DOI:
10.1039/D0RA08819F
(Review Article)
RSC Adv., 2020,
10, 42441-42456
Applications of tert-butanesulfinamide in the synthesis of N-heterocycles via sulfinimines
Received
16th October 2020
, Accepted 16th November 2020
First published on 23rd November 2020
Abstract
Chiral sulfinamides are among the best known chiral auxiliaries in the stereoselective synthesis of amines and their derivatives. The most extensively used enantiopure tert-butanesulfinamide emerged as the gold standard among many others over the last two decades. The present review attempts to provide an overview of tert-butanesulfinamide mediated asymmetric N-heterocycle synthesis via sulfinimines and covers literature from 2010–2020. This methodology offers general access to structurally diverse piperidines, pyrrolidines, azetidines, and their fused derivatives that represent the structural motif of many natural products and therapeutically applicable compounds.
 Rose Mary Philip | Rose Mary Philip was born in Kottayam, Kerala, India. She obtained her dual degree of BSMS (Bachelor of Science and Master of Science) from Indian Institute of Science Education and Research, Thiruvananthapuram in 2019. She has qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Sankaran Radhika | Sankaran Radhika was born in Thodupuzha, Kerala, India, in 1994. She obtained her B. Sc. degree in 2014 and M. Sc. degree in 2016 from St. Joseph's College, Moolamattom (Mahatma Gandhi University). She secured second rank in M. Sc. Pure Chemistry Examination 2016 conducted by Mahatma Gandhi University, Kottayam. She cleared UGC-National Eligibility Test (NET) in 2017. After qualifying KSCSTE fellowship, she is currently doing her doctoral research under the guidance of Dr G. Anilkumar in School of Chemical Sciences, Mahatma Gandhi University. |
 P. V. Saranya | P. V. Saranya was born in 1997 in Kerala, India. She received her B. Sc. degree from Kannur University (Payyanur College, Payyanur) and M. Sc. degree from Cochin University of Science and Technology. She has qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in School of Chemical Sciences, Mahathma Gandhi University. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his PhD in 1996 from Regional Research Laboratory (renamed as National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 100 papers in peer-reviewed journals, 7 patents, 8 book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received Dr S. Vasudev Award from Govt. of Kerala, India for best research (2016) and Evonik research proposal competition award (second prize 2016). His h-index is 29. |
1 Introduction
Chiral auxiliaries serve as vital tools in the synthesis of enantiomerically pure compounds1 including amines and their derivatives with significant bioactivities.2 The development of chiral sulfinamides as efficient chiral auxiliaries occurred over the last two decades. Enantiopure tert-butanesulfinamide, developed by Ellman and co-workers3 represents one of the widely explored chiral sulfinamide reagents attributed to its easy access for large-scale reactions,4 assistance in highly diastereoselective conversions, effortless cleavage,5 and recyclability after reactions.6 The tert-butanesulfinamide condenses directly with a large class of aldehydes and ketones to give the respective enantiopure N-tert-butanesulfinyl aldimines and ketimines that are stable electrophilic species for organometallic additions, cycloadditions, reduction reactions, Mannich-like reactions, etc. with good diastereoselectivities.2,7 Importantly, these sulfinimines are chiral building blocks for the asymmetric synthesis of diverse N-heterocyclic systems.8
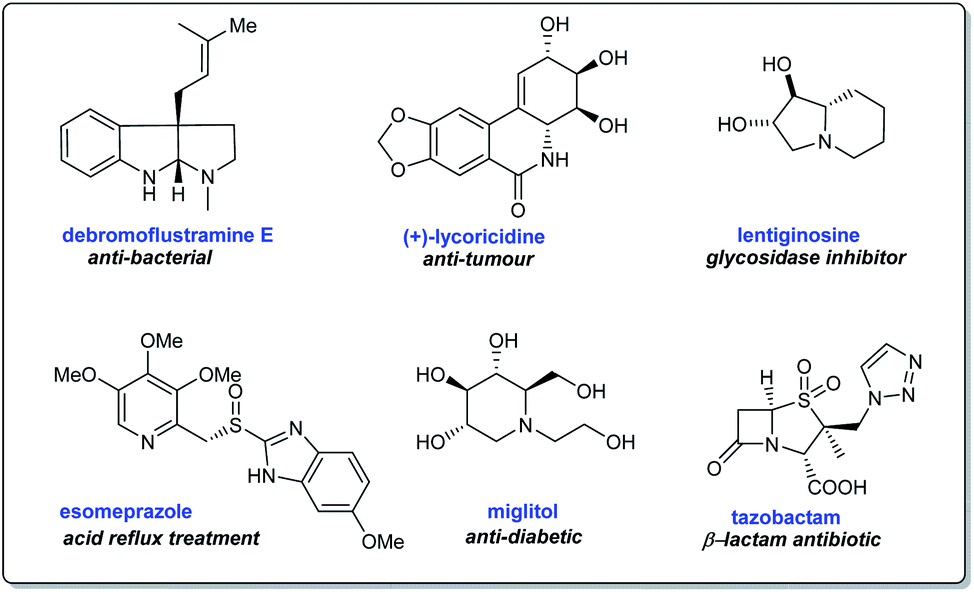
Synthetic methodologies towards N-heterocyclic compounds are of peak demand due to their application in building natural products, bioactive structures, and therapeutic compounds (Scheme 1).9,10 Nitrogen heterocycles present in nature are components of many biologically relevant structures like nucleic acids, vitamins, hormones, agrochemicals, dyes, and several others.11 Besides, a closer look at FDA databases suggests that a major portion of approved drugs with relevant pharmacological properties contain nitrogen heterocycles.11a In this context, the reliable methodology involving N-tert-butanesulfinimines emerged as a significant endeavor offering facile access to enantiopure or highly enantioenriched nitrogen heterocycles comprising of azetidines, pyrrolidines, piperidines, and their polycyclic derivatives.
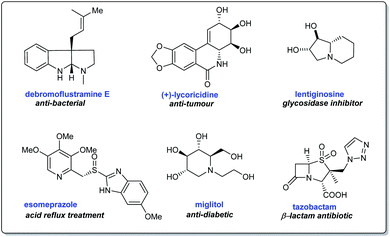 |
| | Scheme 1 Representative bioactive N-heterocycles of interest. | |
In 2010, Ellman et al. extensively reviewed the synthesis and applications of tert-butanesulfinamide.2 The review offered a detailed discussion on all reported preparation methods and reactions based on tert-butanesulfinamide enclosing up to 400 articles. Since then, we witnessed rapid development along these lines and calls for systematic summarization of novel reports in the last decade. Interestingly, a review completely devoted to stereoselective preparative methods of sulfinyl compounds has appeared very recently that included a section on sulfinamide and sulfinimine synthesis.12 Further, the application of chiral sulfinamides as organocatalysts13 and in the synthesis of α-chiral primary amines has been recently reviewed.7
In the current review, we attempt to provide an outline of tert-butanesulfinimine mediated novel methods towards N-heterocycle synthesis and their utility in the synthesis of natural alkaloids and other valuable compounds. The review covers the relevant articles from 2010 to September 2020 and is organized based on the ring size as 4-, 5-, and 6-membered heterocycles, and their derivatives.
2 Four-membered ring
2.1 Azetidine ring
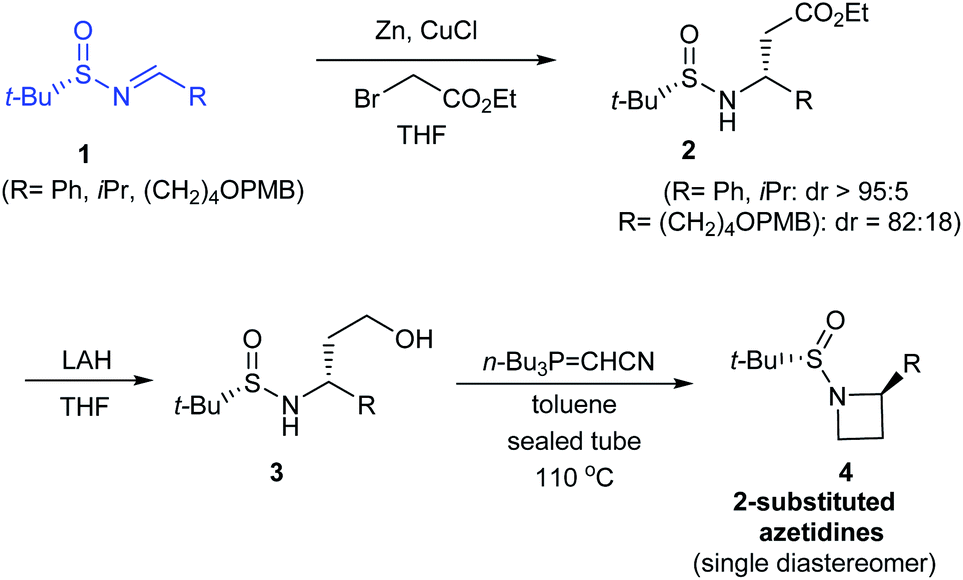
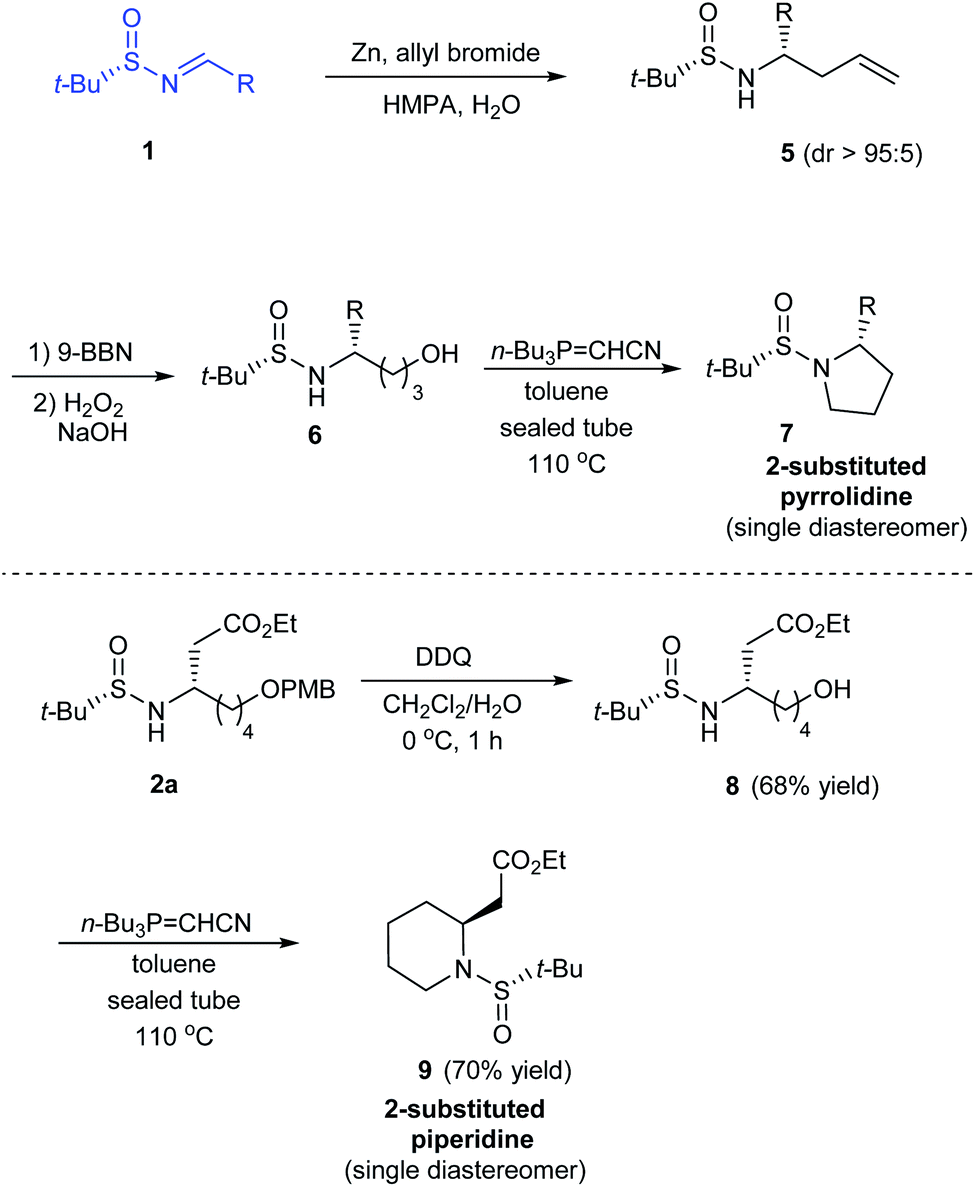
In 2014, Kudale with coworkers disclosed a simple asymmetric method for preparing chiral 2-substituted 4-, 5- and 6-membered cyclic amines by using (S)-tert-butanesulfinamide as a chiral auxiliary.14 From the sulfinimine 1, modified Reformatsky reaction afforded the esters 2 in good diastereoselectivities which in turn underwent LAH reduction followed by cyclization to accomplish 2-substituted azetidines 4 in 66–77% yield (Scheme 2). Similarly, the common precursor 1 underwent a sequence of diastereoselective allylation (dr > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), hydroboration-oxidation of the resultant alkene, and the cyclization to furnish 2-substituted pyrrolidines 7. Lastly, 2-substituted piperidine 9 was synthesised from the intermediate ester 2a (Scheme 3).
:5), hydroboration-oxidation of the resultant alkene, and the cyclization to furnish 2-substituted pyrrolidines 7. Lastly, 2-substituted piperidine 9 was synthesised from the intermediate ester 2a (Scheme 3).
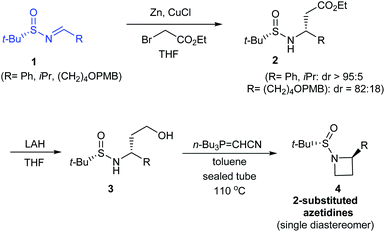 |
| | Scheme 2 Asymmetric synthesis of 2-substituted azetidines. | |
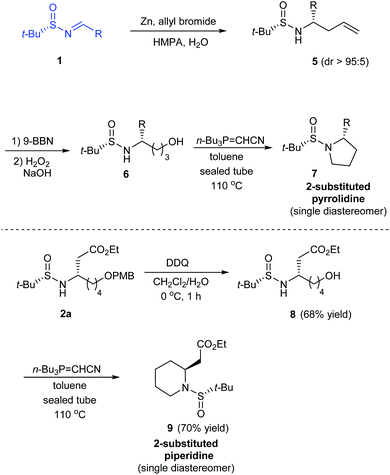 |
| | Scheme 3 Asymmetric synthesis of 2-substituted pyrrolidine and piperidine. | |
In all the above cases, the cyclization was achieved in presence of Tsunoda reagent – cyanomethylenetributylphosphorane, which conducted facile ring closure through C–N bond formation. Notably, the protocol offers general applicability in synthesising 2-substituted cyclic amines in a stereoselective manner.
3 Five-membered ring
3.1 Pyrrolidine ring
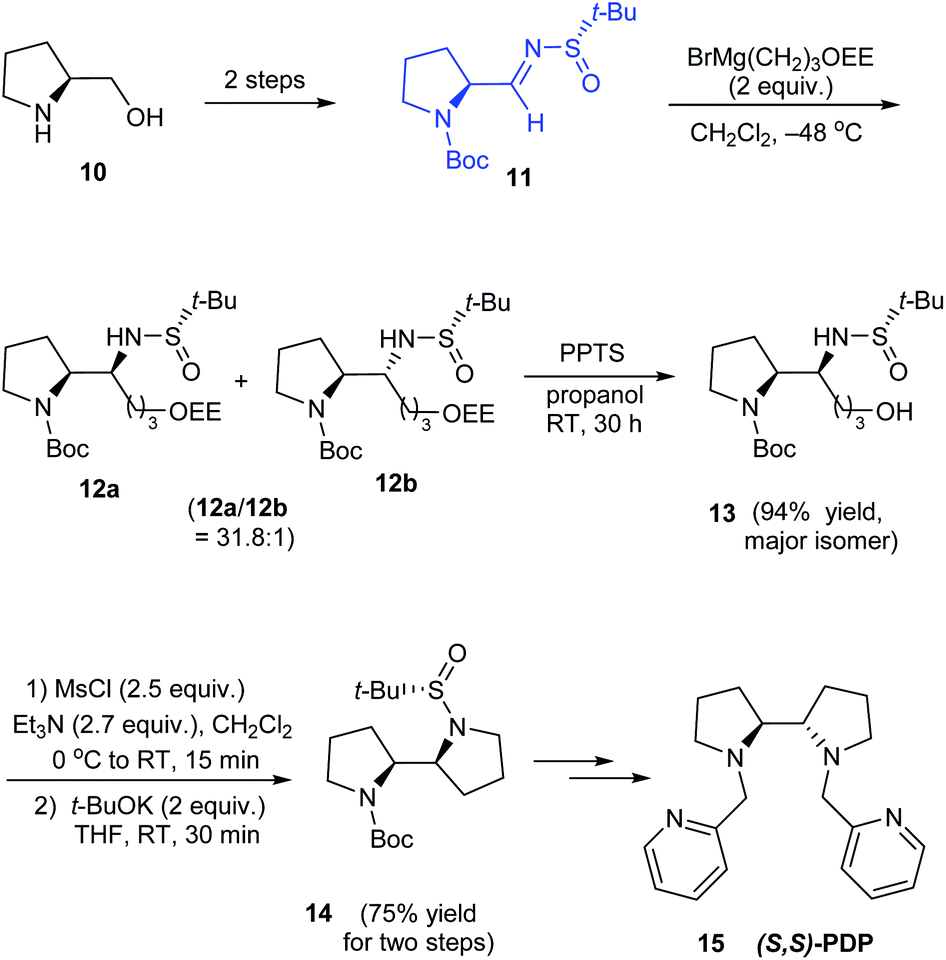
In 2010, Yao and coworkers established a direct route to the stereoselective synthesis of (S,S)-PDP 15, a chiral amine ligand over seven steps starting from L-prolinol 10.15 During the reaction sulfinimine 11 derived from (S)-tert-butanesulfinamide underwent Grignard addition with excellent diastereoselection (up to 94% de). Then, the diastereomeric mixture on reaction with PPTS in propanol yielded the major isomer which subsequently carried out base mediated intramolecular substitution to furnish the bipyrrolidine scaffold in 14 (Scheme 4). The authors envisaged the utility of the same method in synthesising (R,R)-PDP starting from D-proline derivative and employing (R)-tert-butanesulfinamide as the chiral source.
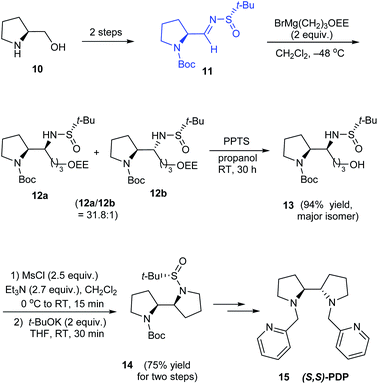 |
| | Scheme 4 Asymmetric synthesis of (S,S)-PDP. | |
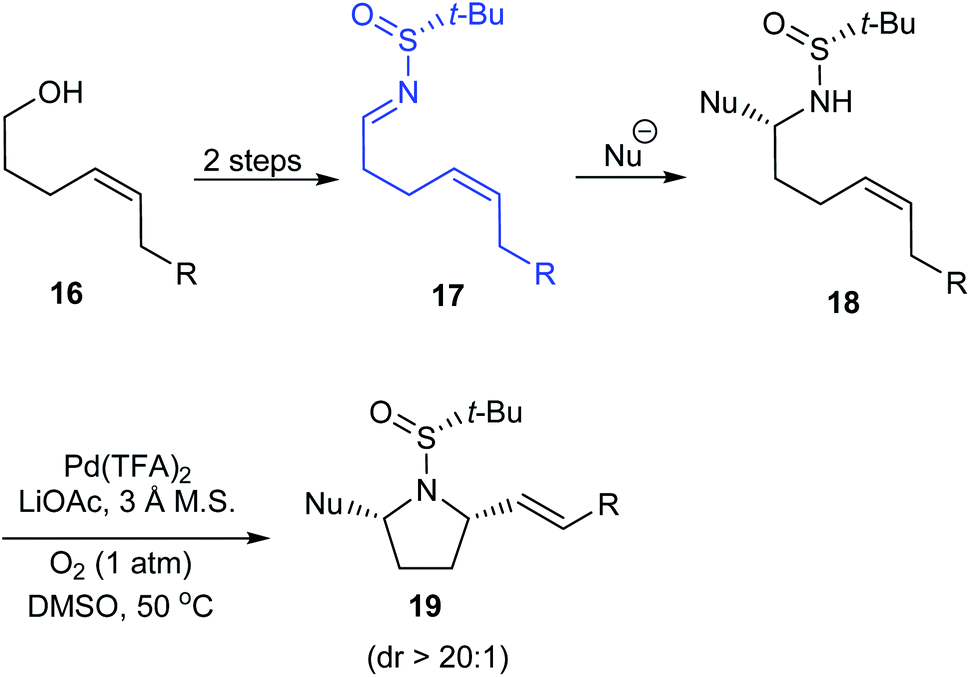
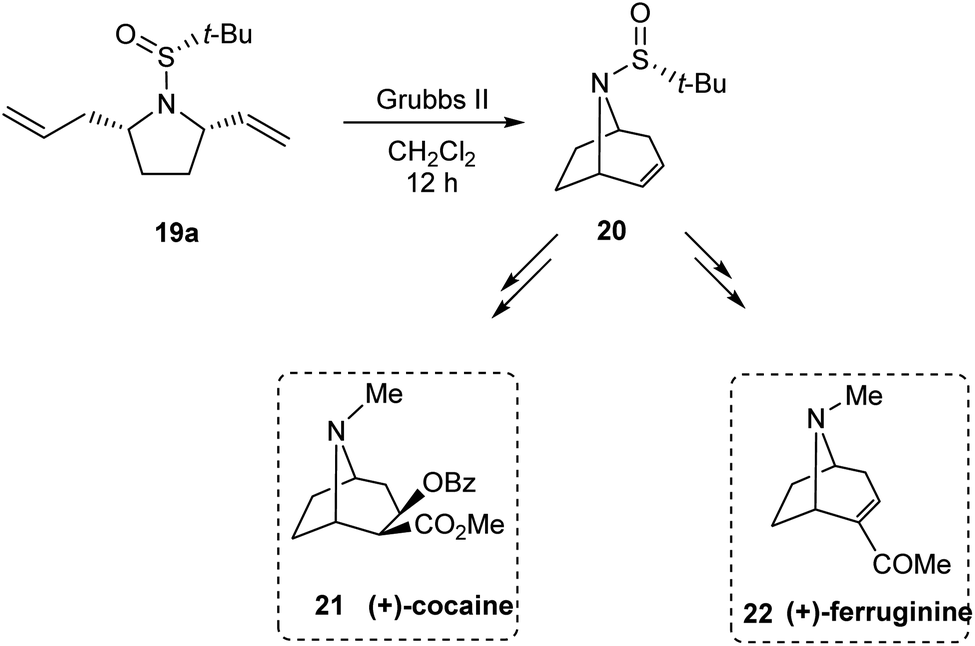
Stahl et al. developed an elegant method to access cis-2,5-disubstituted pyrrolidines in enantiopure form, through a Pd-catalysed oxidative cyclization as the key process.16 Initially, sulfinimine 17 was prepared from cis-4-hexen-1-ol 16 over 2 steps (Scheme 5). Then, sequential diastereoselective nucleophilic addition provided α-substituted sulfinamides 18 which in turn underwent the aerobic oxidative cyclization in presence of Pd(TFA)2 and LiOAc in DMSO to afford the desired pyrrolidines 19. Of note, higher diastereoselectivity (dr > 20:1) in the final step is attributed to the cooperative action of sulphur- and carbon-based stereocenters present in compound 18. Moreover, the applicability of the prepared pyrrolidine 19a in preparing azabicyclic alkaloids 21 and 22 is also discussed (Scheme 6).
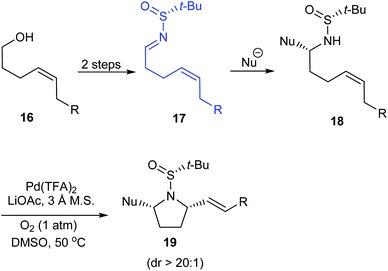 |
| | Scheme 5 Synthesis of cis-2,5-disubstituted pyrrolidines. | |
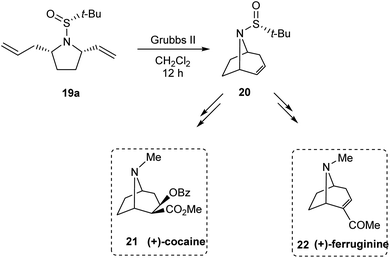 |
| | Scheme 6 Application of cis-2,5-disubstituted pyrrolidine in constructing tropane alkaloids. | |
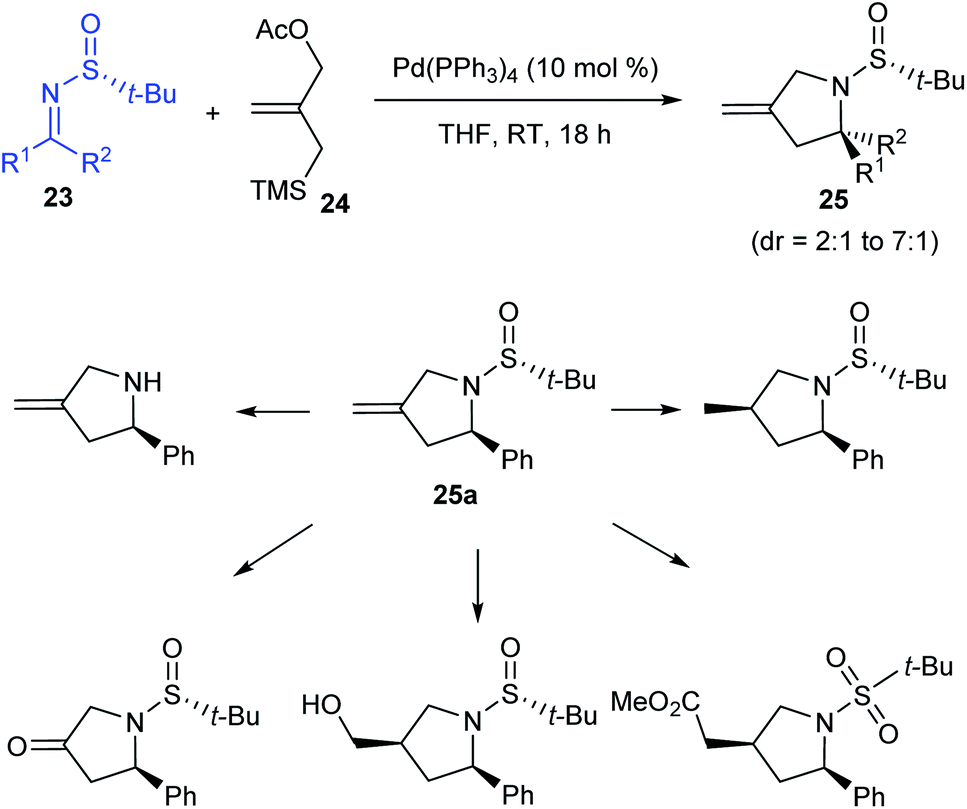
In the following year, the research group of Stockman pioneered in implementing a general protocol for Pd-catalysed [3 + 2] cycloaddition of sulfinimines 23 and trimethylenemethane precursor 24 gaining methylene-pyrrolidines 25 in good yields (up to 100%) and diastereoselectivities (dr = 2:1 to 7:1).17 Working with different sulfinimines, they observed comparatively higher yields and selectivities when aldimines instead of ketimines were used in the reaction. Moreover, they presented the potential functionalization of the methylene-pyrrolidine 25a accomplished through diverse transformations (Scheme 7).
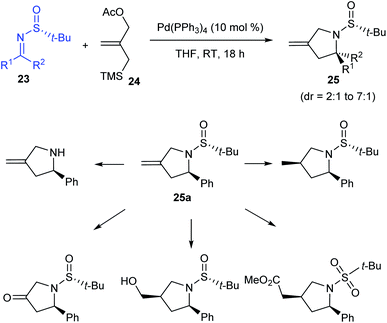 |
| | Scheme 7 Synthesis of methylene-pyrrolidines from sulfinimines. | |
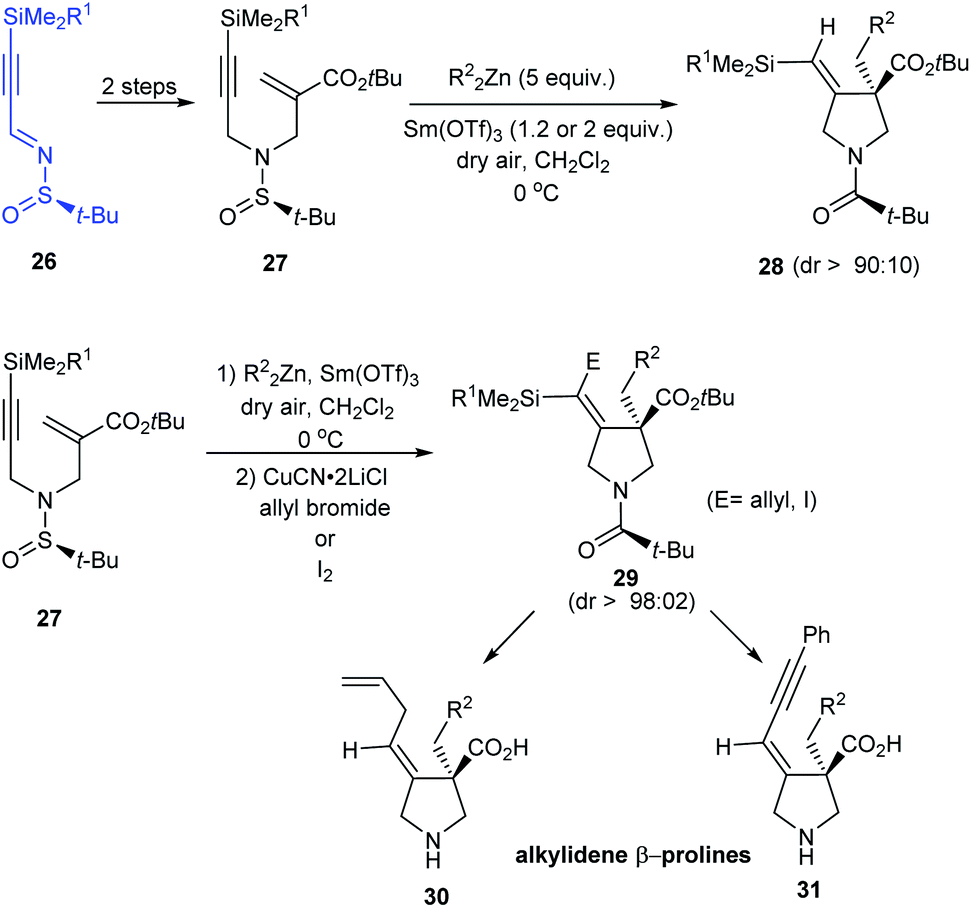
In 2015, enantiomerically pure (E)-alkylidene-β-prolines have been accomplished in good yields from N-tert-butanesulfinimine 26 in three to four steps.18 In the key step, N-tert-butylsulfinyl-α-(aminomethyl)acrylate 27 reacted with the dialkylzinc compounds in a radical zinc transfer based reaction involving 1,4-addition followed by a sequential 5-exo-dig cyclization in presence of Sm(III) triflate (Scheme 8). Herein, high diastereoselection (dr > 90:10) obtained upon cyclization validates N-tert-butylsulfinyl species as an ideal chiral directing group.
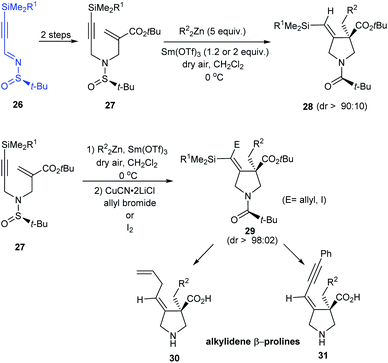 |
| | Scheme 8 Synthesis of enantiopure alkylidene-β-prolines. | |
In addition, functionalized vinyl silanes 29 were generated in situ by an extra step of electrophilic addition to the vinyl zinc intermediates. These iodo- or allyl-vinylsilanes are amenable to further transformations to deliver alkylidene-β-prolines like 30 and 31.
3.2 Benzofused pyrrolidine ring
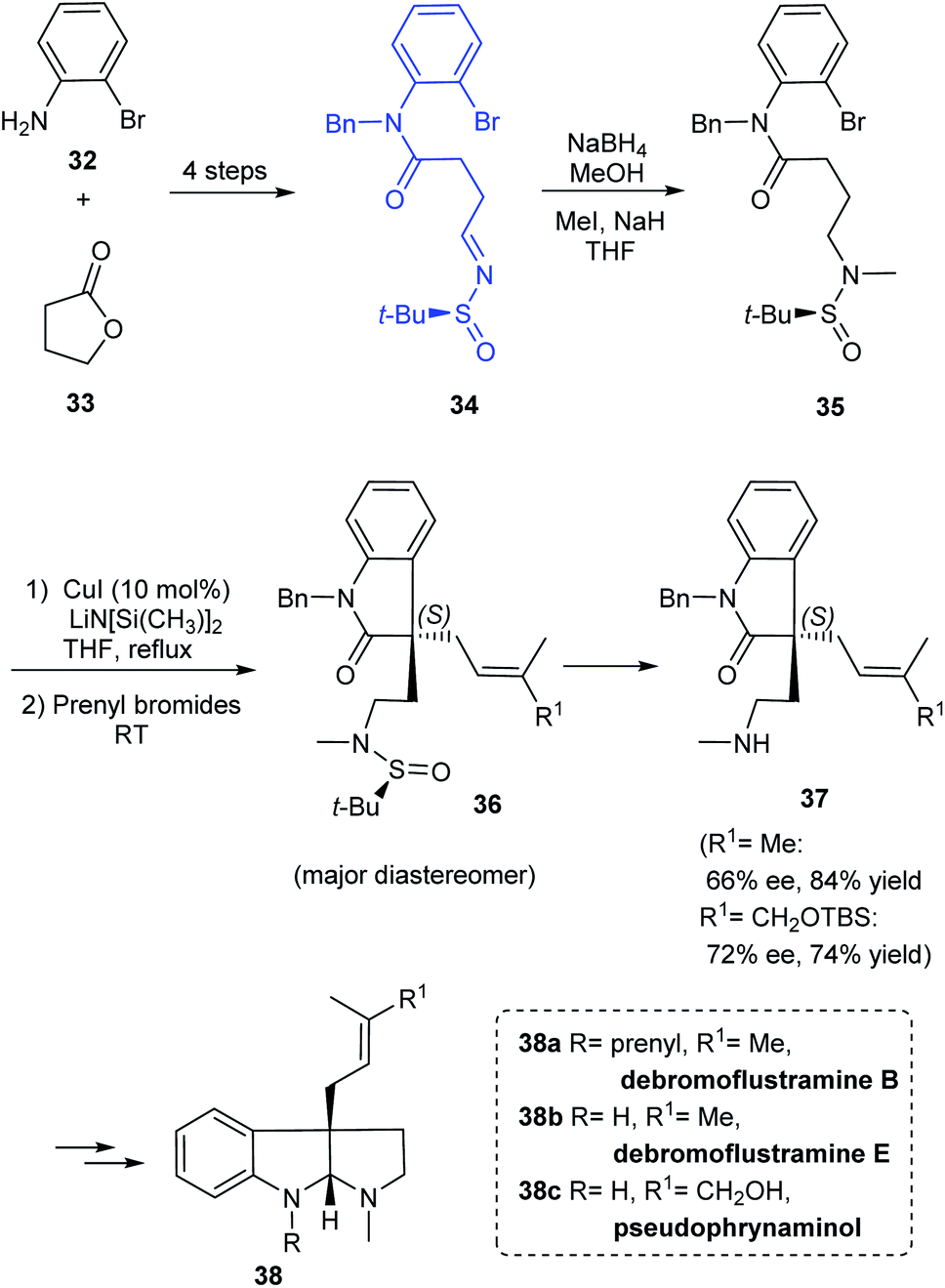
A simple and flexible synthesis of the potent drug candidates, hexahydropyrroloindole alkaloids was established by Zhang et al. in 2012.19 The synthesis commenced with the preparation of sulfinimine 34 which upon reduction and N-methylation provided 35, an o-bromoanilide possessing the chiral tert-butanesulfinamide entity (Scheme 9). The key step in the synthetic strategy involved Cu-catalysed successive arylation–alkylation to give the cyclized product 36 as a non-separable mixture of diastereoisomers. The cleavage of tert-butanesulfinamide group from 36 provided with the enantioenriched amines 37. Furthermore, they demonstrated the utility of the method in preparing alkaloid compounds of interest (−)-debromoflustramine B 38a, (−)-debromoflustramine E 38b, and pseudophrynamine 38c after a sequence of reactions from 37.
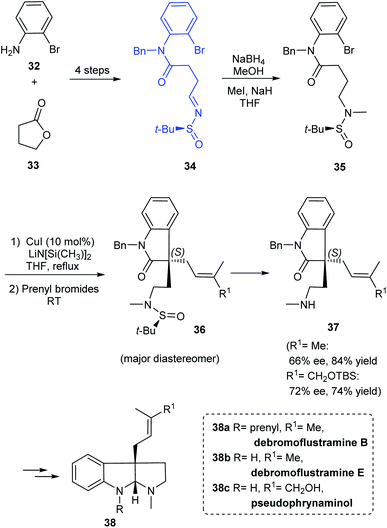 |
| | Scheme 9 General synthetic protocol for hexahydropyrroloindole alkaloids. | |
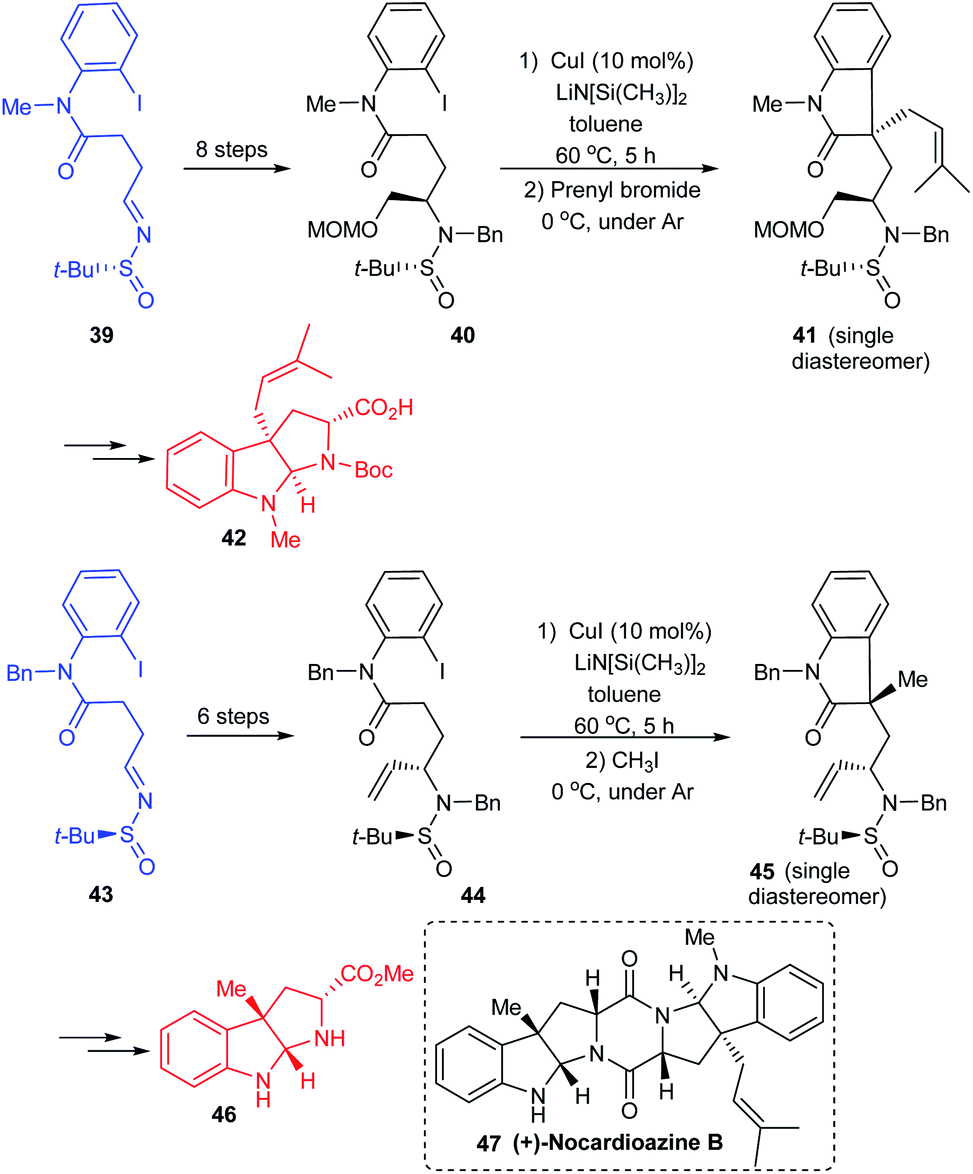
In continuation of their interest in preparing pyrroloindole alkaloids, they successfully achieved the total synthesis of (+)-nocardioazine B utilizing the aforementioned strategy and the final coupling of key fragments 42 and 46.20 Both the fragments were constructed through sulfinimine intermediates 39 and 43 which went through a series of transformations. The compounds 40 and 44 were prepared based on their earlier methods in which the diastereoselective addition of vinyl magnesium bromide to the sulfinimines was assisted by stereo directing tert-butanesulfinyl group. Further, the Cu-catalysed sequential arylation–alkylation represented the key step to render 41 and 45 mostly as a single diastereoisomer bearing quaternary stereocenter at C3a position (Scheme 10). In the final stages of the synthesis, the key intermediates 42 and 46 coupled to give the final alkaloid 47.
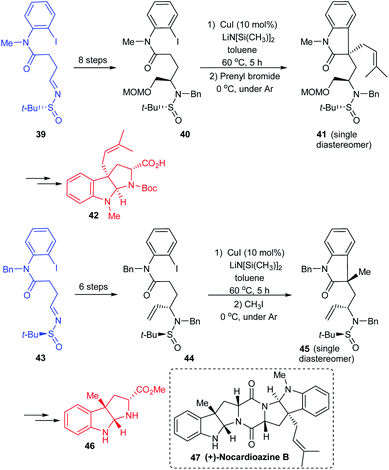 |
| | Scheme 10 Synthetic route towards (+)-nocardioazine B. | |
3.3 γ-Lactam ring
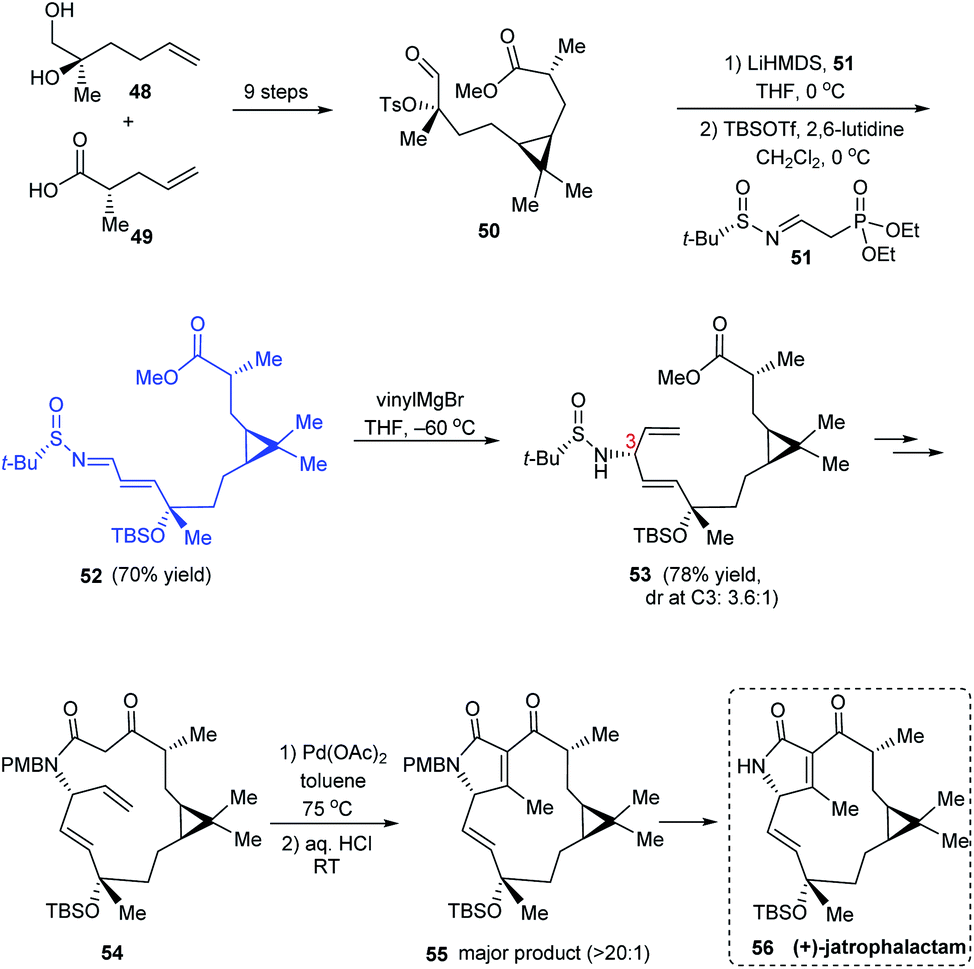
(+)-Jatrophalactam 56 is a diterpenoid lactam that features a unique tricyclic structure comprising of 3, 5 and 10 membered cyclic units. A scalable method for synthesising 56 along with its XRD analysis was reported.21 The revised synthetic route discussed here commences with a set of reactions including the cyclopropanation to construct the ester 50. Thereafter, a Horner's Wittig reaction was performed with 50 using LiHMDS and a novel Horner's reagent 51 prepared from (S)-tert-butanesulfinamide and diethyl (2-oxoethyl)phosphonate, providing the sulfinimine 52 which sequentially undertook vinylation to afford 53 along with its C3 epimer (dr = 3.6:1) (Scheme 11). A sequence of reactions followed wherein the 14 membered lactam formation and the PdII-assisted oxidative cyclization of 54 formed the key conversions to furnish (+)-jatrophalactam 56.
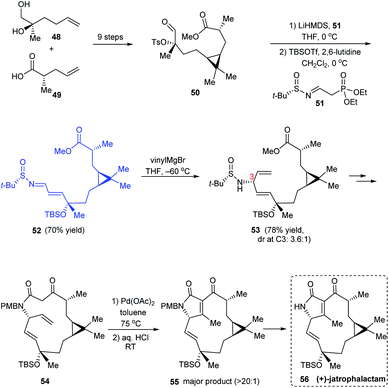 |
| | Scheme 11 Synthesis of (+)-jatrophalactam 56. | |
3.4 Pyrazole ring
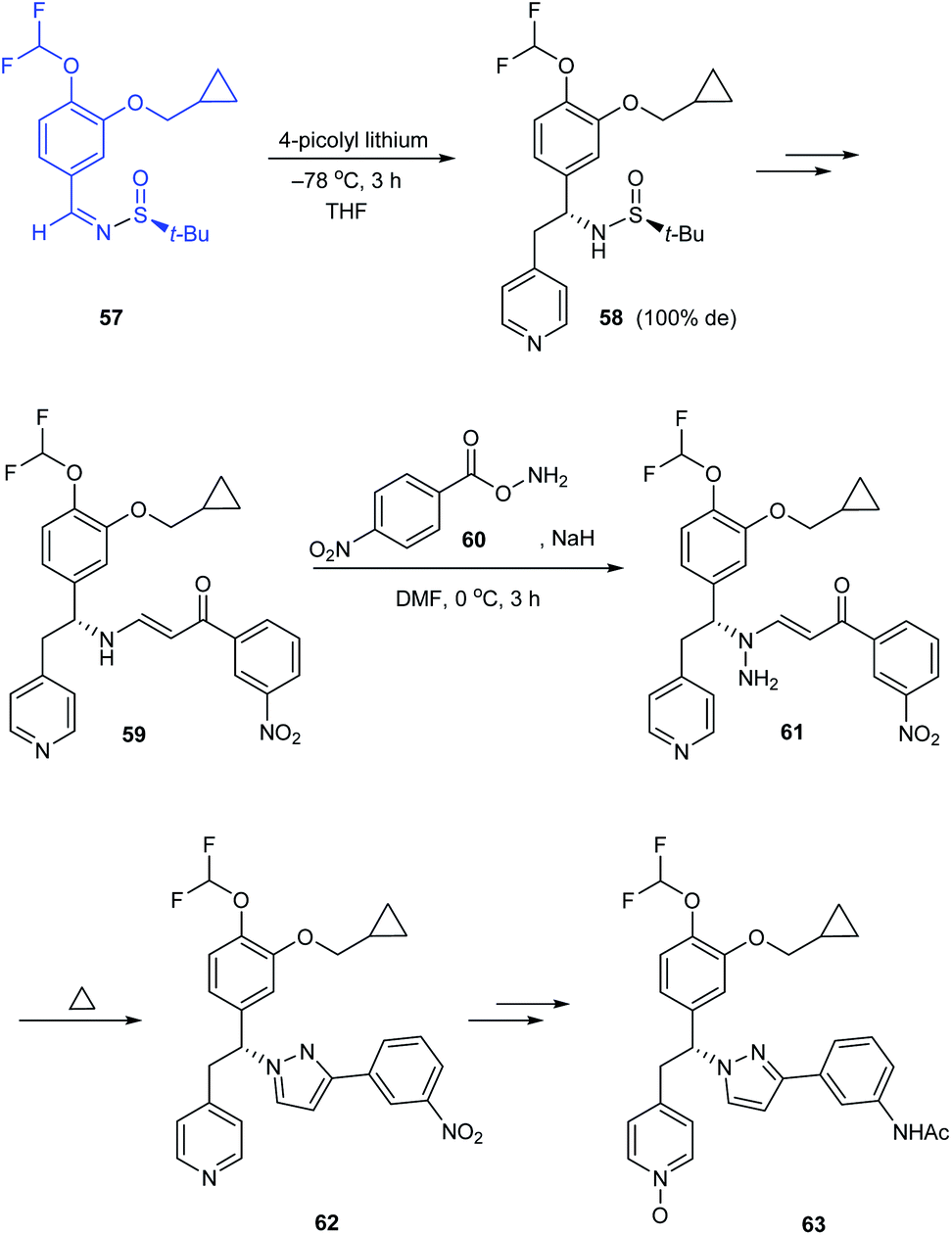
The research group of Jeon disclosed a new pyrazole derivative as an effective PDE4 inhibitor for treating anti-inflammatory diseases.22 In the synthetic route to enantiopure derivatives, the preference to tert-butanesulfinamide over p-toluenesulfinamide has been illustrated based on the resultant yield and diastereoselectivity. While synthesising the (R)-pyrazole derivative 63, sulfinimine 57 derived from (S)-tert-butanesulfinamide was subjected to nucleophilic addition of 4-picolyl lithium to give the addition product 58 in 100% de. Eventually, the pyrazole ring was constructed as the result of one-pot amination and thermal dehydrocyclization of 59 (Scheme 12). Similarly, they prepared (S)-pyrazole derivative starting from (R)-tert-butanesulfinamide.
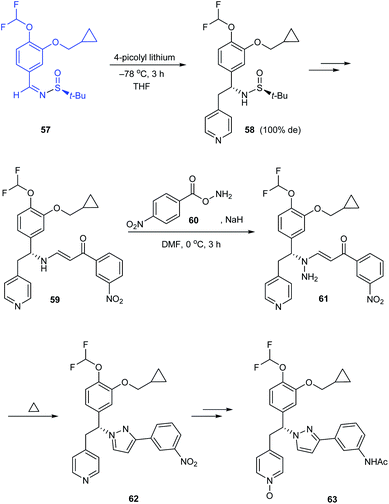 |
| | Scheme 12 Synthesis of (R)-pyrazole derivative through sulfinimine 57. | |
3.5 Isothiazole ring
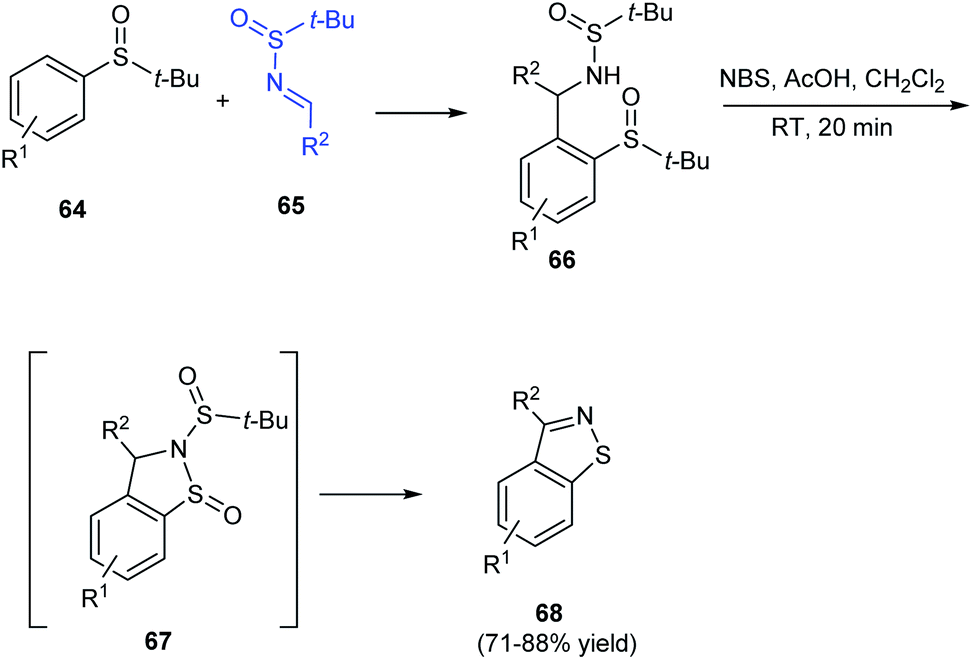
Isothiazoles are recognized as an important group of heterocycles with valuable medicinal applications. In 2016, aryl[4,5]isothiazoles were prepared from tert-butanesulfinamide and tert-butyl sulfoxide as the respective sources of sulfur and nitrogen.23 The synthesis commenced with ortho metalation of aryl sulfoxides 64 and its addition to N-tert-butanesulfinimines 65 to produce the ortho-substituted aryl sulfoxides 66 (Scheme 13). Then, 66 was treated with NBS and acetic acid in DCM to obtain the aryl thiazole 68 through a non-isolated cyclized intermediate 67. Thus using the protocol, differently substituted aryl-thiazoles were synthesised in good yields.
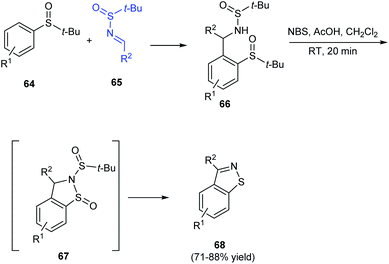 |
| | Scheme 13 Protocol for aryl[4,5]isothiazoles synthesis. | |
4 Six-membered ring
4.1 Piperidine ring
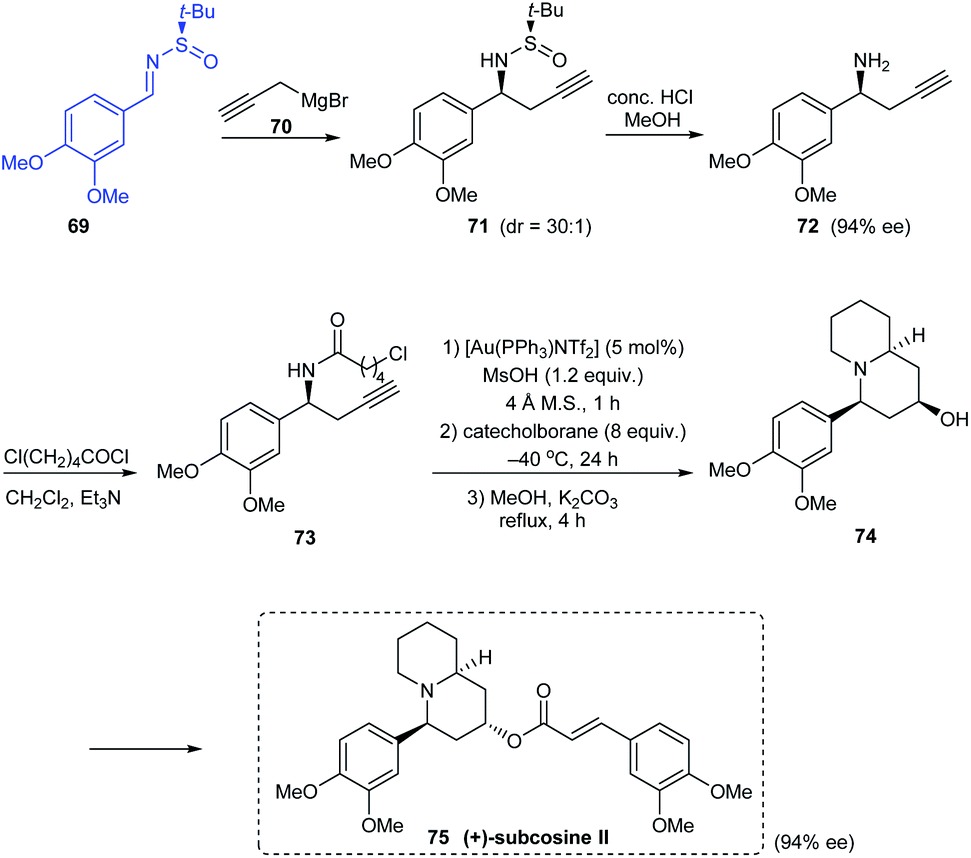
In 2010, an effective asymmetric synthetic route to substituted piperidin-4-ols has been established along with an illustration of its utility in the synthesis of enantiopure (+)-subcosine II 75.24 They introduced a one-pot consecutive reaction of Au-catalysed cyclization, chemoselective reduction, and Ferrier rearrangement to furnish substituted piperidin-4-ols with remarkable diastereoselectivities. The synthesis of (+)-subcosine II began from sulfinimine 69 which smoothly provided the chiral amine 72 in 94% ee. This free amine is readily functionalized for the generation of desired quinolizidine unit in 74 through the proposed one-pot cyclization coupled with an intramolecular alkylation (Scheme 14).
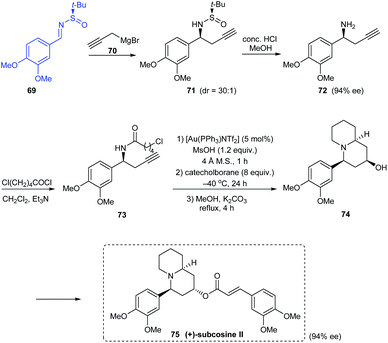 |
| | Scheme 14 Synthesis of enantiopure (+)-subcosine II. | |
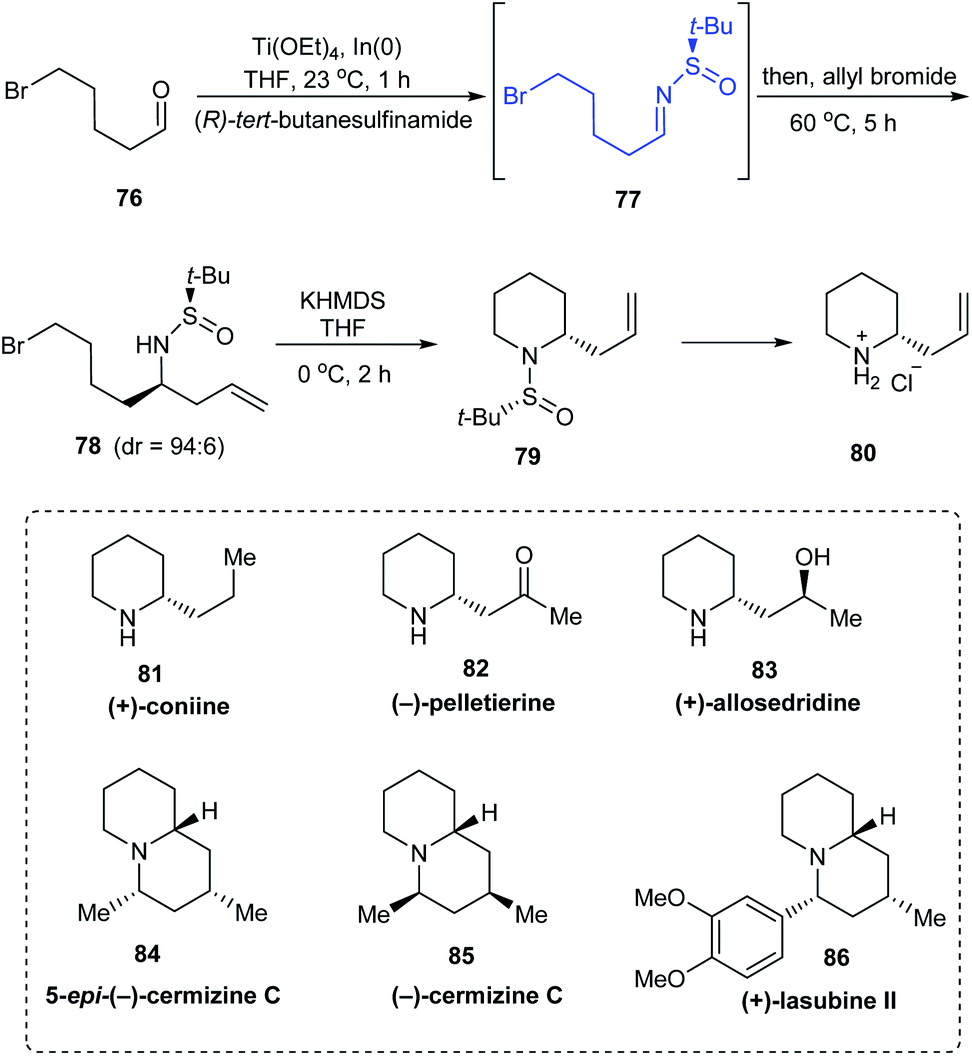
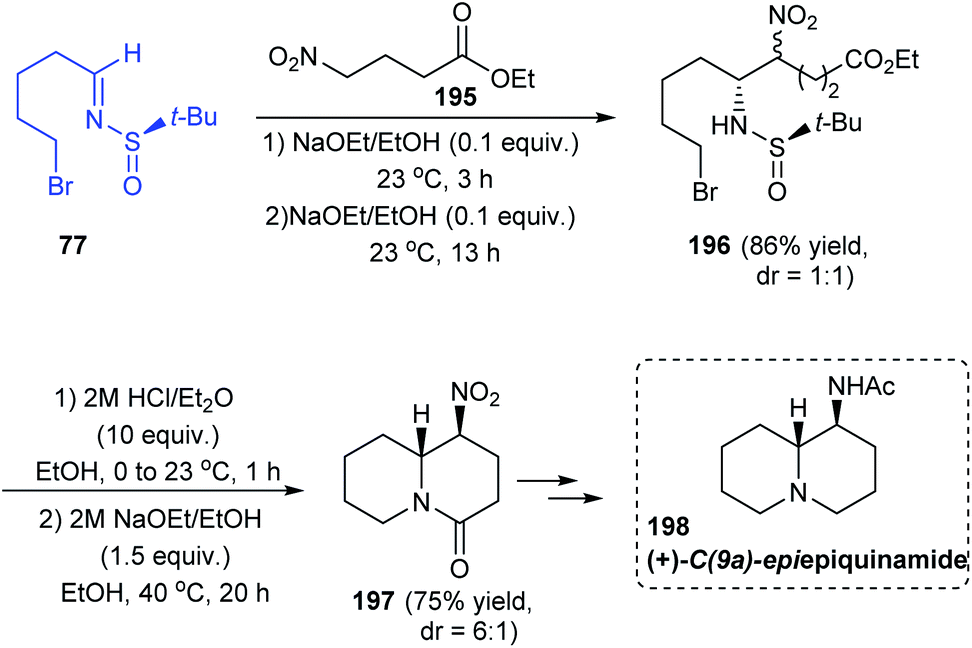
In 2011, a direct and scalable synthetic route to 2-allylpiperidine 79 in enantioenriched form has been developed.25 The route comprises of only two steps from 5-bromo-pentanal 76. Initially, one-pot aminoallylation delivered compound 78 in high diastereoselectivity (dr = 94:6) via the sulfinimine intermediate 77 derived from (R)-tert-butanesulfinamide. Then, base mediated cyclization of the crude compound 78 provided with the desired 2-allylpiperidine 79. A similar method was used in preparing the enantiomer of 79 using (S)-tert-butanesulfinamide as the chiral auxiliary. Compound 80 obtained after the removal of the sulfinyl group formed the fundamental block for providing numerous alkaloids (Scheme 15). The authors discussed the complete synthesis of alkaloids (−)-pelletierine, (+)-coniine, and 5-epi-(+)-cermizine C and also the formal synthetic route to (+)-allosedridine, (−)-cermizine C, and (+)-lasubine II.
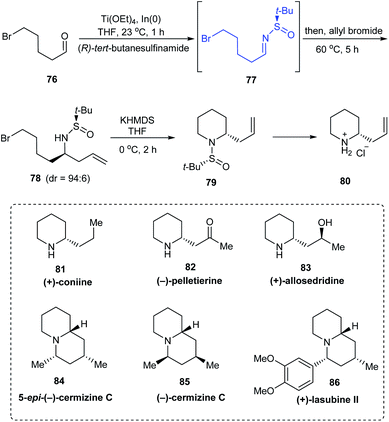 |
| | Scheme 15 Synthesis and applications of 2-allyl piperidines. | |
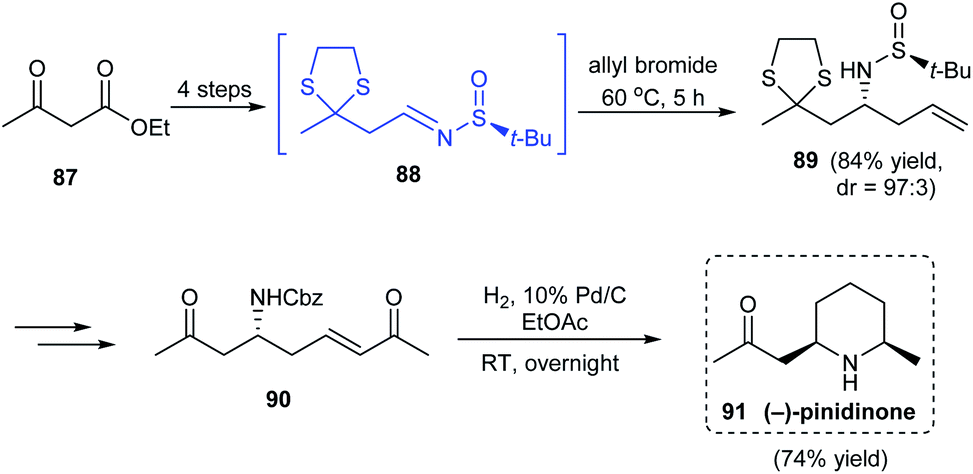
A simple synthetic route to the 2,6-disubstituted derivative of piperidine, (−)-pinidinone 91 was described by the research group of Jun in 2016.26 Treatment of ethyl acetoacetate to successive ketone group protection and ester reduction provided the prerequisite aldehyde which generated sulfinimine intermediate 88 on condensation with (S)-tert-butanesulfinamide. Then, α-aminoallylation of sulfinimine 88 led to the homoallylic amine 89 in good yields and excellent diastereoselectivity (dr = 97:3) (Scheme 16). In the late stage of synthesis, the cross metathesis product 90 conducted hydrogenolysis to deliver the desired cyclized product (−)-pinidinone 91 in good yields.
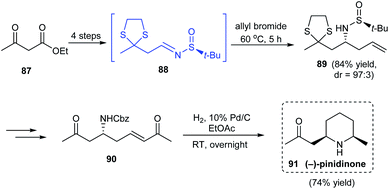 |
| | Scheme 16 Stereoselective synthetic protocol towards enantioenriched (−)-pinidinone. | |
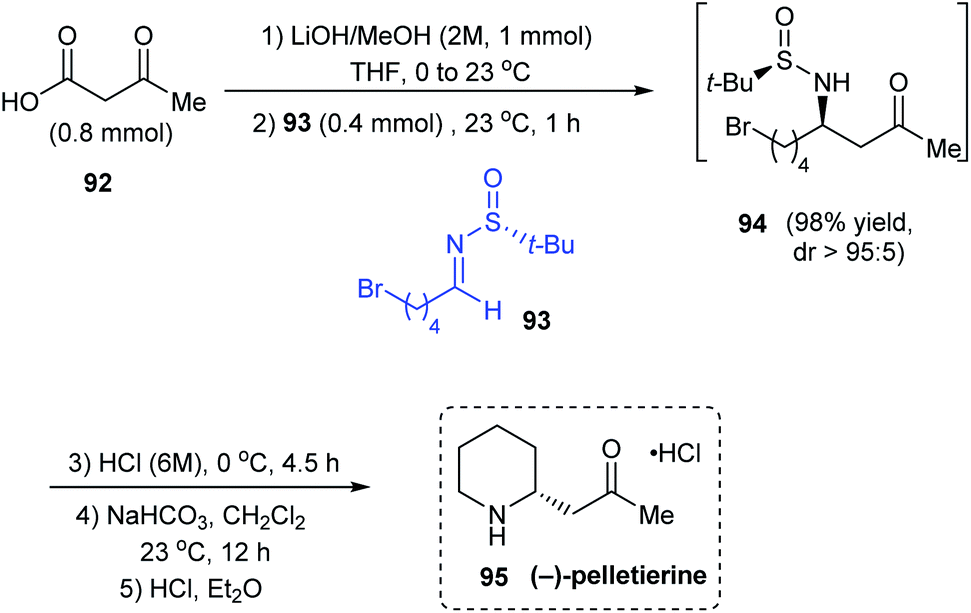
In 2018, the utility of N-tert-butanesulfinyl aldimines as chiral imines in decarboxylative Mannich reactions was investigated to synthesise β-amino ketones with good diastereocontrol and yields.27 Importantly, they proposed an 8-membered cyclic transition state to explain the observed stereoselection. Further, the methodology was applied in synthesising the piperidine based alkaloid (−)-pelletierine. Herein, the first step is the optimized decarboxylative Mannich reaction with the aldimine 93 prepared from 4-bromopentanal, and the β-keto acid 92 under basic conditions in THF to produce β-amino ketone 94 with high diastereoselectivity (dr > 95:5). Without isolating 94, subsequent acid and base treatments accomplished (−)-pelletierine 95 in a column-free method (Scheme 17).
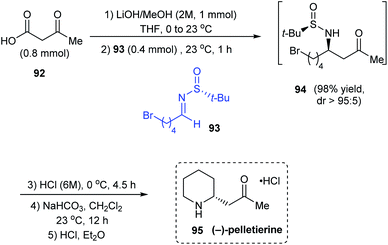 |
| | Scheme 17 Synthesis of (−)-pelletierine commencing from sulfinimine 93. | |
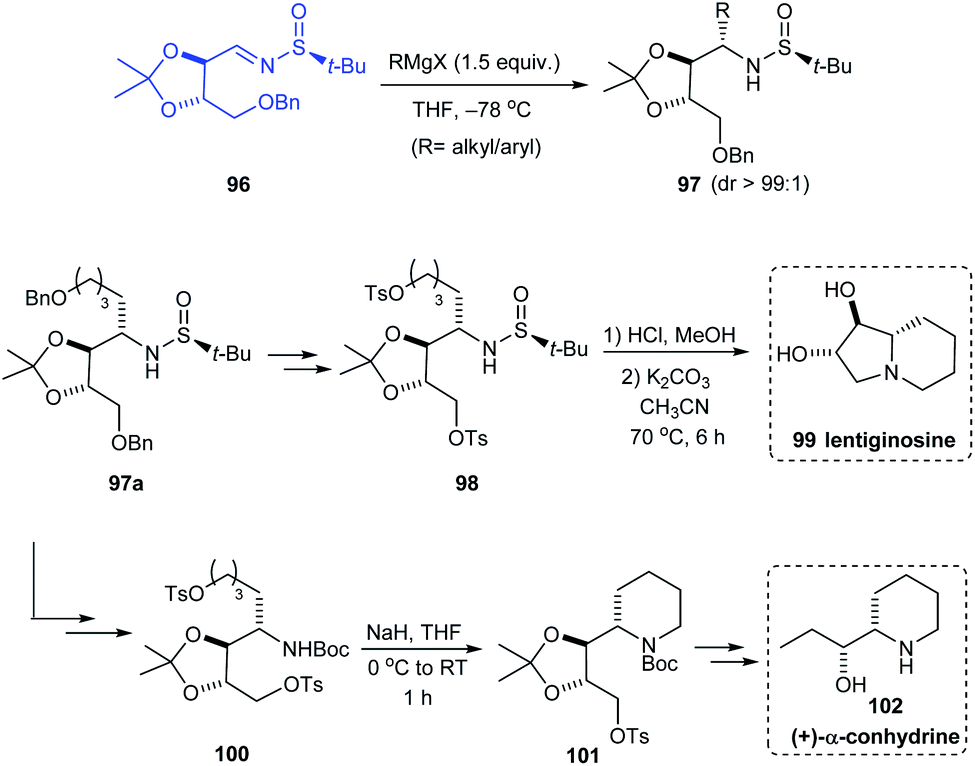
In 2019, Prasad et al. investigated the addition of Grignard reagents (RMgX) to sulfinimine 96 obtained from threitol (tartaric acid diol).28 Alkyl and aryl Grignard reagents upon addition to 96 gave the addition product 97 in good yields and excellent diastereoselectivity (dr > 99:1). Specifically, the addition of benzyloxybutylmagnesium bromide gave the addition product 97a which was then utilized as the precursor for the synthesis of natural products that are glycosidase inhibitors, lentiginosine 99 and (+)-α-conhydrine 102. The bicyclic compound 99 was accomplished via a four-step sequence whereas the synthetic strategy towards 102 comprises of a series of transformations starting from 97a (Scheme 18).
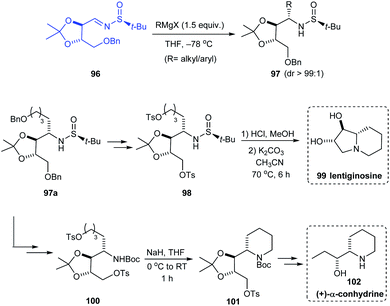 |
| | Scheme 18 Synthesis of lentiginosine and (+)-α-conhydrine by Prasad et al. | |
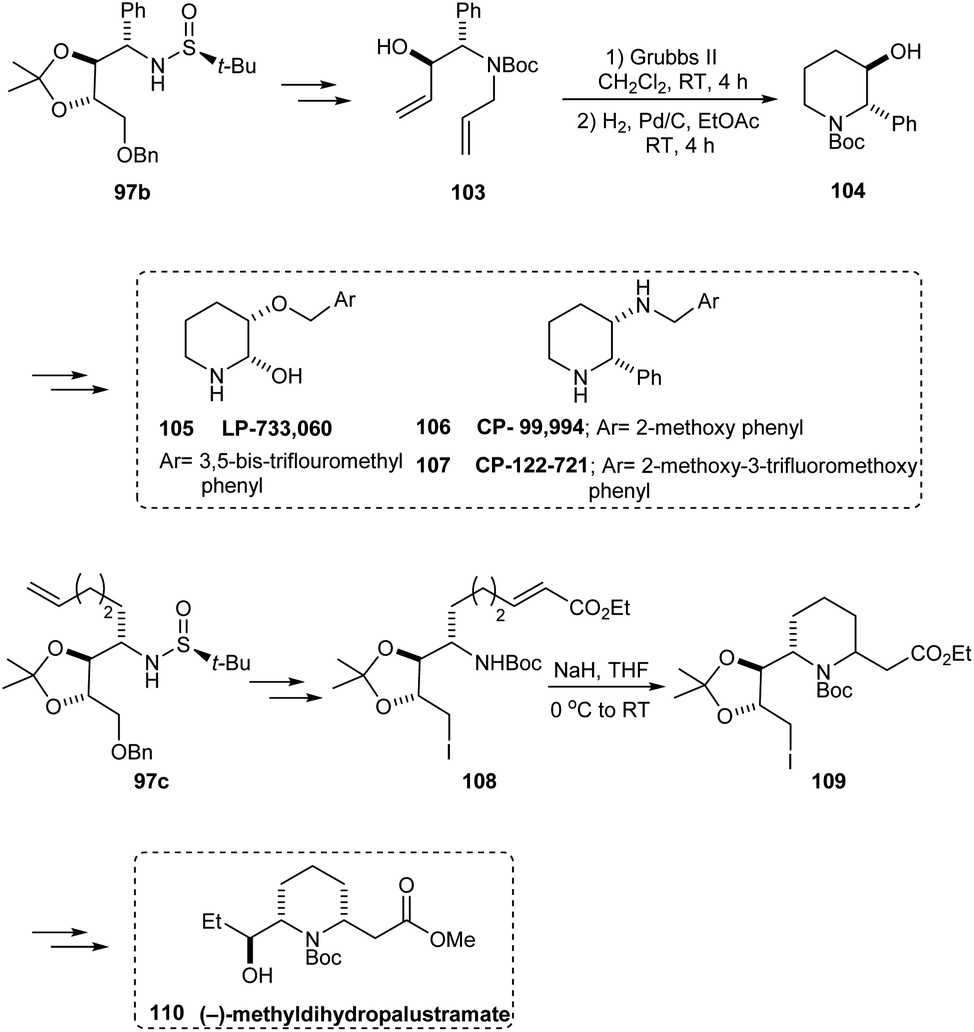
Likewise, the sulfinamide 97b has been applied in synthesising 104, the key intermediate in preparing piperidine derivatives 105–107 that are therapeutically significant. Furthermore, the synthesis of (−)-methyldihydropalustramate 110 was efficiently performed from the sulfinamide 97c (Scheme 19).
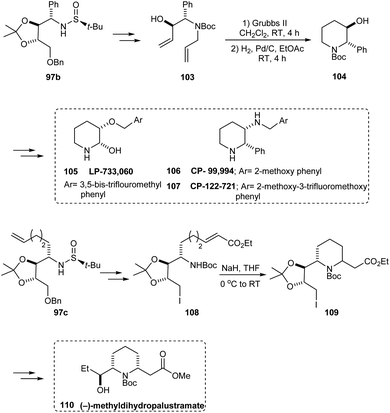 |
| | Scheme 19 Synthetic route to therapeutically relevant piperidine derivatives. | |
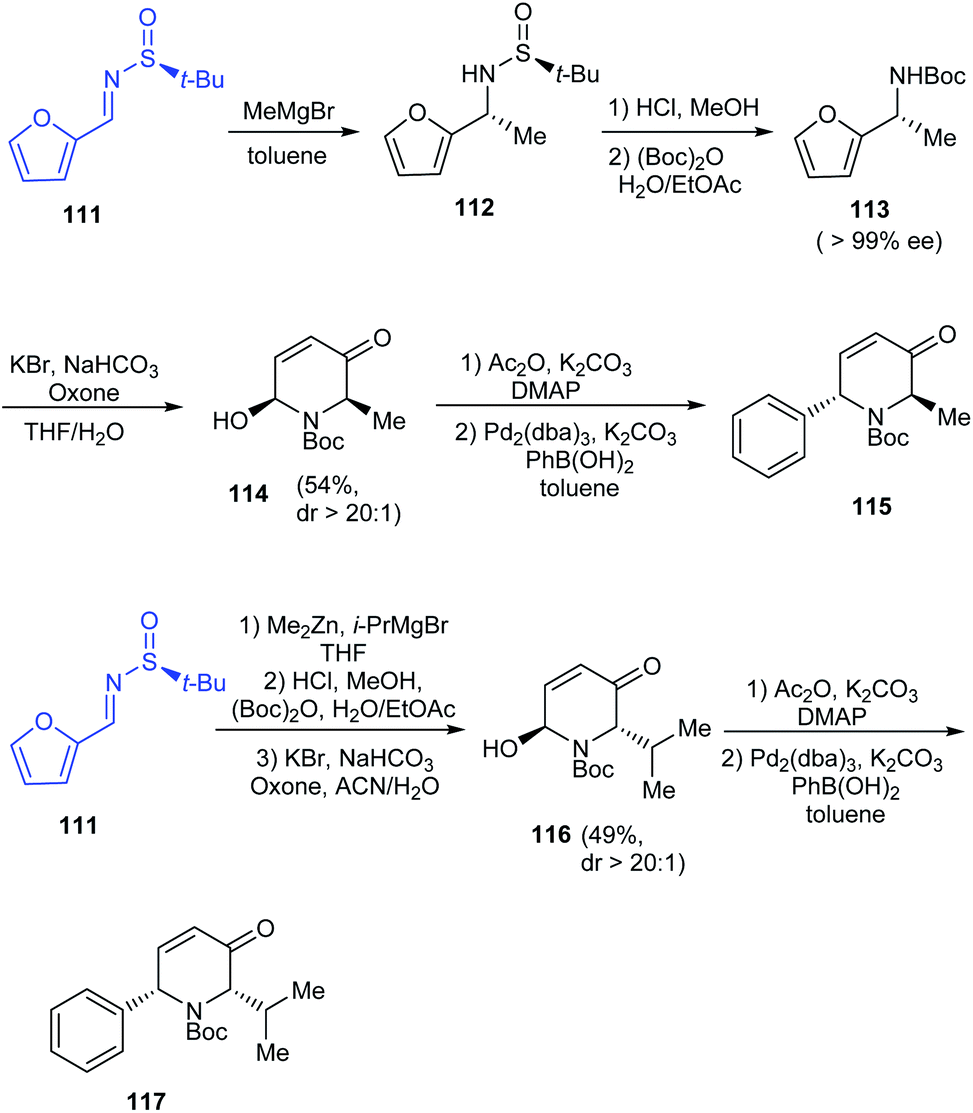
2-Arylpiperidines have shown applications in medicinal chemistry forming vital building blocks of bioactive compounds. Tong et al. introduced a synthetic route to 2-arylpiperidines presenting a sequential aza-Achmatowicz rearrangement and Pd-catalysed arylation using arylboronic acids.29 In their efforts towards enantioselective Pd-catalysed arylation, they utilized (S)-tert-butanesulfinamide to furnish enantioenriched furfuryl carbamates like 113 (>99% ee) that carried out aza-Achmatowicz rearrangement to afford C6-substituted chiral dihydropyridinone 114 and subsequently undertook arylation with high diastereoselectivity (dr > 20:1) even when the yields were lower (Scheme 20). Remarkably, the method can offer access to significant 2-aryl-6-alkyl piperidine derivatives from the dihydropyridinones 115 and 117.
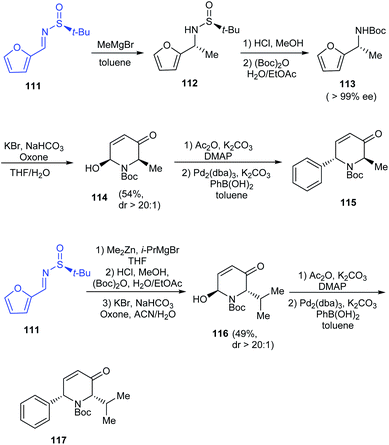 |
| | Scheme 20 The synthetic strategy towards 2-aryl-6-alkyl piperidine derivatives. | |
4.2 Benzofused piperidine ring
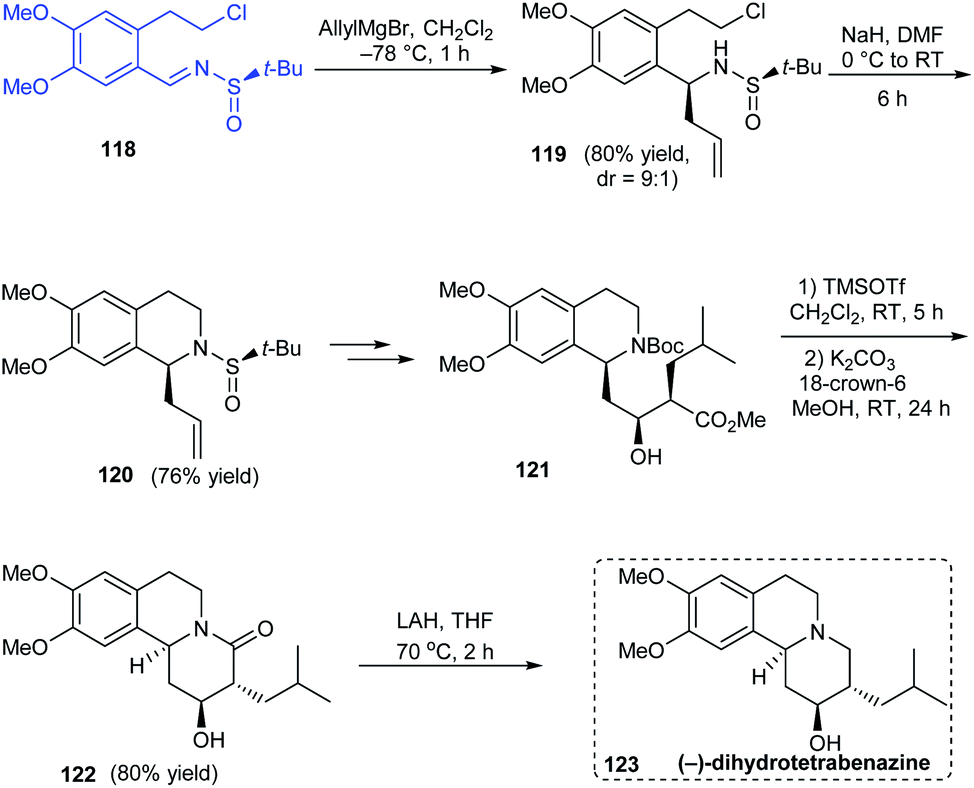
A simple and effective method towards the total synthesis of biologically active alkaloid (−)-dihydrotetrabenazine was disclosed by Reddy's group in 2012 employing (R)-tert-butanesulfinamide as the source of chirality.30 The enantiopure N-tert-butanesulfinimine 118 was exposed to diastereoselective allylation (dr = 9:1) where the major isomer could be isolated and converted to the cyclized sulfinamide 120 via base catalysed ring closure (Scheme 21). Further conversions from 120 including an Evans-aldol reaction rendered the anticipated compound 123 over nine successive steps.
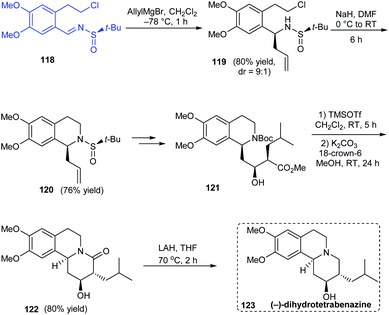 |
| | Scheme 21 Enantioselective total synthesis of (−)-dihydrotetrabenazine. | |
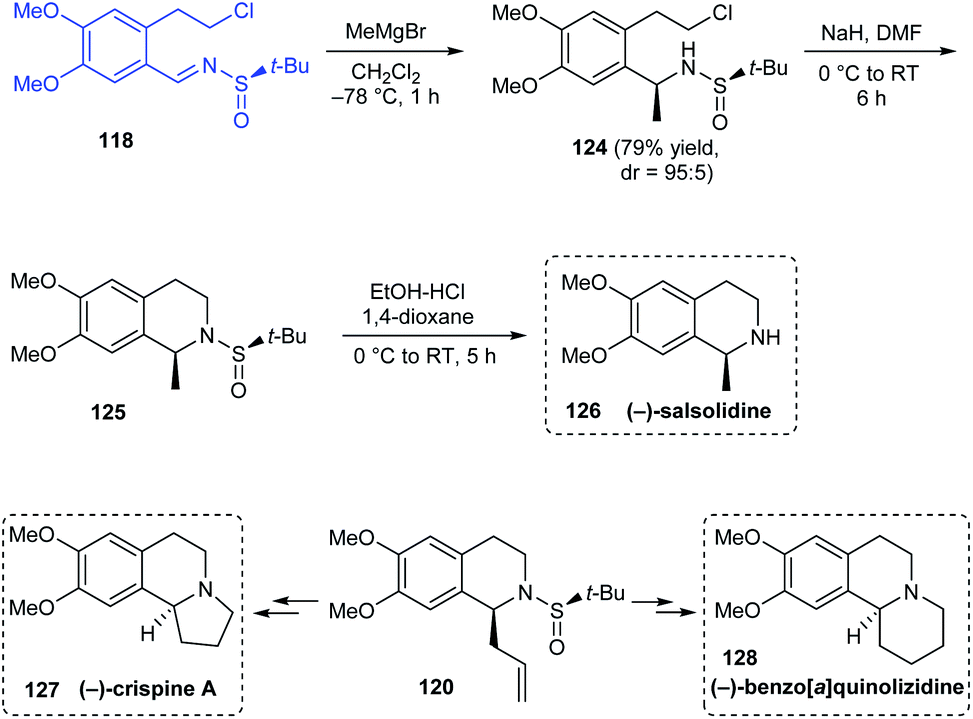
Reddy's group also explored enantiopure tert-butanesulfinamide in the asymmetric synthesis of (−)-crispine A, (−)-salsolidine and (−)-benzo[a]quinolizidine.31 The synthetic route towards (−)-salsolidine 126 started from the aforementioned sulfinimine 118 derived from 2-(3,4-dimethoxyphenyl)ethanol. Subjecting 118 to Grignard reaction provided methyl sulfinamide 124 with good diastereoselectivity (dr = 95:5) which in turn was cyclized under basic conditions to afford the cyclized compound 125. (−)-Salsolidine 118 was obtained straightforward from 125 after the sulfinyl group was removed (Scheme 22). Exposure of previously mentioned cyclized sulfinamide 120 to two different sequence of reactions furnished the desired compounds (−)-crispine A 127 and (−)-benzo[a]quinolizidine 128.
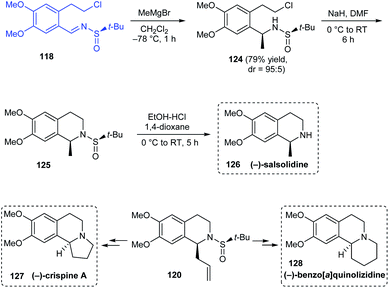 |
| | Scheme 22 Enantioselective synthesis of (−)-crispine A, (−)-benzo[a]quinolizidine, and (−)-salsolidine. | |
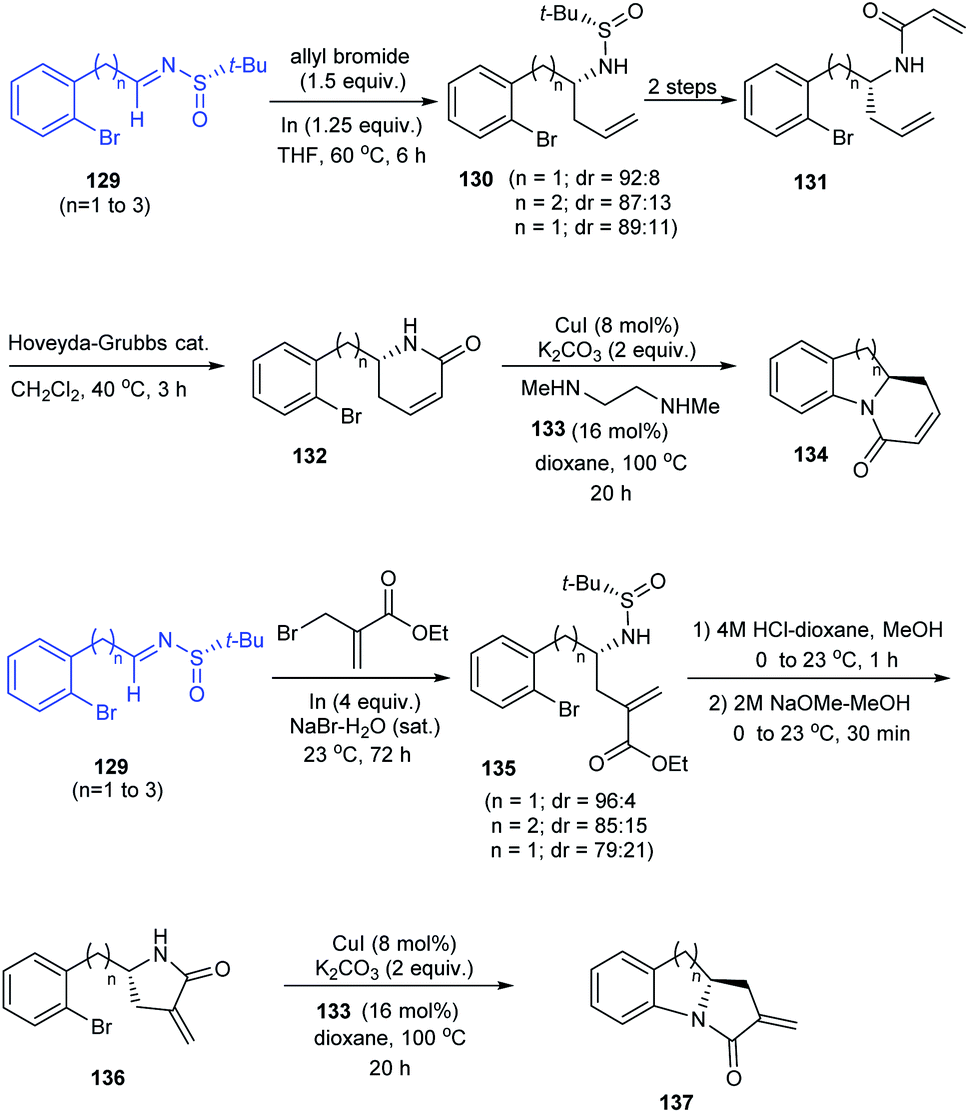
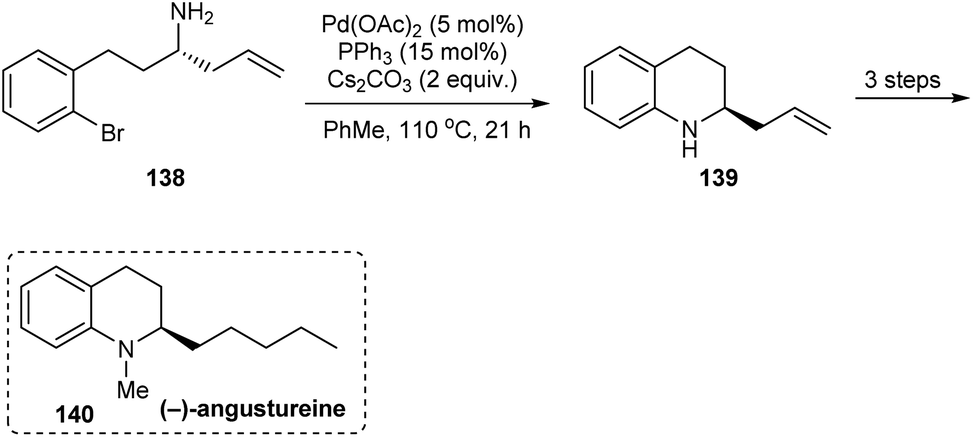
In 2014, an asymmetric synthetic pathway to indolines, tetrahydrobenzazepine and tetrahydroquinoline were accomplished with o-bromophenyl N-tert-butanesulfinimine 129 as a common precursor.32 Initially, derivatives of homoallylic amines generated upon diastereoselective addition on imine 129 successfully formed lactams 132 and 136 which produced the benzo-fused 1-azabicyclo[j.k.0]alkanes 134 and 137 respectively after intramolecular Cu assisted N-arylation (Scheme 23). Besides, Pd-mediated N-arylation led to the construction of benzo-fused 2-allyl substituted N-heterocycle 139, a key intermediate in the synthesis of (−)-angustureine (Scheme 24).
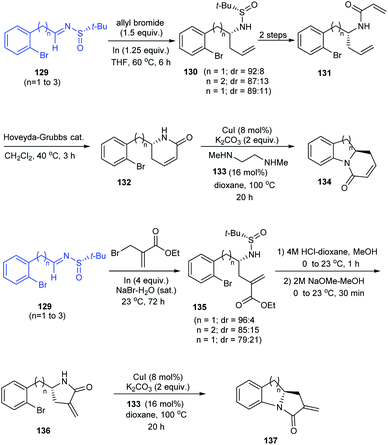 |
| | Scheme 23 Stereoselective synthesis of tetrahydrobenzazepine, tetrahydroquinoline and indolines. | |
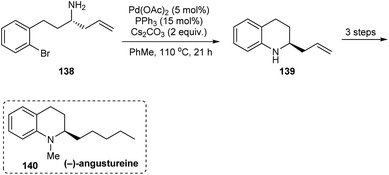 |
| | Scheme 24 Asymmetric synthesis of (−)-angustureine. | |
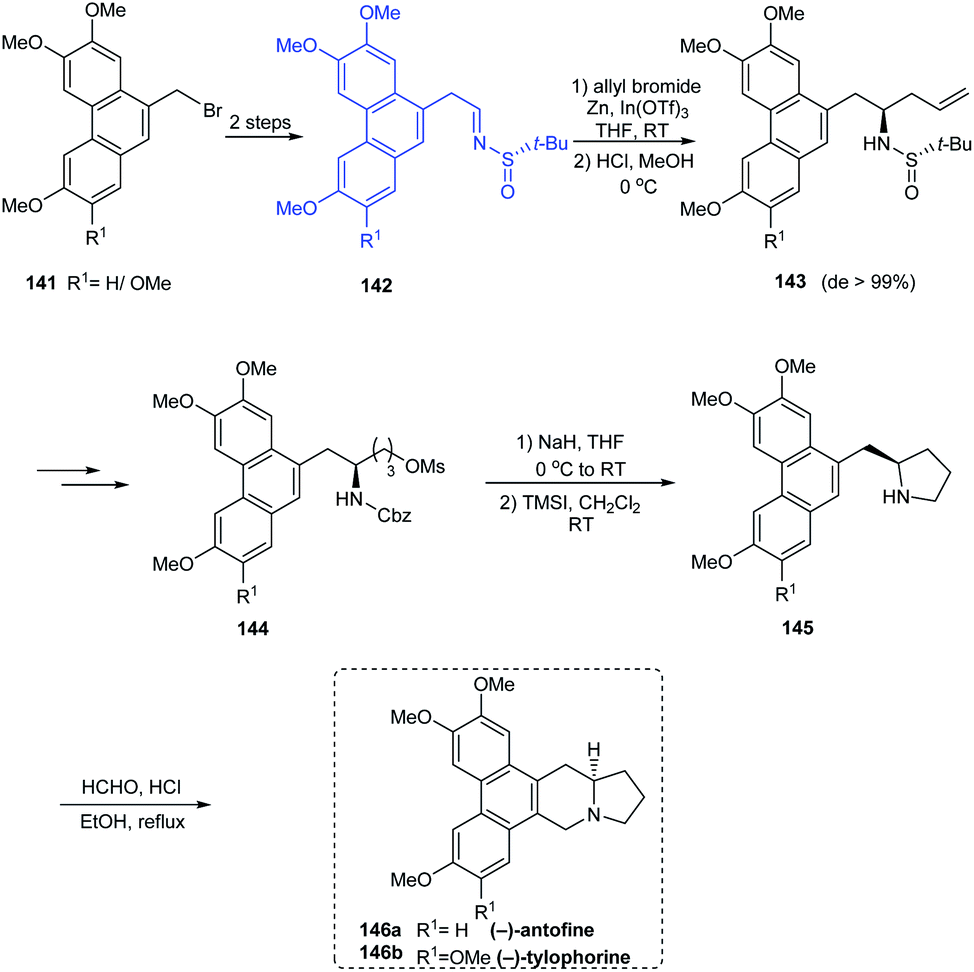
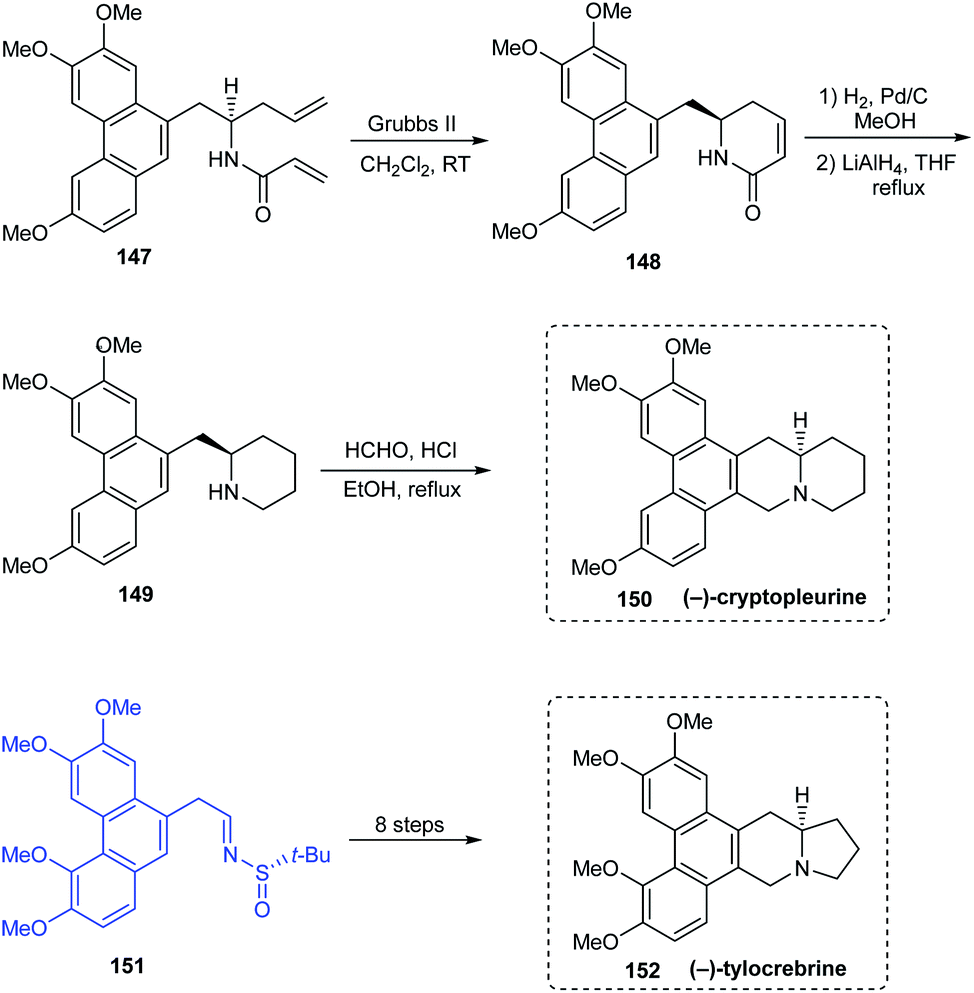
(−)-antofine 146a, (−)-tylophorine 146b, (−)-tylocrebrine 152, and (−)-cryptopleurine 150 belong to phenanthroindolizidine and phenanthroquinolizidine alkaloids whose varied bioactivities prompted the development of novel synthetic methods.33 Wang et al. discussed a collective total synthesis of these four alkaloids mediated by tert-butanesulfinamide as the chiral inductor. The N-tert-butanesulfinimine 142 represents the common precursor in constructing compounds 146a and 146b via a highly diastereoselective allylation (de > 99%), intramolecular SN2 substitution to render the pyrrolidine ring, and finally the Pictet–Spengler annulation (Scheme 25). Subjecting sulfinimine 151, derived from a carefully constructed aldehyde with the C5-methoxy substitution at phenanthrene to a similar set of reactions afforded (−)-tylocrebrine 152 in 8 steps (Scheme 26). Finally, (−)-cryptopleurine 150 was prepared from intermediate 147 obtained after acylation of 143, through a series of ring-closing metathesis reaction, catalytic hydrogenation, lactam reduction and Pictet–Spengler annulation.
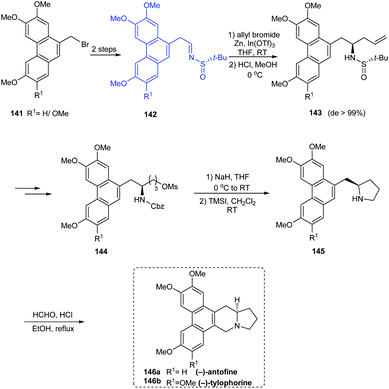 |
| | Scheme 25 Collective synthesis of (−)-antofine and (−)-tylophorine. | |
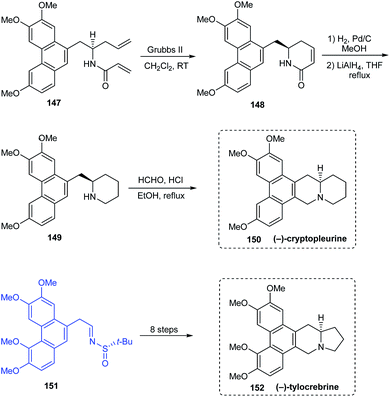 |
| | Scheme 26 Asymmetric total synthesis of (−)-tylocrebrine and (−)-cryptopleurine. | |
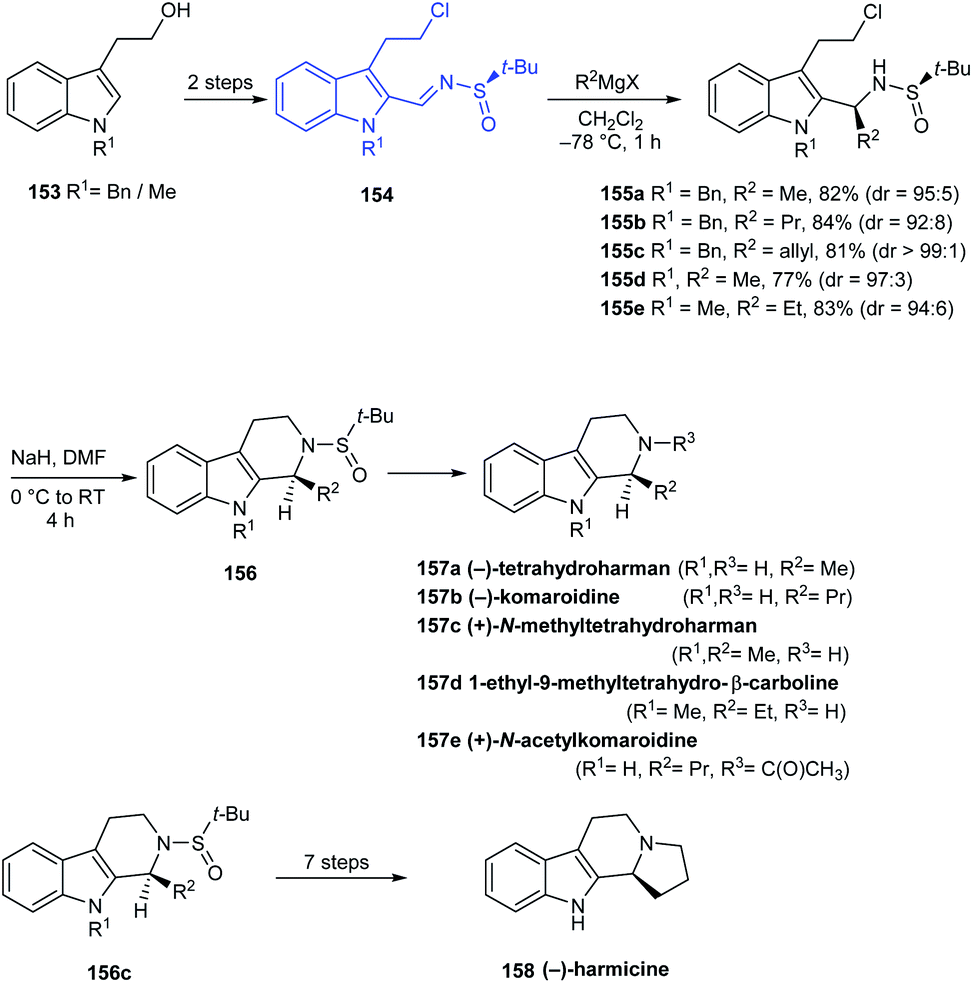
In 2016, Reddy and coworkers reported a facile stereoselective synthetic route to tetrahydro-β-carboline (THBC) alkaloids from a common N-sulfinyimine intermediate 154 where tert-butanesulfinamide acts as a chiral auxiliary.34 The sulfinimine 154 was transformed to the alkyl or allyl derivatives via diastereoselective Grignard addition which was then intramolecularly cyclized, followed by deprotection strategies furnishing the THBC alkaloids (−)-tetrahydroharman 157a, (−)-komaroidine 157b, (+)-N-methyltetrahydroharman 157c, and 1-ethyl-9-methyltetrahydro-β-carboline 157d in good yields (Scheme 27). Meanwhile, (+)-N-acetylkomaroidine 157e was obtained after an additional acetylation step and (−)-harmicine 3 was obtained over seven steps from 155c.
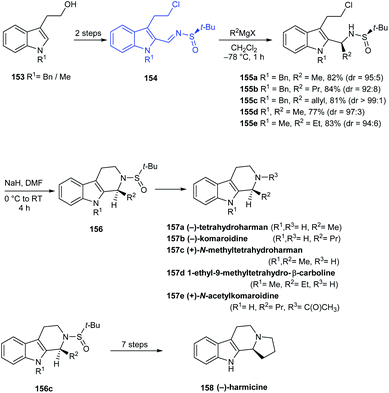 |
| | Scheme 27 Asymmetric synthesis of tetrahydro-β-carboline (THBC) alkaloids from sulfinimine. | |
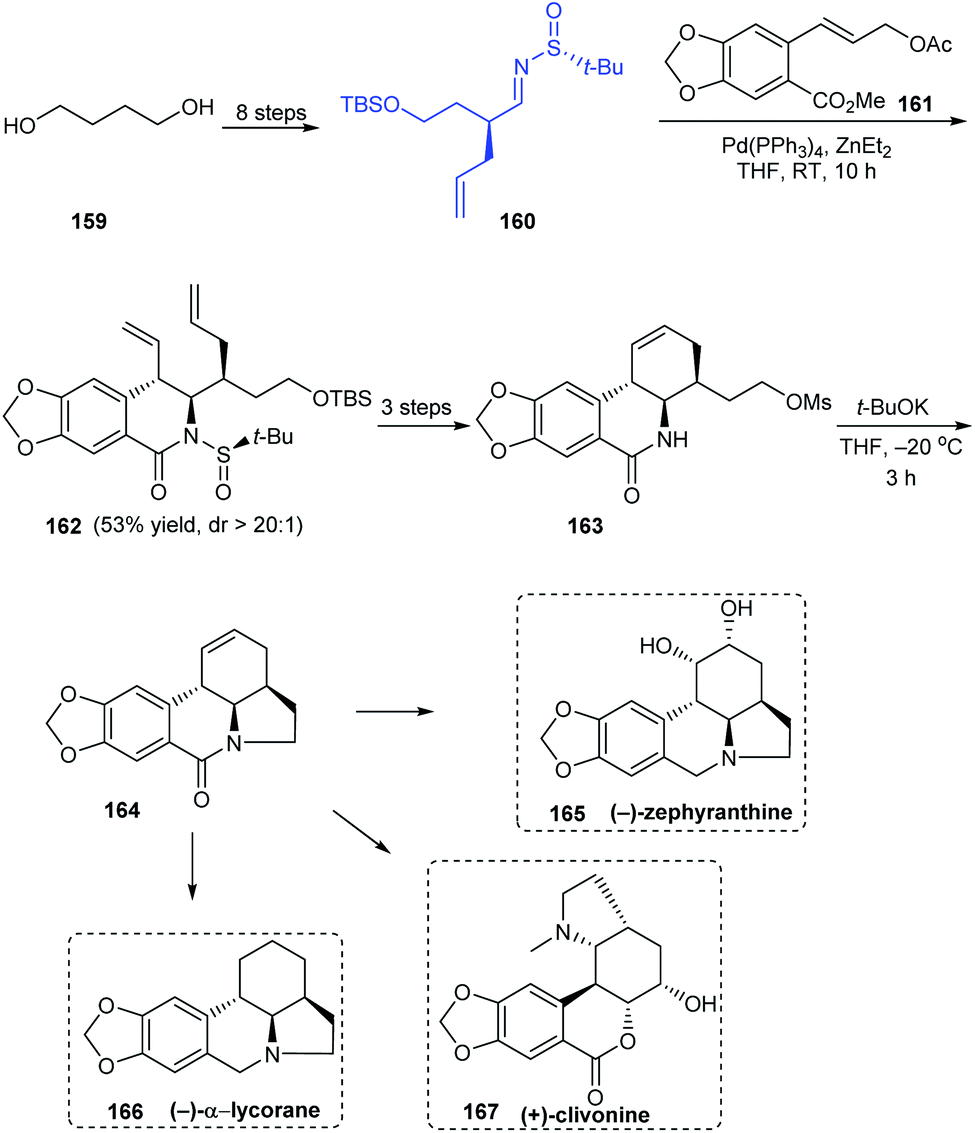
Sun and coworkers were successful in devising an efficient synthetic protocol to Amaryllidaceae alkaloids (−)-α-lycorane, (−)-zephyranthine and (+)-clivonine through divergent synthesis strategy by a common intermediate 164 that can offer access to many alkaloids of the family (Scheme 28).35 In the initial steps, they used their earlier reported procedure in the diastereoselective cinnamylation of imine 160 followed by cyclization to furnish the B ring in a single step. From 162, a facile route to enantiopure cyclic intermediate 164 was established. With 164 in hand, divergent stereoselective total synthesis of 165, 166 and formal synthesis route to 167 were achieved after required conversions.
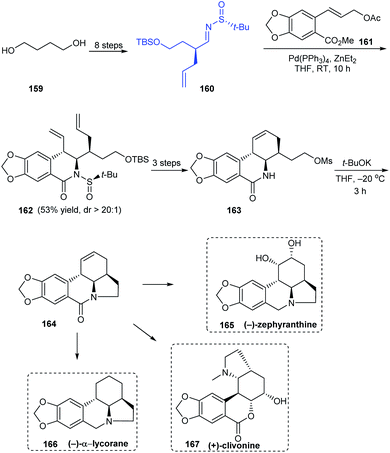 |
| | Scheme 28 Divergent synthesis of three Amaryllidaceae alkaloids. | |
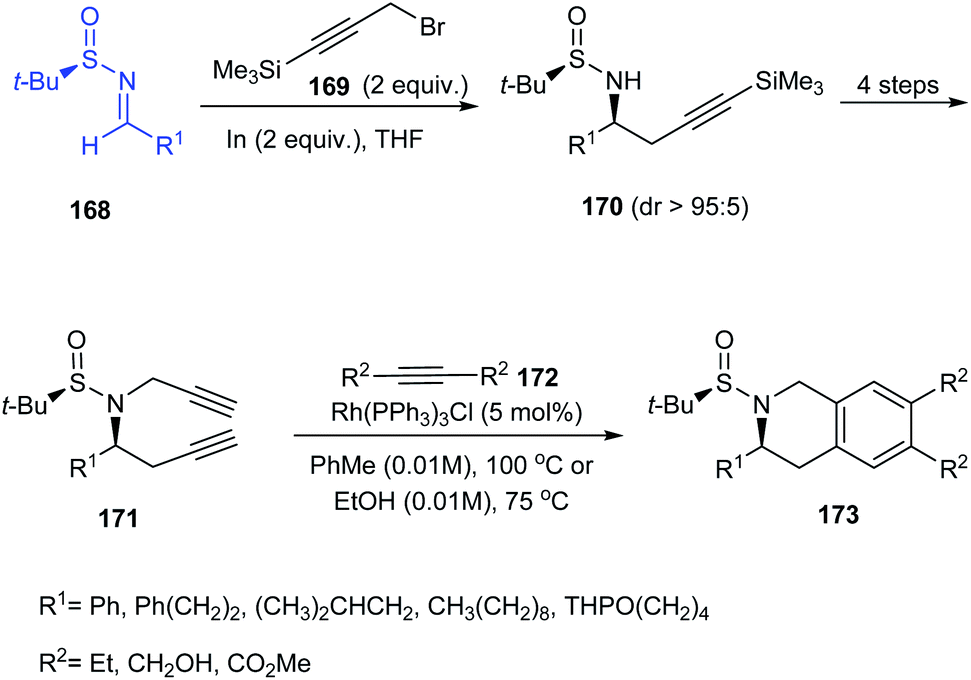
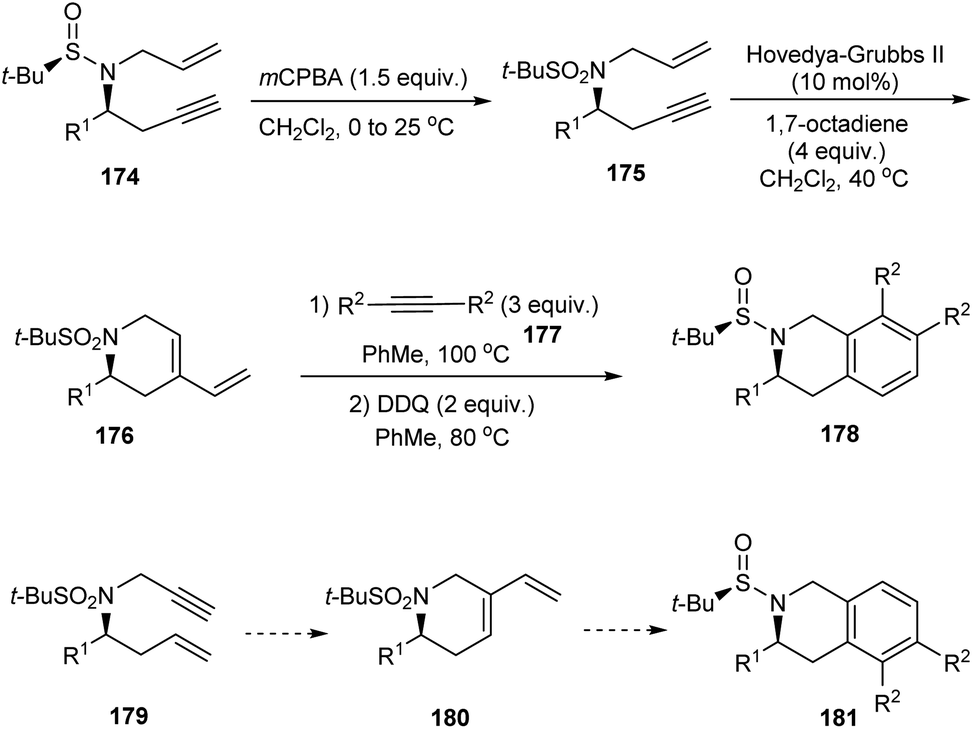
The research group of Foubelo successfully prepared substituted tetrahydroquinolines enantioselectively from N-tert-butanesulfinimines.36 The key synthetic operations in preparing 3,6,7-trisubstituted 1,2,3,4-tetrahydroquinolines 173 include diastereoselective propargylation of the sulfinimine 168 and Rh-catalysed [2 + 2 + 2] cyclotrimerization of alkyne 172 with 4-azaocta-1,7-diyne 171 (Scheme 29). In a similar manner, 168 upon propargylation and allylation gives 4-azaocta-1,7-enynes 174 which subsequently undergoes Ru-catalysed ring closing metathesis forming chiral cyclic diene 176 and thereafter [4 + 2] cycloaddition to yield 7,8-disubstituted 1,2,3,4-tetrahydroquinolines 178. Similarly, 5,6-disubstituted derivative 181 can be synthesised from 5-azaocta-1,7-enynes 179 (Scheme 30).
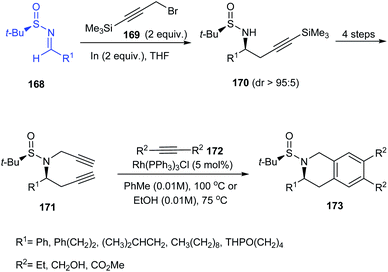 |
| | Scheme 29 The synthetic route to 3,6,7-trisubstituted 1,2,3,4-tetrahydroquinolines. | |
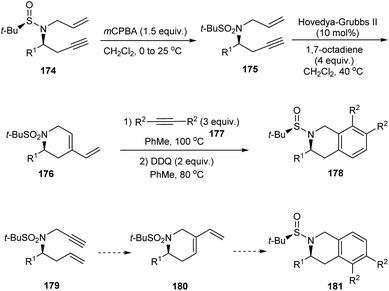 |
| | Scheme 30 The synthetic route to 7,8-disubstituted 1,2,3,4-tetrahydroquinolines. | |
4.3 Other fused rings
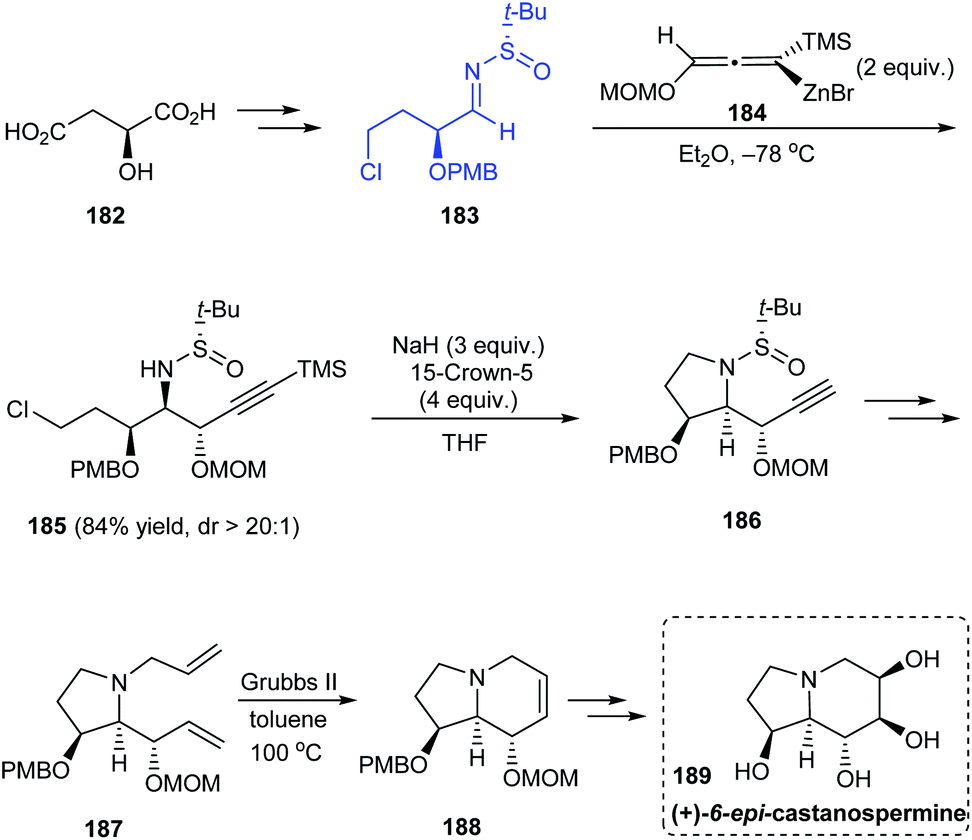
The stereoselective total synthesis of (+)-6-epi-castanospermine 189 was accomplished through an α-chiral sulfinimine precursor 183 readily obtained from (S)-malic acid.37 The highly diastereoselective addition of allenylzinc reagent 184 onto 183 to give acetylenic syn–anti 2-amino-1,3-diether intermediate (dr > 20:1) represents the key step followed by intramolecular substitution reaction to obtain 186, the ring-closure metathesis to generate the piperidine ring and finally the syn-dihydroxylation of olefin to yield the anticipated compound 189 (Scheme 31).
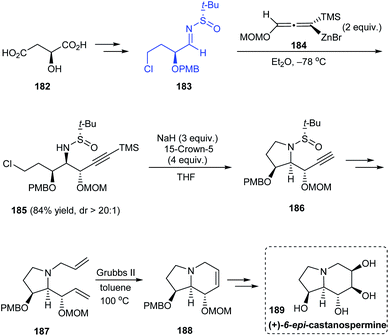 |
| | Scheme 31 Asymmetric total synthesis of (+)-6-epi-castanospermine. | |
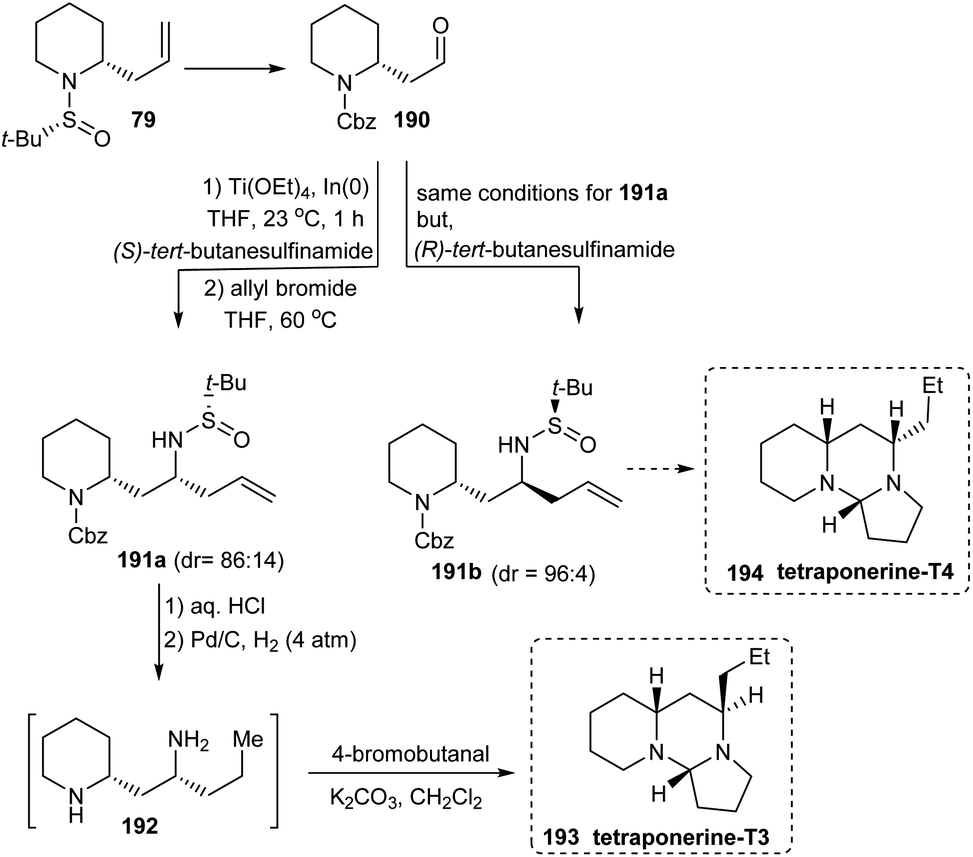
A divergent total synthesis of two tetraponerines T3 193 and T4 194 has been reported in 2012 by the same group of researchers who developed the diastereoselective aminoallylation protocol to deliver 2-allyl piperidines in two steps.38 Here, the first aminoallylation rendered 2-allyl piperidine derivative 79 which was then transformed into aldehyde 190 to conduct the subsequent aminoallylation. From the common aldehyde 190, diamine derivatives 191a and 191b were generated in presence of (S)-tert-butanesulfinamide and (R)-tert-butanesulfinamide respectively in which the latter reaction displayed better diastereoselection (dr = 96:4) when compared to the former one (dr = 86:14) (Scheme 32). Additionally, repeated usage of diastereoselective aminoallylation protocol resulted in enantiomeric improvement which led to productive isolation of 191a (72% yield) and 191b (80% yield) in greater than 99:1 er. With these amine derivatives in hand, tetraponerines T3 193 and T4 194 were prepared over two steps.
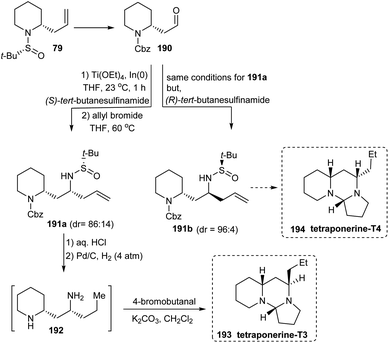 |
| | Scheme 32 Total synthesis of tetraponerines T3 and T4. | |
In 2018, the total synthesis of (+)-C(9a)-epiepiquinamide was accomplished via the N-tert-butanesulfinimine 77 in a six step strategy.39 Initially, 5-bromopentanal was condensed with (R)-tert-butanesulfinamide to give the imine 77. Then, the imine was diastereoselectively coupled with the nitro ester 195 through the aza-Henry reaction to afford the sulfinamide 196 (dr = 1:1). From 196, the double cyclized quinolizidine derivative 197 was achieved as a 6:1 ratio of diastereoisomers upon desulfinylation and intramolecular reactions of the free amine formed (Scheme 33). Finally, the target compound 198 was achieved after a few functional group conversions.
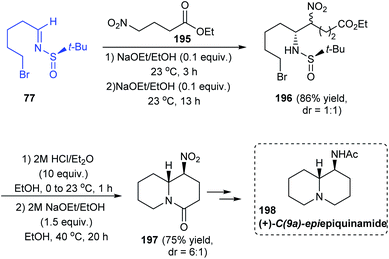 |
| | Scheme 33 Synthetic route towards (+)-C(9a)-epiepiquinamide. | |
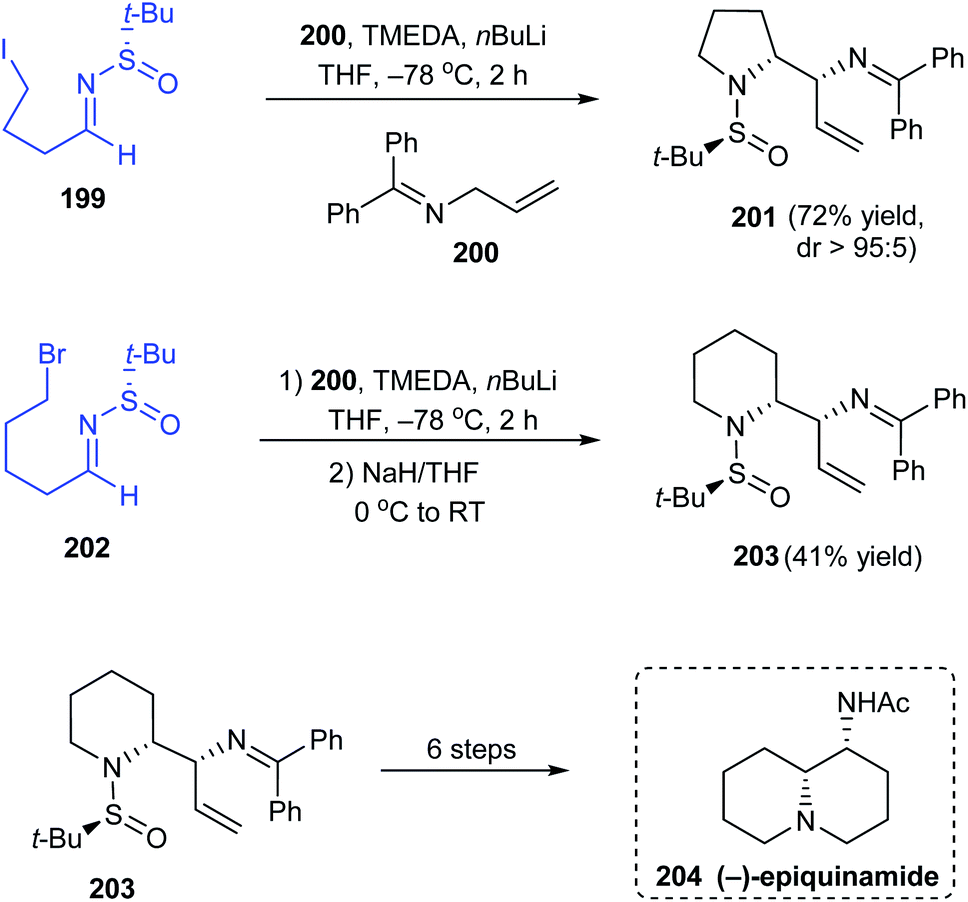
In 2019, Prasad with coworkers examined the addition of lithium anion of diphenylallylimine 200 onto different sulfinimines obtained from aromatic or aliphatic aldehydes.40 They found that the sulfinimines derived from aliphatic aldehydes delivered vicinal diamines as products in good yields and excellent diastereoselectivities. Interestingly, sulfinimine 199 after addition reaction furnished the pyrrolidine sulfinamide 201 in 72% yields and greater than 95:5 dr (Scheme 34). Analogously, the piperidine containing compound 203 was prepared from sulfinimine 202 including an additional step of treatment with NaH to convert the noncyclized products obtained after the addition reaction. The authors successfully applied 203 as a potent starting point towards the synthesis of the alkaloid (−)-epiquinamide 204 via a six step synthetic route.
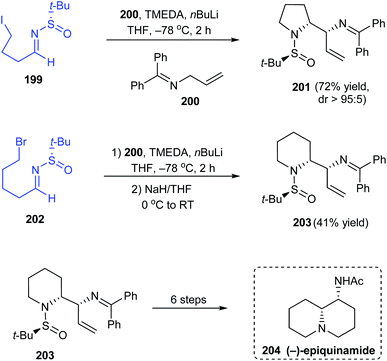 |
| | Scheme 34 Synthesis and application of α-amine bearing pyrrolidine and piperidine sulfinamides. | |
4.4 δ-Lactam
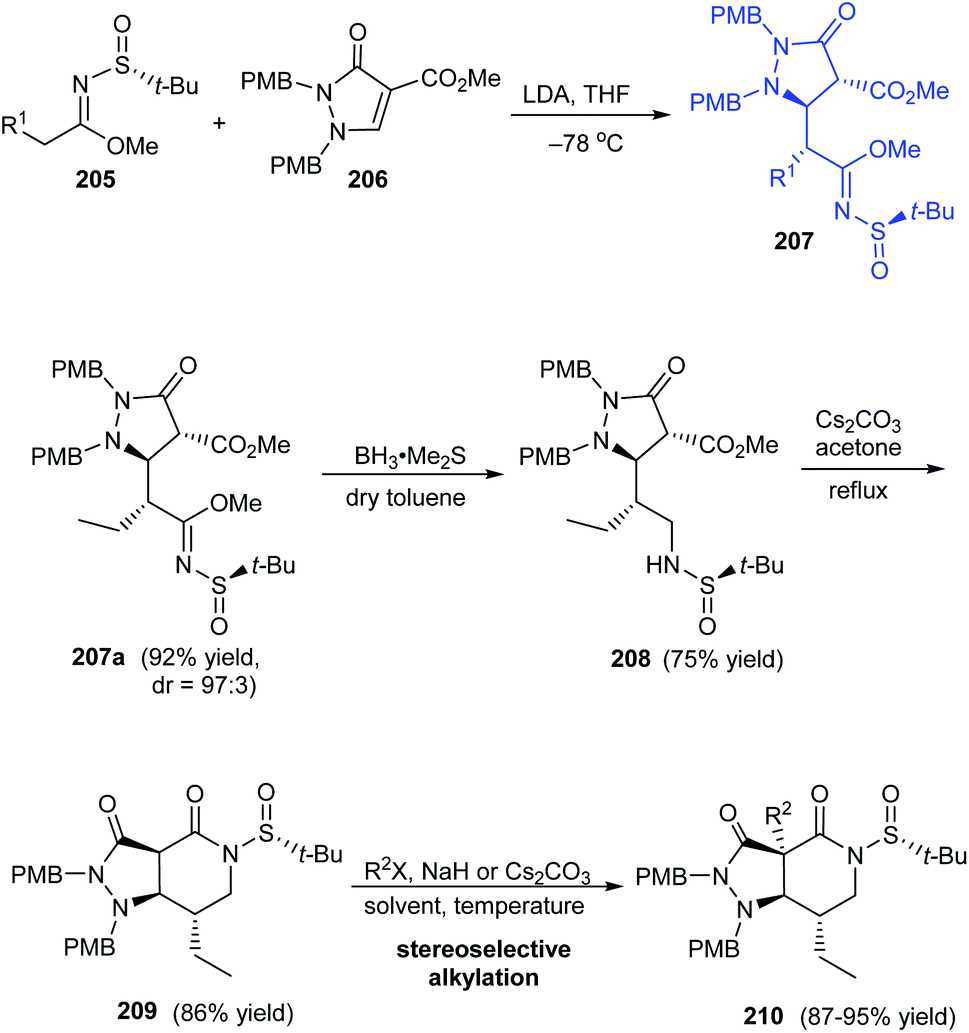
A highly efficient synthetic protocol towards optically active pyrazolopiperidines has been developed by the research group of Song in 2016.41 They devised a highly diastereoselective Michael addition reaction involving α,β-unsaturated pyrazolidinone 206 and (R)-N-tert-butanesulfinyl imidate 205 to provide pyrazolidinones like 207a (dr = 97:3) that can be transformed to pyrazolopiperidine derivative 209 via reduction and cyclization reactions affording good yields of the product (Scheme 35). Further, subjecting the lactam intermediate 209 to stereoselective Michael addition or alkylation will effectively generate various pyrazolopiperidine derivatives like 210 possessing quaternary carbon-center positioned at C3a.
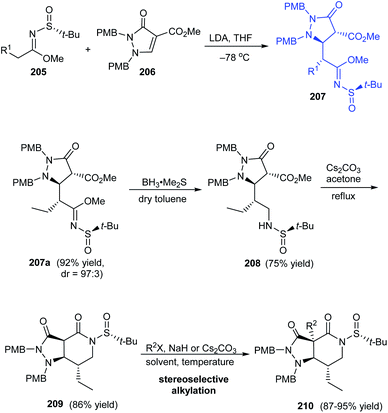 |
| | Scheme 35 Stereoselective synthesis of pyrazolopiperidines. | |
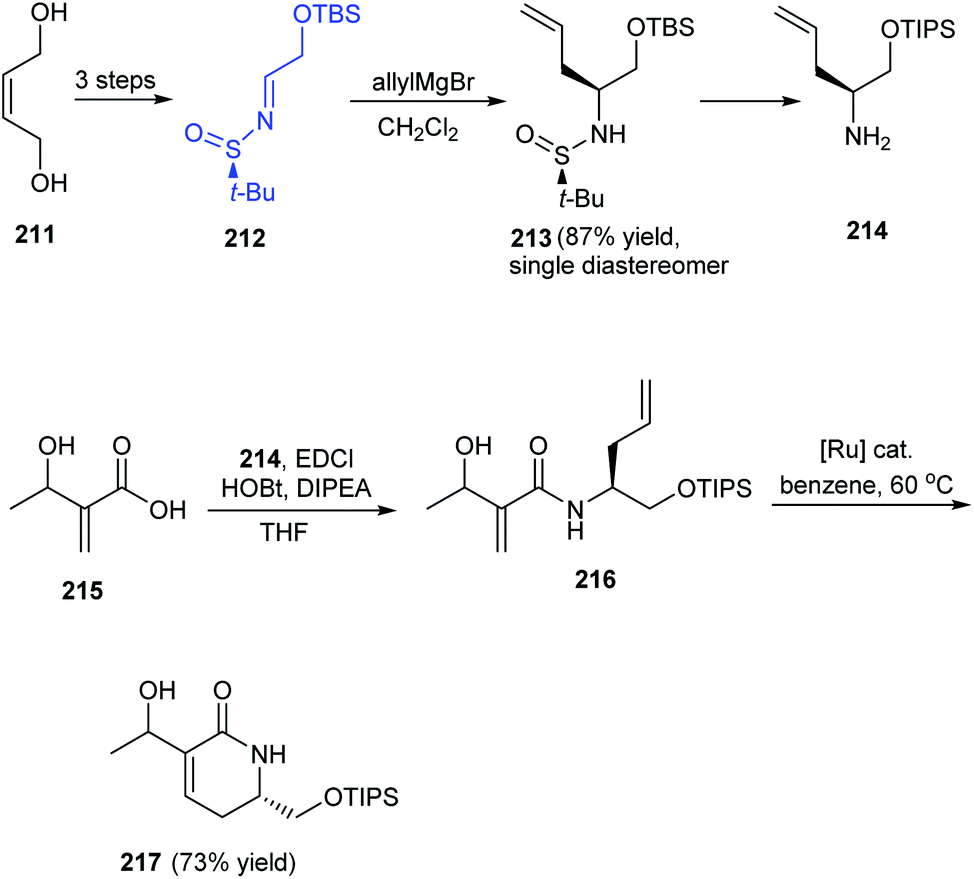
A facile and effective method for synthesis of α-hydroxyethyl α,β-unsaturated δ-lactams in enantioenriched form has been introduced by Stoltz et al. in 2016.42 The initial reactions provided the N-tert-butanesulfinimine 212 which underwent diastereoselective allyl group addition to furnish the δ-stereocenter of the lactam. The coupling of amine 214 with the prepared acid 215 yielded amide 216 which undertook ring-closing metathesis to deliver the desired lactam 217 in 2:3 ratio of diastereomers (Scheme 36). The utility of the method is expected in natural product synthesis and medicinal chemistry wherein α-hydroxyethyl α,β-unsaturated δ-lactams is an important precursor in synthesis.
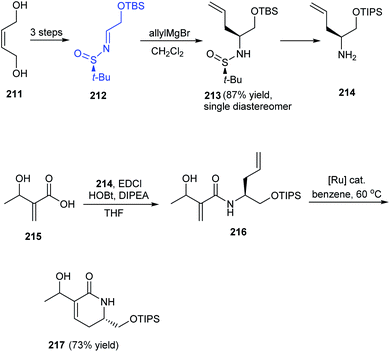 |
| | Scheme 36 Synthetic method towards enantioenriched α-hydroxyethyl α,β-unsaturated δ-lactams. | |
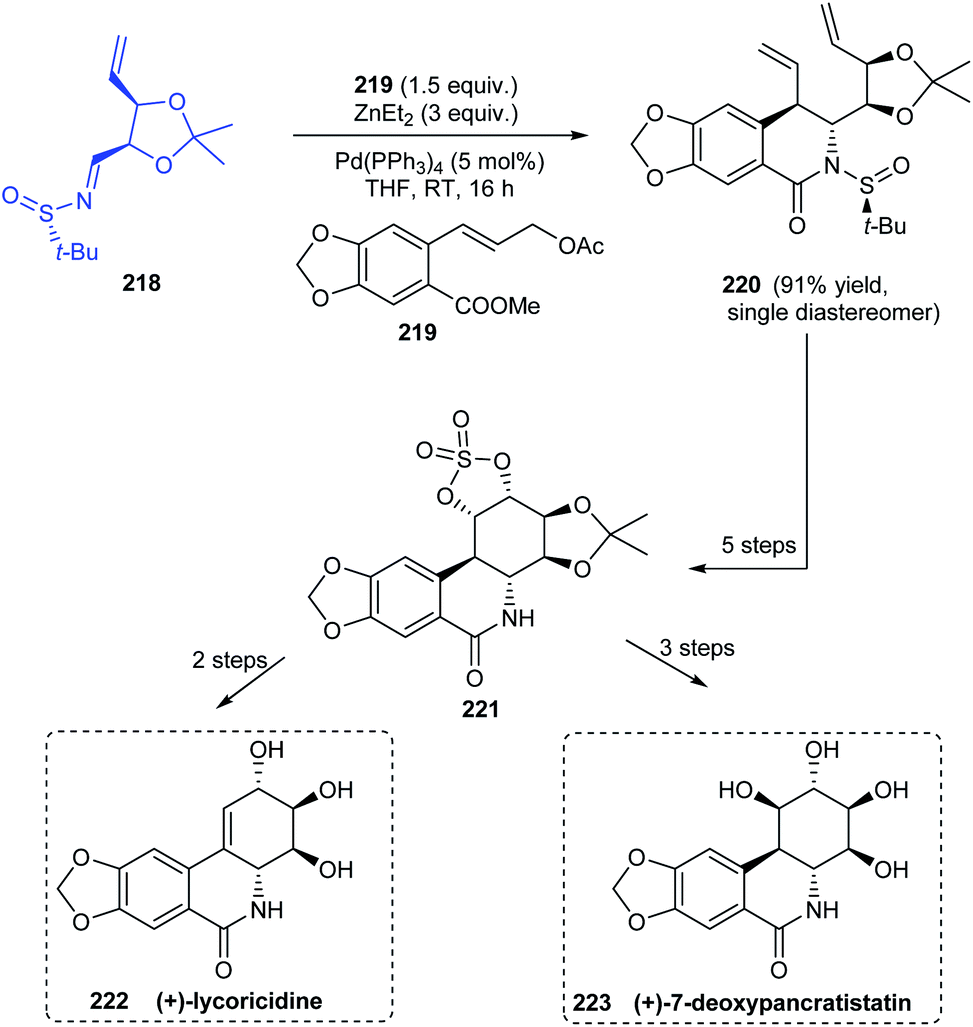
Sun and coworkers established a procedure for diastereoselective cinnamylation of N-tert-butanesulfinimines using cinnamyl acetate and catalysed by palladium.43 Natural products with antitumour properties (+)-lycorcidine 222 and (+)-7-deoxypancratistatin 223 were successfully synthesised using this protocol (Scheme 37). The N-tert-butanesulfinimine 218 derived from an iodoribose derivative underwent the cinnamylation and cyclization continually to afford the lactam 220 as a single diastereomer in 91% yield. Thereafter, a set of reactions followed to obtain the cyclic structure 221 which acted as a common intermediate towards the synthesis of 222 and 223. Moreover, trans-dihydrolycoricidine can be accomplished from 222 through the hydrogenation reaction.
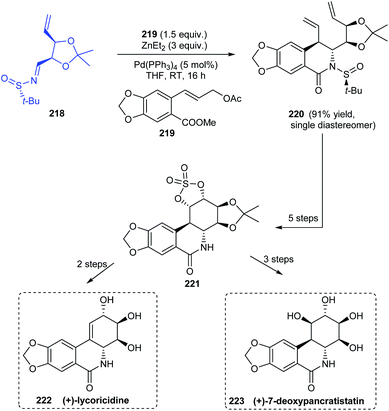 |
| | Scheme 37 Key steps in synthesising (+)-lycorcidine and (+)-7-deoxypancratistatin. | |
5 Miscellaneous
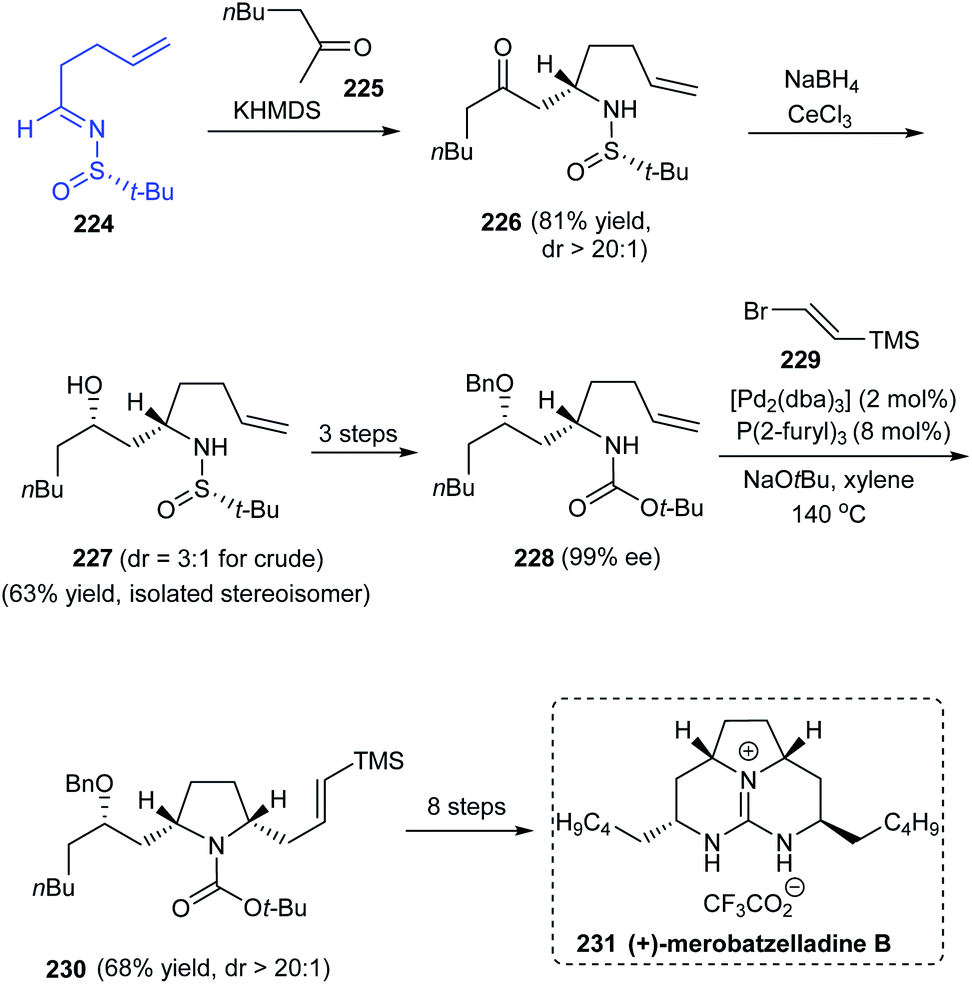
In 2012, Wolfe et al. reported the first total synthesis of the batzelladine alkaloid, (+)-merobatzelladine B 231 in enantiopure form.44 In the initial stages of synthesis, Mannich reaction of sulfinimine 224 provided the ketone 226 with excellent diastereocontrol (dr > 20:1) which in turn conducted a series of reactions to accomplish the functionalized precursor for pyrrolidine synthesis, the γ-aminoalkene. Exposure of 228 to a highly diastereoselective (dr > 20:1) Pd-catalysed carboamination with 229 afforded the pyrrolidine 230. In the subsequent steps, a bicyclic urea intermediate was prepared upon Pd-catalysed carboamination procedure and towards the end, the third ring was achieved via an intramolecular Mitsunobu reaction (Scheme 38).
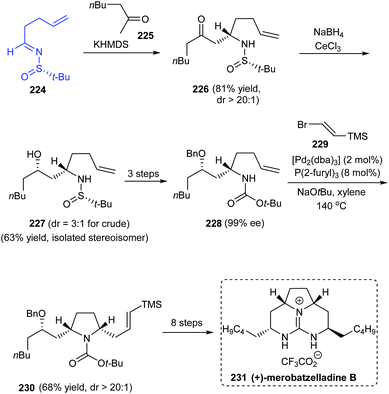 |
| | Scheme 38 Enantioselective total synthesis of (+)-merobatzelladine B. | |
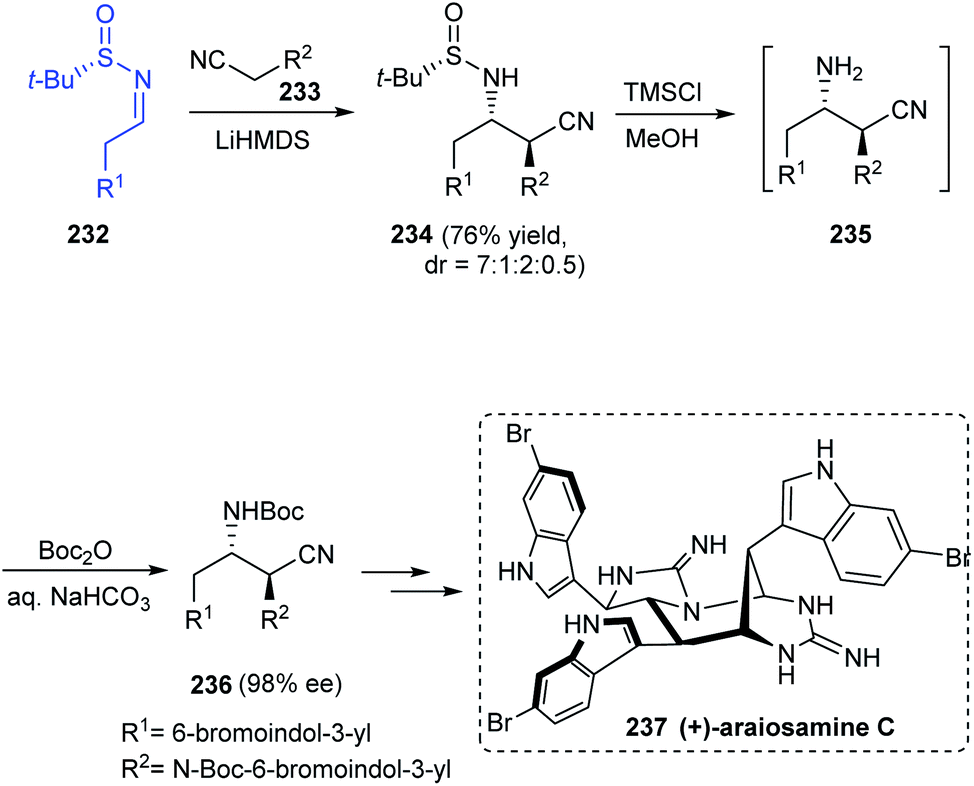
The total synthesis of araiosamines, a group of marine alkaloids with 3 indole and 2 guanidine units has been established via an 11-step process from Baran's lab.45 The synthetic protocol provided with the racemic forms of the target compound and hence to assign the absolute configuration, synthesis of the natural enantiomer was attempted. The synthesis began with a Mannich reaction involving chiral sulfinimine 232 to render the desired Mannich product 234 as a mixture of diastereomers in the ratio 7:1:2:0.5 (Scheme 39). Desulfinylation and carbamoylation followed to generate (S,S)-236 in an enantioenriched form (98% ee) after purification. Further conversions eventually furnished the target compound (+)-araiosamine C 237 in agreement with the optical rotation of its natural enantiomer.
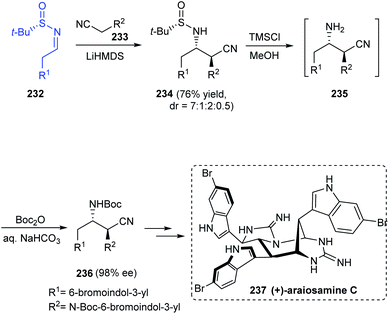 |
| | Scheme 39 Total synthesis of (+)-araiosamine C as a single enantiomer. | |
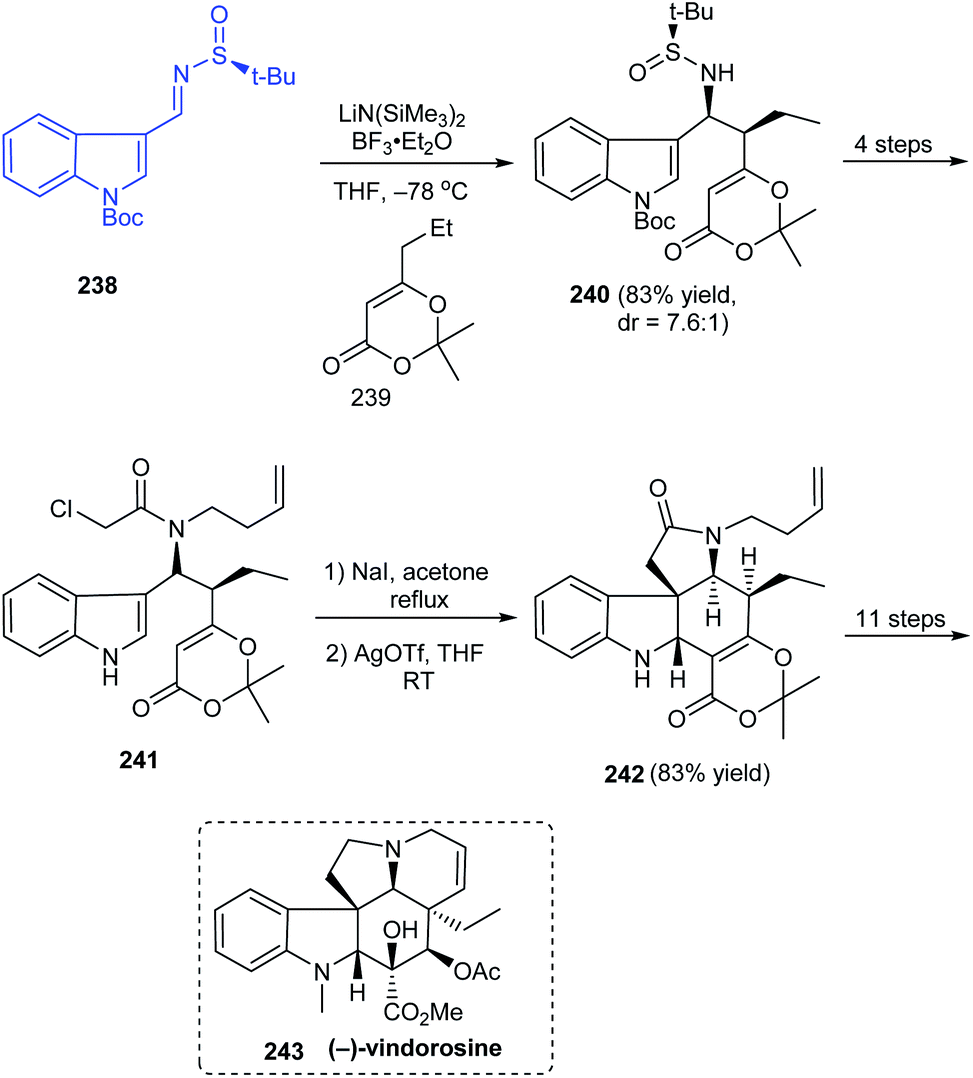
In 2017, Zhang's group described a different route to the total synthesis of the highly challenging alkaloid (−)-vindorosine 243.46 One among the key conversions is a diastereoselective Mannich-type addition onto N-tert-butanesulfinimine 238 to deliver the intermediate 240 in good yields and diastereoselectivity (dr = 7.6:1) (Scheme 40). The other vital transformation employed in the concomitant construction of both C and E rings is the Heathcock/aza-Prins cyclization sequence with 241 to afford the key intermediate 242. Herein, they were successful in completing the pentacyclic (−)-vindorosine 243 and envisioned the utility of intermediate 242 in preparing other related alkaloids.
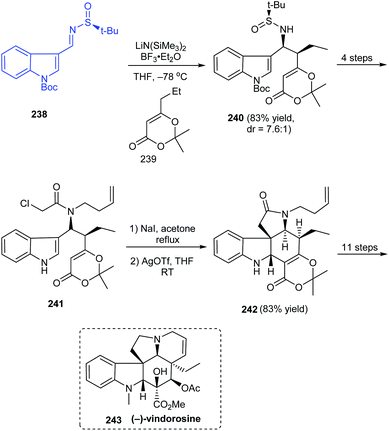 |
| | Scheme 40 Total synthesis of (−)-vindorosine. | |
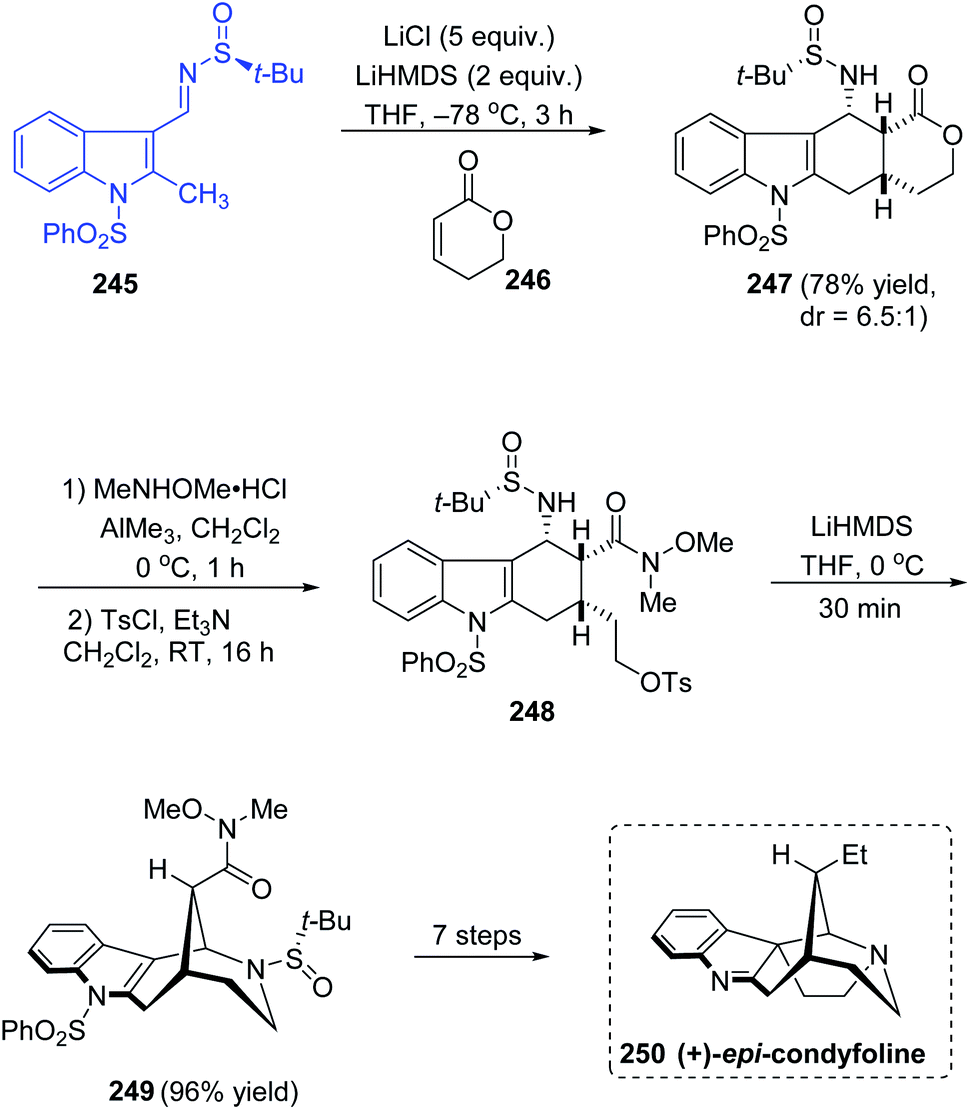
An efficient asymmetric total synthesis of (+)-epi-condyfoline, an aspidospermatan-type indole alkaloid was accomplished in 13 steps commencing from sulfinimine 245 that is formed from 2-methylindole-3-carboxaldehyde.47 The diastereoselective domino Michael/Mannich ring forming reaction with imine 245 and lactone 246 afforded the tetracyclic lactone 247 (dr = 6.5:1), which upon Weinreb amidation left the ring opened and then tosylation of the resultant alcohol followed giving 248. In the next step, the cyclization was carried out with LiHMDS in THF at 0 °C yielding the 2-azabicyclo[3.3.1]nonane structure in 249 and 96% yield was observed in this step (Scheme 41). Then, various transformations succeeded affording the target compound (+)-epi-condyfoline 250.
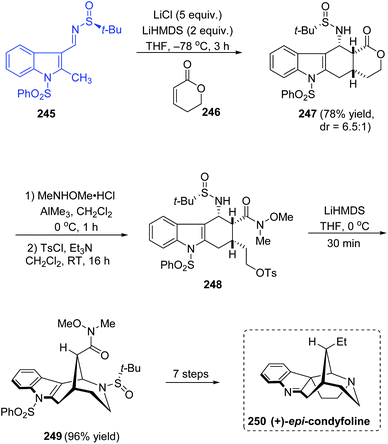 |
| | Scheme 41 Total synthesis of (+)-epi-condyfoline. | |
6 Conclusion
Chiral sulfinamide based methodologies in synthesising nitrogen-containing heterocycles are of great interest due to the biological activity of the products and operational easiness with chiral sulfinamides. Drawn by their efficacy as a chiral inductor, the application of tert-butanesulfinamide in the asymmetric synthesis has been growing in the last two decades. In this review, we have summarized the recent reports on the application of enantiopure tert-butanesulfinamide in the synthesis of N-heterocycles through sulfinimine intermediates. We witnessed sufficient reports in this direction from the past ten years comprising of efficient and scalable protocols.
The extensive use of tert-butanesulfinamide in asymmetric synthesis is ascribed to its low cost and ease of removal from the reaction mixture. More importantly, the highly stereodirecting nature of the tert-butylsulfinyl group is evident from the good to excellent diastereoselectivities obtained during the nucleophilic addition to sulfinimines. In most of the reports, chiral amines formed upon desulfinylation were amenable to other transformations including cyclization to prepare the desired nitrogen-containing cyclic compounds in high optical purity. When certain articles discussed novel synthetic methodologies towards N-heterocycles, several others described the total synthesis of simple or complex polycyclic natural products wherein N-heterocycle formation is a key step. Importantly, the synthesis of anti-tumour, anti-bacterial, and other therapeutically relevant N-heterocyclic compounds is made possible using the method. There are elegant examples that not only accomplished the first time total syntheses of targeted natural products but also are remarkably efficient and can be prepared on a gram-scale without loss in selectivity and yields. Of note, asymmetric synthesis of some of the structurally complex alkaloids like (+)-merobatzelladine B and (−)-vindorosine has been achieved. Furthermore, some reports used efficient divergent strategy in the total synthesis of natural alkaloids wherein the sulfinimine derived intermediates provide access to more than one targets of the family.
Despite a large number of reports, use of tert-butanesulfinamide as the chiral source in the total synthesis of many families of complex natural products is unexplored to large extent. Also, some strategies require harsh and sensitive reaction conditions mostly during organometallic addition procedures. Hence, photoredox-based procedures in the radical generation and addition to sulfinimines might gain more attention in the near future. Further improvement of the prevailing methods and development of new unified strategies will be appreciated.
In summary, based on the developments in the past two decades we believe that the use of tert-butanesulfinamide as an ideal chiral auxiliary will continue to be one of the best methods in N-heterocycle synthesis. We anticipate that the diverse strategies discussed might benefit people across the fields of medicinal chemistry, synthetic chemistry, and agrochemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
RMP and PVS thank the Council of Scientific and Industrial Research (CSIR), New Delhi for a research fellowship. SR and GA thank the Kerala State Council for Science, Technology and Environment (KSCSTE), Trivandrum, for the award of a research fellowship and research grant (Order no. 341/2013/KSCSTE dated 15.03.2013) respectively. We greatly acknowledge the support from the Department of Science and Technology (DST-New Delhi) under the DST-PURSE programme.
References
- G. Diaz-Muñoz, I. L. Miranda, S. K. Sartori, D. C. de Rezende and M. A. N. Diaz, Chirality, 2019, 31, 776–812 CrossRef.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS.
- G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS.
-
(a) D. J. Weix and J. A. Ellman, Org. Synth., 2005, 82, 157–165 CrossRef CAS;
(b) D. J. Weix and J. A. Ellman, Org. Lett., 2003, 5, 1317–1320 CrossRef CAS.
- D. A. Cogan, G. Liu and J. Ellman, Tetrahedron, 1999, 55, 8883–8904 CrossRef CAS.
- M. Wakayama and J. A. Ellman, J. Org. Chem., 2009, 74, 2646–2650 CrossRef CAS.
- C. Achuenu, S. Carret, J.-F. Poisson and F. Berthiol, Eur. J. Org. Chem., 2020, 5901–5916 CAS.
- M. Medjahdi, J. C. Gonzalez-Gomez, F. Foubelo and M. Yus, J. Org. Chem., 2009, 74, 7859–7865 CrossRef CAS.
- M. Henary, C. Kananda, L. Rotolo, B. Savino, E. A. Owens and G. Cravotto, RSC Adv., 2020, 10, 14170–14197 RSC.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
-
(a) N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS;
(b) B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS;
(c) N. Kerru, S. Maddila and S. B. Jonnalagadda, Curr. Org. Chem., 2019, 23, 3156–3192 Search PubMed;
(d) Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS;
(e) D. Z. Zarate, R. Aguilar, R. I. Hernandez-Benitez, E. M. Labarrios, F. Delgado and J. Tamariz, Tetrahedron, 2015, 71, 6961–6978 CrossRef.
- E. Wojaczyńska and J. Wojaczynski, Chem. Rev., 2020, 120, 4578–4611 CrossRef.
- P. Dinér, A. Sadhukhan and B. Blomkvist, ChemCatChem, 2014, 6, 3063–3066 CrossRef.
- A. A. Kudale, P. Anaspure, F. Goswami and M. Yus, Tetrahedron Lett., 2014, 55, 7219–7221 CrossRef CAS.
- X.-N. Song and Z.-J. Yao, Tetrahedron, 2010, 66, 2589–2593 CrossRef CAS.
- J. E. Redford, R. I. McDonald, M. L. Rigsby, J. D. Wiensch and S. S. Stahl, Org. Lett., 2012, 14, 1242–1245 CrossRef CAS.
- G. Procopiou, W. Lewis, G. Harbottle and R. A. Stockman, Org. Lett., 2013, 15, 2030–2033 CrossRef CAS.
- R. Beniazza, E. Romain, F. Chemla, F. Ferreira, O. Jackowski and A. Perez-Luna, Eur. J. Org. Chem., 2015, 7661–7665 CrossRef CAS.
- Y. Zhou, Y. Xi, J. Zhao, X. Sheng, S. Zhang and H. Zhang, Org. Lett., 2012, 14, 3116–3119 CrossRef CAS.
- X. Shen, J. Zhao, Y. Xi, W. Chen, Y. Zhou, X. Yang and H. Zhang, J. Org. Chem., 2018, 83, 14507–14517 CrossRef CAS.
- J. Gao, D. Sun, K. Yu, H. Xie and H. Ding, Org. Lett., 2019, 21, 9603–9607 CrossRef CAS.
- C. M. Park and D. J. Jeon, Org. Biomol. Chem., 2012, 10, 2613–2620 RSC.
- F. Xu, Y. Chen, E. Fan and Z. Sun, Org. Lett., 2016, 18, 2777–2779 CrossRef CAS.
- L. Cui, C. Li and L. Zhang, Angew. Chem., Int. Ed., 2010, 49, 9178–9181 CrossRef CAS.
- I. Bosque, J. C. González-Gómez, F. Foubelo and M. Yus, J. Org. Chem., 2012, 77, 780–784 CrossRef CAS.
- K. Damodar and J.-G. Jun, Bull. Korean Chem. Soc., 2016, 37, 571–575 CrossRef CAS.
- A. Lahosa, T. Soler, A. Arrieta, F. P. Cossio, F. Foubelo and M. Yus, J. Org. Chem., 2017, 82, 7481–7491 CrossRef CAS.
- K. R. Prasad and V. A. Rangari, Tetrahedron, 2019, 75, 130496–130510 CrossRef CAS.
- G. Zhao, D. P. Canterbury, A. P. Taylor, X. Cheng, P. Mikochik, S. W. Bagley and R. Tong, Org. Lett., 2019, 22, 458–463 CrossRef.
- N. S. S. Reddy, A. S. Reddy, J. S. Yadav and B. V. S. Reddy, Tetrahedron Lett., 2012, 53, 6916–6918 CrossRef.
- N. S. S. Reddy, B. J. Reddy and B. V. S. Reddy, Tetrahedron Lett., 2013, 54, 4228–4231 CrossRef CAS.
- J. A. Sirvent, F. Foubelo and M. Yus, J. Org. Chem., 2014, 79, 1356–1367 CrossRef CAS.
- Y. Zheng, Y. Liu and Q. Wang, J. Org. Chem., 2014, 79, 3348–3357 CrossRef CAS.
- N. S. S. Reddy, R. A. Babu and B. V. S. Reddy, Synthesis, 2016, 48, 1079–1086 CrossRef.
- Y.-J. Chen, S.-L. Cai, C.-C. Wang, J.-D. Cheng, S. Kramer and X.-W. Sun, Chem.–Asian J., 2017, 12, 1309–1313 CrossRef CAS.
- A. Sirvent, M. J. Garcia-Munoz, M. Yus and F. Foubelo, Eur. J. Org. Chem., 2019, 113–126 Search PubMed.
- J. Louvel, C. Botuha, F. Chemla, E. Demont, F. Ferreira and A. Perez-Luna, Eur. J. Org. Chem., 2010, 2921–2926 CrossRef CAS.
- I. Bosque, J. C. González-Gómez, A. Guijarro, F. Foubelo and M. Yus, J. Org. Chem., 2012, 77, 10340–10346 CrossRef CAS.
- M. Benlahrech, A. Lahosa, C. Behloul, F. Foubelo and M. Yus, Heterocycles, 2018, 97, 1191–1202 CrossRef CAS.
- M. B. Uphade, A. A. Reddy, S. P. Khandare and K. R. Prasad, Org. Lett., 2019, 21, 9109–9113 CrossRef CAS.
- H.-X. Huang, H.-J. Wang, L. Tan, S.-Q. Wang, P. Tang, H. Song, X.-Y. Liu, D. Zhang and Y. Qin, J. Org. Chem., 2016, 81, 10506–10516 CrossRef CAS.
- S.-J. Han and B. M. Stoltz, Tetrahedron Lett., 2016, 57, 2233–2235 CrossRef CAS.
- S. Cai, B. Yuan, Y. Jiang, G. Lin and X. Sun, Chem. Commun., 2017, 53, 3520–3523 RSC.
- N. R. Babij and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 4128–4130 CrossRef CAS.
- M. Tian, M. Yan and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 14234–14237 CrossRef CAS.
- W. Chen, X.-D. Yang, W.-Y. Tan, X.-Y. Zhang, X.-L. Liao and H. Zhang, Angew. Chem., Int. Ed., 2017, 56, 12327–12331 CrossRef CAS.
- P. Kokkonda and R. B. Andrade, Org. Lett., 2019, 21, 9594–9597 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc