study, five new asymmetric acidiacyclamide analogues [Xxx1: 2-aminobutyric acid (Abu) (8), 2-aminopentyric acid (Nva) (9), tert-butylalanine (Tbu) (10), cyclohexylglycine (Chg) (11) and tert-leucine (Tle) (12)] were synthesized (Fig. 1). We first describe their structural characterization using X-ray diffraction and circular dichroism (CD) spectroscopy, and then discuss the NMR-based quantification of the conformational equilibrium for 1 and eleven asymmetric analogues (the aforementioned six analogues, 2–7, and five new analogues, 8–12).
Results and discussion
Crystal structures
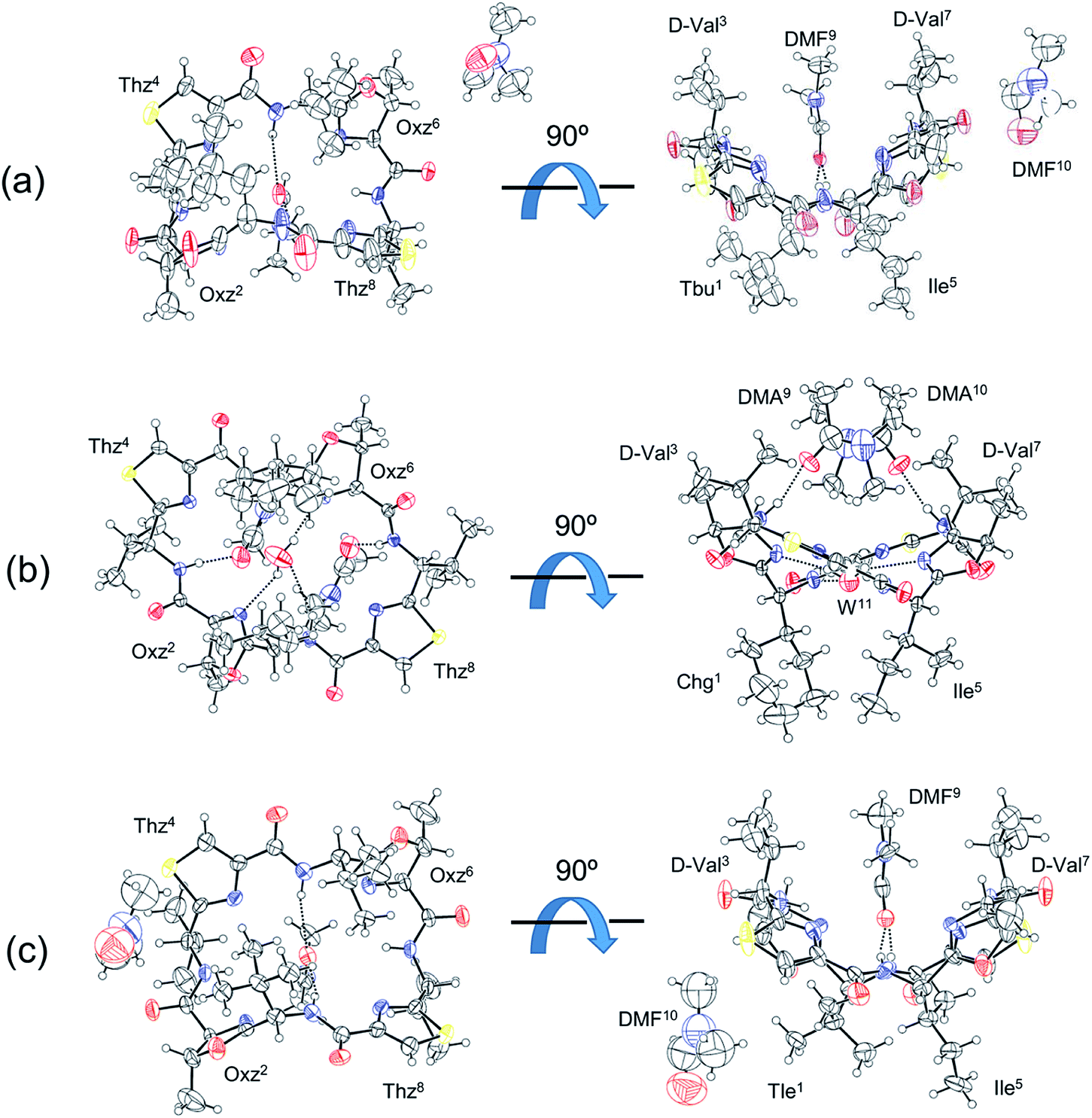
Three of the new five asymmetric analogues, 10, 11 and 12, were crystallized (Fig. 4). Crystals of 10 and 12 were grown in dimethylformamide (DMF) solution. With their crystal structures, each asymmetric unit contained the peptide molecule and two DMF molecules. Each peptide backbone was open, and a DMF molecule was held at the center of the cyclic peptide with two hydrogen bonds: N(Xxx1)–H⋯O(DMF) and N(Ile5)–H⋯O(DMF). The distances and angles of these hydrogen bonds are listed in Table 1. The second DMF molecule was not involved in hydrogen bonding. Peptide 11 was crystallized in a dimethylacetamide (DMA) and water solvent mixture. Within its crystal structure, the asymmetric unit contained the peptide molecule, two DMA molecules and a water molecule. The peptide backbone was open, and a water and two DMA molecules were held at the center of cyclic peptide by a hydrogen bonding network (Fig. 4, Table 1).
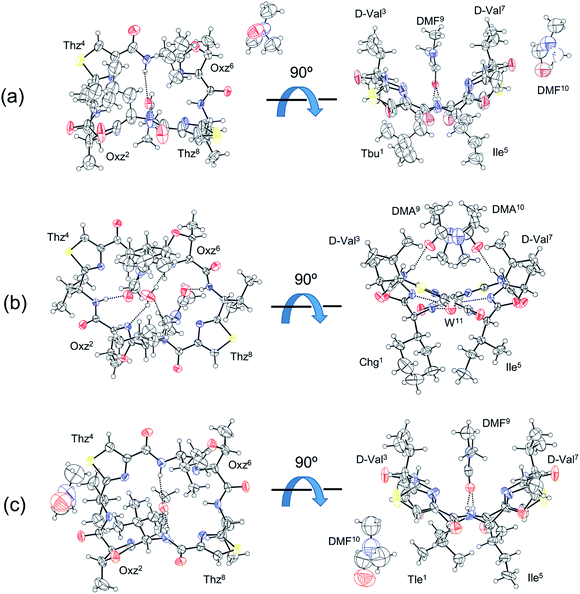 |
| | Fig. 4 Crystal structures of peptides 10, 11 and 12 are shown in (a), (b) and (c), respectively. Shown are top (left) and side (right) views of the peptide rings. The dashed lines represent hydrogen bonds. | |
Table 1 Hydrogen bonds in the crystal structures of 10, 11 and 12a
| Peptide |
Donor |
Acceptor |
Distance (Å) |
Angle (°) |
| D–H |
A |
D⋯A |
D–H⋯A |
| A letter of W represents the oxygen atom of water. |
| 10 |
N(Tbu1)–H |
O(DMF9) |
3.104(2) |
163.2 |
| N(Ile5)–H |
O(DMF10) |
2.928(2) |
163.7 |
| 11 |
N(Chg1)–H |
W |
3.262(7) |
137.3 |
| N(D-Val3)–H |
O(DMA9) |
2.904(6) |
169.2 |
| N(Ile5)–H |
W |
3.247(7) |
137.5 |
| N(D-Val7)–H |
O(DMA10) |
2.916(6) |
168.3 |
| W–H |
N(Oxz2) |
2.835(7) |
165.5 |
| W–H |
N(Oxz6) |
2.831(7) |
137.9 |
| 12 |
N(Tle1)–H |
O(DMF9) |
3.161(6) |
167.4 |
| N(Ile5)–H |
O(DMF10) |
3.161(6) |
167.4 |
We have been using the diagonal distance between the nitrogen atoms of the two Oxz and Thz rings (N(Oxz2)⋯N(Oxz6) and N(Thz4)⋯N(Thz8)) as an index to characterize the square form in crystal.2,7–9 These distances in the crystals of 10, 11 and 12 are listed together with the previously collected data for 1 in Table 2. Whereas the diagonal distances in 10 and 12 are similar to that in 1, the N(Oxz2)⋯N(Oxz6) distance is shorter and the N(Thz4)⋯N(Thz8) distance is longer in 11 than in 1. Thus, 10 and 12 were classified as being in the original square form, while 11 exhibited different diagonal distances but was open. In previous X-ray analyses, the crystal structures of 2 and 5 were classified as being in the folded form,6,7 and the crystal structures of 1, 3, 4, 6 and 7 were classified as being in the square form.2,7–9
Table 2 Comparison of selected nonbonding distances (Å) between the nitrogen atoms of the five-membered rings in 1, 10, 11 and 12
| Peptide |
N(Oxz2)⋯N(Oxz6) |
N(Thz4)⋯N(Thz8) |
| The square structure of 1 (benzene solvated) was previously reported.2 |
| 1a |
6.46 |
6.40 |
| 10 |
6.53 |
6.60 |
| 11 |
5.39 |
8.07 |
| 12 |
6.65 |
6.53 |
CD spectra
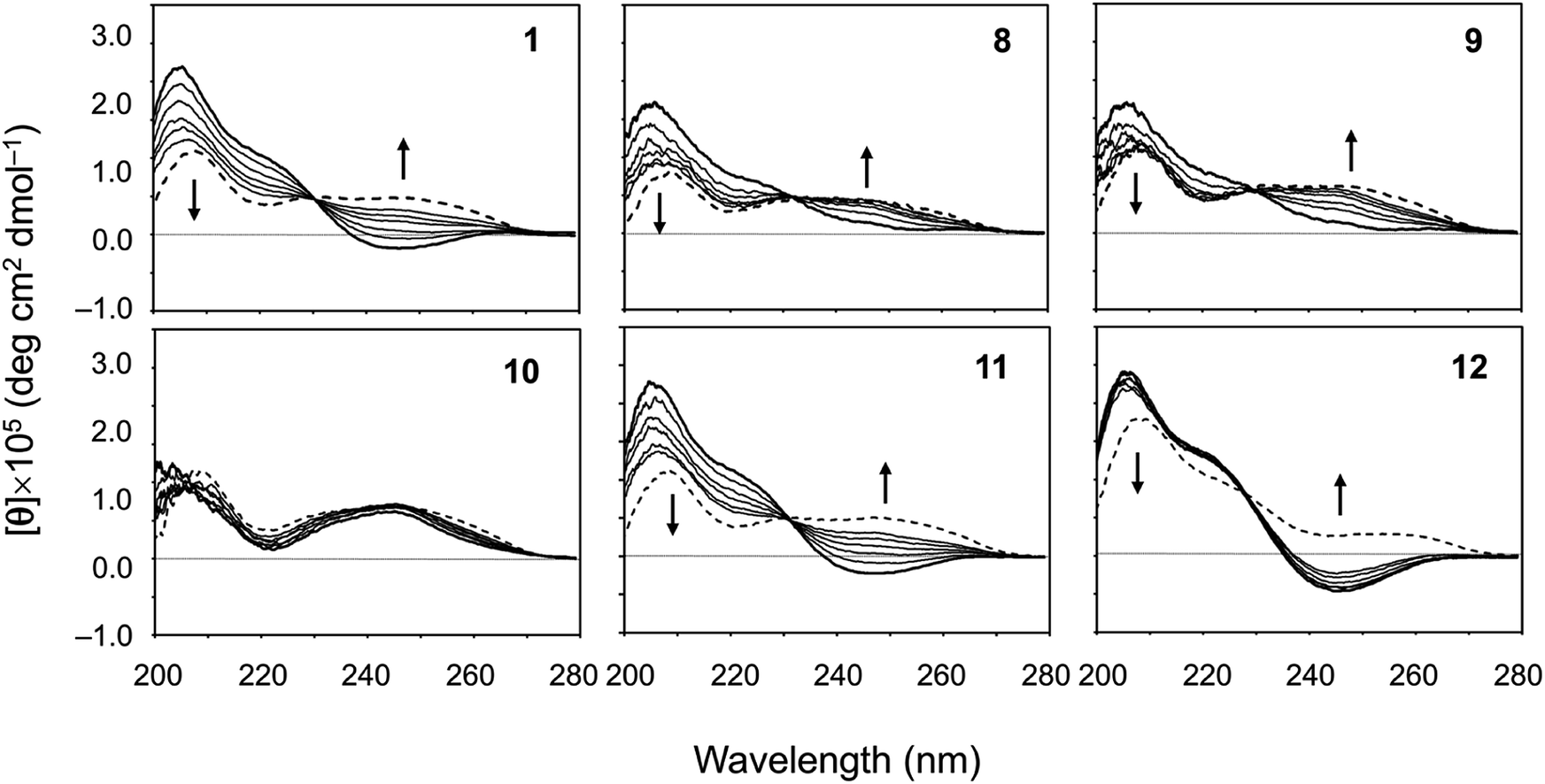
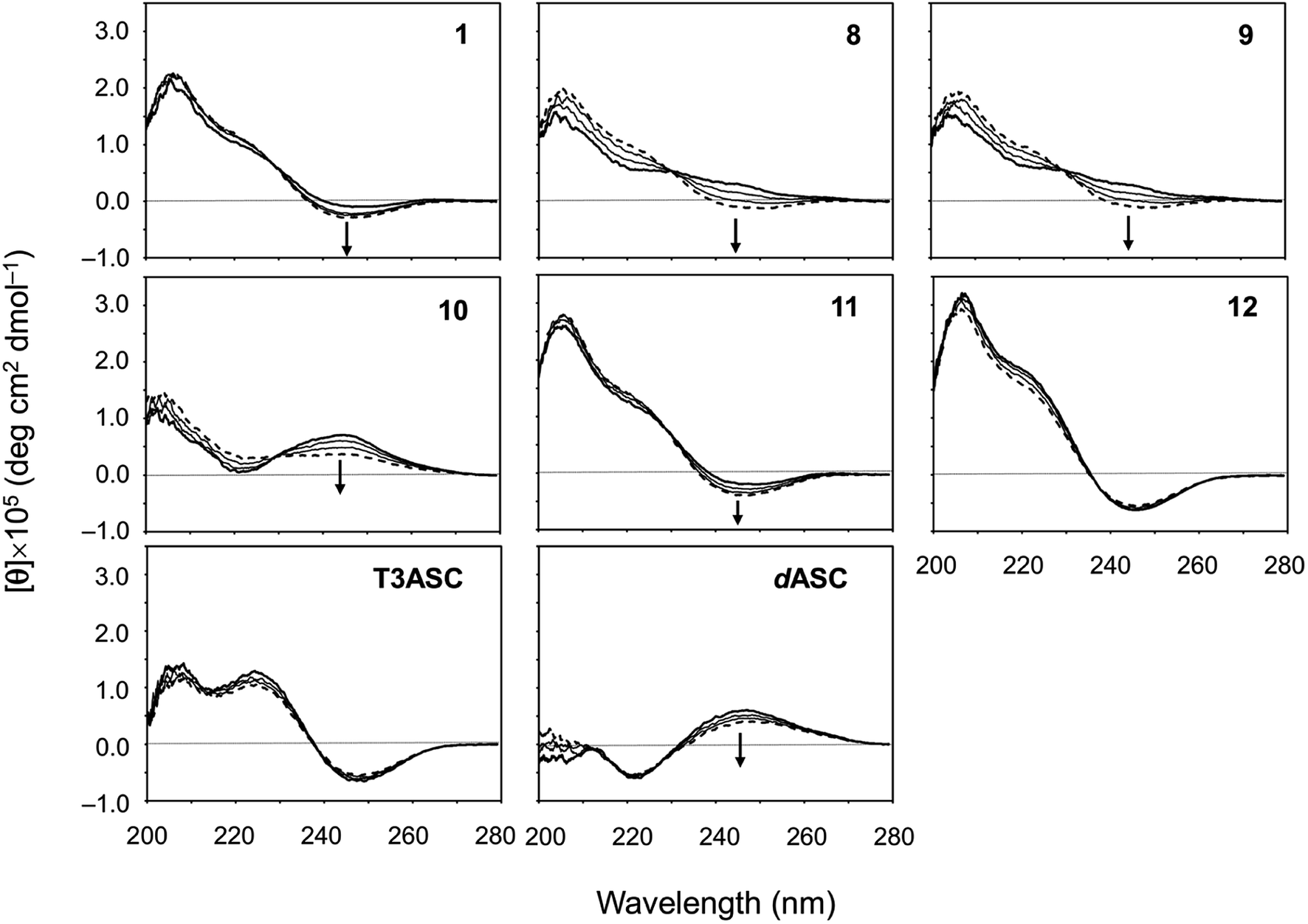
The CD spectral changes for the five new peptides, 8–12, were measured in acetonitrile (CH3CN) during titration of 2,2,2-trifluoroethanol (TFE) (Fig. 5). We have well established that measuring the CD spectral changes in this solvent system is a useful means of monitoring the conformational equilibrium between the square and folded forms of ascidiacyclamides.7–9,20 Previously recorded CD spectra for 1 are also shown in Fig. 5. A positive band at about 205 nm and a small negative band at 245 nm in CH3CN solution (bold line) are characteristic of the square form. TFE titration led to a decrease in [θ]205 and an increase in [θ]245, and a moderate positive band in the range of 230–260 nm was recorded in 100% TFE solution (dashed line). These spectral features are characteristic of the folded form. Moreover, the presence of a single isodichroic point at around 230 nm suggests conformational equilibrium.
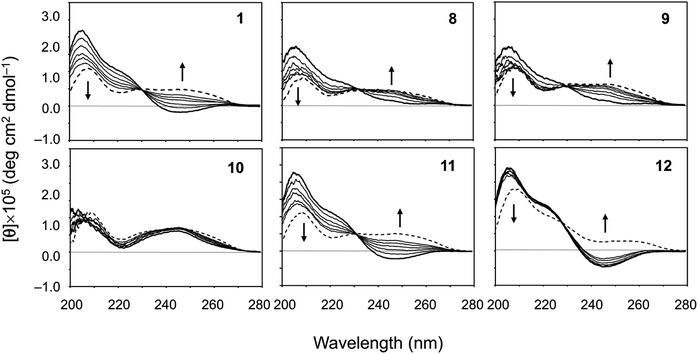 |
| | Fig. 5 CD spectral changes elicited by titration of TFE for peptides 1 and 8–12. The CD spectra for 1 were taken from a previous report.7 The spectra were measured in CH3CN solution while changing the TFE concentration (10%, 20%, 30%, 40%, 50% and 100%). The spectra in 100% CH3CN and 100% TFE solution are drawn in bold and dashed lines, respectively. | |
The spectral changes observed with 11 showed the same features as 1 in this solvent system. The conformation of 11 in CH3CN solution was the square form, which was converted to the folded form through TFE titration. The spectra for 3 had similar features.7 The spectrum of 12 in CH3CN solution had the same shape as that of 1, but the spectral changes elicited by TFE titration were very small and were very similar to those of 7.9 The spectrum of 10 exhibited the features of the folded form, even in CH3CN solution, and TFE titration elicited little change. The spectra of 2, 4, 5 and 6 have also been exhibited folded form.7,8 In crystal, however, 2 and 5 were in the folded form, while 4, 6 and 10 assumed the square form. The spectral features of 8 and 9 were very similar to each other. They did not exhibit a clear negative band around 245 nm in CH3CN solution, as 1 did, but a single isodichroic point at around 230 nm was observed. Previously recorded CD spectra for 2–7 are shown in Fig. S41.†
We also evaluated the temperature dependencies of the CD spectra of ascidiacyclamides in CH3CN solution at 273, 293, 313 and 333 K. The temperature dependencies of 1, T3ASC, dASC and 8–12 are shown in Fig. 6; those of peptides 2–7 are shown in Fig. S42.† The [θ]245 values decreased as temperature increased in all ascidiacyclamides spectra, except those of T3ASC, 7 and 12. Peptides 1, 3 and 11, which exhibited a negative band at 245 nm at low temperature, showed a slight decrease in [θ]245 with increasing temperature. By contrast, peptides 2, 4, 5, 6, 8, 9 and 10 all showed strong temperature dependencies, with a positive band in the range of 230–260 at low temperature. In particular, a clear negative band at 245 nm appeared for 8 and 9 as the temperature increased. The spectrum of dASC also showed a positive band in the range of 230–260 nm at low temperature. However, the decrease in [θ]245 with increasing temperature of dASC was slight compared with these peptides. The spectra of T3ASC, 7 and 12 were little affected by increasing temperature. These observations suggest the following:
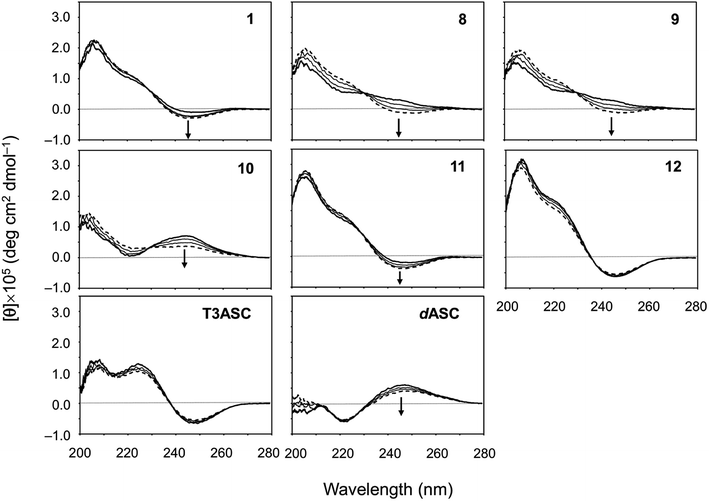 |
| | Fig. 6 Temperature dependence of the CD spectra of peptides 1, 8–12, T3ASC and dASC. The CD spectra for 1 are taken from a previous report.29 The spectra were measured every 20 K in CH3CN solution at 273–333 K. The CD spectra at 273 and 333 K are drawn in bold and dashed lines, respectively. | |
(1) The conformation of ascidiacyclamide changes from the folded form to the square form with increasing temperature.
(2) T3ASC, 7 and 12 in CH3CN solution form the most stable square form against temperature increases.
(3) dASC in CH3CN solution forms the most stable folded form against temperature increases.
VT-1H NMR
All ascidiacyclamides were examined using variable temperature 1H NMR spectroscopy (VT-1H NMR). The spectra were recorded every 10 K from 273 K to 333 K in CH3CN-d3 solution. The temperature coefficients (Δδ/ΔT (ppb K−1)) of all amide and Thz protons are listed in Table 3. Although the temperature coefficients of 1–5 were reported previously,7 we examined them again because different NMR equipment was used in the present study. In 1, the chemical shifts of the amide protons of Ile1,5 residues were shifted to lower fields with a temperature coefficient of 2.0 ppb K−1, whereas the chemical shifts of the amide protons of D-Val3,7 residues showed no temperature dependence. The temperature coefficients of all amide protons in 2–6 and 8–11 were positive, though they differed slightly among the 10 peptides. By contrast, none of the amide protons in T3ASC or 12 showed any temperature dependence, and the temperature coefficients of all amide protons in dASC and 7 were negative. The behavior of the chemical shifts of amide protons elicited by temperature changes are generally discussed as a phenomenon caused by perturbation of intra/intermolecular hydrogen bonds, exposure to the solvent or shielding from the solvent,23 but it is difficult to clearly conclude that from the present results. On the other hand, the behavior of the chemical shifts of Thz protons can be clearly explained. When the Thz rings face each other in the folded form, the chemical shifts of the Thz4 and Thz8 protons appear in a higher field than those in the square form due to the ring-current effects of the Thz8 and Thz4 rings, respectively. In other words, the chemical shifts of Thz protons are very sensitive to backbone conformational changes. The temperature coefficients of the Thz protons were positive in dASC, 1–6 and 8–11. These observations suggest that the conformation of ASCs change from the folded form to the square form with increasing temperature. The Thz protons in T3ASC, 7 and 12 exhibited no temperature dependence. The temperature coefficients of the Thz protons of dASC were not zero, but their temperature dependence was the lowest. There was thus good agreement between the temperature dependencies of the Thz protons chemical shifts and CD spectral changes.
Table 3 Amide proton and Thz proton temperature coefficients Δδ/ΔT (ppb K−1) of peptides 1–12, T3ASC and dASC in CH3CN-d3 solution
| Peptide |
Xxx1NH |
D-Val3NH |
Ile5NH |
D-Val7NH |
ThzHa |
ThzHa |
| The chemical shifts of Thz4H and Thz8H were indistinguishable. The temperature coefficients of these peptides have already been reported.7 However, they were measured again because different NMR equipment was used in this study. dASC also includes allo-Thr2NH and allo-Thr6NH. The temperature coefficient for both was −2.3 ppb K−1. The correlation coefficients were less than 0.9. The chemical shifts of D-Val3NH and D-Val7NH were indistinguishable. |
| 1b |
2.0 |
0.0 |
2.0 |
0.0 |
1.9 |
1.9 |
| 2b |
2.0 |
1.6 |
2.2 |
1.0 |
3.9 |
3.9 |
| 3b |
2.3 |
0.6 |
2.9 |
0.9 |
2.9 |
3.1 |
| 4b |
1.9 |
0.7 |
2.4 |
1.5 |
3.3 |
3.3 |
| 5b |
1.1 |
0.7 |
2.1 |
1.0 |
3.5 |
3.2 |
| 6 |
2.0 |
0.9 |
2.5 |
1.5 |
3.5 |
3.5 |
| 7 |
—d |
−2.2 |
−0.7 |
−1.4 |
0.0 |
—d |
| 8 |
2.2 |
—d |
2.7 |
1.0 |
3.3 |
3.3 |
| 9 |
2.0 |
—d,e |
2.5 |
0.9e |
3.0 |
3.2 |
| 10 |
—d |
0.6 |
2.0 |
1.4 |
2.5 |
2.7 |
| 11 |
2.5 |
—d,e |
2.1 |
0.5e |
2.0 |
2.0 |
| 12 |
0.0 |
—d |
0.0 |
—d |
0.0 |
0.0 |
| T3ASC |
0.0 |
—d |
0.0 |
—d |
0.0 |
0.0 |
| dASCc |
−1.1 |
−1.3 |
−1.1 |
−1.3 |
1.3 |
1.3 |
NMR-based quantification of the conformational equilibrium
The conformational equilibrium constants (K) (Fig. 2) of ascidiacyclamides were determined using data from VT-1H NMR studies in CH3CN-d3 solution. Based on structural analyses, T3ASC and dASC were used as reference peptides to provide reference chemical shifts for the fully square and folded forms, respectively. K can be estimated for ascidiacyclamides from conformationally sensitive proton chemical shifts via eqn (1), where δobs is the chemical shift of the Thz proton within the equilibrating peptide, δS (= 8.09 ppm) is the chemical shift of the Thz proton in the fully square form (T3ASC), and δF (=7.35 ppm) is the chemical shift of the Thz proton in the fully folded form (dASC). The chemical shift of the Thz proton in dASC moved slightly to a lower field with increasing temperature, so the chemical shift at the lowest temperature (273 K) was used as the δF value.| | |
K = (δS − δobs)/(δobs − δF)
| (1) |
For 1–12, values of K and the Gibbs free energy (ΔG°) were measured every 10 K from 237 K to 333 K (Tables S30, S32, S34, S36, S38, S40, S42, S44, S46, S48, S50 and S51†). In addition, ΔG° at 298 K was estimated from the thermodynamic parameters of ascidiacyclamide and the asymmetric analogues. The enthalpy (ΔH°) and entropy (ΔS°) were determined through a linear van't Hoff plot (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K versus 1/T), and then ΔG° at 298 K was calculated from eqn (2). For 7 and 12, we could not obtain their values of ΔH° and ΔS° by a van't Hoff plot because their K values showed no temperature dependencies.
K versus 1/T), and then ΔG° at 298 K was calculated from eqn (2). For 7 and 12, we could not obtain their values of ΔH° and ΔS° by a van't Hoff plot because their K values showed no temperature dependencies.
| | |
ΔG° = ΔH° − TΔS° = −RTlnK
| (2) |
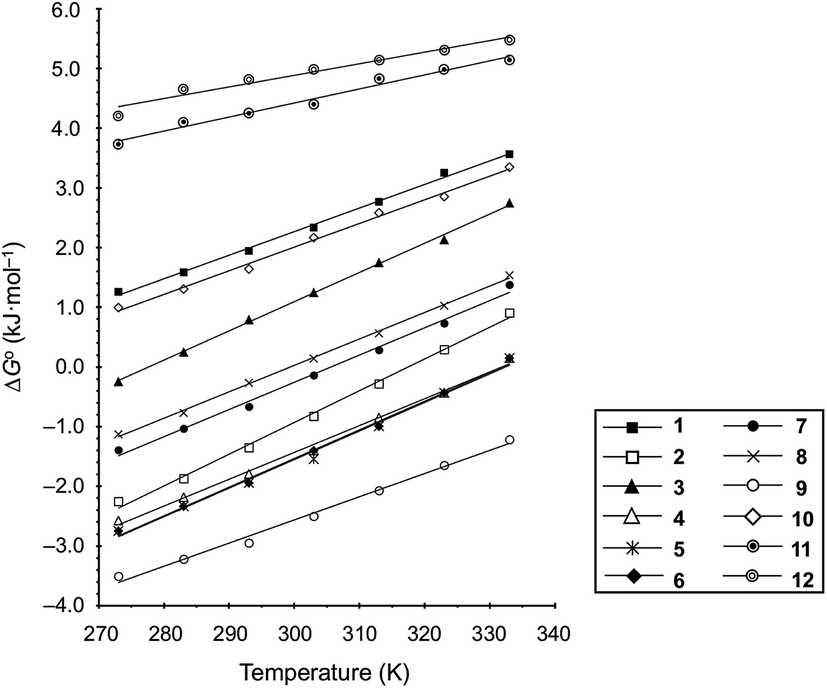
These values are listed in Table 4. Folding of ascidiacyclamides was enthalpically favorable and entropically unfavorable. This thermodynamic profile is considered reasonable because four hydrogen bonds are formed by folding: N(Xxx1)H⋯Oγ(Oxz6), N(D-Val3)H⋯O(Thz8), N(Ile5)H⋯Oγ(Oxz2) and N(D-Val7)H⋯O(Thz4). The average values of ΔH° and ΔS° were respectively determined to be −13.55 (2.06) kJ mol−1 and −43.72 (4.71) J K−1 mol−1, and there were no significant differences in these values among ascidiacyclamides. The ΔG° values for both 7 and 12 were positive and very large, which means that the conformational equilibrium positions of 7 and 12 are significantly shifted in the opposite direction. As shown in Fig. 7, within the measured temperature range, the ΔG° values for 1 and 11 are positive at every temperature, suggesting that there is no spontaneous folding. The ΔG° values for 3 are positive at higher temperatures but negative below 278 K. Peptides 8 and 9 will fold spontaneously at temperatures below 303 K. Peptides 4, 5 and 6 will fold spontaneously at temperatures below 333 K, and the ΔG° values are near zero at around 333 K. Peptide 10 appears to spontaneously fold at any temperature.
Table 4 Thermodynamic parameters of peptides 1–12
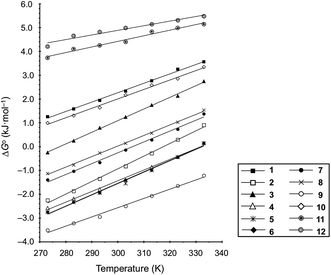 |
| | Fig. 7 Plots of the temperature dependences of the free energies for peptides 1–12. | |
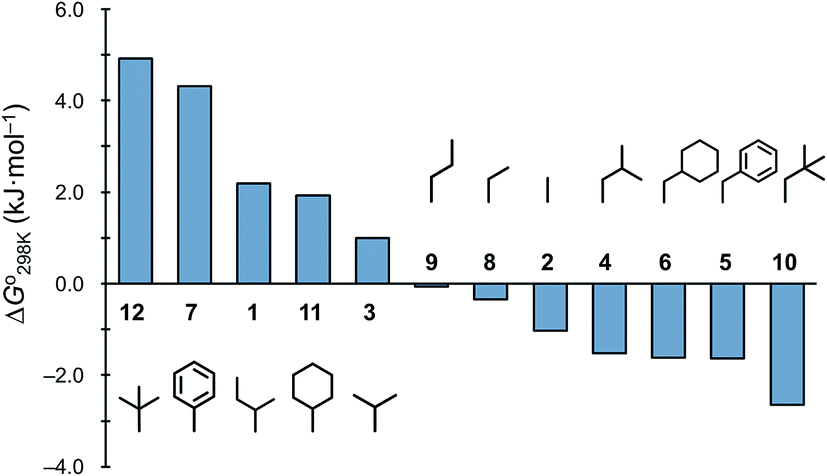
The ΔG° values at 298 K for all ascidiacyclamides are arranged in descending order in Fig. 8. These results allow for a logical discussion that takes into account the counterpoise between the bulkiness of substituents in the Xxx1 and Ile5 residues. Symmetric peptide (1) showed a preference for the square form in CH3CN-d3 solution at 298 K, with a ΔG° value of 2.19 kJ mol−1, while 11, which contains a cyclohexyl group, had nearly the same ΔG° value as 1. Comparison among peptides 1, 2, 3, 8 and 9 revealed that the ΔG° value decreased with a decrease in the carbon number of the substituent. In addition, although peptides 3 and 9 have the same number of carbon atoms in their side chains, the ΔG° value was smaller for 9, which has a linear alkyl group, than for 3, which has a branched alkyl group (sec-butyl (1) > isopropyl (3) > n-propyl (9) > ethyl (8) > methyl (2)). The ΔG° value for 9 was nearly zero, and those of 2 and 8 were negative, indicating a preference for the folded form. In the square form, the side chain of the Xxx1 residue is close to the sec-butyl group of the Ile5 residue, and we suggest that the attractive force between these two functional groups is greater than the repulsive force. Consequently, with a decrease in the bulkiness of the substituent in the Xxx1 residue, the attractive force will be weakened, and the peptide will be easier to fold. Peptides 4, 5, 6 and 10 had even greater preferences for the folded form. The ΔG° value for 10 was −2.64 kJ mol−1, which was the minimum value, but those of 4, 5 and 6 were nearly the same. The common feature for the substituents of 4, 5, 6 and 10 is a β-methylene. This spacer may reduce contacts between the Xxx1 and Ile5 residues within these peptides. By contrast, peptides 7 and 12 had strong preferences for the square form. Although the tert-butyl group (12) and sec-butyl group (1) are both four carbon alkyl groups, the former is branched and therefore may exert a greater dispersion force. While it is apparent that the van der Waals space occupied by a cyclohexyl group (11) is larger than that occupied by a planarity phenyl group (7), the ΔG° for 7 was nevertheless significantly greater than that for 11. A dispersion force like the CH–π interaction may be acting between the phenyl group of the Phg1 residue and sec-butyl group of the Ile5 residue. The additional attractive force may increase the stability of the square form, though that could not be verified here. Still, the very high stabilities of square forms of 7 and 12 are noteworthy.
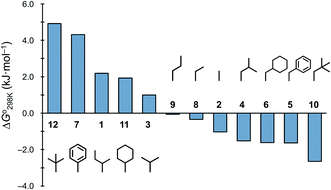 |
| | Fig. 8 Plots of the free energies for peptides 1–12 at 298 K in descending order. The chemical structures in the figure are the side chains of the Xxx1 residues. | |
Structure–cytotoxicity relationship
We assessed the cytotoxicity of the five new peptides, 8–12, by determining the ED50 against P388 mouse lymphocytic leukemia cells. Our findings, including data collected previously, are summarized in Table 5 with their structures. Peptides 3, 7, 11 and 12, which have the same conformation as the parent peptide (1) in both the crystal and solution, showed cytotoxicity equivalent to or stronger than peptide 1. Peptide 9, with a 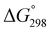 value close to zero, exhibited half the cytotoxicity of 1, while peptides 2 and 8, with negative
value close to zero, exhibited half the cytotoxicity of 1, while peptides 2 and 8, with negative 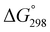 values, showed one-fifth the cytotoxicity and no cytotoxicity, respectively. These results suggest that a structural change from the square to the folded form decreases the cytotoxicity. Consistent with those ideas, the reference peptide for the square form (T3ASC) showed the strongest cytotoxicity, while the reference for the folded form (dASC) showed no cytotoxicity. Peptides 4, 5, 6 and 10 all had negative ΔG° values. The cytotoxicity of 4 was reduced to one-third that of 1, and 10 showed no cytotoxicity. One the other hand, peptides 5 and 6 showed cytotoxicity equal to or stronger than 1. Thus, peptides 5 and 6 did not adhere to the structure–cytotoxicity relationship observed with the others.
values, showed one-fifth the cytotoxicity and no cytotoxicity, respectively. These results suggest that a structural change from the square to the folded form decreases the cytotoxicity. Consistent with those ideas, the reference peptide for the square form (T3ASC) showed the strongest cytotoxicity, while the reference for the folded form (dASC) showed no cytotoxicity. Peptides 4, 5, 6 and 10 all had negative ΔG° values. The cytotoxicity of 4 was reduced to one-third that of 1, and 10 showed no cytotoxicity. One the other hand, peptides 5 and 6 showed cytotoxicity equal to or stronger than 1. Thus, peptides 5 and 6 did not adhere to the structure–cytotoxicity relationship observed with the others.
Table 5 Summary of cytotoxicities and structures of peptides 1–12, T3ASC and dASC
| Peptide |
ED50 (μg mL−1) |
Structure in solid |
Structure in solution |
| These are taken from a previous report.7 This datum is taken from a previous report.2 This datum is taken from a previous report.6 These data are taken from a previous report.8 These data are taken from a previous report.9 These crystal structures have not obtained yet. These data are taken from a previous report.20 This datum is taken from a previous report.22 This datum is taken from a previous report.21 |
| 1 |
10.5a |
Squareb |
 |
| 2 |
49.0a |
Foldeda |
 |
| 3 |
7.4a |
Squarea |
 |
| 4 |
29.5a |
Squarec |
 |
| 5 |
11.8a |
Foldeda |
 |
| 6 |
5.6d |
Squared |
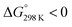 |
| 7 |
12.4e |
Squaree |
 |
| 8 |
>100 |
—f |
 |
| 9 |
18.7 |
—f |
 |
| 10 |
>100 |
Square |
 |
| 11 |
3.4 |
Square |
 |
| 12 |
5.5 |
Square |
 |
| T3ASC |
0.93g |
Squareg |
— |
| dASC |
>100h |
Foldedi |
— |
Conclusions
NMR-based experiments making use of two reference peptides (T3ASC and dASC) enabled highly precise determination of equilibrium constants and then thermodynamic parameters (ΔG°, ΔH° and ΔS°). For peptides 1, 2, 3, 8, 9 and 11, there was a correlation between the bulkiness of their substituents and their ΔG° value. This suggests that the dispersion force between the substituent in the Xxx1 residue and sec-butyl group of the Ile5 residue contributed to the stabilization of the square form. Peptides 7 and 12 showed large ΔG° values independently of temperature. This suggests the stability of the square forms of 7 and 12 may involve other factors in addition to the dispersive force. Peptides 4, 5, 6 and 10 had negative ΔG° values, despite of their bulky substituents. The β-methylene bearing on substituents may weaken the dispersion force between the side chains of Xxx1 and Ile5 residues. However, the crystal structures of these peptides were square form, except for 5. Considering the structure–cytotoxicity relationship based on the estimated ΔG° values, good correlation was exhibited for ten peptides except for 5 and 6. The estimated parameters in the present study allowed for a more detailed discussion of the ascidiacyclamide conformational equilibrium.
Experimental
Synthesis
First, the Thz unit (Boc-D-Val(Thz)-OMe) was synthesized as previously described.24,25 Peptides were synthesized using a conventional liquid-phase method. The linear peptide Boc-Xxx-allo-Thr-D-Val-Thz-Ile-allo-Thr-D-Val-Thz-OMe was synthesized using 1-hydroxybenzotiazole (Watanabe Chemical lnd. Ltd., Hiroshima, Japan) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Watanabe Chemical lnd. Ltd., Hiroshima, Japan). Then, macro-cyclization to cyclo(-Xxx-allo-Thr-D-Val-Thz-Ile-allo-Thr-D-Val-Thz-) was accomplished using benzotriazolyloxy-tris (pyrrolidino)-phosphonium hexafluorophosphate (Watanabe Chemical lnd. Ltd., Hiroshima, Japan) in the presence of 4-dimethylaminopyridine (Nacalai Tesque, Kyoto, Japan). Thereafter, the Oxz rings were formed by reacting the Ile-allo-Thr moieties with bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) (Fujifilm Wako Pure Chemical Corp., Osaka, Japan).26 The synthesis and characterization of 8–12 are detailed in ESI.†
1H NMR spectra
1H NMR spectra were recorded on an Agilent DD2 600 MHz NMR spectrometer (Agilent Technologies, California, USA). Peptide concentrations were about 5.0 mM in CH3CN-d3. Chemical shifts were measured relative to internal trimethylsilane at 0.00 ppm. The protons were assigned using two dimensional correlated spectroscopy (2D-COSY) and rotating-frame Overhauser effect spectroscopy (ROESY; mixing time = 500 ms). The assignment lists and 1H NMR spectra for all peptides are given in ESI.†
X-ray diffraction
Peptides 10 and 12 were crystallized in DMF, and peptide 11 was crystallized in DMA. X-ray diffraction data for 10 were collected with a Rigaku CrysAlis Pro (Rigaku Corp., Tokyo, Japan). The structure was solved using SHELXT27 and refined using SHELXL2016/4.28 X-ray diffraction data for 11 and 12 were collected with a Bruker Smart APEXII (Bruker Corp., Massachusetts, USA). The structures were solved using SHELX-97 (ref. 29) and refined using SHELXL-97.29 The crystal data for 10, 11 and 12 have been deposited with the Cambridge Crystallographic Data Center under deposition numbers 2007056, 2007057 and 2007058,† respectively. The crystal data for these three peptides are given in ESI.†
CD spectra
The CD spectra were measured using a JASCO spectrapolarimeter J-820 (JASCO ltd., Tokyo, Japan). The peptide concentrations were about 0.04 mM in CH3CN solution, and the path length was 1 cm. The experiments with TFE titration were performed at room temperature. The temperature dependencies of the CD spectra were measured at 273, 293, 313 and 333 K. The spectra were scanned at a speed of 5 nm min−1 with 0.1 nm interval uptake to a computer. Data were averaged over each 1 nm and plotted.
Assay for cytotoxicity
The cytotoxicity of the peptides was assessed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method as described previously with some modifications.30,31 388 mouse lymphocytic leukemia cells were cultured in RPMI 1640 medium (10% fetal calf serum) at 37 °C under 5% CO2. The test materials were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM, and the solution was diluted with Essential Medium to concentrations of 200, 20, and 2 μM, respectively. Each solution was combined with cells suspended (1 × 105 cells per mL) in the same medium. After incubating at 37 °C for 72 h under 5% CO2, the cells were labeled with 5 mg mL−1 MTT in phosphate-buffered saline (PBS), and the absorbance of formazan dissolved in 20% sodium dodecyl sulfate (SDS) in 0.1 N HCl was measured at 540 nm with a microplate reader MTP-310 (Corona electric Co. Ltd, Hitachinaka, Japan). The absorbance values were expressed as percentages relative to that of the control cell suspension that was prepared using the same procedure described above but without a test substance. All assays were performed three times, semilogarithmic plots were constructed from the averaged data, and the effective dose of a substance required to inhibit cell growth by 50% (ED50) was determined.
Conflicts of interest
There are no conflicts to declare.
References
- Y. Hamamoto, M. Endo, M. Nakagawa, T. Nakanishi and K. Mizukawa, Chem. Commun., 1983, 323–324 RSC
 .
. - T. Ishida, M. Inoue, Y. Hamada, S. Kato and T. Shioiri, Chem. Commun., 1987, 370–371 RSC .
- T. Ishida, Y. In, M. Inoue, Y. Hamada and T. Shioiri, Biopolymers, 1992, 32, 131–143 CrossRef CAS .
- Y. In, M. Doi, M. Inoue, Y. Hamada and T. Shioiri, Chem. Pharm. Bull., 1993, 41, 1686–1690 CrossRef CAS .
- T. Ishida, M. Tanaka, M. Nabae, M. Inoue, S. Kato, Y. Hamada and T. Shioiri, J. Org. Chem., 1988, 53, 107–112 CrossRef CAS .
- M. Doi, F. Shinozaki, Y. In, T. Ishida, D. Yamamoto, M. Kamigauchi, M. Sugiura, Y. Hamada, K. Kohda and T. Shioiri, Biopolymers, 1999, 49, 459–469 CrossRef CAS .
- A. Asano, K. Minoura, T. Yamada, A. Numata, T. Ishida, Y. Katsuya, Y. Mezaki, M. Sasaki, T. Taniguchi, M. Nakai, H. Hasegawa, A. Terashima and M. Doi, J. Pept. Res., 2002, 60, 10–22 CrossRef CAS .
- A. Asano, T. Yamada, A. Numata and M. Doi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o488–o490 CrossRef .
- A. Asano, T. Yamada and M. Doi, Bioorg. Med. Chem., 2011, 19, 3372–3377 CrossRef CAS .
- A. Asano, K. Minoura, T. Yamada and M. Doi, J. Pept. Sci., 2016, 22, 156–165 CrossRef CAS .
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of organic compounds, Wily & Sons, New York, 1994 Search PubMed .
- E. L. Eliel, N. L. Allinger, S. J. Angyal and G. A. Morrison, Conformational Analysis, Wiley & Sons, New York, 1965 Search PubMed .
-
(a) N. L. Allinger and J. Allinger, J. Am. Chem. Soc., 1958, 80, 5476–5480 CrossRef CAS ;
(b) N. L. Allinger, J. Allinger, L. A. Freiberg, R. F. Czaja and N. A. LeBel, J. Am. Chem. Soc., 1960, 82, 5876–5882 CrossRef CAS .
-
(a) N. L. Allinger and H. M. Blatter, J. Am. Chem. Soc., 1961, 83, 994–995 CrossRef CAS ;
(b) N. L. Allinger and L. A. Freiberg, J. Am. Chem. Soc., 1962, 84, 2201–2203 CrossRef CAS ;
(c) B. Rickborn, J. Am. Chem. Soc., 1962, 84, 2414–2417 CrossRef CAS ;
(d) W. D. Cotterill and M. J. T. Robinson, Tetrahedron, 1964, 20, 777–790 CrossRef CAS .
-
(a) C. Djerassi, E. J. Warawa, J. M. Berdahl and E. J. Eisenbraun, J. Am. Chem. Soc., 1961, 83, 3334–3336 CrossRef CAS ;
(b) W. D. Cotterill and M. J. T. Robinson, Tetrahedron, 1964, 20, 765–775 CrossRef CAS .
-
(a) O. Takahashi, K. Yamasaki, Y. Kohno, K. Ueda, H. Suezawa and M. Nishio, Chem.–Asian J., 2006, 1, 852–859 CrossRef CAS ;
(b) O. Takahashi, K. Yamasaki, Y. Kohno, K. Ueda, Y. Umezawa, H. Suezawa and M. Nishio, Tetrahedron, 2008, 64, 2433–2440 CrossRef CAS .
-
(a) Y. Carcenac, P. Diter, C. Wakselman and M. Tordeux, New J. Chem., 2006, 30, 442–446 RSC ;
(b) Y. Carcenac, M. Tordeux, C. Wakselman and P. Diter, New J. Chem., 2006, 30, 447–457 RSC .
- F. A. Syud, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 11577–11578 CrossRef CAS .
- C. D. Tatko and M. L. Waters, J. Am. Chem. Soc., 2004, 126, 2028–2034 CrossRef CAS .
- A. Asano, T. Yamada, T. Taniguchi, M. Sasaki, K. Yoza and M. Doi, J. Pept. Sci., 2018, e3120 CrossRef .
- A. Asano, M. Doi, K. Kobayashi, M. Arimoto, T. Ishida, Y. Katsuya, Y. Mezaki, H. Hasegawa, M. Nakai, M. Sasaki, T. Taniguchi and A. Terashima, Biopolymers, 2001, 58, 295–304 CrossRef CAS .
- A. Asano, T. Yamada, Y. Katsuya, T. Taniguchi, M. Sasaki and M. Doi, J. Pept. Res., 2006, 66(suppl. 1), 90–98 CrossRef .
-
(a) M. Ohnishi and D. W. Urry, Biochem. Biophys. Res. Commun., 1969, 36, 194–202 CrossRef CAS ;
(b) N. Sugawara, E. S. Stevens, G. M. Bonora and C. Tonioro, J. Am. Chem. Soc., 1980, 102, 7044–7047 CrossRef CAS ;
(c) E. S. Stevens, N. Sugawara, G. M. Bonora and C. Tonioro, J. Am. Chem. Soc., 1980, 102, 7044–7047 CrossRef ;
(d) R. R. Wilkening, E. S. Stevens, G. M. Bonora and C. Tonioro, J. Am. Chem. Soc., 1983, 105, 2560–2561 CrossRef CAS ;
(e) G. M. Bonora, C. Mapelli, C. Toniolo, R. R. Wilkening and E. S. Stevens, Int. J. Biol. Macromol., 1984, 6, 179–188 CrossRef CAS .
- Y. Hamada, S. Kato and T. Shioiri, Tetrahedron Lett., 1985, 26, 3223–3226 CrossRef CAS .
- Y. Hamada, M. Shibata, T. Sugiura, S. Kato and T. Shioiri, J. Org. Chem., 1987, 52, 1252–1255 CrossRef CAS .
- A. J. Phillips, Y. Uto, P. Wipf, M. J. Reno and D. R. Williams, Org. Lett., 2000, 2, 1165–1168 CrossRef CAS .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS .
- K. Kohda, Y. Ohta, Y. Yokoyama, T. Kato, Y. Suzumura, Y. Hamada and T. Shioiri, Biochem. Pharmacol., 1989, 38, 4497–4500 CrossRef CAS .
- K. Kohda, Y. Ohta, Y. Kawazoe, T. Kato, Y. Suzumura, Y. Hamada and T. Shioiri, Biochem. Pharmacol., 1989, 38, 4500–4502 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2007056–2007058. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07396b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Katsuhiko Minoura,
Yuki Kojima,
Taishi Yoshii,
Ryoya Ito,
Takeshi Yamada
*,
Katsuhiko Minoura,
Yuki Kojima,
Taishi Yoshii,
Ryoya Ito,
Takeshi Yamada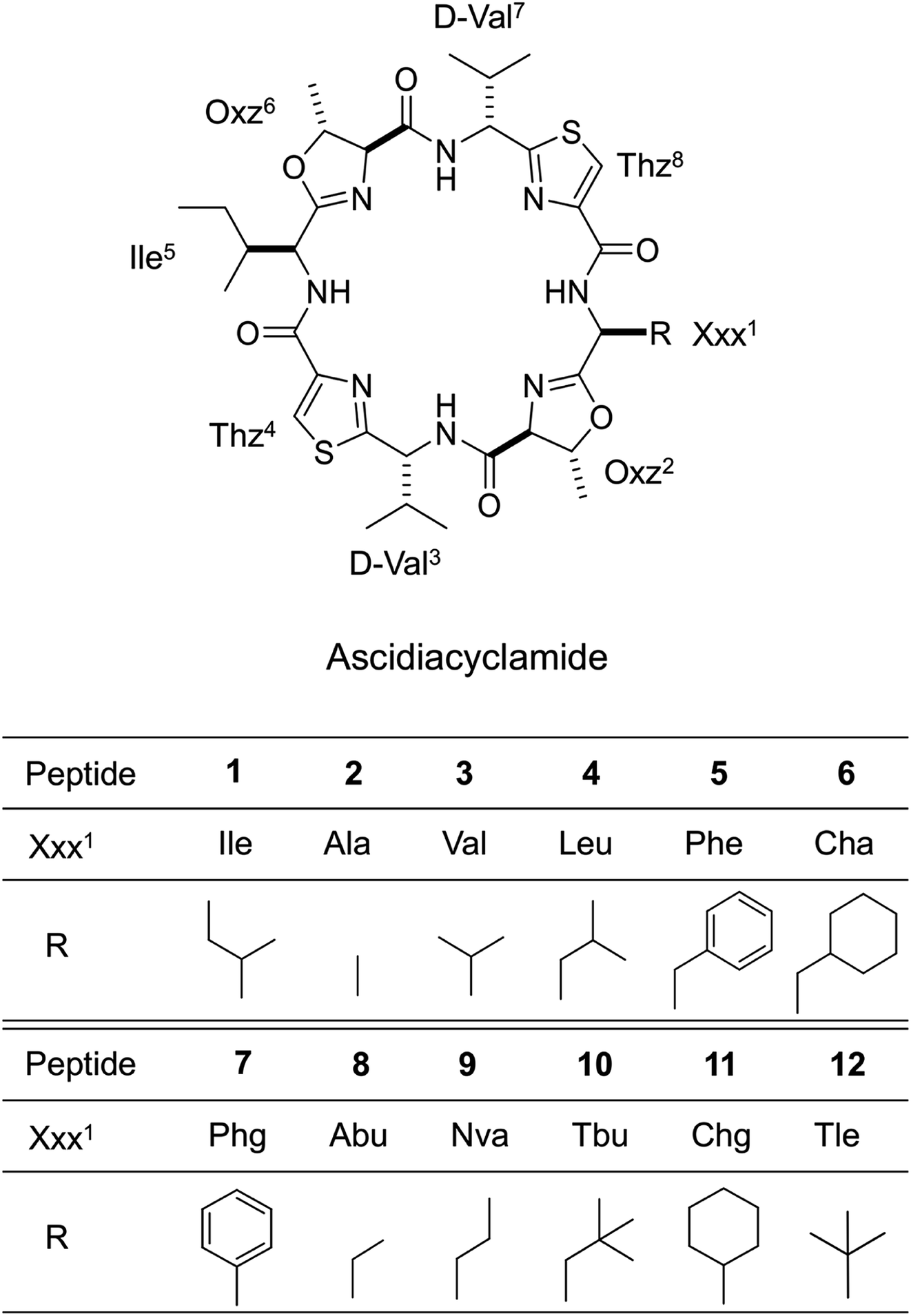
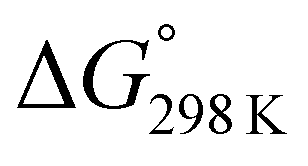 value was calculated from ΔG° = −RT
value was calculated from ΔG° = −RT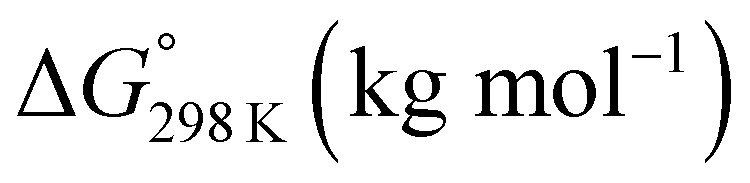
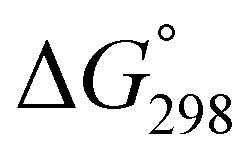 value close to zero, exhibited half the cytotoxicity of 1, while peptides 2 and 8, with negative
value close to zero, exhibited half the cytotoxicity of 1, while peptides 2 and 8, with negative 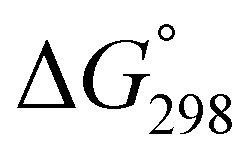 values, showed one-fifth the cytotoxicity and no cytotoxicity, respectively. These results suggest that a structural change from the square to the folded form decreases the cytotoxicity. Consistent with those ideas, the reference peptide for the square form (T3ASC) showed the strongest cytotoxicity, while the reference for the folded form (dASC) showed no cytotoxicity. Peptides 4, 5, 6 and 10 all had negative ΔG° values. The cytotoxicity of 4 was reduced to one-third that of 1, and 10 showed no cytotoxicity. One the other hand, peptides 5 and 6 showed cytotoxicity equal to or stronger than 1. Thus, peptides 5 and 6 did not adhere to the structure–cytotoxicity relationship observed with the others.
values, showed one-fifth the cytotoxicity and no cytotoxicity, respectively. These results suggest that a structural change from the square to the folded form decreases the cytotoxicity. Consistent with those ideas, the reference peptide for the square form (T3ASC) showed the strongest cytotoxicity, while the reference for the folded form (dASC) showed no cytotoxicity. Peptides 4, 5, 6 and 10 all had negative ΔG° values. The cytotoxicity of 4 was reduced to one-third that of 1, and 10 showed no cytotoxicity. One the other hand, peptides 5 and 6 showed cytotoxicity equal to or stronger than 1. Thus, peptides 5 and 6 did not adhere to the structure–cytotoxicity relationship observed with the others.