
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreening the synthesis of peptide therapeutics: an industrial perspective
Vincent Martin
,
Peter H. G. Egelund
,
Henrik Johansson
 ,
Sebastian Thordal Le Quement
,
Felix Wojcik
and
Daniel Sejer Pedersen
*
,
Sebastian Thordal Le Quement
,
Felix Wojcik
and
Daniel Sejer Pedersen
*
Novo Nordisk A/S, CMC API Development, Smørmosevej 17-19, DK-2880 Bagsværd, Denmark. E-mail: dsps@novonordisk.com; Tel: +45 4444 8888
First published on 24th November 2020
Abstract
Solid-phase peptide synthesis (SPPS) is generally the method of choice for the chemical synthesis of peptides, allowing routine synthesis of virtually any type of peptide sequence, including complex or cyclic peptide products. Importantly, SPPS can be automated and is scalable, which has led to its widespread adoption in the pharmaceutical industry, and a variety of marketed peptide-based drugs are now manufactured using this approach. However, SPPS-based synthetic strategies suffer from a negative environmental footprint mainly due to extensive solvent use. Moreover, most of the solvents used in peptide chemistry are classified as problematic by environmental agencies around the world and will soon need to be replaced, which in recent years has spurred a movement in academia and industry to make peptide synthesis greener. These efforts have been centred around solvent substitution, recycling and reduction, as well as exploring alternative synthetic methods. In this review, we focus on methods pertaining to solvent substitution and reduction with large-scale industrial production in mind, and further outline emerging technologies for peptide synthesis. Specifically, the technical requirements for large-scale manufacturing of peptide therapeutics are addressed.
 Vincent Martin | Vincent Martin obtained his MSc and PhD degrees in organic chemistry from the University of Montpellier (France). After his PhD, he joined the laboratory of Prof. Krishnamurthy at the Scripps Research Institute as a Research Associate, working on origin of life and oligonucleotides chemistry from 2015 to 2017. Since 2018, he is a postdoctoral fellow at Novo Nordisk, focusing on the development of new methods for green solid-phase synthesis of peptides. |
 Peter H. G. Egelund | Peter H. G. Egelund obtained his BSc and MSc degrees in chemistry from the University of Southern Denmark (Denmark). He has performed several projects in organic synthesis, including potential antibiotics and anti-cancer medicines. Since 2020 he is a Development Scientist, focusing on finding green alternatives for DMF in solid-phase peptide synthesis. |
 Henrik Johansson | Henrik Johansson obtained his BSc and MSc degrees in organic chemistry from Lund University (Sweden), and his PhD in medicinal chemistry from The University of Copenhagen (Denmark). In 2015 he joined GSK and The Francis Crick Institute (UK) as a Postdoctoral Research Associate in chemical biology with the Crick-GSK Biomedical LinkLabs, working on Cys-reactive fragment screening and E3 ubiquitin ligases in the group of Katrin Rittinger. Since 2020 he is a Development Scientist at Novo Nordisk working with protein ligation. |
 Sebastian Thordal Le Quement | Sebastian Thordal Le Quement obtained his MSc and PhD degrees in synthetic organic chemistry from the Technical University of Denmark and the University of Copenhagen. After his PhD, he has held several postdoctoral and senior scientist positions in academia, including a stay at the Broad Institute of MIT and Harvard from 2012–2014. Since 2015, he has been employed at Novo Nordisk, and is now a Team Leader in chemical development mainly focusing on early-stage API development and manufacturing. |
 Felix Wojcik | Felix Wojcik studied chemistry at Heidelberg University (Germany) and obtained his doctor of natural sciences degree from the Free University of Berlin (Germany) for his work at the Max Planck Institute of Colloids and Interfaces (Biomolecular Systems Department). From 2014–2019 he performed his postdoctoral studies in chemical biology at Princeton University (USA) with the group of Tom Muir. Since 2019 he holds a Development Scientist position at Novo Nordisk mainly focusing on early-stage chemical API processes including synthetic, semi-recombinant and enzymatic processes. |
 Daniel Sejer Pedersen | Daniel Sejer Pedersen obtained his MSc and PhD degrees in synthetic organic chemistry from the University of Copenhagen (Denmark) and Cambridge University (UK), respectively. Daniel has held several positions at Universities and in industry and was an Associate Professor at the University of Copenhagen from 2010 to 2017. Since 2017 he has been employed as a Senior Development Scientist in Chemical Development at Novo Nordisk focusing on GMP manufacturing for phase 3 clinical trials. |
1. Introduction
The first synthetic peptide therapeutic oxytocin was introduced in 1962, and as of 2017 over 60 peptide drugs have been approved in the US, Europe and Japan, more than 150 drugs are currently in active clinical development, and >260 have been tested in human clinical trials.1 An often vented concern about peptide therapeutics is their poor oral bioavailability, which seriously hampers oral administration. This drawback is usually circumvented by alternative routes of administration, such as subcutaneous injection or inhalation,2 but advances in peptide formulation such as using permeation enhancers for increased oral absorption will undoubtedly accelerate the growth of this important class of therapeutic molecules.3,4 Tellingly, the market for peptide therapeutics is currently valued at USD 23 billion, but is anticipated to increase to USD 57 billion by 2027.5 Peptide and protein active pharmaceutical ingredients (APIs) can be prepared by either chemical or biological routes, where chemical synthesis is the current standard for preparation of peptides (especially those bearing unnatural amino acids or particular functional groups), while biological routes such as recombinant expression and fermentation approaches, enzymatic or semisynthetic approaches are more advantageous for large peptides and proteins.6 Chemical synthesis of peptides can be achieved either in solution- or by solid-phase, both strategies having their advantages and disadvantages.7 For short peptides (≤10–15 amino acids), solution-phase synthesis is usually the strategy of choice.6 On the other hand, the landmark invention of solid-phase peptide synthesis (SPPS) by Merrifield, i.e. the anchoring of a peptide to an insoluble solid support composed of a polymer (e.g. polystyrene),8 has enabled the synthesis of longer peptides that were previously unobtainable via traditional solution-phase chemistry. Steady improvements in protecting groups, coupling reagents, and peptide synthesis conditions have enabled routine access to peptides of high purity, in a scalable manner.9–11 However, the SPPS synthetic cycle, comprised of a series of repetitive cycles of coupling, washing and deprotection steps with easy separation of reagents from the solid support by filtration, often employs super-stoichiometric amounts of reagents to push the reaction to completion, generally with poor atom economy (Fig. 1). Furthermore, owing to the major contribution of solvents to the mass balance of a typical SPPS process,12,13 more environmentally benign alternatives to the most frequently used solvents are needed, as well as technologies that promote reduced solvent use and recycling. Today, most of the reagents and solvents applied in peptide chemistry are classified as environmentally problematic substances by the ECHA (European Chemicals Agency) under the REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals) regulation. Current and impending regulation by REACH has the classic SPPS solvents dimethylformamide (DMF) and dichloromethane (CH2Cl2) as well as N-methyl-2-pyrrolidone (NMP) and dimethylacetamide (DMAc) heading for restriction (ECHA Annex XVII) and/or authorization for use (ECHA Annex XIV),14 necessitating that alternative SPPS solvents are identified in the immediate future to avoid disruption of industrial production of therapeutic peptides.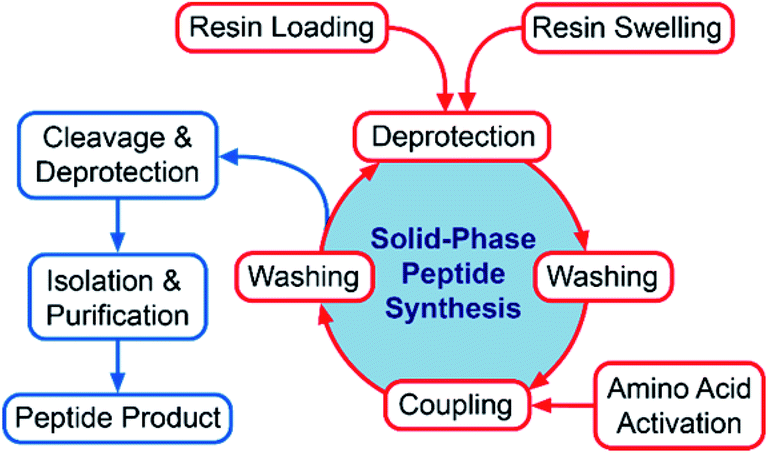 | ||
| Fig. 1 Overview of the SPPS cycle and subsequent deprotection, work-up and purification steps. The present review focuses on technical aspects of the SPPS cycle (shown in red) while cleavage, deprotection, isolation and purification steps (shown in blue), are only briefly touched upon. | ||
The greening of peptide synthesis has been reviewed from various perspectives in recent years. Albericio and co-workers have published two reviews primarily discussing the SPPS cycle, loading and cleavage and deprotection, water-based SPPS approaches as well as alternative synthetic approaches for peptide synthesis mostly of relevance to academic research.15,16 Isidro-Llobet et al. have given a high-level overview on the status of green peptide synthesis and purification discussing all aspects of peptide synthesis and new technological platforms for peptide synthesis and purification from an industrial viewpoint.17 Lax and Shah have reviewed the economic and environmental factors affecting the sustainability of peptide manufacturing from an industrial perspective, comparing solution and solid phase approaches with recombinant, semi-synthetic and ligation methods.18 To date no reviews have delved on the specific technical requirements for a green peptide synthesis protocol from an industrial large-scale manufacturing perspective. The aim of the present review is therefore to outline and discuss the technical requirements that the pharmaceutical industry has for SPPS and a green peptide synthesis protocol, and to put the progress made to date into an API manufacturing context. In relation to SPPS this review will focus on greener improvements pertaining to the SPPS cycle, including swelling, coupling, Fmoc-removal and washing steps (Fig. 1). A detailed analysis of peptide cleavage and deprotection, precipitation, intermediate processing (e.g. cyclisations, oxidation etc.), downstream purification, isolation, and the synthesis of raw materials for SPPS (e.g. amino acid building blocks and coupling reagents) is beyond the scope of the present review and is only briefly touched upon. In addition to SPPS, a selected number of emerging technologies for peptide synthesis (e.g. water-based SPPS, protein ligation and peptide synthesis in continuous flow) with potential use for future large-scale manufacturing of peptide APIs are highlighted and discussed.
2. Peptide synthesis in an industrial setting
From the pharmaceutical industry perspective, the least disruptive short-term scenario for the continued manufacture of therapeutic peptides would be to adjust current SPPS protocols and transition to non-hazardous, green SPPS solvents. This would be greatly advantageous from a procurement point of view, as the necessary supply chain for a variety of raw materials (e.g. 9-fluorenylmethoxycarbonyl (Fmoc)-protected amino acids, activating reagents, resins or linkers) is fully established and cost-efficient building blocks can be easily procured from several independent suppliers in both non-GMP and GMP qualities.18,19 In the longer term, the development of a completely new platform for the synthesis of peptides, which would utilize raw materials with lower environmental impact, is attractive. For example, Fmoc-protected amino acids and coupling reagents are used in excess during SPPS processes (poor atom economy) and are additionally manufactured by classical organic synthesis means, often involving the use of processes with a large negative environmental footprint. Thus, transitioning from the current Fmoc-based chemistry to emerging peptide synthesis technologies appears desirable from an environmental perspective and should be pursued by both academia and industry. For example, this can be done in consortiums such as the American Chemical Society Green Chemistry Institute, where pharmaceutical companies are working together with academia to promote greener chemistries, including SPPS.20 Of crucial importance to this discussion, are considerations about the definition of a green solvent, especially in the context of organic synthesis. Several big pharmaceutical companies have published solvent selection guides which are very helpful when faced with the choice of process solvents.21,22 However, these guides are generally not aligned, and it can sometimes be difficult to deduce which criteria the classification of the solvents have been based upon. Indeed, the classification can be influenced by the area of expertise of the company and what would be considered as a good solvent for substitution for one company, could prove less pertinent for another. As there is no such thing as a perfectly green solvent, it is often a balance between benefits and drawbacks for a specific application. The IMI-CHEM21, a European consortium which promotes sustainable biological and chemical methodologies has also published a guide which is based on a survey of publicly available solvent selection guides.23 This consortium proposes a set of Safety, Health and Environment criteria aligned with the Global Harmonized System (GHS) and EU regulations. Finally, the volumes of solvents employed during manufacturing should also be kept in mind. For example, switching from a problematic solvent to a green solvent may have a larger negative environmental impact if the volumes are multiplied by several digits in the process, emphasizing the necessity for a holistic assessment of the process at hand. From a technical perspective, it may also not be possible to carry out the desired synthesis in a pilot plant beyond a certain volume (maximum capacity of reactors). At Novo Nordisk we have decided to focus on the ICH (International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) classification,24 and REACH status of solvents when conducting solvent substitution, taking both environmental and safety factors into account. Currently, we operate with three solvent categories: (a) REACH compliant, (b) REACH affected, (c) Incomplete data (currently REACH compliant but a significant amount of data is not available). The classification of solvents is clearly much more complex than reflected by these three categories, but we have judged that it is better to keep the classification simple and easy-to-use by our project teams. These teams can then proceed to make an in-depth analysis for individual projects taking all relevant aspects into consideration (e.g. recyclability, cost, supply chain, CO2-foot print etc.).It should be noted that any new methodology for the manufacturing of peptides is unlikely to be applied to already marketed products. A seemingly simple change such as changing the solvent system will most likely affect the purity profile and critical quality attributes associated with the API. New impurities generated in the manufacturing process will have to be qualified in new toxicology studies etc. requiring extensive experimental and regulatory paper work.19 Typically, such changes are only made if there is a substantial financial incentive or if required by the authorities. Regulatory requirements can be dispensed for due to socio-economic considerations, to prevent a cost increase for the patient/society and to ensure that patients have uninterrupted access to medicines. The present review will hence discuss the greening of peptide synthesis from the perspective of new chemical entities and not for marketed drugs. In addition, a new SPPS solvent must fulfil several basic technical requirements to qualify as a reasonable substitute for DMF/NMP. Listed below, we have identified a set of criteria and considerations which would allow for the implementation of a process comparable to our current capability when using DMF or NMP.
2.1 SPPS large-scale synthesis equipment
SPPS on scale is normally performed in batch mode. Reactor volumes up to 1000 litres are common and reactors are often jacketed to allow heating/cooling. Large-scale SPPS reactors are fitted with a filter at the bottom and mixing is performed by mechanical agitation rather than bubbling of gas from the bottom of the reactor, shaking or similar means that are common for laboratory scale SPPS. The resin from which the peptide chain is growing during the SPPS cycle remains inside the reactor, and between coupling and deprotection steps the solvent is drained through the bottom filter and the resin is washed with solvents to remove excess reagents and by-products. Unlike in a laboratory setting, all operations for synthesis such as heating, filtering and addition of reagents and solvents as well as isolation and purification are time consuming and thus present engineering challenges. Solvents are typically stored in tank farms outside the facility and must be pumped in, filtering can be slow due to clogging or insufficient vacuum, reagents are dissolved in separate vessels and are pumped into the reactor with the resin etc. These are all engineering aspects that must be considered during process development to ensure a relatively smooth transfer to the pilot plant that take the plant's limitations into consideration. For the same reason the stability of reagents, building blocks and peptide resins in solution must be well understood to ensure that no significant decomposition occurs during extended holding times and to allow the preparation of solutions well in advance to increase efficiency.2.2 SPPS solvent viscosity and melting point
The room temperature within a production plant will typically vary between 15–35 °C depending on the geographical location and the season. Moreover, for practical and safety reasons solvents are usually stored outside the production plant in tank farms in the terrain (or underground) exposed to the surrounding climate. Although measures are taken in modern facilities to insulate tank farms and ensure heating of piping for solvent transfer this is by no means the industry standard and solvents can, depending on the geographical location and season, experience temperatures well below room temperature. Consequently, to prevent technical challenges at low temperatures, it is preferable that solvents have melting points of ≤10 °C. From a recycling perspective it is preferable that solvents do not have very high boiling points. That said, cost-efficient recycling of for example DMF from SPPS processes is performed by some companies and used for cleaning of process equipment. The financial incentive for recycling will have to be evaluated case by case but ideally a boiling point below 100 °C should be aimed for. The viscosity of solvents should also be considered before implementing a new solvent system. Solvents with high viscosity can present challenges during solvent transfer and can complicate purging and cleaning of reactors and piping during and after production. This may necessitate elaborate and time-consuming cleaning procedures and large volumes of cleaning solvent, and in the worst case could lead to cross contamination. For pilot plant production in general we have found that a viscosity of ≤4 mPa s is acceptable, however, in the context of SPPS a viscosity nearer that of DMF (0.8 mPa s) may be required to ensure appropriate swelling of the resin and diffusion of reagents in commonly used solid supports.132.3 Scalability
To ensure that an SPPS process can be performed efficiently at a reasonable production scale in terms of kg-output of API and in a minimum of batches the following considerations regarding solubility of reagents, building blocks and by-products, reagent and solvent stability and resin swelling should be considered:• Dissolution of building blocks, reagents and by-products: amino acid building blocks, reagents, by-products etc. should be soluble at a concentration of ≥0.25 M (ideally up to 0.40 M) to prevent excessive solvent volumes and clogging of filters. It can be tolerated that some building blocks are soluble at a lower concentration or that special conditions are applied for single couplings or deprotections but overall a concentration ≥0.25 M should be aimed for. Similarly, poor solubility of certain by-products may be compensated for by utilising alternative solvents for washing. For example, in standard SPPS the removal of diisopropylurea (DIU), a by-product from diisopropylcarbodimimide (DIC) mediated couplings, is complicated by the low solubility of DIU in many polar aprotic solvents. As a result, DIU can precipitate during couplings and lead to clogging of filters during draining and washing operations. To dissolve and remove precipitated DIU, 2-propanol (in which DIU has a high solubility) is commonly employed for washing steps.
• Reagent and solvent stability: during production, solutions of amino acids, coupling reagents and additives may be exposed to extended holding times at room temperature. Good stability for at least one week at room temperature is desirable for standard amino acid building blocks. Moreover, the solvent itself must also be stable throughout the peptide synthesis cycle, in particular with respect to possible side-reactions with the solid-supported peptide in order to prevent the formation of undesired acylated/alkylated peptide-related species that may be difficult to remove during purification (resulting in a yield reduction and/or increased solvent consumption during isolation and purification).
• Resin swelling: the resin swelling capability is essential for the process performance during synthesis, but it is important to avoid excessive swelling because it will limit the possible production scale. In general, we recommend swelling in the range of approximately 4–7 mL g−1 for the starting resin (resin with linker or preloaded with the first amino acid), which balances good swelling to facilitate the coupling and deprotection reactions during SPPS, while not swelling excessively. However, the acceptable maximum swelling should be evaluated case by case. If for example the yearly API demand is low and suitable equipment is available high swelling can be tolerated, in particular if it improves process robustness and product quality.
2.4 Process performance
We have found consideration of the following parameters critical to maximise output and optimise process time:• Resins with the highest possible loading that does not impede process performance nor reduce product quality should be aimed for (ideally >1 mmol g−1). During development stages, the process will be optimized for the highest possible loading and it is not uncommon that high loadings cannot be achieved due to aggregation or other factors. We aim for a starting loading of minimum 0.30 mmol g−1 but ideally significantly higher.
• For a standard amino acid coupling cycle, the reaction time for completion of coupling at room temperature should not exceed 60–90 min and stay below 30–40 min for Fmoc-removal. In practise, this should make it possible to maintain a cycle time of ≤3 hours per amino acid residue, operations included.
• The loading of the first amino acid on polystyrene resin at room temperature should not exceed 120 min.
• During washings of the resin after coupling and deprotection, the solvent should remain efficient at solubilizing by-products and excess reagents in order to minimize the overall solvent volumes.
2.5 Cleavage, deprotection, purification and isolation
After completed elongation of the peptide chain, cleavage from the resin and removal of the side chain protecting groups to release the crude peptide, are most frequently carried out simultaneously. The reaction is typically performed using a cleavage cocktail comprised of trifluoroacetic acid (TFA), water and scavengers. Replacement of DMF, NMP or CH2Cl2, is unlikely to have a direct impact on this step since washing with a volatile solvent and thorough drying would remove any residual SPPS solvent that might influence the cleavage reaction or lead to generation of solvent-related impurities. Alternative final cleavage protocols such as using 0.1 N HCl in hexafluoro-2-propanol (HFIP) or trifluoroethanol (TFE) have been proposed to avoid using TFA,25 which in itself is hazardous and environmentally damaging, yet fluorinated solvents such as HFIP and TFE are REACH impacted. Soft cleavages allowing for release of fully protected peptide fragments (C-terminal acids) from the resin are usually conducted by treatment with dilute TFA or HFIP in CH2Cl2. CH2Cl2 is an ICH class 2 solvent and has been flagged with an SVHC profile in REACH and a substitution for an environmentally more benign solvent should be pursued. Post cleavage, several strategies for the isolation of peptide products are available. The exact strategy is usually evaluated on a case-by-case basis and is very dependent on the nature of the API. The various approaches all have pros and cons with respect to purity, yield and chemical waste generation. The most common strategy for work-up after cleavage involves removal of the resin by filtration, followed by precipitation of the cleaved peptide with an anti-solvent such as diisopropyl ether (DIPE) or t-butylmethyl ether (TBME). Ethers can be problematic due to their peroxide-forming propensity but can be handled if the appropriate safety precautions are taken. Precipitation of the peptide together with the resin is a viable alternative and may facilitate filtration and washing of the peptide product. An appropriate precipitation solvent should be able to dissolve all by-products (mainly scavenged side chain protecting groups). An extractive work-up process can also be utilised, in which the peptide product is solubilized in an aqueous buffer followed by washing with an appropriate organic solvent in order to remove by-products. Depending on the purity of the isolated crude peptide, several purification steps typically using reverse-phase HPLC and ion-exchange chromatography are usually applied for purification of the crude peptide. The purification process can consume significant quantities of solvent, usually acetonitrile (MeCN) and water. However, MeCN recycling is feasible and consequently can reduce the amount of waste drastically.26 After salt exchange to obtain the desired salt-form product isolation is typically performed by freeze-drying (lyophilisation). However, the use of spray-drying is becoming increasingly wide-spread because of high turnover resulting from a continuous process, and due to relatively small equipment size when compared to more traditional freeze-drying equipment. In the case of lyophilisation, the solution containing the API is often concentrated to increase throughput by for example rotary evaporation, thin-film evaporation or UF-DF (ultrafiltration–diafiltration).192.6 Cost, supply and intellectual property rights (IPR)
Ideally a new platform for peptide manufacturing should not increase the cost of the API compared to the current cost of manufacturing by SPPS. That said, scientist developing new methodology should not be discouraged by a higher cost of a new methodology as it is expected that a widely adapted technological breakthrough will result in a significant reduction in cost over time. Nevertheless, a “reality-check” of a new process or process solvent to assess if it is reasonable to expect that it can reach a cost similar to that for current SPPS should be performed. As a part of this exercise it is important to assess if there are any IPR related issues on the production or use of a new solvent system to ensure competitiveness between solvent manufacturers, a more robust supply chain and avoid obstacles in relation to licensing and royalties. In addition to the cost for a new solvent, the quality is essential to avoid potentially reactive impurities arising from the solvent manufacturing process or stemming from solvent decomposition. Moreover, a supply chain with multiple independent suppliers is necessary to ensure a reliable supply and to decrease the bulk price of the solvent. That said, it should be kept in mind that the greening of peptide synthesis in fact has the potential to reduce production cost. A change to new solvent systems may reduce the required solvent volumes, facilitate recycling of solvents, provide improved purity profiles and increased yields, avoid costly and time consuming problems pertaining to new environmental regulatory requirements etc.17,18 Benchmarking new processes using available green chemistry metrics such as atom economy, complete E-factor (cEF), and Process Mass Intensity (PMI) may provide useful input for reagent and solvent selection and reduction,27–29 and ultimately cost reduction. The cost of a peptide manufacturing process is the sum of the required: (i) FTEs (full-time employees), (ii) pilot plant time, and (iii) raw materials. The contribution from each of these categories varies between projects and is influenced by many factors such as scale and length of the peptide target and will inevitably affect the development programme. In 2018, CMC at Novo Nordisk assessed the cost of raw materials for the SPPS campaign of a 39-mer peptide as part of a clinical phase 2 programme (data not shown). From this analysis the cost distribution for raw materials revealed that solvents accounted for 27%, resin for 15%, coupling reagents for 23%, and amino acid building blocks and other reagents for the remaining 35% (three non-standard amino acid building blocks accounted for 16% of the 35%). During process optimisation towards phase 3 the cost contribution from solvents would likely increase even further when the equivalents of building blocks and reagents are adjusted. In conclusion, solvents represent one of the major raw material cost drivers in solid-phase peptide synthesis, and in addition to addressing environmental concerns and adhering to regulatory requirements there is a clear financial incentive to replace, reduce and recycle process solvents if possible.3. SPPS in alternative solvent systems
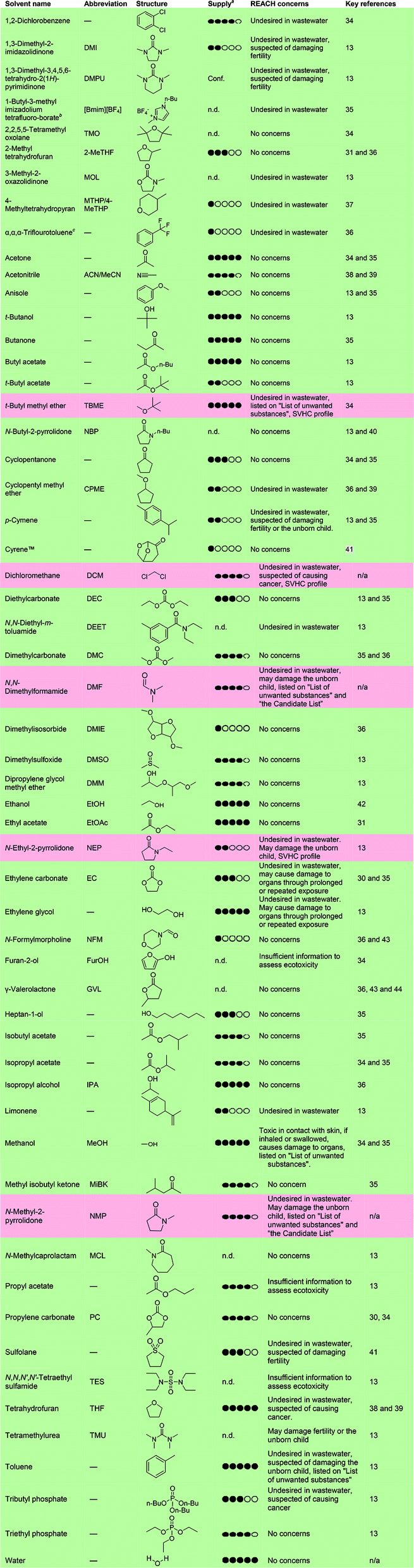
The concept of “greening” SPPS through replacement of problematic solvents has been gaining momentum in recent years and has been pioneered by a handful of research groups.30,31 The following subsections deal with the state-of-the-art in solvent replacement for resin swelling and the SPPS cycle as outlined in Fig. 1 (vide supra). A comprehensive overview of solvents previously described in the literature in relation to green peptide synthesis, and standard SPPS solvents (e.g. DMF, NMP and CH2Cl2), is outlined below along with key references and notes on supply based on the amounts manufactured and/or imported to the European Economic Area (EEA) annually as reported by ECHA (Table 1).14 The solvents have been assessed according to the Novo Nordisk internal EcoChem criteria, which takes into account both environmental and safety aspects of chemicals based on ECHA REACH data, and are divided into two categories; REACH compliant (green) and REACH affected (red). It should be noted that currently REACH compliant solvents may be suspected of having environmental or safety problems and could be under active investigation. This aspect should also be taken into consideration when selecting process solvents. Major REACH concerns for all solvents have been listed in Table 1.a 1 black circle indicates an annual supply in the EEA of <100 tonnes, 2 circles = 100–1000 tonnes, 3 circles = 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes, 4 circles = 10000–100 000 tonnes, and 5 black circles indicates >100000 tonnes; n.d. = no data available; conf. = confidential; n/a = not applicable for standard solvents.b Imidazolium-based ionic liquids are associated with concerns regarding their ecotoxicity and thermal decomposition.45,46c Per- and polyfluoroalkyl substances are currently under investigation by ECHA REACH because these substances are considered to be persistent. The use of per- and polyfluoroalkyl substances will likely be restricted in the future. 000 tonnes, 4 circles = 10000–100 000 tonnes, and 5 black circles indicates >100000 tonnes; n.d. = no data available; conf. = confidential; n/a = not applicable for standard solvents.b Imidazolium-based ionic liquids are associated with concerns regarding their ecotoxicity and thermal decomposition.45,46c Per- and polyfluoroalkyl substances are currently under investigation by ECHA REACH because these substances are considered to be persistent. The use of per- and polyfluoroalkyl substances will likely be restricted in the future. |
|---|
 |
3.1 Swelling, dissolution and stability
Good resin swelling is adamant to achieve efficient SPPS but at the same time should not be excessive to ensure high product output per production batch. At the on-set of SPPS, resin swelling is dominated by the properties of the resin itself but as the peptide chain grows it will gradually affect the swelling properties of the peptide resin. We have seen examples of peptide resins where the peptide induces a dramatic increase in swelling or conversely makes the resin shrink. Every peptide target will affect the swelling properties of the peptide-resin uniquely and this must be addressed during development to optimise all coupling and deprotection steps. Consequently, most of the scientific literature that investigates resin swelling look at the resin-linker system alone or with the first amino acid pre-loaded, which is a good starting point if it is kept in mind that this will change during the course of the synthesis. A compilation of swelling data was recently published by Albericio et al.,15 but in this section swelling data from many research teams are presented and discussed in full to give the reader the complete picture and possibility to make their own analysis of the data at hand.Lawrenson et al. studied the swelling properties of nine different starting resins in 25 solvents as outlined in Table 2.35 The solvents covered a wide range of polarities as well as hydrogen bond donor/acceptor properties, and were divided into three categories based on their resin swelling properties; good (swelling >4.0 mL g−1), moderate (swelling from 2 to 4 mL g−1), and poor (swelling <2 mL g−1). Significant differences between the different resins and solvents were observed. The most versatile resins were ChemMatrix® and ArgoGel™ (a crosslinked polyethylene glycol (PEG) resin and a PEG grafted onto polystyrene resin, respectively), which were able to swell >4 mL g−1 in a wide variety of solvents, and the least versatile resin was the polyamide SpheriTide™ which was only able to swell >4 mL g−1 in DMF, NMP and CH2Cl2. Swelling of the ChemMatrix® resin was sometimes excessive, which can be problematic from a production perspective as a higher swelling volume limits the batch size. The polystyrene-based resins showed different swelling properties with the Merrifield resin swelling well in some of the moderately polar solvents containing oxygen atoms, whereas the ParaMax resin swelled well in all of them. The JandaJel™ resin swelled particularly well in non-polar solvents such as limonene or p-cymene. Cyclopentanone and dimethyl isosorbide were found to be good solvents for almost all the resins, while moderate swelling was observed for the SpheriTide™ resin. Lawrenson et al. proposed that resin swelling is dependent on the composition of the resin rather than its functionalisation, although the structurally comparable Merrifield and ParaMax resins (ParaMax resin being a Merrifield resin functionalized exclusively on the para position of the aromatic ring) displayed differences in their swelling properties. Functionalising resins with different linkers and studying their swelling properties could shed more light on this topic.
| a Green: good swelling (>4.0 mL g−1). Orange: moderate swelling (2.0 to 4.0 mL g−1). Red: poor swelling (<2.0 mL g−1).b The resin (100 mg) was weighed into a 2 mL syringe fitted with a polypropylene fritted disc (void volume = 0.12 mL). Solvent (2 mL) was added and the syringe was agitated for 1 hour at room temperature. The solvent was removed by compressing the syringe piston. The resin was then allowed to return to its maximum volume by slowly withdrawing the piston. The volume was recorded, and the degree of swelling calculated from the following formula: degree of swelling (mL g−1) = 10 × (measured volume − 0.12). |
|---|
 |
A computational model was applied to predict the swelling of resins in different solvents and solvent mixtures, suggesting that some solvent mixtures would achieve better swelling compared to neat solvents. Changing the physical properties by using solvent mixtures could increase the solubility of reagents or decrease melting point and/or viscosity of the solvent. Although a single solvent system might be preferred from a handling and recycling perspective, a major advantage of a mixed solvent system is the ability to adjust the solvent ratio. This flexibility could be beneficial throughout the SPPS cycle, when polarities of various functionalized peptide resins change, or to adapt to the physical properties of the vast number of reagents and resins. The influence of different ratios of solvent mixtures on swelling was further described by North and co-workers.34 Both in silico and laboratory experiments were carried out on Merrifield and HypoGel 200® resins (Table 3). Resin swelling was shown not to correlate linearly with solvent composition. Moreover, some solvent mixtures showed increased swelling volumes compared to the corresponding neat solvents. For example, propylene carbonate (PC) and ethyl acetate (EtOAc) swelled Merrifield resins at 1.8 and 3.8 mL g−1 respectively, while a 1:9 PC/EtOAc mixture swelled the same resin with a 20% increase compared to neat EtOAc. A 1:9 PC/EtOAc mixture and neat EtOAc were compared for the synthesis of the tripeptide H-Leu-Ala-Phe-OH on Merrifield resin. The 1:9 PC/EtOAc mixture gave a 5% increase in yield compared to neat EtOAc, but a higher purity was obtained in EtOAc (95.2% and 96.9% purity, respectively).
| a The solvents were not miscible in this ratio.b For mixtures of DMF and water, addition of >10% water reduced the swelling to the same level as observed for pure water (approximately 2 mL g−1).c The resin (100 mg) was weighed into a 2 mL syringe fitted with a polypropylene fritted disc (void volume = 0.12 mL). Solvent (2 mL) was added and the syringe agitated for 1 hour at room temperature. The solvent was removed by compressing the syringe piston. The resin was then allowed to return to its maximum volume by slowly withdrawing the piston. The volume was recorded, and the degree of swelling calculated from the following formula: degree of swelling (mL g−1) = 10 × (measured volume − 0.12). |
|---|
 |
Ferrazzano et al. reported the combinations of Cyrene™ or sulfolane with DEC, and anisole with DMC in varying ratios as green alternatives for SPPS (Table 4),41 although sulfolane is undesired in waste water and suspected of reproductive toxicity, and anisole is suspected of causing genetic defects (see Table 1).14 The swelling properties of Merrifield, ChemMatrix® and Tentagel™ resins in combination with either Wang or Rink Amide linkers were evaluated.
| a Green: good swelling (>4.0 mL g−1). Orange: moderate swelling (2.0 to 4.0 mL g−1). Red: poor swelling (<2.0 mL g−1).b The resin (100 mg) was weighed into a 3 mL syringe fitted with a polypropylene fritted disc (void volume = 0.2 mL). Solvent (2 mL) was added and the syringe agitated for 30 min at room temperature + 5 min equilibration. The solvent was removed under vacuum and the volume of the dry resin was recorded. The degree of swelling was calculated from the following formula: degree of swelling (mL g−1) = 10 × (measured volume − 0.2). |
|---|
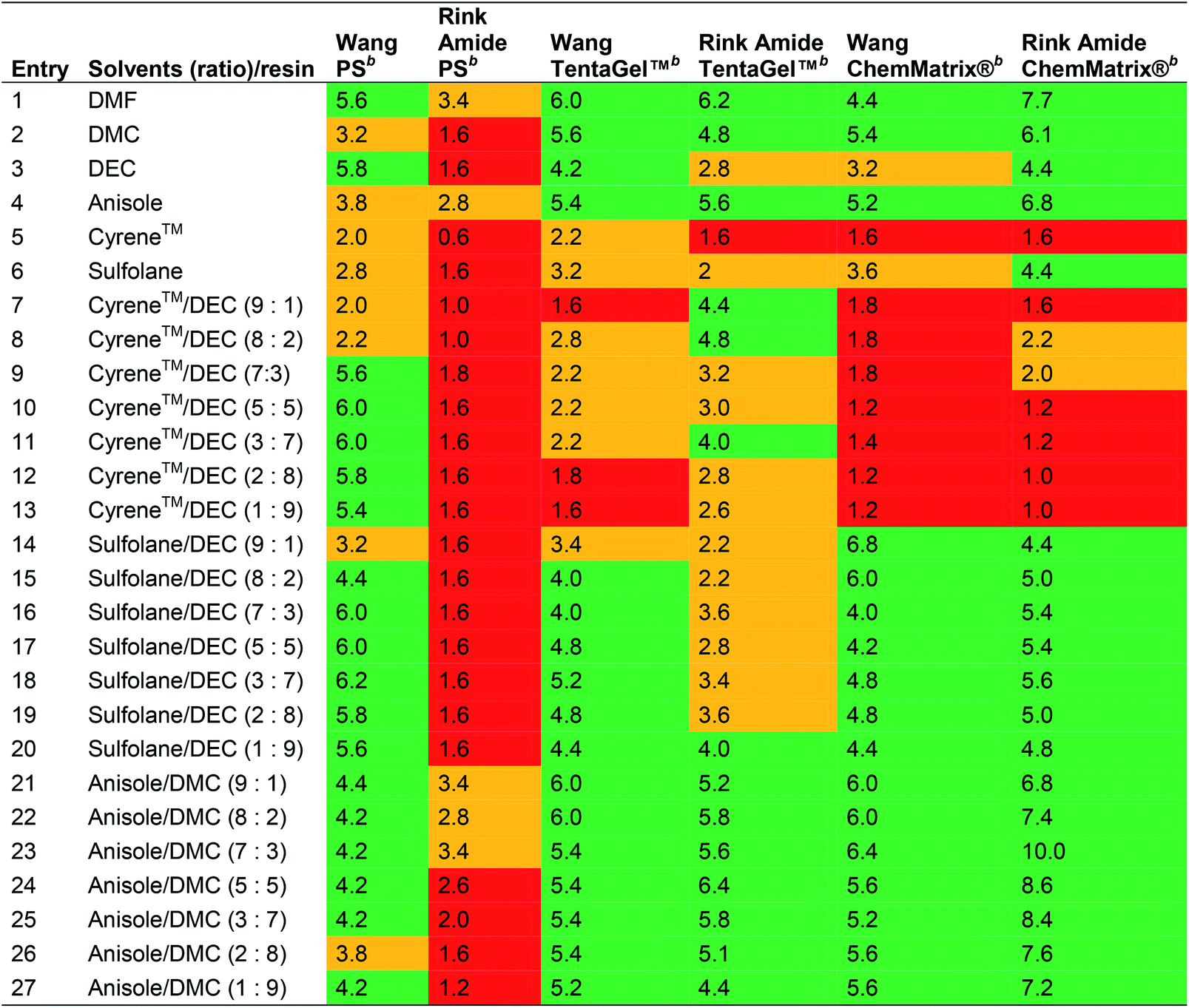 |
In line with the observations of Lawrenson et al. (vide supra), solvent mixtures generally gave improved swelling compared to neat solvents, and furthermore, the linker appeared to affect resin swelling. For the PS resin for example, more swelling was observed for the Wang over the Rink Amide linker, while for the ChemMatrix® resin the opposite was generally observed. When comparing Wang and Rink Amide linkers on TentaGel™ resin, most swelling was observed in 8:2 and 9:1 anisole/DMC (6.0 mL g−1, entries 21 and 22), and 5:5 anisole/DMC (6.4 mL g−1, entry 24) respectively. The best swelling for the Wang ChemMatrix® resin was obtained with 9:1 sulfolane/DEC (6.8 mL g−1, entry 14) while the Rink Amide ChemMatrix® resin had the highest swelling volume in 7:3 anisole/DMC (10.0 mL g−1, entry 23). Overall, Ferrazzano et al. found, as anticipated, that DMF display good swelling properties, but also that mixtures of anisole/DMC were able to match the swelling profile of DMF very well (entries 21 and 1, respectively). In addition to swelling, Ferrazzano et al. performed solubility experiments with the most promising solvent mixtures (Cyrene™/DEC (3:7), sulfolane/DEC (3:7) and anisole/DMC (7:3)). Dissolving Fmoc-Val-OH at a concentration of 0.2 M was only possible in sulfolane/DEC (3:7). Activated amino acids are typically more soluble, and so the solubility of Fmoc-Val-OH was reassessed with the activating reagents (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU®)/N,N-diisopropylethylamine (DIPEA), N,N′-diisopropylcarbodiimide (DIC)/ethyl (hydroxyimino)cyanoacetate (Oxyma Pure), DIC/hydroxybenzotriazole (HOBt), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), and 2-(1I-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU). The best result was obtained with DIC/Oxyma Pure that was able to dissolve Fmoc-Val-OH in all solvent systems in 5 min. Next, DIC/Oxyma Pure in the three different solvent systems was evaluated for its ability to solubilise 18 representative Fmoc-protected amino acid building blocks. In all cases, the activated Fmoc-amino acid was fully soluble at a concentration of 0.2 M.41
In another study, Lopez et al. studied the swelling and solubilisation properties of a wide range of solvents, including many environmentally problematic or poorly characterised ones (e.g. DMPU, tetramethyl urea and sulfolane), using an aminomethyl-polystyrene resin (Table 5). Initially, only solvents able to induce a resin swelling ±30% with respect to DMF were considered of interest (i.e. swelling in the range 4.4–8.2 mL g−1), however, later this was changed to solvents providing a swelling of ≥6 mL g−1.13
| a Green: good swelling (>4.0 mL g−1). Orange: moderate swelling (2.0 to 4.0 mL g−1). Red: poor swelling (<2.0 mL g−1).b One gram of amino methyl resin was placed in a 10 mL graduated cylinder and the solvent was added to give a final volume of 10 mL. The suspension was gently stirred and allowed to settle for 3.0 h. The final swelling volume was measured in mL g−1. |
|---|
 |
Among the solvents studied, 14 solvents were found to swell the resin ≥6 mL g−1, and were studied further for their ability to dissolve two amino acids (Fmoc-Gln(Trt)-OH and Fmoc-Gly-OH), Oxyma, N,N′-diisopropylcarbodiimide (DIC) and N,N′-diisopropylurea (DIU) at a concentration of 0.2 M (Table 6). Out of the 14 solvents, five (anisole, propyl acetate, diethyl carbonate, toluene and ethyl acetate) were unable to dissolve the Fmoc-amino acids and/or DIU. With exception of propyl acetate and diethyl carbonate that were considered too poor, all solvents were evaluated for coupling and Fmoc-removal (vide infra). The partial solubility of DIU in THF, 2-MeTHF and DMF was accepted by the authors because these are expected to be removed by extensive washing after coupling. From a green SPPS perspective this is undesirable as the washing steps have the biggest negative impact in terms of solvent consumption. Moreover, out of 14 solvents studied in depth, seven are problematic from an environmental perspective (NEP, DMPU, THF, TMU, toluene, DMF and NMP, see Table 1), and should not be in scope for green SPPS.
| Solvent/reagent | Fmoc-Gln(Trt)-OH | Fmoc-Gly-OH | Oxyma | DIC | DIU |
|---|---|---|---|---|---|
| a The indicated solvent (10 mL) was added to 2 mmol of each reagent. All mixtures were stirred at the same speed and temperature (22–23 °C) for 10 min. Subsequent visual inspection was reported as soluble (S), partially soluble (PS), or insoluble (I).b Poorly characterised solvent.c Environmentally problematic solvent. | |||||
| Anisoleb | I | I | S | S | I |
| DMIE | S | S | S | S | S |
| NEPc | S | S | S | S | S |
| DMPUc | S | S | S | S | S |
| THFb | S | S | S | S | PS |
| NBP | S | S | S | S | S |
| 2-MeTHF | S | S | S | S | PS |
| Propyl acetate | S | S | S | S | I |
| TMUc | S | S | S | S | S |
| DEC | I | I | S | S | I |
| Toluenec | I | I | I | S | I |
| DMFc | S | PS | S | S | PS |
| NMPc | S | S | S | S | S |
| EtOAc | S | I | S | S | I |
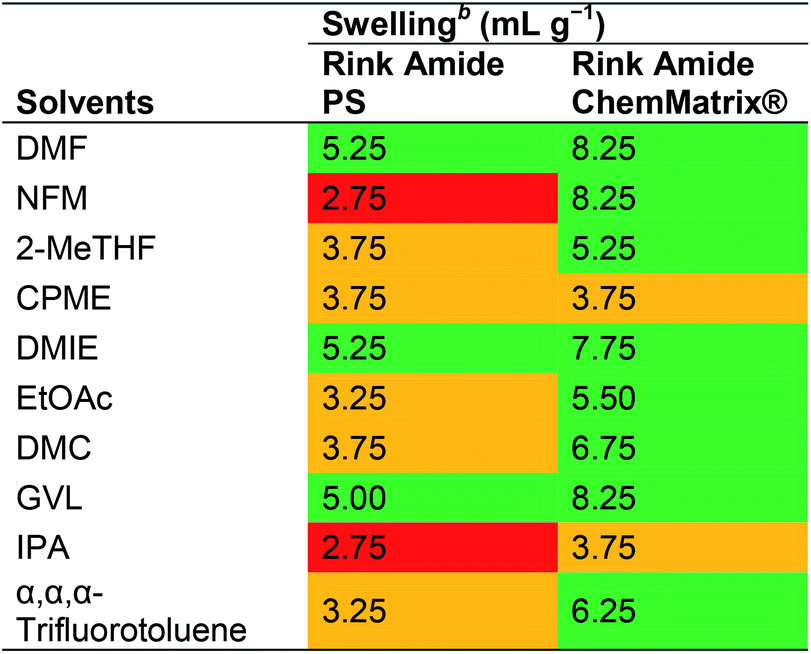
Albericio and co-workers reported a swelling study using PS and ChemMatrix® resins in nine green solvents selected based on the GSK solvents classification list.21,36 The resin swelling properties of NFM, 2-MeTHF, CPME, DMIE, EtOAc, DMC, GVL, IPA and α,α,α-trifluorotoluene were compared to DMF (Table 7). While the ChemMatrix® resin appeared to swell well in all solvents except CPME and IPA (3.75 mL g−1 in both cases), the PS resin only swelled above 4 mL g−1 in DMIE and GVL. In a more recent report focusing on NBP, Albericio and co-workers demonstrated that NBP is able to dissolve all Fmoc-protected amino acids and common coupling reagents at concentrations well above what is usually employed in SPPS, but that it is also overall slightly inferior to DMF in swelling of aminomethyl polystyrene and H-Rink Amide ChemMatrix resins.40
| a Green: good swelling (>4.0 mL g−1), orange: moderate (2.0 to 4.0 mL g−1), and red: low (<2.0 mL g−1).b The resin (200 mg) was placed in a 5 mL syringe and enough solvent to swell the resin and allowed to stand for 5 min. The swelled resin was compressed with the piston until no more solvent could be removed. The piston was pulled slowly until the resin recuperated its maximum volume and the resin volume was recorded. The swelling was calculated according to the following formula: degree of swelling (mL g−1) = (volume of the swelled resin + 0.15 mL)/0.2 g. |
|---|
 |
In a production facility, amino acid building blocks and reagents may be exposed to prolonged holding times in solution. It is therefore important to investigate the stability of Fmoc-amino acids and reagents in solution in any new solvent introduced. For example, although unintentional partial Fmoc-removal may occur during storage (resulting in double incorporation of amino acids during coupling), few reports have looked into this important aspect of SPPS, and only for coupling reagents. Tofteng et al. examined the influence of microwave heating and water content in DMF on the stability and performance of a range of coupling reagents, and concluded that COMU® has poor hydrolytic stability in DMF at room temperature (<5 h), while HATU and especially DIC-Oxyma were more suitable for automated SPPS.47 Kumar et al. addressed the poor stability of COMU® in DMF by investigating COMU® in MeCN, GVL and NFM, and found it to be relatively stable in MeCN and GVL (89% and 84% of COMU® remaining after 48 h, respectively), whereas it completely degraded in DMF and NFM after 48 hours (0 and 3% of COMU® remaining, respectively).48
3.2 The peptide synthesis cycle
When investigating alternative solvents and reagents for SPPS, we suggest that reaction times at room temperature should ideally not exceed 90 min for couplings and 40 min for Fmoc-removal. This is typically the case for standard SPPS processes taking place in DMF or NMP, and within this timeframe full conversion should be achieved with minimal side-reactions. In addition, the process safety of coupling reagents should be considered carefully because these often contain high energy functional groups. Reagents derived from HOBt/HOAt should be used with care as these are known shock-sensitive and explosive substances.49–51 In a recent thermal stability study reported by Pfizer, 45 commonly employed peptide coupling reagents were evaluated.49 In general, coupling reagents based on the HOBt motif were found to be less safe.Albericio and co-workers have published several papers on the use of various solvents for green SPPS. In one study, MeCN and THF were used as DMF substitutes, in combination with DIC and various additives (HOAt, HOBt, Oxyma Pure and Oxyma-B) on an H-Rink Amide AM ChemMatrix® resin, resulting in improved yields and suppression of racemisation compared to DMF. The methodology proved successful both in solution and on solid support for several peptide sequences with up to 10 amino acid residues, including difficult to couple amino acids such as 2-aminoisobutyric acid (Aib).38 The best yields were obtained with THF and Oxyma Pure as coupling reagent, but as the authors note, THF is not a green solvent and thus further work was required.14 In a follow-up study, the synthesis of the Aib-enkephalin pentapeptide was investigated on PS and PEG (ChemMatrix®) resins using 2-MeTHF or CPME in combination with DIC and an additive (HOBt, HOAt, Oxyma Pure, Oxyma-B or K-Oxyma) or with HBTU, HATU or COMU® (Fig. 2 and Table 8). The best results were obtained with DIC/Oxyma Pure in 2-MeTHF both on PS and PEG resins (PS: 97.0%, PEG: 81.9% yield) with a purity superior to the one obtained in DMF (PS: 42.8%, PEG: 53.0%). Even with K-Oxyma/DIC, which gave the best result in DMF (PS: 71.0%, PEG: 85.6% purity), the purity was still lower when compared to the synthesis in 2-MeTHF. With DIC/Oxyma Pure, CPME was superior to DMF on PEG resin (PS: 29.2%, PEG: 61.1% purity).
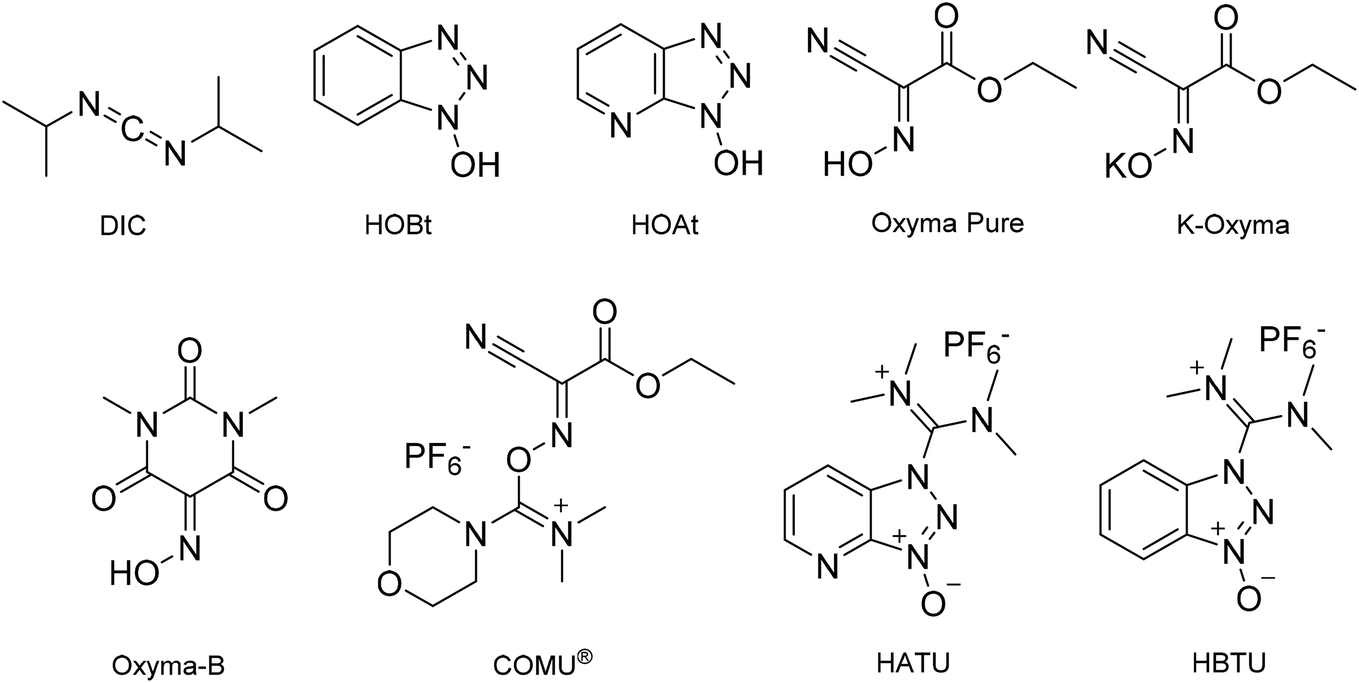 | ||
| Fig. 2 Chemical structures of peptide coupling reagents and additives commonly employed in solid-phase peptide synthesis. | ||
| Resin | Coupling reagent | Solvent | Purityb (%) |
|---|---|---|---|
| a 1 h couplings were applied except for the Aib residues that were coupled twice (2 × 1 h).b Purity was determined by RP-HPLC analysis. | |||
| PS | DIC/Oxyma Pure | 2-MeTHF | 97.0 |
| DIC/Oxyma Pure | DMF | 42.8 | |
| DIC/Oxyma Pure | CPME | 29.2 | |
| DIC/K-Oxyma | DMF | 71.0 | |
| ChemMatrix® | DIC/Oxyma Pure | 2-MeTHF | 81.9 |
| DIC/Oxyma Pure | DMF | 53.0 | |
| DIC/Oxyma Pure | CPME | 61.1 | |
| DIC/K-Oxyma | DMF | 85.6 | |
The uronium-based coupling reagents gave poor purities both in 2-MeTHF and CPME on both resins.39 In another study by Albericio and co-workers, the potential of IPA, EtOAc and 2-MeTHF for washing and deprotection, and 2-MeTHF for coupling was evaluated.31 Different protocols for the synthesis of the Aib-enkephalin pentapeptide and the Aib-ACP decapeptide on Rink Amide ChemMatrix® and Rink Amide PS resins were evaluated (Table 9).
| Resin | Step | Method 1 (reference) | Method 2 | Method 3 | Method 4 | Method 5 |
|---|---|---|---|---|---|---|
| a Fmoc-removal performed at 40 °C.b Fmoc-removal and coupling performed at 40 °C. | ||||||
| — | Washing | 1 × DMF | 1 × DMF | 1 × IPA | 1 × EtOAc | 1 × 2-MeTHF |
| 1 × CH2Cl2 | 1 × CH2Cl2 | 3 × EtOAc | ||||
| 1 × DMF | 1 × DMF | 1 × 2-MeTHF | ||||
| — | Deprotection | 20% piperidine in DMF (7 min) | 20% piperidine in DMF (7 min) | 20% piperidine in IPA (7 min) | 20% piperidine in EtOAc (7 min) | 20% piperidine in 2-MeTHF (7 min) |
| — | Washing | 1 × DMF | 1 × DMF | 1 × IPA | 1 × EtOAc | 1 × 2-MeTHF |
| 1 × CH2Cl2 | 1 × CH2Cl2 | 3 × EtOAc | ||||
| 1 × DMF | 1 × DMF | 1 × 2-MeTHF | ||||
| — | Coupling | Fmoc-AA-OH/DIC/Oxyma Pure (3:3:3 equiv.) 1 h |
||||
| DMF | 2-MeTHF | 2-MeTHF | 2-MeTHF | 2-MeTHF | ||
| Chem-Matrix resin | Purity (%), Aib-enkephalin | 53.0 | 81.9 | 30.8 | 91.9 | 95.0 |
| Purity (%), Aib-ACP | 37.8 | 70.0 | No data | 30.4 | 37.3, 42.8a, 87.1b | |
| PS resin | Purity (%), Aib-enkephalin | 71.8 | 97.0 | No data | 38.6, 54.6a | 41.6, 88.6a |
| Purity (%), Aib-ACP | No data | No data | No data | No data | 7.1a, 25.4b | |
The synthesis of Aib-enkephalin on Rink Amide ChemMatrix® resin resulted in a much higher purity when using 2-MeTHF for couplings (81.9% and 95.0%, methods 2 and 5) instead of DMF (53.0%, method 1), while employing IPA for washings and deprotection (method 3) gave the worst outcome (30.8%). Next, the different conditions were evaluated for the synthesis of the Aib-ACP decapeptide on Rink Amide ChemMatrix® resin. In this case, the best yield was obtained applying method 2 (70.0%), while methods 1 and 5 gave similar results (37.8% and 37.3% respectively) and method 4 gave a low purity (30.4%). The outcome using method 5 was improved significantly by heating the deprotection and coupling steps to 40 °C (87.1%), yet none of the other methods were evaluated at 40 °C, which would have been interesting for comparison purposes. The Aib-ACP peptide synthesis was not reported for methods 1–4 on the Rink Amide PS resin, and was overall inferior on this resin, while the best yield for the pentapeptide Aib-enkephalin on PS resin was obtained with method 2 (97.0%).
Albericio and co-workers have also reported a study using the green solvent GVL for SPPS and successfully applied it to coupling and deprotection under microwave irradiation with both PS and PEG-based resins.44 However, it was found that when using terminal glycine residues, GVL could undergo ring-opening acylation of the α-amino functionality. The extent of acylation for H-Gly-Phe-Leu-NH2 on solid support was evaluated by submitting the peptidyl bound resin in GVL to three stress conditions: room temperature for 12 hours, microwave irradiation at 90 °C for 10 min and room temperature for 12 hours in the presence of a 20% piperidine in GVL, yielding 15%, 25% and 70% acylation respectively. It should be noted that no acylation was detected during the synthesis of H-Leu-Phe-Gly-NH2 or the challenging Jung-Rademann and 65-74ACP (containing one Gly residue) decapeptides under similar conditions, indicating that side reaction is sequence dependent. It was subsequently found that GVL ring-opening could be mitigated by employing freshly prepared solutions of 4-Me-piperidine for Fmoc-removal,54 and the acylation could be eliminated by using dipeptides Fmoc-AA-Gly-OH for SPPS assembly of the peptide backbone to avoid N-terminal glycine in the peptide sequence.55 The need for using dipeptides appears rather cumbersome and may limit the use of GVL for SPPS on large scale.
As described earlier Lopez et al. studied a variety of solvents for their swelling properties and ability to dissolve Fmoc-amino acids and SPPS reagents. In the same paper, the most promising solvents were evaluated for peptide coupling and Fmoc-removal (Table 10).13 Coupling was performed in solution and the rate of amide bond formation between Fmoc-Leu-OH and H-Phe-OMe (1 equiv. each) in the presence of DIC and Oxyma Pure (1 equiv. each) was monitored over two hours. In less polar solvents such as anisole, toluene, 2-MeTHF, THF and ethyl acetate, the coupling reaction was fast, but precipitation of DIU and/or the dipeptide was also observed. In contrast, in the polar aprotic solvents DMF, DMPU, DMSO, NBP, NEP, NMP and TMU the reaction was slower, but no precipitation was detected. One should be careful with translating these results to SPPS though, as the choice of solvent will impact resin swelling and diffusion of reagents on solid support, and consequently reaction rates.
| Solvent/time (min) | Coupling reaction (% remaining S.M.)a | ||||
|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | 120 | |
| a Fmoc-Leu-OH (1 equiv.) and Oxyma Pure (1 equiv.) were dissolved and DIC (1 equiv.) was added. The mixture was stirred at room temperature for 10 min, and H-Phe-OMe (1 equiv.) was added. The reaction was monitored by RP-HPLC.b Not measured. Assumed to have reached full conversion. | |||||
| Anisole | 100.0 | 4.0 | 3.4 | 3.3 | N/Ab |
| DMF | 100.0 | 53.8 | 42.5 | 36.8 | 22.9 |
| DMI | 100.0 | 58.9 | 49.8 | 42.7 | 27.2 |
| DMPU | 100.0 | 76.5 | 66.4 | 56.4 | 42.2 |
| DMSO | 100.0 | 82.1 | 75.9 | 70.7 | 54.0 |
| EtOAc | 100.0 | 4.8 | 2.9 | 3.0 | N/Ab |
| 2-MeTHF | 100.0 | 13.4 | 9.8 | 7.9 | 4.4 |
| NBP | 100.0 | 58.7 | 50.7 | 45.9 | 30.6 |
| NEP | 100.0 | 65.1 | 56.7 | 50.7 | 33.5 |
| NMP | 100.0 | 67.1 | 58.7 | 51.7 | 34.9 |
| THF | 100.0 | 16.1 | 11.9 | 9.8 | 5.7 |
| TMU | 100.0 | 57.8 | 50.1 | 43.7 | 27.8 |
| Toluene | 100.0 | 5.6 | 4.7 | 4.3 | N/Ab |
Based on the results outlined in Table 10, five solvents (TMU, DMI, NBP, DMPU and DMSO) were selected for SPPS of a model octapeptide used in the manufacturing process of octreotide. It should be stressed that of the five selected solvents, three (TMU, DMI and DMPU) are problematic in relation to human and environmental toxicity and cannot be considered meaningful substitutes for DMF and NMP.14 SPPS was carried out on an automated peptide synthesiser and the crude purity of the peptide was compared head-to-head with SPPS in DMF. DMF provided the highest purity (86%) followed by NBP (80%), TMU (78%), DMI (78%), DMSO (52%) and DMPU (51%). However, because TMU and DMI show reproductive toxicity, these were deselected and NBP was the recommended solvent for SPPS. NBP gave a slightly lower crude yield compared to DMF (93% vs. 100% mass gain), but from an API production viewpoint it was more concerning that 1.5% epimerisation was observed in NBP (vs. 0% in DMF).13 The authors discussed that the lower yield and purity could be caused by the significantly higher viscosity of NBP compared to DMF (4 cP vs. 0.8 cP), giving poorer solvent and reagent diffusion in the resin and consequently inferior swelling, coupling, deprotection and washing. In order to generalise the use of NBP for peptide synthesis, it was suggested that increasing the temperature during synthesis may alleviate this problem, and a special protocol at 45 °C for problematic Fmoc-Arg(Pbf)-OH couplings was developed after it was found that δ-lactamisation of Arg was a particularly favoured side-reaction in NBP.56 Further work regarding amino acid racemisation and aspartimide formation showed that NBP performed on par with DMF in terms of racemisation but was superior to DMF at suppressing aspartimide formation at both 45 °C and room temperature.40 Still, further studies involving longer and more complex peptides, as well as a variety of linkers and resins would be beneficial to assess the use of NBP as a general substitute for DMF.
Another potential solution for greener SPPS is the use of mixtures instead of neat solvents. As it was demonstrated by North and co-workers, a mixture can offer better swelling properties than neat solvents as well as better solubility or chemical properties in general (vide supra). North and co-workers reported the synthesis of H-Leu-Ala-Phe-OH on Merrifield resin with DIC/Oxyma Pure in four different solvent mixtures and compared these to the synthesis performed in the neat solvents (Table 11).34 A small improvement in crude yield (approx. 5%) between neat EtOAc and the PC/EtOAc (1:9) mixture was observed (292 mg g−1 and 307 mg g−1, respectively). Both neat TMO and neat PC gave poor results (0 mg g−1 and 8 mg g−1 of resin respectively) while 132 mg g−1 were obtained in a TMO/PC (3:7) mixture and up to 200 mg g−1 was obtained for the TMO/PC (2:3) mixture, despite the immiscibility of the two solvents in this ratio. It should be noted that a rather low concentration was used for all syntheses (0.1 M) and that no studies on the solubility of reagents was reported. Moreover, without a head-to-head comparison with DMF it is difficult to judge the potential of these mixtures for application in SPPS.
| Solvent | Yieldb (mg g−1) | Purity (RP-HPLC) |
|---|---|---|
| a Fmoc-removal was carried out with 20% piperidine in solvent (v/v) for 20 min. Couplings were performed for 1 h at room temperature. Fmoc-amino acid (3.0 equiv.), HBTU (3.0 equiv.), HOBt (3.0 equiv.) and DIPEA (6.0 equiv.) were dissolved at a 0.1 M concentration and stirred for 20 min before adding this mixture to the resin. For the synthesis in PC, after the final Fmoc-removal, the resin was also washed with EtOAc (5 mL).b Yields are given in mg g−1 of resin as the starting loading for the Merrifield resin was imprecise (0.4–0.8 mmol g−1).c Immiscible in this ratio. | ||
| PC | 8 | 3.1% |
| EtOAc | 292 | 96.9% |
| TMO | 0 | — |
| EtOAc/PC (9:1) |
307 | 95.2% |
| TMO/PC (9:1) |
124 | 100% |
| TMO/PC (3:7) |
132 | 96.7% |
| TMO/PC (2:3)c |
200 | 94.2% |
Pawlas et al. have reported the synthesis of the melanocortin receptor agonist peptide in green solvent mixtures on a Ramage aminomethylstyrene (RMG-AMS) resin.37 The synthesis required SPPS assembly of the peptide backbone followed by the selective deprotection of a lysine and an aspartic acid followed by on-resin macro-lactamisation. For the SPPS assembly of the peptide backbone a 1:1 mixture of NBP and EtOAc was employed, followed by selective sidechain deprotection in a 1:1 mixture of MeCN and EtOAc, and final on-resin lactamisation in a 1:1 mixture of NBP and EtOAc. The desired peptide was obtained in 62% yield and 95% purity by RP-HPLC. By mixing the relatively expensive green solvent NBP with the cost-efficient solvent EtOAc, they managed to reduce production cost. Head-to-head comparison with the same synthesis in DMF was not disclosed, however, the yield is significantly better than the one reported by Bradley and co-workers (47%),57 using an alternative protecting group strategy for the Lys and Asp residues on a Rink Amide ChemMatrix® resin. In a follow-up report, Pawlas et al. reported an optimized protocol for the synthesis of the Aib-ACP decapeptide on RMG-AMS resin using a mixture of DMSO/EtOAc (1:9) as solvent for the coupling and deprotection steps.58 Couplings were carried out with 1.3 equiv. of Fmoc-amino acid and Oxyma Pure, and 3.25 equiv. DIC at 45–50 °C for 30 min (except for the Aib–Aib coupling that was coupled twice with 1 equiv. amino acid/Oxyma Pure/DIC). The peptide was obtained with a purity of 76% and a crude yield of 84% after cleavage and precipitation. Notably, Pawlas et al. discussed cost and supply chain and introduced the concept ReGreen in which EtOAc, DMSO and Oxyma Pure are recycled. To illustrate the benefits of recycling, a comparison of the solvent cost per AA coupling cycle was made for SPPS using either DMF, NBP/EtOAc (1:1), DMSO/EtOAc (1:9) or DMSO/EtOAc (1:9) with recycling steps. The cheapest AA coupling cycle was achieved using DMSO/EtOAc (1:9) with recycling (151 € per AA cycle) followed by DMF (297 € per AA cycle), and NBP/EtOAc (1:1) (406 € per AA cycle). By contrast, if DMSO/EtOAc (1:9) was utilised without recycling, the cost was estimated to 431 € per AA cycle.58 Furthermore, in a recent report Pawlas and co-workers also addressed the occurrence of hydrogen cyanide (HCN) formation during amide bond-forming reactions mediated by DIC and Oxyma.59,60 Binary solvent mixtures of NBP and EtOAc (1:1 and 1:4) gave significantly lower amounts of HCN and faster amide coupling than observed in DMF or neat NBP, and in addition the authors showed that HCN can be successfully scavenged during the coupling reaction by dimethyl trisulfide (DMTS) with no impact on amide bond formation.60
Recently, Ferrazzano et al. reported the synthesis of the Aib-enkephalin pentapeptide, Aib-ACP decapeptide, and the octreotide octapeptide in solvent mixtures that were found to display good swelling and solubilising properties (vide supra), including Cyrene™/DEC (3:7), sulfolane/DEC (3:7) and anisole/DMC (7:3) (Table 12).41 The synthesis of the Aib-enkephalin peptide in DMF gave very low to medium purity on Wang PS and Rink Amide ChemMatrix® resins respectively (Table 12, entries 1 and 2). In stark contrast, using the sulfolane/DEC (3:7) solvent system resulted in much higher purities (entries 3 and 4). Similarly, higher purity compared to DMF was obtained when using Cyrene™/DEC (3:7) on Wang PS resin (entry 5) and anisole/DMC (7:3) on Rink Amide ChemMatrix® resin (entry 6). Synthesis of the Aib-ACP peptide was only evaluated on Rink Amide ChemMatrix® resin, with DMF giving the best, albeit still low, purity (entries 7–9). Finally, the synthesis of the octreotide peptide was evaluated in DMF and in anisole/DMC (7:3) on a 2-chlorotrityl resin (entries 10 and 11). Again, DMF was found to be superior providing good purity (88%). However, by implementing a double coupling of Fmoc-Cys-OH or alternatively performing all couplings at 40 °C, a comparable purity could be obtained in the anisole/DMC (7:3) mixture.
| Entry | Peptide | Solvent | Resin | Purity (%) |
|---|---|---|---|---|
| a Coupling was performed for 1 h at a 0.2 M concentration using 3 equiv. of Fmoc-protected amino acid, 3 equiv. of DIC and 3 equiv. of Oxyma Pure.b Double coupling 2 × 1 h of the Fmoc-Leu-OH amino acid.c Coupling of Fmoc-Cys-OH at 40 °C.d Coupling for all steps at 40 °C. | ||||
| 1 | Aib-enkephalin | DMF | Wang PS | 13.5 |
| 2 | Aib-enkephalin | DMF | Rink Amide ChemMatrix® | 53.0 |
| 3 | Aib-enkephalin | Sulfolane/DEC (3:7) |
Wang PS | 72.8, 69.1b |
| 4 | Aib-enkephalin | Sulfolane/DEC (3:7) |
Rink Amide ChemMatrix® | 62 |
| 5 | Aib-enkephalin | Cyrene™/DEC (3:7) |
Wang PS | 37.3, 72.0b |
| 6 | Aib-enkephalin | Anisole/DMC (7:3) |
Rink Amide ChemMatrix® | 72.1 |
| 7 | Aib-ACP | DMF | Rink Amide ChemMatrix® | 37.8 |
| 8 | Aib-ACP | Sulfolane/DEC (3:7) |
Rink Amide ChemMatrix® | 10.0 |
| 9 | Aib-ACP | Anisole/DMC (7:3) |
Rink Amide ChemMatrix® | 31.0 |
| 10 | Octreotide | DMF | H-Thr(tBu)-ol-2CT-PS | 88 |
| 11 | Octreotide | Anisole/DMC (7:3) |
H-Thr(tBu)-ol-2CT-PS | 64.6, 82.3c, 84.9d |
| a Colour code for Fmoc-removal performance: green: good; orange: moderate; red: poor.b Fmoc-Phe-OH (0.1 mmol) was dissolved in the solvent to be studied and toluene was added as an internal standard. Piperidine was added, and the reaction was monitored by RP-HPLC.c Time required to achieve complete Fmoc-removal.d The resin was swelled in the solvent to be studied followed by addition of 20% piperidine in the same solvent for 45 s or 7 min. The resin was washed and coupled with Boc-Gly-OH, COMU® and DIPEA in DMF. Fmoc-removal of any remaining Fmoc-protected peptide was performed by treatment with 20% piperidine in DMF, followed by coupling with Boc-Leu-OH, COMU®, and DIPEA in DMF. The yield was calculated by the ratio of H-Gly-Ile-Asp-Tyr-Ile-Asn-Gly-NH2 vs. H-Leu-Ile-Asp-Tyr-Ile-Asn-Gly-NH2.e 16.6% of starting materials after stirring for 30 min. |
|---|
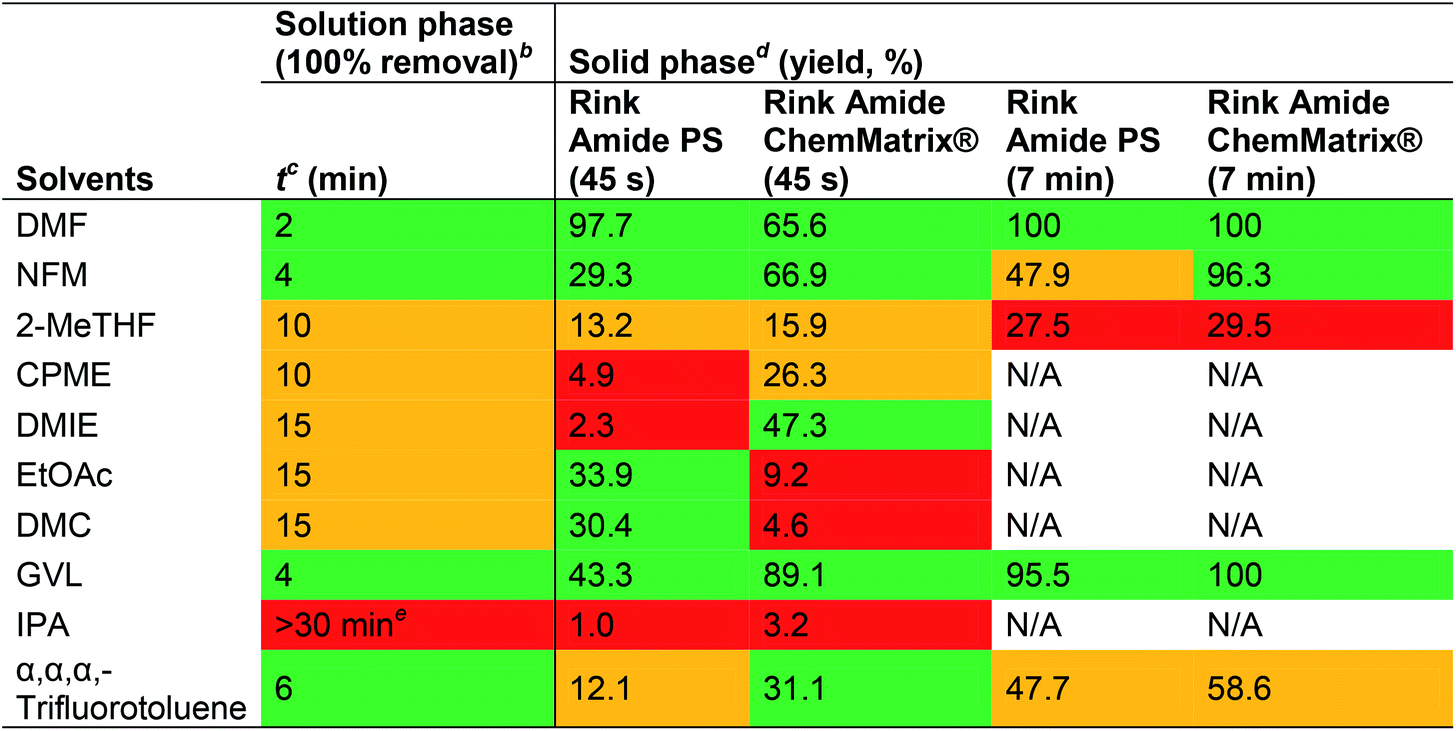 |
First, the Fmoc-removal properties in various solvents were evaluated in solution using Fmoc-Phe-OH as a model system. NFM, GVL and α,α,α-trifluorotoluene gave the most promising results, affording complete Fmoc-removal in 4–6 min, although none of the evaluated solvents could match the deprotection kinetics observed in DMF (2 min). For the SPPS, the peptide Fmoc-Ile-Asp(OtBu)-Tyr(tBu)-Ile-Asn(Trt)-Gly (intermediate of the 65-74ACP peptide) on Rink Amide PS or Rink Amide ChemMatrix® resins was utilised as a model system. The efficacy of Fmoc-removal was evaluated in all solvents using a 45 s deprotection protocol. In general, Fmoc-deprotection was more efficient on the ChemMatrix® resin than on the PS resin (except for EtOAc and DMC). Notably, Fmoc-removal on the ChemMatrix® resin using GVL performed significantly better than in DMF (89.1% vs. 65.6%). Next, the solvents which performed well on both resins were evaluated using a 7 min deprotection protocol (NFM, 2-MeTHF, GVL and α,α,α-trifluorotoluene). GVL was found to cleave 95.5% of the Fmoc-group on the PS resin and 100% on the ChemMatrix® resin, almost on par with DMF (100% on both resins). NFM performed well with the ChemMatrix® resin (96.3%) but was inefficient on the PS resin (47.9%). α,α,α-Trifluorotoluene gave mediocre results on both resins (47.7% and 58.6%) and 2-MeTHF gave poor results on both resins (<30%). Heating of the Fmoc-removal was not evaluated in this study, although the authors had previously demonstrated that this could significantly improve the deprotection kinetics and the purity of the final product.31 Overall, GVL appeared most promising for Fmoc-removal although the solvent has potential drawbacks with ring-opening acylation of N-terminal glycine residues (vide supra).44 Lawrenson et al. used PC for SPPS on a ChemMatrix® resin with success and reported that Fmoc-removal with 20% piperidine in PC was comparable to using 20% piperidine in DMF (vide supra).30 A double 10 + 20 min deprotection protocol was used and afforded purities of 77% (PC) and 79% (DMF).
Zinieris et al. have studied the impact of the piperidine concentration during Fmoc-removal by comparing the rate of Fmoc-removal from Fmoc-Gln-, Fmoc-Val-, Fmoc-Pro- and Fmoc-Lys(Boc)- on a 2-chlorotrityl resin.61 Solutions of 5, 10 and 20% piperidine in DMF were compared by measuring the UV absorbance of the deprotection solution (Table 14).
| Resin-bound amino acid | 20% piperidine in DMF | 10% piperidine in DMF | 5% piperidine in DMF | |||
|---|---|---|---|---|---|---|
| t1/2 (s) | Deprotection ≥ 99.99%b (min) | t1/2 (s) | Deprotection ≥ 99.99%b (min) | t1/2 (s) | Deprotection ≥ 99.99%b (min) | |
| a The deprotection solution was added to the resin and after 10 s was filtered and the UV-absorption of the filtrate was measured. The procedure was repeated until the UV absorption of the filtrate <0.05 AU. The percentage of the Fmoc-removal was expressed as a percentage of the absorption measured when the deprotection solution was added to the same amount of resin sample for 20 min. The time needed for 50% Fmoc-removal corresponds to the half-life of the deprotection step.b Time needed for 99.99% deprotection = −ln 0.0001 t1/2/0.693. | ||||||
| Fmoc-Gln | 20.5 | 4.5 | 23.2 | 5.1 | 53.1 | 11.8 |
| Fmoc-Val | 41.5 | 9.2 | 44.6 | 9.9 | 47.1 | 10.4 |
| Fmoc-Pro | 25.4 | 5.6 | 27.0 | 5.9 | 32.0 | 7.1 |
| Fmoc-Lys(Boc) | 10.3 | 2.3 | 22.3 | 4.9 | 27.4 | 6.1 |
As expected, the required time to reach full deprotection increased with decreasing piperidine concentration. Interestingly, the difference for the sterically hindered Fmoc-Val was small and all deprotections were found to go to completion in <15 min. Zinieries et al. synthesised a sterically hindered 11-mer peptide (H-Ala10-Lys-OH) using deprotection mixtures containing 5, 10 or 20% piperidine in DMF,61 and showed that all three deprotection mixtures were suitable for Fmoc-removal with acceptable deprotection times (the last four alanine residues required longer deprotection times to reach completion). Next, the effect of piperidine concentration on SPPS was evaluated on 2-chlorotrityl resin comparing 5 and 20% piperidine in DMF for the synthesis of the ACP decapeptide and the pentamer Leu-enkephalin. As both peptides were obtained in comparable purities and yields, it appears that a reduction of the piperidine concentration from 20% to 5–10% is feasible for Fmoc-removal without compromising the crude purity. This could potentially have implications for large-scale manufacturing as fewer washes would be required to remove residual piperidine.
One approach for greening Fmoc-removal could be to identify an alternative base that works well in a green solvent and possibly at low concentrations. Several research groups have studied the substitution of piperidine with for example piperazine, 4-Me-piperidine and morpholine. Although most of these studies were conducted in DMF, they clearly demonstrated that piperidine substitution is feasible (vide infra).64–66 Lopez et al. studied the rate of Fmoc-removal on-resin using 4-Me-piperidine in various solvents (Table 15 and Fig. 3).13 A linear peptide derived from octreotide was treated with a 20% solution of 4-Me-piperidine in the desired solvent and aliquots of resin were removed for analysis after 5, 10 and 30 min. Unlike for the coupling reaction (vide supra), Fmoc-removal was observed to be slow in nonpolar solvents (e.g. anisole, EtOAc, 2-MeTHF, THF and toluene) and fast in polar aprotic solvent (e.g. DMF, DMSO, DMPU, NEP, DMI and NBP), giving full conversion in ≤5 min.
| Solvent/time | Fmoc-removala (% conversion)b | ||
|---|---|---|---|
| 5 min | 10 min | 30 min | |
| a Peptide resin aliquots were cleaved from the solid support with a 3% TFA solution in CH2Cl2 and the %-conversion to Fmoc-deprotected peptide was measured by RP-HPLC.b Relative area-% of deprotected peptide measured by RP-HPLC. | |||
| Anisole | 18.1 | 30.0 | 55.5 |
| DMF | 100.0 | 100.0 | 100.0 |
| DMI | 99.5 | 100.0 | 100.0 |
| DMPU | 100.0 | 100.0 | 100.0 |
| DMSO | 100.0 | 100.0 | 100.0 |
| EtOAc | 11.8 | 23.0 | 49.7 |
| 2-MeTHF | 11.4 | 21.0 | 58.6 |
| NBP | 100.0 | 100.0 | 100.0 |
| NEP | 100.0 | 100.0 | 100.0 |
| NMP | 100.0 | 100.0 | 100.0 |
| THF | 36.6 | 60.5 | 98.5 |
| TMU | 99.0 | 100.0 | 100.0 |
| Toluene | 16.5 | 25.3 | 50.4 |
 | ||
| Fig. 3 Chemical structures of organic bases investigated for Fmoc-removal in SPPS. | ||
Lebl and co-workers have studied 2-, 3-, 4-Me-piperidine and 4-Me-piperazine as potential replacements for piperidine.65 The kinetics of the Fmoc-removal of the Fmoc-Ile-chlorotrityl resin were monitored by measuring the UV absorbance of the resulting dibenzofulvene-base adduct. A comparison of 25% piperidine in DMF with 25% 2-, 3-, 4-Me-piperidine and 4-Me-piperazine was conducted and the half-time for Fmoc-removal was determined (Fig. 3 and Table 16). The study showed that both 3- and 4-Me-piperidine were comparable to piperidine. Furthermore, 4-Me-piperidine is in the same price range as piperidine and was further evaluated in the synthesis of four 10-mer peptides: (enkephalin)2, the luteinizing hormone-releasing hormone (LHRH), the ACP peptide (65–74) and the β-amyloid peptide (25–34). Both manual SPPS and a tilted plate centrifugation-based peptide synthesizer (96-wellplate, 24 copies of each sequences synthesised) were employed (Table 17).
No significant difference between the two bases could be observed, thus demonstrating that 4-Me-piperidine is a good alternative to piperidine. The concentration of the base was not explicitly stated but most likely the 25% solution reported earlier was employed. The authors stated that they had employed 4-Me-piperidine over a 2 year period for the synthesis of thousands of peptides ranging from 5 to 24 residues, without observing any difficulties that could be attributed to the nature of the base. Unlike piperidine, 4-Me-piperidine has the advantage of not being a controlled substance, but from a human and environmental toxicity perspective, 4-Me-piperidine does not represent a more benign substance.67 Luna et al. have reported a head-to-head comparison of piperidine, piperazine and 4-Me-piperidine for the synthesis of four peptides under microwave irradiation (Table 18).64
| Peptidea/base | Crude yield (%) | Purity (%) | Purified yield (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4-Me-piperidine | Piperidine | Piperazine | 4-Me-piperidine | Piperidine | Piperazine | 4-Me-piperidine | Piperidine | Piperazine | |
| a Coupling: 0.125 M concentration in DMF with DIC/Oxyma Pure, Rink Amide aminomethyl PS resin on a Liberty Blue™ (90 °C, 30 W, 110 s). Fmoc-removal: 20% 4-Me-piperidine (v/v) in DMF, or 20% piperidine in DMF (v/v), or 10% piperazine in 9:1 DMF/EtOH (w/v) on a Liberty Blue™ (90 °C, 30 W, 50 s). Prior to analysis peptides were cleaved from the resin and deprotected, precipitated with cold DEE, centrifuged, washed with cold DEE five times, dried, dissolved in water, lyophilized and analysed. |
|||||||||
| FISEAIIHVLHSR | 73.2 | 72.0 | 53.6 | 47.6 | 43.6 | 57.7 | 34.9 | 31.3 | 31.0 |
| TLEEFSAKL | 99.1 | 98.4 | 99.4 | 65.1 | 83.0 | 74.8 | 64.5 | 81.7 | 74.3 |
| KKWRWWLKALAKK | 93.6 | 81.6 | 74.7 | 50.4 | 59.1 | 55.6 | 44.4 | 48.2 | 41.5 |
| VAPIAKYLATALAKW-ALKQGFAKLKS | 68.5 | 79.3 | 68.2 | 20.6 | 29.0 | 21.4 | 14.1 | 23.8 | 14.6 |
Piperazine is not soluble at 20% w/v in DMF, thus only 10% w/v was evaluated with concomitant addition of 10% ethanol to improve the solubility. For three of the four peptides the highest purity and isolated yields were obtained with piperidine, although in some instances 4-Me-piperidine and piperazine performed in the same range as piperidine.64 Pribylka et al. studied the Fmoc-removal of Fmoc-Ala bound to a Rink Amide resin and reported successful deprotection using both 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU, Fig. 3) in 2-MeTHF (from 0.1% to 5% DBU), and NaOH in a mixture of 2-MeTHF/MeOH (1:1) (from 0.02 M to 0.5 M). Complete deprotection after 15 min was achieved with 0.5% DBU in 2-MeTHF and 0.1 M NaOH in 2-MeTHF/MeOH (1:1).68 From a large-scale manufacturing perspective, DBU is undesirable due to a significantly higher cost compared to piperidine and its susceptibility to induce aspartimide formation.69 However, because of the low DBU concentration, it may be a viable alternative to piperidine if aspartimide formation can be controlled. The use of NaOH in 2-MeTHF/MeOH is an interesting and inexpensive alternative to piperidine and hence warrants further investigation.
3.3 Precipitation
The use of diethyl ether (DEE) and diisopropyl ether (DIPE) remains the industry standard for the precipitation of peptides after cleavage and global deprotection. Both anti-solvents are considered unproblematic in relation to external environment, but the volatility of DEE and the peroxide-forming propensity of both solvents have sparked the search for alternatives. Although tert-butyl methyl ether (TBME) has been a popular alternative to DEE and DIPE, it is impacted by REACH, included on the ECHA SVHC list, and should be avoided.14 Albericio and co-workers have reported the use of cyclopentyl methyl ether (CPME) and 2-MeTHF as green alternatives for peptide precipitation.52,70,71 The precipitation of 5-, 10-, 16-, 20- and 28-mer peptides was compared in CPME, 2-MeTHF, DEE and TBME, including both C-terminal amides and carboxylic acids (Fig. 4 and Table 19). Apart from the pentapeptide Leu-enkephalin that was soluble in CPME, 2-MeTHF and TBME, all peptides were recovered in similar yields and purities in all solvents. Moreover, supported by scanning electron microscopy (SEM) they showed that CPME, unlike 2-MeTHF and TBME, prevents osmotic shock of 2-chlorotrityl PS resin during peptide precipitation, allowing recycling and re-use of the resin after re-activation.71 | ||
| Fig. 4 Chemical structures of ethers investigated as anti-solvents for post-cleavage peptide precipitation. | ||
| Peptidea | Resin | Loading (mmol g−1) | Crude yield (%) | |||
|---|---|---|---|---|---|---|
| 2-MeTHF | CPME | DEE | TBME | |||
| a The peptide was cleaved and deprotected using TFA/TIPS/H2O (95:2.5:2.5) (1 mL/100 mg) with shaking (1 h). The same volume of each chilled ether was added (5 × cleavage solution volume), and the solution was kept in an ice bath for 30 min. The solution was centrifuged for 5 min at 5000 rpm, and the supernatant was decanted. The procedure was repeated with a second batch of the same solvent. Residual ether was removed under a stream of N2, the solid was dissolved in water and lyophilized to determine the crude yield. |
||||||
| Leu-enkephalin (5-mer) | CTC-PS | 1.0 | 0 | 0 | 79 | 15 |
| ACP-OH (10-mer) | Wang-PS | 0.6 | 65 | 54 | 59 | 59 |
| ACP-NH2 (10-mer) | AM-Rink Amide-PS | 0.6 | 57 | 90 | 75 | 60 |
| ABRF 1992 (16-mer) | AM-Rink Amide-PS | 0.6 | 94 | 93 | 93 | 94 |
| ABRF 1992 (16-mer) | MBHA-Wang-PS | 1.1 | 100 | 95 | 100 | 100 |
| ABRF 1992 (16-mer) | MBHA-Rink Amide-PS | 1.1 | 100 | 97 | 100 | 100 |
| ABRF 1992 (16-mer) | Rink Amide-CM | 0.45 | 30 | 32 | 32 | 32 |
| ABC (20-mer) | Rink Amide-CM | 0.45 | 34 | 27 | 27 | 33 |
| Thymosin (28-mer) | AM-Rink Amide-PS | 0.6 | 74 | 75 | 75 | 72 |
Pawlas et al. have reported the use of a 20% 4-methyltetrahydropyrane (4-MeTHP, Fig. 4) in heptane for peptide precipitation.37 Crude recovery yields after cleavage and precipitation of an acetylated heptapeptide were compared in DEE, CPME, 2-MeTHF, 4-MeTHP and 20% 4-MeTHP in heptane. The 4-MeTHP/heptane mixture gave a result comparable to DEE (both 91%) and both DEE and 4-MeTHP/heptane mixtures were superior to TBME (81%), 2-MeTHF (72%) and 4-MeTHP (76%).
Taking all different aspects of SPPS into account, the most promising results for green solvent replacement of DMF have been obtained using GVL, NBP, PC, 2-MeTHF, and solvent combinations such as NBP/EtOAc (1:1), Anisole/DMC (7:3) and DMSO/EtOAc (1:9). Still, several of the solvents above have drawbacks when compared to DMF and finding a suitable direct alternative that performs on equal terms in swelling, coupling, and Fmoc-removal has proven difficult. In-depth analysis of side-product formation could provide insight into how to mitigate drawbacks, but this is often overlooked. Of crucial importance for API production is control over peptide epimerisation, which can cause severe issues with downstream purification, and therefore should be scrutinised when evaluating new solvents for SPPS. Of the solvents listed here, GVL has been shown to be reactive during SPPS, while the viscosity of NBP can result in sub-optimal diffusion and thus lower yields and (stereochemical) purity. Its high viscosity may also lead to technical challenges in production during filtrations and transfer of solutions. PC only showed good results at low concentration and appeared to be limited to the relatively costly ChemMatrix® resins. 2-MeTHF is promising but the lack of data regarding toxicity could become a complication in the future. A similar problem could arise for anisole, which likewise is poorly characterised and furthermore is suspected of causing genetic defects. Thus, we think that the use of solvent mixtures currently holds the biggest promise to replace DMF, where NBP/EtOAc (1:1) and DMSO/EtOAc (1:9) are particularly promising as the use of these mixtures mitigates drawbacks observed for neat NBP and DMSO such as viscosity, melting point and cost. The use of solvent mixtures is not a problem for large-scale production, and a solvent mixture with properties that mimic those of DMF more closely could be envisioned. In general, the cost of many green solvents such as GVL, DMSO and NBP is higher than for DMF. However, widespread use and resulting large-scale production is likely to bring the cost of new SPPS solvents down to a level more comparable to the cost of DMF.
4. Alternative manufacturing approaches and emerging technologies
4.1 Water-based SPPS
In PMI (process mass intensity) calculations,72,73 water is treated on par with organic solvents. Nevertheless, water may offer interesting possibilities for the greening of peptide synthesis as outlined below. The use of water for SPPS has been investigated by several research groups. Key problems of this strategy include poor solubility of reagents and Fmoc-protected amino acids as well as poor swelling properties of polystyrene-based resins in particular. These challenges have typically been tackled by developing alternative amino acid building blocks and SPPS reagents.:1 water/EtOH. The coupling reagent 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) was used in combination with N-methyl morpholine and the surfactant Triton X (2.0%) in order to increase swelling and reagent solubility. The purified peptide Leu-enkephalin was isolated in 61% overall yield.42 The 1,1-dioxobenzo[b]thiophene-2-ylmethyloxycarbonyl (Bsmoc) protecting group was originally developed by Carpino et al. for continuous solution-phase synthesis86,87 and was later adopted by the CEM corporation as a suitable protecting group for water-based SPPS.88 The Bsmoc protecting group and related 2-(tert-butylsulfonyl)-2-propenyloxycarbonyl (Bspoc)87,89 and 1,1-dioxonaphtho[1,2-b]thiophene-2-methyloxycarbonyl (α-Nsmoc)90 groups offer some advantages over Fmoc-chemistry, undergoing a different mechanism for deprotection that allows efficient removal of the protection group and scavenging even at low amine concentrations.86,87,89,91 The Bsmoc strategy has been applied for the synthesis of several peptide targets88,92,93 but the approach has not found widespread use, most likely due to a significant difference in price and availability between Bsmoc and Fmoc-protected amino acids. The price and current lack of supply chain for GMP and non-GMP Bsmoc-protected amino acids alike makes the strategy unlikely to be adopted for peptide API production in the foreseeable future.
 | ||
| Fig. 5 Water-soluble sulfone-derived amino acid protecting groups developed by Hojo et al.42,82,84 and Carpino et al.86,89,90,94 | ||
The research group of Kolmar has reported a class of sulfonate derived protecting groups that mimic the classic SPPS protecting groups closely and are soluble in water, alcohols and mixtures of water and alcohol (commercialised by the company Sulfotools) (Fig. 6).95,96 The Fmoc-analogue Smoc is removed under basic conditions (e.g. aqueous ammonia, ethanolamine or ethylenediamine, or piperidine), whereas the SBoc, tBuS, StBuS, sulfo-Trt and BOMS are removed under acidic conditions (e.g. phosphoric acid, hydrochloric acid, trifluoroacetic acid). The Cbz-analogues SulfoCbz and BzS were also reported. The use of Smoc-protected amino acids for peptide synthesis, a strategy named aqueous solid-phase peptide synthesis (ASPPS), have several attractive features. The charged sulfonate moiety enhances the aqueous solubility of the reagents but also renders the protecting group fluorescent, allowing real-time monitoring of coupling and washing steps. Furthermore, by using sulfoacetic acid or sulfobenzoic acid as capping agents during the synthesis cycle, deletion sequences become sulfonated and can be removed from the desired peptide along with any cleaved protecting groups by separation using ion-exchange chromatography. Kolmar and co-workers performed the synthesis of 22 short (<10 AA) peptides on commercially available resins (e.g. ChemMatrix Rink amide) in aqueous NaHCO3 mixtures with organic co-solvents (MeCN or EtOH), using Smoc-AA (3 equiv.), EDC-HCl (5.5 equiv.) and Oxyma (3 equiv.). Furthermore, they determined epimerisation to be <0.5% for most studied amino acids in model peptides (up to 2% for Asp/Asn) and showed that side reactions such as aspartimide formation were sequence and temperature dependent.96 Overall, ASPPS is an interesting technology which requires further development. It is currently not clear if the approach would be generally successful when applied to longer and more complex peptide sequences. Furthermore, the Sulfo-tag aspect was shown as a proof-of-concept using high amounts of excess reagents (50–60 equiv. of each carboxylic acid and coupling agent) but not applied to the general synthesis of peptides and would require more development to be applicable to large-scale synthesis. Lastly, there is currently no supply chain for Smoc-protected amino acids, and being a proprietary platform, it would require licensing for wider commercial use.
 | ||
| Fig. 6 Sulfonate-derived protecting groups developed by Kolmar and co-workers and commercialised by the company Sulfotools.95,96 | ||
:1), followed by determination of the substrate scope and an evaluation of aqueous stability. Overall, the use of DIC in combination with 2-hydroxypyridine-N-oxide (HOPO) was found to be the most effective coupling reagent. The stability of the activated species was evaluated by activating benzoic and hydrocinnamic acid with the activating agents DIC-HOPO, DIC, COMU®, TPTU, CDI, DMT-NN BF4 and EEDQ. The activated carboxylic acid was kept in a mixture of MeCN and water for 15 min at 30 °C, followed by coupling to n-butylamine, before final analysis of the reaction by RP-HPLC. Among the different coupling agents, the DIC-HOPO combination was found to be the most stable affording 99% conversion for benzoic acid and 94% conversion for hydrocinnamic acid. Although the synthesis of peptides was not a focal point of the investigation, the study still provides information regarding the stability of coupling reagents and their potential use in SPPS. Kolmar and co-workers have patented several sulfonate-based coupling reagents derived from DIC, DCC, EDC, HBTU, PyBOB and others (Fig. 7).95 In line with the work described above, better aqueous solubility and easier purification by ion exchange chromatography can be expected but no data on their efficiency as coupling reagents has been disclosed making it unclear how well this technology works in practice, and for their recently published protocol, EDC-HCl and Oxyma was used.96
 | ||
| Fig. 7 Examples of water-soluble sulfonate-derived coupling agents as reported by Kolmar and co-workers.95 | ||
A water-based SPPS strategy using N-carboxyanhydride (NCA) derivatives on a ChemMatrix® resin was reported by Gentilucci and co-workers.98 The NCA amino acids for coupling were synthesised by microwave irradiation of the amino acid in the presence of triphosgene under solvent free conditions. Yields in the range 90–95% with a purity of 80–85% were reported for the NCA amino acids. With no further purification, the NCA amino acids were used for coupling on solid support. To prevent polymerization of the NCA building blocks, the pH had to be maintained at ∼10.5 and the temperature at 5 °C. After washing of unreacted NCA amino acids, the pH was adjusted to 3.0, which induced decarboxylation of the carbamic acid, yielding the free amine. Applying this strategy, the tetrapeptide H-Phe-Pro-Trp-Phe-NH2 was synthesised using various ChemMatrix® linker systems (Rink Amide, Pal and Ramage) to give 68%, 66% and 52% yield, respectively, which is significantly lower than standard SPPS conducted in organic solvents.98
Although some interesting approaches for performing SPPS in water have been reported, current state-of-the-art is still in its infancy. Except for resins, most reagents and building blocks are not available on scale, which poses questions about cost and reliability of the supply chain. Moreover, it should be noted that aqueous waste contaminated with organic material should be disposed of as organic waste (water should be treated as organic waste in PMI calculations).103
4.2 Liquid-phase and membrane-enhanced peptide synthesis
The use of soluble anchors for the solution-phase synthesis of peptides is an emerging technology which has been termed liquid-phase peptide synthesis (LPPS). This technology, which is essentially a further elaboration of peptide synthesis on PEG chains,104 is currently being promoted by two companies (Jitsubo and Ajinomoto Bio-Pharma Services). The technology is based on the attachment of the peptide to a highly lipophilic and soluble tag, which ensures retention of the growing peptide in an organic layer, during aqueous work-up. Both companies have demonstrated that the technique can be applied to the synthesis of a wide range of peptide sequences normally synthesized by SPPS (vide infra), but the technology usually requires convergent synthesis by coupling of peptide fragments to access medium to long peptides (>20 amino acids). Takahashi et al. used fluorene-based105 or diphenylmethane-based soluble anchors that have been commercialised under the name Ajiphase® (Fig. 8).106 Analogously to SPPS, the anchor can be loaded with an amino acid and then elongated by coupling/deprotection cycles with Fmoc-protected amino acids. However, unlike SPPS the synthesis cycle is executed in an organic solvent (originally chloroform) with isolation of intermediates by precipitation with MeOH or MeCN. In order to improve the LPPS methodology and reduce the use of halogenated solvents, Takahashi et al. developed a 2nd generation of anchors consisting of benzylalcohol- and diphenyl-methane-based anchors bearing phytyl groups in order to allow liquid–liquid extractions instead of precipitation.107 This new class of anchors allows a one-pot synthetic strategy reducing process time and providing a scalable method. Of real interest is that LPPS uses standard building blocks and common reagents. The first amino acid is loaded onto the anchor using either an ester- or amine-functionalised anchor for the synthesis of C-terminal carboxylic acids or amides, respectively. Fmoc-amino acids are coupled using EDC and the intermediate products are worked up in a series of aqueous extractions to remove by-products and excess reagents, followed by Fmoc-removal and coupling of the next amino acid derivative (Fig. 8). In one report, the methodology was applied to the synthesis of the 10-mer degarelix and the 20-mer bivalirudin to give both peptides in good crude purity (89% and 84%, respectively). Takahashi et al. estimated that the solvent consumption was reduced >10-fold when comparing their LPPS methodology to standard SPPS (174 mL g−1 compared to 2183 mL g−1). | ||
| Fig. 8 Ajiphase® soluble anchors for liquid-phase peptide synthesis (LPPS) developed by Takahashi et al. and commercialised by Ajinomoto Bio-Pharma Services.105–107 | ||
A similar LPPS methodology has been reported by Chiba and co-workers using benzylalcohol-based anchors, which have been commercialised by Jitsubo (Fig. 9).108 Using standard protected amino acids and coupling reagents, the peptide is assembled via a coupling and Fmoc-removal cycle, followed by precipitation of the tagged peptide to remove by-products and excess reagents. Differently substituted anchors that allow for the synthesis of peptides with C-terminal acids and amides have been developed (1–3). An anchor that can be cleaved with TFA (3), as well as an acid stable anchor suitable for Boc-chemistry (2), and an anchor that can be cleaved with a 1% TFA solution (1) have been reported.109 The methodology was applied to the synthesis of the cyclic heptapeptide mahafacyclin B110 and the 16-mer α-conotoxin MII111 to give the products in good yields (40 and 43%, respectively), albeit the purity of the crude products was not disclosed. The methodology was reported to be problematic with longer peptides (>20 amino acid residues), which necessitated the development of a fragment-based coupling strategy. Using this fragment-based approach, the 20-mer peptides bivalirudin, and the 28-mer h-ghrelin were synthesised (fragment coupling yields of 85% and 68%, respectively).108 Recently, an improved protocol utilising propylamine as a sacrificial amine to scavenge excess activated amino acid after coupling has been reported.109 By applying this principle it was possible to proceed directly to Fmoc-removal without the intermediate precipitation step, resulting in a faster process and reduced solvent consumption. The methodology was successfully applied to the synthesis of the 10-mer peptide icatibant on a 100 g scale (86% purity, 65% yield after purification).
 | ||
| Fig. 9 Soluble anchors for liquid phase peptide synthesis developed by Chiba and co-workers.108,112 | ||
Alternatively, LPPS with group-assisted purification (GAP) based on much smaller phosphine oxide- or phosphate-based benzylic scaffolds have also been reported. Seifert et al. developed a soluble and re-usable diphenylphosphine oxide-benzyl tag (HOBndpp, Fig. 10A) for LPPS of a pentamer in excellent yield and purity without chromatography,113 while Li et al. recently reported LPPS using recyclable benzyl alcohol anchors based on tri(4-formylphenyl) phosphonate (TFP),114 tri(4-benzoylphenyl) phosphate (TBP),115 and diphenylphosphinyloxyl diphenyl ketone (DDK)116 scaffolds (Fig. 10A), enabling Boc and Fmoc LPPS of up to 16-mer peptides in good yields without intermediate HPLC purification.114–116 The LPPS strategy has also been combined with organic solvent nanofiltration (OSN) for peptide synthesis in an approach termed Membrane-Enhanced Peptide Synthesis (MEPS). The first reports of MEPS by Livingston and co-workers described Fmoc-LPPS in DMF on a soluble 5 kDa methoxy-amino-PEG support in combination with post-reaction constant volume diafiltration (10 and 12 diavolumes post coupling and Fmoc-removal respectively) over an Inopor ZrO2 membrane (2 kDa molecular weight cut-off, MWCO) to remove excess reagents while retaining the peptide, yielding two pentapeptides on gram scale in higher purity than achieved by SPPS.117,118 In follow-up work, an Inopor TiO2/Al3O3 ceramic membrane (450 Da MWCO) was used for MEPS of a hexapeptide (anchor identity not disclosed) in THF. Fmoc-AAs and HBTU/HOBt (all 1.05 equiv.) and DIEA (2 equiv.) were used for coupling, with 4 and 14 diavolumes of fresh THF used for diafiltration post coupling and Fmoc-removal respectively.119 Further work by Castro et al. provided an expansion of the globular supports for MEPS, showing that branched, multi-functionalised polymer anchors such as 6.2 kDa PyPEG (Fig. 10B) were efficiently retained by ceramic membranes during filtration, and could be employed for the MEPS of a tetrapeptide with higher crude purity than was obtained by SPPS, albeit using the problematic solvents CH2Cl2 and DMF.120
 | ||
| Fig. 10 Chemical structures of (A) Benzyl alcohol anchors for LPPS with group-assisted purification based on diphenylphosphine oxide (Bndpp),113 tri(4-formylphenyl) phosphonate (TFP),114 tri(4-benzoylphenyl) phosphate (TBP),115 and diphenylphosphinyloxyl diphenyl ketone (DDK)116 scaffolds, and (B) branched 6.2 kDa PEG-based anchor PyPEG for membrane-enhanced peptide synthesis (MEPS).120 | ||
From an industrial perspective, LPPS and MEPS are promising technologies for several reasons. They use standard amino acid derivatives and coupling reagents and are amenable to large scale production without the need for specially designed or dedicated SPPS reactors (apart from a flow setup and suitable membranes in MEPS). The reactions are carried out in solution, which has the advantage of more efficient mass transfer, faster reaction rates and less impact of protein folding and steric hindrance compared to SPPS, allowing efficient coupling with only slight excess of reagents. Hence, LPPS and MEPS have the potential to significantly reduce reagent consumption compared to standard SPPS. The volumes of solvent typically required for filtration in MEPS are currently high and would need further optimisation to reduce overall solvent use, but solvent nanofiltration can also facilitate organic solvent recycling in peptide API production,121 which is especially important if problematic solvents, such as DMF, chloroform or THF are used, and would further remove the need for repeated precipitation/wash cycles in LPPS that might be unpractical on scale. Initial efforts for solvent replacement in LPPS have been made, e.g. Takahashi et al. have suggested that chloroform could be replaced with CPME during Fmoc-removal,107 but further work on green solvent substitution aimed at large-scale industrial production would benefit LPPS and MEPS.
4.3 Peptide synthesis by fragment ligation
SPPS is generally not a feasible approach for the synthesis of large peptides (>50 AA) on scale, yet the size and complexity of peptide therapeutics in development and under clinical evaluation is increasing, posing new challenges for cost-efficient high-quality manufacture of such APIs. One promising approach to circumvent this limitation is convergent synthesis of peptides and proteins using unprotected peptide fragments, through ligation. The potential of combining peptides of synthetic and recombinant origin is especially attractive, bridging already well-established synthetic and recombinant tracks in pharmaceutical development, while providing larger freedom for introduction of unnatural amino acids or site-specific modifications. Over the last decades, extensive development of chemical and enzymatic ligation methods on research lab scale has been reported, enabling the synthesis of a wide array of peptides and proteins.122–124 In chemical peptide ligation, a C-terminally functionalised peptide is chemo-selectively merged with a second peptide bearing a suitable N-terminal reactive group. Significant advances in the field include traceless Staudinger ligation,125,126 ketoacid-hydroxylamine (KAHA) ligation,127,128 Ser/Thr ligation (STL),129 and diselenide–selenoester ligation (DSL),130 but the by far most explored methodology is Native Chemical Ligation (NCL).131 NCL features the trans-thioesterification and subsequent S-to-N-acyl shift of C-terminal peptide thioesters with N-terminal cysteine-containing ligation partners (Fig. 11A). From a process development point-of-view, all chemical peptide ligation methods come with their own challenges such as the use/generation of potentially hazardous chemicals, or challenges with sourcing and cost of bespoke building blocks. Furthermore, while NCL is typically carried out under denaturing conditions such as aqueous 6 M guanidine hydrochloride or 8 M urea buffers, limited solubility of unprotected peptide ligation partners and consequently low volume yields is still a recurring challenge. It can sometimes be alleviated with organic co-solvents, detergents or other additives.122 Still, with peptide concentrations of 1–5 mM common in NCL,131,132 high volumes of solvent would be required for large-scale synthesis. Consequently, examples of industrial adoption of chemical peptide ligation for the manufacture of peptide therapeutics are still very limited. | ||
| Fig. 11 Selected protein ligation approaches with potential for large-scale production of therapeutic peptides. (A) In Native Chemical Ligation a peptide thioester reacts with a second peptide bearing an N-terminal Cys residue through trans-thioesterification and subsequent S-to-N-acyl shift. Potential sources for peptide thioester generation include (i) hydrazides,165,166 (ii) N-acylureas,139,140 (iii) bis(2-sulfanylethyl)amido (SEA) amides,141,167 (iv) peptides fused with intein proteins,145,168 and (v) peptides with C-terminal enzymatic recognition motifs or tags.146,147,169 (B) Protein trans-splicing involves peptides fused with split intein sequences that self-assemble to form an active intein unit. The intein is then autocatalytically excised while ligating the peptides of interest. PTS requires a Cys or Ser residue N-terminal on the first split intein partner (IntN), typically an Asn residue C-terminal on the second split intein partner (IntC) which cyclises to a succinimide moiety upon excision, and a Cys, Ser, or Thr residue N-terminal on the C-extein peptide.123,124 (C) Transpeptidase and peptidyl ligase enzymes catalyse amide bond formation between peptides (chemoenzymatic transamidation/ligation), each with their own peptide recognition motifs and requirement for efficient catalysis. | ||
Fortunately, recent development in mitigating some of these challenges may prove fruitful. For example, flow chemistry offers a higher degree in control of reaction parameters such as reaction temperature, volume and mixing compared to batch chemistry (vide infra).133,134 Chisholm et al. recently reported a protocol for tandem NCL and Cys photodesulfurisation from trifluoroethyl (TFET) thioester peptides using continuous flow giving significantly shorter reaction times for the synthesis of example peptide therapeutics enfurtivide and somatorelin,135 while Ollivier et al. developed a protocol using S-(2-((2-sulfanylethyl)amino)ethyl)peptidyl (SEAE) thioesters for efficient macrocyclization of peptides even with difficult NCL junctions such as Pro.136 As these authors note, continuous-flow solutions hold the potential for efficient scale-up, automation and rapid synthesis of therapeutic peptides, all attractive from an industrial perspective. One challenge in NCL, batch or flow, is efficient access to peptide thioesters. To this end, various resins for SPPS of thioester precursors are now commercially available (e.g. pyruvic acid hydrazone,137 and trityl hydrazine,138 o-amino(methyl)aniline ((Me)Dbz),139,140 and trityl bis(2-sulfanylethyl)amido (SEA)141 resins for the synthesis of peptide hydrazides, (Me)Nbz N-acylureas, and SEA amides respectively), but they are also complemented by a variety of methods for the production of thioester precursors by recombinant means (Fig. 11A). The use of thioesters generated from recombinant sources for ligation is usually referred to as Expressed Protein Ligation (EPL).142 Expressed peptide intein fusions (internal protein domains that induce protein splicing and re-ligation of the flanking extein sequences under certain conditions) or other C-terminal peptide tags can undergo thiolysis or hydrazinolysis to generate peptide C-terminal thioesters or hydrazides (thioester precursors) respectively.124,142–145 Such C-terminal tags have been reduced to minimal sequences as Komiya et al. recently developed an elegant method of hydrazinolysis of peptides bearing a cleavable C-terminal three amino acid motif (Cys-Pro-Leu) using biocatalysis and DKP formation with commercially available carboxypeptidase Y.146 Similarly, Qiao et al. used the cyanide donor 2-nitro-5-cyanatobenzoic acid (NTCB) to activate Cys residues on C-terminally tagged expressed proteins, with subsequent hydrazinolysis to generated peptide hydrazides, in a process denoted activated cysteine-based protein ligation (ACPL).147 Such advances in C-terminal activation strategies are interesting from a manufacturing perspective as they appear to facilitate access to peptide thioesters from recombinant sources on large scale.
In addition to NCL and EPL, protein trans-splicing (PTS) and enzymatic transamidations or ligations hold potential for efficient large-scale production of peptides. PTS is a naturally occurring phenomenon where two separate, split intein sequences self-associate with high affinity to form an active intein complex that is autocatalytically excised, while ligating the flanking extein sequences (Fig. 11B).148 A wide variety of naturally occurring and bioengineered split inteins have been studied,124 with some N- or C-terminal split intein partners (IntN or IntC) as short as 6–16 AA,149–153 which opens up for synthetic production if desired. Challenges with this approach are that splicing efficiency is often dependent on the nature of the flanking extein sequences, and the extent of premature cleavage of the intein-fusion protein. Furthermore, some bioengineered split inteins may require IP rights for commercial use. Similar challenges exist for fully enzymatic ligations, where supply chains for large scale sourcing of enzymes with reproducible activity and purity are not yet established. Nevertheless, transpeptidases such as sortase A,154,155 the peptide asparaginyl ligases (PAL) butelase 1 (ref. 156) and OaAEP1b,157 and peptidyl ligases subtiligase,158–160 peptiligase,161 omniligase,162 and trypsiligase163 are all well characterised enzymes offering different recognition motifs and sequence preferences (Fig. 11C). In fact, it was recently demonstrated that chemoenzymatic peptide synthesis can be a sustainable approach for the production of therapeutic peptides on scale as Pawlas et al. reported the medium scale (53 gram) synthesis of the antidiabetic peptide therapeutic exenatide using a two-fragment ligation catalysed by omniligase.164 Importantly, the authors showed that the scaled-up chemoenzymatic process was more cost-efficient and environmentally sustainable compared to conventional SPPS and a lab-scale exenatide benchmark process, and with the ability to purify the ligation partners prior to ligation, the purity profile of isolated API could also be significantly improved compared to when using conventional chemical approaches.164 Peptide synthesis by fragment ligation is thus already a viable approach for the production of complex peptide APIs with improved environmental impact, and while further development will be required before ligation is well established in industrial process development and production, it will likely be of increasing importance for therapeutic peptide manufacture in the near future.
4.4 Peptide synthesis in flow
Not long after the development of SPPS it was recognised that applying continuous flow to the solid support could facilitate some aspects of the SPPS cycle such as speeding up removal of excess reagents and washing, allowing for high degree of reaction control and monitoring, and potentially automation.170–173 Since then, major technological advances in solid supports (resins), pumps and reactors have led to a surge in potential for peptide synthesis in flow, slashing reaction times, allowing for precise control of reaction conditions and increasing throughput.134,174,175 Even so, examples of large-scale peptide synthesis by continuous flow are still scarce.176,177 Enabled by flow chemistry, some research groups have approached this challenge by introducing novel methods for peptide coupling. In a series of papers,134,178–180 Fuse et al. have showed that by using continuous flow microreactors, strongly activated amino acids such as acid anhydrides can undergo amide coupling in seconds with efficient suppression of racemisation, due to low reaction volumes and efficient mixing. The carboxylic acids were activated to symmetrical anhydrides or mixed carbonic anhydrides in flow using solutions of triphosgene178,179 or isobutyl chloroformate180 in MeCN and coupled with protected or unprotected amino acids respectively (in DMF or MeCN), to afford di-, tri- and tetrapeptides in good yields (Scheme 1A). Furthermore, Fuse and co-workers have also reported efficient synthesis of amino acid N-carboxy anhydrides (NCAs) using triphosgene in a microflow reactor.181 Controlled polymerisation of NCAs has emerged as a viable method for polypeptide synthesis yet it requires precise control of reaction parameters such as pH to limit side reactions.182,183 Knudsen et al. have described automated oligopeptide synthesis by flow chemistry using N-Cbz protected NCAs,184 and recently Jolley et al. demonstrated that NCAs can be used for large scale synthesis of peptides using continuous flow. A custom-made continuous stirred tank reactor (CSTR) with shear mixing, continuous solids addition and automated pH control was developed to tightly control the reaction parameters and allow production of di- and tripeptides with high conversion and a rate up to 535 g L−1 h−1 (Scheme 1B).185 Collectively, these methods demonstrate that flow chemistry can enable the use of reagents and synthetic approaches that are not suited for batch synthesis, and is useful for efficient synthesis of short peptides. However, as previously discussed, new technology is more likely to be adapted in industry on the short term if it can be incorporated into existing infrastructure, thereby reducing hurdles related to investment cost and workflow. Consequently, technological advances based on Fmoc chemistry, such as automated fast-flow peptide synthesis (AFPS) developed by Pentelute and co-workers,186,187 are more likely to be impactful in industry as well-established supply chains and infrastructure for storage and use already exist. The development of AFPS has been described in a series of papers over the last few years. The first-generation setup consisted of a disposable fluoroalkoxy-tube reactor coupled with an HPLC pump that delivered DMF and 50% piperidine solution for wash and Fmoc-removal respectively, and a syringe pump that delivered 0.34 M solutions of activated amino acids to the reaction vessel via a heat exchange loop. The reaction vessel and heat exchanger were immersed in a water bath held at 60 °C, and the flow-through was monitored by UV absorbance at 304 nm.186 By expanding the inner diameter of the reaction vessel and incorporating stainless steel components, the flow rates were increased while keeping back pressure low in a second-generation setup, which enabled peptide synthesis to be carried out with 200 mg resin, flow rates up to 60 mL min−1 and 1.8 min cycle time for each amino acid incorporated (Scheme 1C).186,188 Further improvements have involved full automation and the introduction of a mixing module, where amino acid, coupling reagents, and base were mixed at 90 °C prior to addition to the reaction vessel, which was also held at 90 °C. High flow rates (80 mL min−1) allowed for short mixing times, which suppressed epimerisation of sensitive activated amino acids such as Cys and His and brought the entire cycle time down to 40 seconds for each amino acid incorporated, using 6–20 equivalents of amino acid per coupling.189 The latest developments have led to a fully automated system with settings for reagent addition, coupling times and coupling reagents, and heating tailored to the individual amino acids, leading to peptide synthesis in unprecedented speed and fidelity. Moreover, this technology has expanded the realm for which peptides and proteins are accessible by synthetic means, as demonstrated by the synthesis of nine different single-domain proteins ranging 86–164 AA, up to three times the length accessible by SPPS, in 3.5–6.5 h.187 The main current limitations for large-scale use are excessive use of reagents and reactor capacity, as it currently holds up to 200 mg of resin with a maximum loading of 0.49 mmol g−1. Designing parallel and/or larger reaction chambers could increase throughput but would invariably require additional optimisation of the reaction parameters. Furthermore, AFPS has so far been developed based on DMF as solvent, which is problematic from a sustainability perspective. Thus, integrating the AFPS platform with green solvents, and reducing the amounts of reagents needed for efficient coupling would greatly benefit the technology, and facilitate its adoption for use beyond research scale. | ||
| Scheme 1 (A) Examples of microflow peptide synthesis with strong carboxylic acid activation by triphosgene178,179 or isobutyl chloroformate.180 (B) Large scale peptide synthesis through controlled continuous flow polymerisation of N-carboxy anhydrides (NCAs).185 Unspecified counter ions are illustrated as M+ and X− respectively. (C) Example protocol for automated fast-flow peptide synthesis at 60 °C to enable each amino acid coupling cycle within 1.8 min.186,188 | ||
4.5 Peptide synthesis under solvent-free conditions
Several academic research groups have reported the synthesis of short peptides under solvent-free conditions, which dramatically reduces waste generation.190,191 One such method is ball milling that promotes chemical reactions under solvent-free conditions by mechanical milling/grinding of reagents in a stainless steel ball mill.192 Declerck et al. reported the synthesis of the tripeptide aspartame by ball milling in a yield of 84% using NCA-amino acid derivatives.193 To further improve the methodology, the use of small amounts of solvent such as EtOAc and GVL to facilitate mixing, has also been explored for the synthesis of di-, tri- and tetra-peptides194 and longer peptides such as the 5-mer Leu-enkephalin (46% overall yield).195 A head-to-head comparison between SPPS, solution-phase synthesis and ball milling for the synthesis of the tetrapeptide H-Val-Val-Ile-Ala-OH was recently published.196 The best purity was obtained by SPPS (96%), whereas solution-phase synthesis and ball-milling gave similar purities (88% and 85%, respectively). It was however noted that ball milling gave a higher yield (59%) compared to SPPS (54%) and solution-phase synthesis (43%). Further application of the ball milling approach by Hérnandez et al. involved amide bond formation by bio-catalysis (mechanoenzymatic synthesis). Cbz- or Boc-protected amino acid ethyl esters were milled with amino amides in the presence of the cysteine protease papain, with sodium carbonate decahydrate as base and L-cysteine as additive to afford dipeptides in good yields,197 and using N-unprotected amino acid esters (e.g. Phe and Gly) achieved homo-oligomerisation and polypeptide products of various degree of polymerisation.198 Apart from generating such homo-oligomers, ball milling has, to the best of our knowledge, not been applied for the synthesis of peptides longer than 5 amino acid residues, nor been scaled up, and the technology would benefit from further development targeting longer and more complex peptide sequences.Lamaty and co-workers have also reported the use of a reactive extrusion process for peptide coupling.199 Extruders are composed of a barrel/pipe containing one or two rotating screws that allow efficient transport and mixing of reagents through compression and shearing forces. It is possible to control the temperature of the barrel and thus induce melting to facilitate mixing. The addition of a small amount of a solvent can be beneficial to promote better extrusion. In this way, protected di- and tripeptides were synthesised on gram scale, coupling NHS-activated Boc-protected amino acids with methyl ester amino acids or dipeptides, NaHCO3 and acetone (approximately 0.15 mL of acetone/1 g of solid reagents). The reaction products were obtained as pastes that were purified by simple dissolution in EtOAc and aqueous washes. Using this method, an 81% yield for the synthesis of aspartame was reported.199 The output efficiency for the three studied synthetic strategies was calculated using a space-time yield (STY) approach (Table 20). The STY was determined for three dipeptides and the reactive extrusion method was found to be far superior to ball-milling and solution-phase synthesis.
| Dipeptide | STY (g per cm3 per day−1) | ||
|---|---|---|---|
| Reactive extrusion | Ball-milling | Solution-phasea | |
| a Reaction performed with Et3N as base and DMF as solvent.b Reaction performed with NaHCO3 as base and acetone as solvent. | |||
| Boc-Trp-Gly-OMe | 48.0 | 2.6 | 1.8 |
| Boc-Trp-Phe-OMe | 37.0 | 2.1 | 1.1 |
| Boc-Asp(OBn)-Phe-OMe | 471.2 | 2.2 | 4.9 (0.2)b |
In another solvent free approach, Jain and co-workers have reported the use of microwave irradiation for the synthesis of short peptides (Table 21).200 Optimisation of reaction parameters and screening of coupling reagents identified the use of DIC/HONB in combination with DIPEA as the best conditions. A Boc-protected amino acid and an amino acid methyl ester (or peptide methyl ester) were coupled at 60 °C for 15 min (microwave irradiation). The method was applied to synthesis of di-, tri-, tetra- and pentapeptides, with yields ranging from 55% to 99% and purities >90%. The method was amenable to gram scale synthesis with no loss in purity or yield.
| Peptide | Yielda (%) | Purityb (%) |
|---|---|---|
| a Reaction conditions: coupling: AA (1.2 equiv.), DIPEA (3 equiv.), DIC (1.2 equiv.), HONB (1.2 equiv.), 60 °C, 15 min, 40 W, deprotection: 6 N HCl, 25 °C, 15 min.b Purity was determined by RP-HPLC analysis. | ||
| Ser(Bn)-Ile | 99 | 96 |
| Pro-Met-Ile | 70 | 98 |
| Ala-Thr(Bn)-Phe-Ile | 60 | 97 |
| Arg-Lys(Z)-Asp(Bn)-Val-Tyr(Bn) | 55 | 95 |
| Carnosine | 78 | 95 |
| Aspartame | 92 | 98 |
| Thyrotropin releasing hormone | 82 | 94 |
| Thymopentin | 53 | 97 |
| Leu-enkephalin | 62 | 99 |
Reactions under solvent-free or quasi-solvent-free conditions can be an efficient method for the assembly of short peptides in good yields and purities. However, the development of such methods is still in its early days and it remains to be demonstrated if these can be scaled up and applied to the synthesis of longer peptide sequences. Unlike ball milling where scaling-up is challenging, reactive extrusion offers some interesting opportunities for scaling up in a flow setup where reagents are continuously added to the reactive extrusion pipe. Similar to solution-phase flow chemistry it should be possible to achieve a high throughput of material in a relatively small reactive extrusion reactor. Moreover, one could imagine a reactive extrusion design where new activated amino acids and reagents are fed into the reactor at different points along the pipe to achieve a multi-step synthesis in a continuous flow solid state system.
5. Conclusion and outlook
In recent years a growing number of publications has been addressing the substitution of environmentally problematic solvents such as CH2Cl2, DMF and NMP in SPPS. However, in order to ensure that newly developed methods will be amenable to large scale production, an industrial perspective on this issue has been missing. As outlined above a wide range of physical characteristics of the different components of an SPPS process, i.e. amino acid building blocks, reagents, resins and solvents, must meet current requirements dictated by the peptide production plants. In addition, published literature often lack the purity assessments, characterisation of side-products and reporting of crude yields/mass gain that would greatly improve their industrial utility. For the benefit of the environment it is our hope that academic research groups will take up-scaling into consideration when developing greener peptide synthesis methodologies, and that the pharmaceutical industry shows willingness to invest in greener approaches. Actions for greening the synthesis of peptide pharmaceuticals should be taken with both short- and long term in mind. In the short term, replacing DMF with a green solvent or mixture of green solvents is necessary to allow the continued manufacture of peptides with the current set of building blocks and reagents. However, if not changed significantly, SPPS will continue to be a wasteful process regardless of the solvent used. Thus, in the long term it would be of great interest if an entirely new platform for peptide synthesis that is amenable for large scale production could be developed, and several emerging technologies have been described herein, including water-based SPPS, LPPS, protein ligation and peptide synthesis in flow. The water-based ASPPS approach and the LPPS/MEPS methods are promising alternatives, and it will be interesting to follow the maturation of these platforms over time. In general, these methods have mostly dealt with linear assembly of shorter peptides, where one amino acid at the time is attached to the growing peptide chain. However, as the size and complexity of peptide pharmaceuticals keeps growing it is likely that more convergent strategies where smaller peptide fragments are merged using ligation techniques such as expressed protein ligation or chemoenzymatic ligation will be of increased importance to meet the manufacturing demand. Although examples of large-scale synthesis of peptides using such ligation approaches are still few, there are green and cost incentives for further development. Overall, the prospects of greening peptide synthesis in general, and SPPS in particular, by solvent substitution looks bright. In addition to the encouraging work of academic groups, major industrial players are actively publishing their findings in this area and lectures/posters on the topic are frequently presented at major international conferences. At Novo Nordisk, our new environmental strategy sets out targets and milestones to attain company-wide carbon-neutrality within the next 10 years. In order to achieve this goal, we are investing considerable resources into the development of a short-term greener platform for SPPS and are investigating a range of alternative platforms for peptide synthesis that will be reported in due course.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Per Vedsø from Novo Nordisk for project support, and Dr Frank Dettner, Dr Raphael Liffert and Dr Ralph O. Schönleber from Bachem AG for fruitful discussions during the preparation of this manuscript. Jesper Grossman from Novo Nordisk H&S Consulting is thanked for his kind assistance with REACH classification of solvents. Dr Rune Severinsen and Dr Sebastian Brandes from Novo Nordisk, and an anonymous reviewer are all thanked for their critical review of the manuscript. Dr Vincent Martin is funded by the Novo Nordisk R&D STAR Fellowship Programme.References
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- W. Al Bakri, M. D. Donovan, M. Cueto, Y. Wu, C. Orekie and Z. Yang, Expert Opin. Drug Delivery, 2018, 15, 991–1005 CrossRef CAS.
- M. Davies, T. R. Pieber, M. L. Hartoft-Nielsen, O. K. H. Hansen, S. Jabbour and J. Rosenstock, JAMA, J. Am. Med. Assoc., 2017, 318, 1460–1470 CrossRef CAS.
- D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS.
- Peptide Therapeutics Market: Global Forecast to 2027 in Coherent Market insight, https://www.coherentmarketinsights.com/press-release/global-peptide-therapeutics-market-to-surpass-us-450-billion-by-2026-951.
- A. A. Zompra, A. S. Galanis, O. Werbitzky and F. Albericio, Future Med. Chem., 2009, 1, 361–377 CrossRef CAS.
- L. Andersson, L. Blomberg, M. Flegel, L. Lepsa, B. Nilsson and M. Verlander, Biopolymers, 2000, 55, 227–250 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- A. Isidro-Llobet, M. Alvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 CrossRef CAS.
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS.
- J. Lopez, S. Pletscher, A. Aemissegger, C. Bucher and F. Gallou, Org. Process Res. Dev., 2018, 22, 494–503 CrossRef CAS.
- European Chemicals Agency Candidate List of substances of very high concern for Authorisation, http://echa.europa.eu/candidate-list-table.
- F. Albericio, O. Al Musaimi and B. G. de la Torre, Green Chem., 2020, 22, 996–1018 RSC.
- Y. E. Jad, A. Kumar, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2019, 7, 3671–3683 CrossRef CAS.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS.
- E. R. Lax and T. Shah, in Peptide Therapeutics: Strategy and Tactics for Chemistry, Manufacturing, and Controls, The Royal Society of Chemistry, 2019, pp. 151–193 Search PubMed.
- J. H. Rasmussen, Bioorg. Med. Chem., 2018, 26, 2914–2918 CrossRef CAS.
- American Chemical Society Green Chemistry Institute, https://www.acs.org/content/acs/en/greenchemistry.html.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- International Council For Harmonisation Of Technical Requirements For Pharmaceuticals For Human Use, ICH Harmonised Guideline, Impurities: Guideline for Residual Solvents Q3C(R6), 2016 Search PubMed.
- P. Palladino and D. A. Stetsenko, Org. Lett., 2012, 14, 6346–6349 CrossRef CAS.
- A. Van Gysel, F. Soeterbroeck and G. Vermeulen, Acetonitrile Recycling Process, WO/2005/044783, 2005 Search PubMed.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- V. R. Veleva, B. W. Cue and S. Todorova, ACS Sustainable Chem. Eng., 2018, 6, 2–14 CrossRef CAS.
- S. G. Koenig, C. Bee, A. Borovika, C. Briddell, J. Colberg, G. R. Humphrey, M. E. Kopach, I. Martinez, S. Nambiar, S. V. Plummer, S. D. Ribe, F. Roschangar, J. P. Scott and H. F. Sneddon, ACS Sustainable Chem. Eng., 2019, 7, 16937–16951 CrossRef CAS.
- S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685–1691 RSC.
- Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814 CrossRef CAS.
- The Public Activities Coordination Tool, https://echa.europa.eu/pact.
- European Chemicals Agency, https://echa.europa.eu.
- Y. Ran, F. Byrne, I. D. V. Ingram and M. North, Chem.–Eur. J., 2019, 25, 4951–4964 CrossRef CAS.
- S. Lawrenson, M. North, F. Peigneguy and A. Routledge, Green Chem., 2017, 19, 952–962 RSC.
- Y. E. Jad, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2017, 21, 365–369 CrossRef CAS.
- J. Pawlas, B. Antonic, M. Lundqvist, T. Svensson, J. Finnman and J. H. Rasmussen, Green Chem., 2019, 21, 2594–2600 RSC.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393–2398 RSC.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419–426 CrossRef CAS.
- A. Kumar, M. Alhassan, J. Lopez, F. Albericio and B. G. de la Torre, ChemSusChem, 2020, 13, 5288 CrossRef CAS.
- L. Ferrazzano, D. Corbisiero, G. Martelli, A. Tolomelli, A. Viola, A. Ricci and W. Cabri, ACS Sustainable Chem. Eng., 2019, 7(15), 12867–12877 CrossRef CAS.
- K. Hojo, M. Maeda, N. Tanakamaru, K. Mochida and K. Kawasaki, Protein Pept. Lett., 2006, 13, 189–192 CrossRef CAS.
- A. Kumar, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2986–2988 CrossRef CAS.
- A. Kumar, Y. E. Jad, J. M. Collins, F. Albericio and B. G. de la Torre, ACS Sustainable Chem. Eng., 2018, 6, 8034–8039 CrossRef CAS.
- A. Bado-Nilles, A.-O. Diallo, G. Marlair, P. Pandard, L. Chabot, A. Geffard, C. Len, J.-M. Porcher and W. Sanchez, J. Hazard. Mater., 2015, 283, 202–210 CrossRef CAS.
- A.-O. Diallo, G. Fayet, C. Len and G. Marlair, Ind. Eng. Chem. Res., 2012, 51, 3149–3156 CrossRef CAS.
- A. Pernille Tofteng, S. L. Pedersen, D. Staerk and K. J. Jensen, Chem.–Eur. J., 2012, 18, 9024–9031 CrossRef CAS.
- A. Kumar, Y. E. Jad, B. G. de la Torre, A. El-Faham and F. Albericio, J. Pept. Sci., 2017, 23, 763–768 CrossRef CAS.
- J. B. Sperry, C. J. Minteer, J. Y. Tao, R. Johnson, R. Duzguner, M. Hawksworth, S. Oke, P. F. Richardson, R. Barnhart, D. R. Bill, R. A. Giusto and J. D. Weaver, Org. Process Res. Dev., 2018, 22, 1262–1275 CrossRef CAS.
- R. Subiros-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem.–Eur. J., 2009, 15, 9394–9403 CrossRef CAS.
- K. D. Wehrstedt, P. A. Wandrey and D. Heitkamp, J. Hazard. Mater., 2005, 126, 1–7 CrossRef CAS.
- O. Al Musaimi, Y. E. Jad, A. Kumar, A. El-Faham, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2018, 22, 1809–1816 CrossRef CAS.
- O. Al Musaimi, A. El-Faham, A. Basso, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2019, 60, 151058 CrossRef CAS.
- A. Kumar, A. Sharma, B. G. de la Torre and F. Albericio, Molecules, 2019, 24, 809 CrossRef.
- A. Kumar, A. Thompson-Adewumi, K. P. Nandhini, J. M. Collins, F. Albericio and B. G. de la Torre, Org. Process Res. Dev., 2019, 23, 1096–1100 CrossRef CAS.
- B. G. de la Torre, A. Kumar, M. Alhassan, C. Bucher, F. Albericio and J. Lopez, Green Chem., 2020, 22, 3162–3169 RSC.
- M. Staderini, A. Gambardella, A. Lilienkampf and M. Bradley, Org. Lett., 2018, 20, 3170–3173 CrossRef CAS.
- J. Pawlas and J. H. Rasmussen, Green Chem., 2019, 21, 5990–5998 RSC.
- A. D. McFarland, J. Y. Buser, M. C. Embry, C. B. Held and S. P. Kolis, Org. Process Res. Dev., 2019, 23, 2099–2105 CrossRef CAS.
- M. Erny, M. Lundqvist, J. H. Rasmussen, O. Ludemann-Hombourger, F. Bihel and J. Pawlas, Org. Process Res. Dev., 2020, 24, 1341–1349 CrossRef CAS.
- N. Zinieris, L. Leondiadis and N. Ferderigos, J. Comb. Chem., 2005, 7, 4–6 CrossRef CAS.
- European Chemicals Agency - Piperidine infocard, https://echa.europa.eu/substance-information/-/substanceinfo/100.003.467.
- US Department of Justice, Drug Enforcement Administration, Diversion Control Division, https:// www.deadiversion.usdoj.gov/21cfr/cfr/1310/1310_02.htm..
- O. F. Luna, J. Gomez, C. Cardenas, F. Albericio, S. H. Marshall and F. Guzman, Molecules, 2016, 21, 1542 CrossRef.
- J. Hachmann and M. Lebl, J. Comb. Chem., 2006, 8, 149 CrossRef CAS.
- G. B. Fields, in Peptide Synthesis Protocols, ed. M. W. Pennington and B. M. Dunn, Humana Press, Totowa, NJ, 1995, pp. 17–27 Search PubMed.
- European Chemicals Agency - 4-methylPiperidine infocard, https://echa.europa.eu/substance-information/-/substanceinfo/100.009.959.
- A. Pribylka, V. Krchnak and E. Schutznerova, Green Chem., 2019, 21, 775–779 RSC.
- K. Ralhan, V. G. KrishnaKumar and S. Gupta, RSC Adv., 2015, 5, 104417–104425 RSC.
- O. Al Musaimi, Y. E. Jad, A. Kumar, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Current Opinion Green Sustainable Chemistry, 2018, 11, 99–103 CrossRef.
- O. Al Musaimi, A. El-Faham, Z. Almarhoon, A. Basso, B. G. de la Torre and F. Albericio, Polymers, 2019, 11(5), 874 CrossRef CAS.
- J. Li, J. Albrecht, A. Borovika and M. D. Eastgate, ACS Sustainable Chem. Eng., 2018, 6, 1121–1132 CrossRef CAS.
- J. Li, E. M. Simmons and M. D. Eastgate, Green Chem., 2017, 19, 127–139 RSC.
- F. I. Auzanneau, M. Meldal and K. Bock, J. Pept. Sci., 1995, 1, 31–44 CrossRef CAS.
- F. Garcia-Martin, M. Quintanar-Audelo, Y. Garcia-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Cote, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef CAS.
- M. Meldal, Tetrahedron Lett., 1992, 33, 3077–3080 CrossRef CAS.
- G. I. Tesser and I. C. Balvert-Geers, Int. J. Pept. Protein Res., 1975, 7, 295–305 CrossRef CAS.
- H. Kunz and G. Schaumlöffel, Liebigs Ann. Chem., 1985, 1985, 1784–1793 CrossRef.
- H. Kunz, Angew. Chem., Int. Ed., 1978, 17, 67–68 CrossRef.
- H. Kunz, Chem. Ber., 1976, 109, 2670–2683 CrossRef CAS.
- R. B. Merrifield and A. E. Bach, J. Org. Chem., 1978, 43, 4808–4816 CrossRef CAS.
- K. Hojo, M. Maeda, T. J. Smith, E. Kita, F. Yamaguchi, S. Yamamoto and K. Kawasaki, Chem. Pharm. Bull., 2004, 52, 422–427 CrossRef CAS.
- K. Hojo, M. Maeda and K. Kawasaki, J. Pept. Sci., 2001, 7, 615–618 CrossRef CAS.
- K. Hojo, M. Maeda, Y. Takahara, S. Yamamoto and K. Kawasaki, Tetrahedron Lett., 2003, 44, 2849–2851 CrossRef CAS.
- K. Hojo, M. Maeda and K. Kawasaki, Tetrahedron Lett., 2004, 45, 9293–9295 CrossRef CAS.
- L. A. Carpino, M. Ismail, G. A. Truran, E. M. E. Mansour, S. Iguchi, D. Ionescu, A. El-Faham, C. Riemer and R. Warrass, J. Org. Chem., 1999, 64, 4324–4338 CrossRef CAS.
- L. A. Carpino, M. Philbin, M. Ismail, G. A. Truran, E. M. E. Mansour, S. Iguchi, D. Ionescu, A. El-Faham, C. Riemer, R. Warrass and M. S. Weiss, J. Am. Chem. Soc., 1997, 119, 9915–9916 CrossRef CAS.
- J. C. Collins, Water Soluble Solid Phase Peptide Synthesis, US Pat., 2012/0157563 A1, 2012 Search PubMed.
- L. A. Carpino and M. Philbin, J. Org. Chem., 1999, 64, 4315–4323 CrossRef CAS.
- L. A. Carpino, A. A. Abdel-Maksoud, D. Ionescu, E. M. Mansour and M. A. Zewail, J. Org. Chem., 2007, 72, 1729–1736 CrossRef CAS.
- A. El-Faham and F. Albericio, Pept. Sci., 2020, 112, e24164 CAS.
- V. V. Suresh Babu, N. S. Sudarshan and S. A. Naik, Int. J. Pept. Res. Ther., 2008, 14, 105–112 CrossRef CAS.
- L. A. Carpino, S. Ghassemi, D. Ionescu, M. Ismail, D. Sadat-Aalaee, G. A. Truran, E. M. E. Mansour, G. A. Siwruk, J. S. Eynon and B. Morgan, Org. Process Res. Dev., 2003, 7, 28–37 CrossRef CAS.
- L. A. Carpino and E. M. Mansour, J. Org. Chem., 1999, 64, 8399–8401 CrossRef CAS.
- S. Knauer, T. M. L. Roese, O. Avrutina, H. Kolmar and C. Uth, Method For Peptide Synthesis And Apparatus For Carrying Out A Method For Solid Phase Synthesis Of Peptides, WO/2016/050764, 2016 Search PubMed.
- S. Knauer, N. Koch, C. Uth, R. Meusinger, O. Avrutina and H. Kolmar, Angew. Chem., Int. Ed., 2020, 59, 12984–12990 CrossRef CAS.
- M. Badland, R. Crook, B. Delayre, S. J. Fussell, I. Gladwell, M. Hawksworth, R. M. Howard, R. Walton and G. A. Weisenburger, Tetrahedron Lett., 2017, 58, 4391–4394 CrossRef CAS.
- R. De Marco, A. Tolomelli, A. Greco and L. Gentilucci, ACS Sustainable Chem. Eng., 2013, 1, 566–569 CrossRef CAS.
- K. Hojo, N. Shinozaki, K. Hidaka, Y. Tsuda, Y. Fukumori, H. Ichikawa and J. D. Wade, Amino Acids, 2014, 46, 2347–2354 CrossRef CAS.
- A. Mahindra, K. Nooney, S. Uraon, K. K. Sharma and R. Jain, RSC Adv., 2013, 3, 16810–16816 RSC.
- A. S. Galanis, F. Albericio and M. Grotli, Org. Lett., 2009, 11, 4488–4491 CrossRef CAS.
- K. Hojo, A. Hara, H. Kitai, M. Onishi, H. Ichikawa, Y. Fukumori and K. Kawasaki, Chem. Cent. J., 2011, 5, 49 CrossRef CAS.
- A. Borovika, J. Albrecht, J. Li, A. S. Wells, C. Briddell, B. R. Dillon, L. J. Diorazio, J. R. Gage, F. Gallou, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Martinez, M. Olbrich, J. L. Piper, F. Roschangar, E. C. Sherer and M. D. Eastgate, Nat. Sustain., 2019, 2, 1034–1040 CrossRef.
- V. N. R. Pillai, M. Mutter, E. Bayer and I. Gatfield, J. Org. Chem., 1980, 45, 5364–5370 CrossRef CAS.
- D. Takahashi and T. Yamamoto, Tetrahedron Lett., 2012, 53, 1936–1939 CrossRef CAS.
- D. Takahashi, T. Yano and T. Fukui, Org. Lett., 2012, 14, 4514–4517 CrossRef CAS.
- D. Takahashi, T. Inomata and T. Fukui, Angew. Chem., Int. Ed., 2017, 129, 7911–7915 CrossRef.
- Y. Okada, H. Suzuki, T. Nakae, S. Fujita, H. Abe, K. Nagano, T. Yamada, N. Ebata, S. Kim and K. Chiba, J. Org. Chem., 2013, 78, 320–327 CrossRef CAS.
- Y. Okada, R. Takasawa, D. Kubo, N. Iwanaga, S. Fujita, K. Suzuki, H. Suzuki, H. Kamiya and K. Chiba, Org. Process Res. Dev., 2019, 23(11), 2576–2581 CrossRef CAS.
- Y. Fujita, S. Fujita, Y. Okada and K. Chiba, Org. Lett., 2013, 15, 1155–1157 CrossRef CAS.
- S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Tetrahedron, 2013, 69, 2555–2559 CrossRef CAS.
- E. Matsumoto, Y. Fujita, Y. Okada, E. I. Kauppinen, H. Kamiya and K. Chiba, J. Pept. Sci., 2015, 21, 691–695 CrossRef CAS.
- C. W. Seifert, A. Paniagua, G. A. White, L. Cai and G. Li, Eur. J. Org. Chem., 2016, 2016, 1714–1719 CrossRef CAS.
- H. Li, J. Chao, J. Hasan, G. Tian, Y. Jin, Z. Zhang and C. Qin, J. Org. Chem., 2020, 85, 6271–6280 CrossRef CAS.
- H. Li, J. Chao, G. Tian, J. Hasan, Y. Jin, Z. Zhang and C. Qin, Org. Chem. Front., 2020, 7, 689–696 RSC.
- H. Li, J. Chao, Z. Zhang, G. Tian, J. Li, N. Chang and C. Qin, Org. Lett., 2020, 22, 3323–3328 CrossRef CAS.
- S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrow and A. G. Livingston, Chem. Commun., 2010, 46, 2808–2810 RSC.
- S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrow and A. G. Livingston, Org. Process Res. Dev., 2010, 14, 1313–1325 CrossRef CAS.
- W. Chen, M. Sharifzadeh, N. Shah and A. G. Livingston, Ind. Eng. Chem. Res., 2017, 56, 6796–6804 CrossRef CAS.
- V. Castro, C. Noti, W. Chen, M. Cristau, A. Livignston, H. Rodríguez and F. Albericio, Macromolecules, 2017, 50, 1626–1634 CrossRef CAS.
- D. Ormerod, B. Noten, M. Dorbec, L. Andersson, A. Buekenhoudt and L. Goetelen, Org. Process Res. Dev., 2015, 19, 841–848 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2020, 120(6), 3051–3126 CrossRef CAS.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2001, 3, 9–12 CrossRef CAS.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS.
- J. W. Bode, Acc. Chem. Res., 2017, 50, 2104–2115 CrossRef CAS.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- R. E. Thompson, X. Liu, N. Alonso-García, P. J. B. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- S. Fuse, Y. Otake and H. Nakamura, Chem.–Asian J., 2018, 13, 3818–3832 CrossRef CAS.
- T. S. Chisholm, D. Clayton, L. J. Dowman, J. Sayers and R. J. Payne, J. Am. Chem. Soc., 2018, 140, 9020–9024 CrossRef CAS.
- N. Ollivier, T. Toupy, R. C. Hartkoorn, R. Desmet, J.-C. M. Monbaliu and O. Melnyk, Nat. Commun., 2018, 9, 2847 CrossRef.
- P. S. Chelushkin, K. V. Polyanichko, M. V. Leko, M. Y. Dorosh, T. Bruckdorfer and S. V. Burov, Tetrahedron Lett., 2015, 56, 619–622 CrossRef CAS.
- G. Stavropoulos, D. Gatos, V. Magafa and K. Barlos, Lett. Pept. Sci., 1996, 2, 315–318 CrossRef.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS.
- N. Ollivier, L. Raibaut, A. Blanpain, R. Desmet, J. Dheur, R. Mhidia, E. Boll, H. Drobecq, S. L. Pira and O. Melnyk, J. Pept. Sci., 2014, 20, 92–97 CrossRef CAS.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- T. C. Evans Jr, J. Benner and M. Q. Xu, Protein Sci., 1998, 7, 2256–2264 CrossRef CAS.
- J. Thom, D. Anderson, J. McGregor and G. Cotton, Bioconjugate Chem., 2011, 22, 1017–1020 CrossRef CAS.
- M. Vila-Perelló, Z. Liu, N. H. Shah, J. A. Willis, J. Idoyaga and T. W. Muir, J. Am. Chem. Soc., 2013, 135, 286–292 CrossRef.
- C. Komiya, A. Shigenaga, J. Tsukimoto, M. Ueda, T. Morisaki, T. Inokuma, K. Itoh and A. Otaka, Chem. Commun., 2019, 55, 7029–7032 RSC.
- Y. Qiao, G. Yu, K. C. Kratch, X. A. Wang, W. W. Wang, S. Z. Leeuwon, S. Xu, J. S. Morse and W. R. Liu, J. Am. Chem. Soc., 2020, 142, 7047–7054 CrossRef CAS.
- N. H. Shah and T. W. Muir, Chem. Sci., 2014, 5, 446–461 RSC.
- J. H. Appleby, K. Zhou, G. Volkmann and X.-Q. Liu, J. Biol. Chem., 2009, 284, 6194–6199 CrossRef CAS.
- Y. Lin, M. Li, H. Song, L. Xu, Q. Meng and X. Q. Liu, PLoS One, 2013, 8, e59516 CrossRef CAS.
- J. H. Appleby-Tagoe, I. V. Thiel, Y. Wang, Y. Wang, H. D. Mootz and X.-Q. Liu, J. Biol. Chem., 2011, 286, 34440 CrossRef CAS.
- M. Neugebauer, J. K. Böcker, J. C. J. Matern, S. Pietrokovski and H. D. Mootz, Biol. Chem., 2017, 398, 57–67 CAS.
- W. Sun, J. Yang and X.-Q. Liu, J. Biol. Chem., 2004, 279, 35281–35286 CrossRef CAS.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS.
- X. Huang, A. Aulabaugh, W. Ding, B. Kapoor, L. Alksne, K. Tabei and G. Ellestad, Biochemistry, 2003, 42, 11307–11315 CrossRef CAS.
- G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Nat. Commun., 2015, 6, 10199 CrossRef CAS.
- T. K. Chang, D. Y. Jackson, J. P. Burnier and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12544–12548 CrossRef CAS.
- D. Jackson, J. Burnier, C. Quan, M. Stanley, J. Tom and J. Wells, Science, 1994, 266, 243–247 CrossRef CAS.
- A. M. Weeks and J. A. Wells, Chem. Rev., 2020, 120, 3127–3160 CrossRef CAS.
- A. Toplak, T. Nuijens, P. J. L. M. Quaedflieg, B. Wu and D. B. Janssen, Adv. Synth. Catal., 2016, 358, 2140–2147 CrossRef CAS.
- M. Schmidt, A. Toplak, P. J. L. M. Quaedflieg, H. Ippel, G. J. J. Richelle, T. M. Hackeng, J. H. van Maarseveen and T. Nuijens, Adv. Synth. Catal., 2017, 359, 2050–2055 CrossRef CAS.
- S. Liebscher, M. Schopfel, T. Aumuller, A. Sharkhuukhen, A. Pech, E. Hoss, C. Parthier, G. Jahreis, M. T. Stubbs and F. Bordusa, Angew. Chem., Int. Ed., 2014, 53, 3024–3028 CrossRef CAS.
- J. Pawlas, T. Nuijens, J. Persson, T. Svensson, M. Schmidt, A. Toplak, M. Nilsson and J. H. Rasmussen, Green Chem., 2019, 21, 6451–6457 RSC.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS.
- T. C. Evans Jr, J. Benner and M.-Q. Xu, Protein Sci., 1998, 7, 2256–2264 CrossRef CAS.
- J. J. Ling, R. L. Policarpo, A. E. Rabideau, X. Liao and B. L. Pentelute, J. Am. Chem. Soc., 2012, 134, 10749–10752 CrossRef CAS.
- R. P. W. Scott, K. K. Chan, P. Kucera and S. Zolty, J. Chromatogr. Sci., 1971, 9, 577–591 CrossRef CAS.
- E. Bayer, G. Jung, I. Halász and I. Sebestian, Tetrahedron Lett., 1970, 11, 4503–4505 CrossRef.
- E. Atherton, E. Brown, R. C. Sheppard and A. Rosevear, J. Chem. Soc., Chem. Commun., 1981, 1151–1152 RSC.
- T. J. Lukas, M. B. Prystowsky and B. W. Erickson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2791–2795 CrossRef CAS.
- C. P. Gordon, Org. Biomol. Chem., 2018, 16, 180–196 RSC.
- N. Ahmed, Chem. Biol. Drug Des., 2018, 91, 647–650 CrossRef CAS.
- V. Caciagli, F. Cardinali, F. Bonelli and P. Lombardi, J. Pept. Sci., 1998, 4, 327–334 CrossRef CAS.
- V. Caciagli, F. Cardinali, A. Hänsicke, G. Tuchalski and P. Lombardi, J. Pept. Sci., 1997, 3, 224–230 CrossRef CAS.
- S. Fuse, Y. Mifune and T. Takahashi, Angew. Chem., Int. Ed., 2014, 53, 851–855 CrossRef CAS.
- S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491 CrossRef CAS.
- S. Fuse, K. Masuda, Y. Otake and H. Nakamura, Chem.–Eur. J., 2019, 25, 15091–15097 CrossRef CAS.
- Y. Otake, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2018, 57, 11389–11393 CrossRef CAS.
- R. F. Hirschmann, J. Org. Chem., 1967, 32, 3415–3425 CrossRef CAS.
- K. Klinker and M. Barz, Macromol. Rapid Commun., 2015, 36, 1943–1957 CrossRef CAS.
- K. R. Knudsen, M. Ladlow, Z. Bandpey and S. V. Ley, J. Flow Chem., 2014, 4, 18–21 CrossRef CAS.
- K. E. Jolley, W. Nye, C. G. Nino, N. Kapur, A. Rabion, K. Rossen and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1557–1565 CrossRef CAS.
- M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, ChemBioChem, 2014, 15, 713–720 CrossRef CAS.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 CrossRef CAS.
- S. K. Mong, A. A. Vinogradov, M. D. Simon and B. L. Pentelute, ChemBioChem, 2014, 15, 721–733 CrossRef CAS.
- A. J. Mijalis, D. A. Thomas 3rd, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, Nat. Chem. Biol., 2017, 13, 464–466 CrossRef CAS.
- C. Bolm and J. G. Hernandez, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS.
- M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318–9321 CrossRef CAS.
- V. Porte, M. Thioloy, T. Pigoux, T.-X. Métro, J. Martinez and F. Lamaty, Eur. J. Org. Chem., 2016, 2016, 3505–3508 CrossRef CAS.
- J. Bonnamour, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2013, 15, 1116–1120 RSC.
- O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T.-X. Métro, Beilstein J. Org. Chem., 2017, 13, 2087–2093 CrossRef CAS.
- J. G. Hernandez, K. J. Ardila-Fierro, D. Crawford, S. L. James and C. Bolm, Green Chem., 2017, 19, 2620–2625 RSC.
- K. J. Ardila-Fierro, D. E. Crawford, A. Körner, S. L. James, C. Bolm and J. G. Hernández, Green Chem., 2018, 20, 1262–1269 RSC.
- Y. Yeboue, B. Gallard, N. Le Moigne, M. Jean, F. Lamaty, J. Martinez and T. X. Metro, ACS Sustainable Chem. Eng., 2018, 6, 16001–16004 CrossRef CAS.
- A. Mahindra, N. Patel, N. Bagra and R. Jain, RSC Adv., 2014, 4, 3065–3069 RSC.
| This journal is © The Royal Society of Chemistry 2020 |