
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA brain glioma gene delivery strategy by angiopep-2 and TAT-modified magnetic lipid-polymer hybrid nanoparticles
Lanxin Qiao†
a,
Yu Qin† b,
Yaxin Wang†a,
Yi Lianga,
Dunwan Zhub,
Wei Xionga,
Lu Lia,
Di Baoa,
Linhua Zhangb and
Xu Jin*a
b,
Yaxin Wang†a,
Yi Lianga,
Dunwan Zhub,
Wei Xionga,
Lu Lia,
Di Baoa,
Linhua Zhangb and
Xu Jin*a
aDepartment of Anesthesiology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China. E-mail: jxsys2020@gmail.com
bChinese Academy of Medical Sciences, Peking Union Medical College, Institute of Biomedical Engineering, Tianjin 300192, China
First published on 13th November 2020
Abstract
Owing to the existence of the blood–brain barrier (BBB), most treatments cannot achieve significant effects on gliomas. In this study, synergistic multitarget Ang-TAT-Fe3O4-pDNA-(ss)373 lipid-polymer hybrid nanoparticles (LPNPs) were designed to penetrate the BBB and deliver therapeutic genes to glioma cells. The basic material of the nanoparticles was PCL3750-ss-PEG7500-ss-PCL3750, and is called (ss)373 herein. (ss)373 NPs, Fe3O4 magnetic nanoparticles (MNPs), DOTAP, and DSPE-PEG-MAL formed the basic structure of LPNPs by self-assembly. The Fe3O4 MNPs were wrapped in (ss)373 NPs to implement magnetic targeting. Then, the Angiopep-2 peptide (Ang) and transactivator of transcription (TAT) were coupled with DSPE-PEG-MAL. Both can enhance BBB penetration and tumor targeting. Finally, the pDNA was compressed on DOTAP to form the complete gene delivery system. The results indicated that the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs were 302.33 nm in size. In addition, their zeta potential was 4.66 mV, and they had good biocompatibility. The optimal nanoparticles/pDNA ratio was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as shown by gel retardation assay. In this characterization, compared with other LPNPs, the modified single Ang or without the addition of the Fe3O4 MNPs, the penetration efficiency of the BBB model formed by hCMEC/D3 cells, and the transfection efficiency of C6 cells using pEGFP-C1 as the reporter gene were significantly improved with Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs in the magnetic field.
:1, as shown by gel retardation assay. In this characterization, compared with other LPNPs, the modified single Ang or without the addition of the Fe3O4 MNPs, the penetration efficiency of the BBB model formed by hCMEC/D3 cells, and the transfection efficiency of C6 cells using pEGFP-C1 as the reporter gene were significantly improved with Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs in the magnetic field.
Introduction
Glioma is a kind of malignant tumor derived from the glial tissue of the central nervous system, which grows invasively and is highly aggressive.1 The treatment of glioma is a comprehensive treatment based on surgical resection; however, the recurrence and metastasis after surgery remain difficult and intractable for clinical treatment.2 Currently, there is an urgent need for further treatment to eliminate the residual tumor cells, and achieve a better therapeutic effect in the long term. Gene therapy has been considered a therapeutic method with high potential and promise for glioma treatment. The two important factors for implementing gene therapy successfully are penetrating the blood–brain barrier (BBB) efficiently, and achieving gene transfection of the glioma cells effectively.3 In order to achieve these two aims, we designed and synthetized the Ang-TAT-Fe3O4-DOTAP-(ss)373 lipid-polymer hybrid nanoparticle (LPNPs) as the gene delivery system.The gene delivery system includes a viral vector and non-viral vector.4 Various viral vectors have been proven to be able to deliver genes into the central nervous system cells with large capacity and high efficiency. However, their application is limited owing to their biological specificity, such as immunogenicity and toxicity. By contrast, non-viral vectors with low immunogenicity and relative safety have attracted extensive attention.5,6 However, the disadvantages of non-viral vectors are poorer brain targeting and lower transfection efficiency for therapy of central nervous system diseases.7
To improve brain targeting, the penetration efficiency of the BBB should be improved first. The BBB mainly comprises the brain capillary endothelial cells and other different types of cells, like pericytes and astrocytes, that also contribute to the formation of the BBB.8,9 The main function of the BBB is the separation of the blood circulation system from the brain tissue, prevention of harmful substances from entering the brain, and providing a relatively stable internal environment for the brain tissue to maintain its normal function.10 While the BBB restricts harmful substances from entering the brain, it is also a barrier for gene delivery.11 There is currently no safe and efficient method to circumvent the BBB, and work directly in the brain.12 Several studies have shown that cell-penetrating peptides (CPPs), protein engineering, and magnetic targeting can effectively penetrate the BBB.13–15
Receptor-mediated endocytosis plays an important role in BBB penetration. Receptor-mediated transporters are highly expressed in brain capillary endothelial cells, including the insulin receptor, transferrin receptor protein 1, and low-density lipoprotein receptor-related protein 1.16 They can be used as targets for active brain-targeted gene therapy. Angiopep-2 peptide (Ang), an active target molecule with high affinity for lipoprotein receptor-related protein 1, has been proven to significantly improve the ability of nano-carriers to penetrate the BBB. It exhibits a much higher BBB transcytosis efficacy than transferrin and aprotinin.17–19 CPPs can penetrate the plasma membrane and the nucleus of almost any live cells.20,21 As a kind of CPP, the transactivator of transcription (TAT) peptide has shown fast endosomal or lysosomal escape and quick nuclear localization, and can easily penetrate through biofilm barriers like the BBB.22,23 TAT and Ang can bind to DSPE-PEG2000-MAL with the cysteine structure through the MAL molecule group. Magnetic nanoparticles (MNPs) are increasingly used in brain-targeted therapy. In particular, Fe3O4 MNPs exhibit promising tumor targeting ability under magnetic fields,24–26 and exert a brilliant magnetic resonance imaging performance in a noninvasive manner.27
Previous studies, which focused more on single or double target molecules, did not focus on multiple brain-targeted gene delivery systems like in this study.28,29 Considering the introduction of additional target molecules, Fe3O4 MNPs in particular, the size and instability of the nanoparticles will increase.25 The aim of this study was to improve the brain targeting of the gene delivery system, while maintaining the stability of the carrier and the particle size being within the safe range.
Another problem to be solved is the improvement of the transfection efficiency of glioma cells. Therefore, PCL3750-ss-PEG7500-ss-PCL3750, which is abbreviated as (ss)373, was chosen as the basic material, and was used to form the basic structure of the LPNPs by self-assembly with DOTAP and DSPE-PEG-MAL. Considering that the low loading rate of pDNA is one of the reasons for the low transfection efficiency, it is necessary to increase the pDNA loading rate as much as possible. (ss)373 NPs have amphiphilicity, which indicates the hydrophobic core constituted by polycaprolactone (PCL) and hydrophilic shell constituted by polyethylene glycol (PEG). The hydrophobic core can carry Fe3O4 MNPs, and the hydrophilic shell can connect with DOTAP and DSPE-PEG-MAL efficiently. DOTAP with the positive charge can compress pDNA with the negative charge, and this may increase the loading rate of pDNA.30
Another reason for the low transfection efficiency is that the vector cannot completely decompose and release enough pDNA in glioma cells. The reduction in the sensitivity of LPNPs may solve this problem. The concentration of glutathione in the cytosol, mitochondria, and cell nucleus is 100 to 1000 times higher than that in the extracellular fluids and circulation,31 and with a higher concentration in glioma cells.32 The high concentration of glutathione in glioma cells can cause a reductive cleavage of the disulfide-linked PEG shell in (ss)373 NPs, and then decompose the whole LPNPs associated with the rapid release of the encapsulated contents.33 The dissociation of LPNPs in glioma cells can completely release the pDNA, which may further increase the transfection efficiency of the glioma cells.
In this study, we focused on the construction and application of multiple brain-targeted Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs. Compared with other single or double molecule brain-targeted gene delivery systems, the multiple brain-targeted magnetic LPNPs may have better ability to penetrate the BBB and transfect glioma cells.
Experimental section
Materials
Py-ss-PEG7500-ss-Py was purchased from Biomatrik Inc. (Zhejiang, China). ε-Hexanolipid was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N [maleimide (poly(ethylene glycol)-2000)] (DSPE-PEG2000-MAL) were purchased from Nanocs Biological Technology (Beijing, China). Oleic acid-modified Fe3O4 MNPs were synthesized by Xi'an Ruixi Biological Technology Co., Ltd. (Xi'an, China). Cell-penetrating peptide (TAT) with terminal cysteine (98%, YGRKKRRQRRRC) and Angiopep-2 peptide (Ang) with terminal cysteine (95%, TFFYGGSRGKRNNFKTEEYC) were synthesized by China Peptides Co., Ltd. (Shanghai, China). The plasmid DNA (pEGFP-C1) was provided by Hailing Zhang, a professor from the Institute of Medical Biology Chinese Academy of Medical Sciences. Tryptone and yeast extract were purchased from the OXOID Biological Technology Company (Shanghai, China). The Genopure Plasmid Maxi Kit was purchased from Shanghai Roche Pharmaceuticals Limited (Shanghai, China). 6× DNA loading buffer and 50× TAE were purchased from Solarbio Life Sciences Company (Beijing, China). The cy5-labeling kit for pDNA was obtained from Mirus Bio (Madison, WI, USA).Preparation of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs
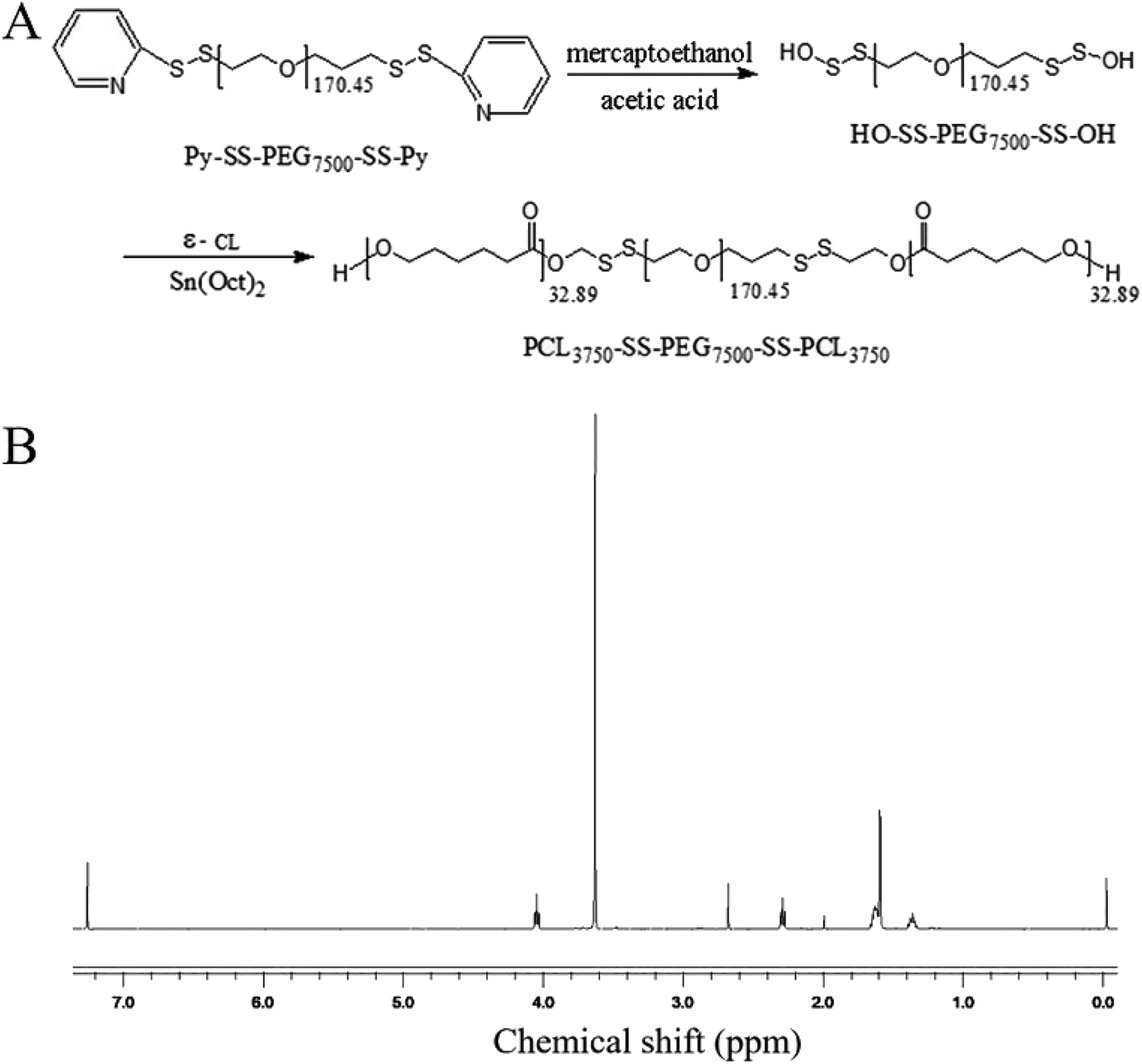
000) and placed in distilled water for 24 h. After freeze-drying, HO-ss-PEG7500-ss-OH was obtained as a pure white powder product.HO-ss-PEG7500-ss-OH (1 g) and purified ε-hexanolipid monomer (1 g) were added to a polystyrene tube. The ε-hexanolipid monomer was completely mixed with HO-ss-PEG7500-ss-OH in the oven at 50–60 °C. A drop of the catalyst stannous caprylate was added to the reaction system, then vacuumed for 5 min, and filled with nitrogen for 5 min. This process was repeated thrice. Finally, the polymerization tube was sealed under the protection of nitrogen and reacted for 24 h in an oil bath at 100 °C. At the end of the reaction, the resulting solution was dissolved in 5–10 mL dichloromethane, and then poured into 250 mL anhydrous ether. After extraction and precipitation, the pure white product was purified thrice and dried in vacuum at 350 °C. Finally, the pure white granular product, PCL3750-ss-PEG7500-ss-PCL3750, was obtained, and the product was characterized by 1H NMR and Gel Permeation Chromatography (GPC) spectrum. PCL3750-ss-PEG7500-ss-PCL3750 is abbreviated to (ss)373 according to the first digit of the molecular weight of each group and the special disulfide bond structure linking each molecular group.
:TAT is 1–2:1.28 Ang-cys (0.6 mg) and TAT-cys (0.4 mg) (mole ratio = 1:1) were dissolved and mixed well in PBS containing the prepared DOTAP-Fe3O4-(ss)373 LPNPs, and the resulting solution was allowed to react completely at 4 °C for 12 h. Then, the reaction products were dialyzed with PBS for 2 h, and the dialysate was changed thrice during dialysis. Eventually, the Ang-TAT-Fe3O4-DOTAP-(ss)373 LPNPs were prepared.In the other control groups, the prepared DOTAP-(ss)373 LPNPs were modified with either Ang or both Ang and TAT. By adding only 0.6 mg Ang-cys, 0.6 mg Ang-cys and 0.4 mg TAT-cys, and neither of them, the Ang-DOTAP-(ss)373 LPNPs, Ang-TAT-DOTAP-(ss)373 LPNPs, and DOTAP-(ss)373 LPNPs were obtained, respectively. Subsequently, the process was the same as that followed for the preparation of the Ang-TAT-Fe3O4-DOTAP-(ss)373 LPNPs.
:1, 5:1, 10:1, and 20:1, and were fully complexed by shaking at 20 °C for 30 min.35 Then, the carrier was dialyzed with 500 mL PBS for 2 h, and the dialysate was changed thrice during dialysis. Eventually, the different N/P ratios of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs were prepared. The prepared systems were directly analyzed qualitatively by agarose-gel electrophoresis (Tanon 1600/1600R, Ewell Bio-Technology Co., Ltd., Guangzhou, China) to determine the optimal N/P ratio. The N/P ratio = 5:1 was chosen for subsequent experiments.We used two fluorescent strategies to detect the position of pDNA in the LPNPs and cells. One was using Cy5 to label pDNA, which directly carried fluorescent molecules, and the labeling process was consistent with the description available in the literature.36 Another was to use pEGFP-C1 as the reporter gene and GFP as the fluorescent tracer to measure the gene transfection efficiency of the C6 cells. Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs were prepared using Cy5-labeled pDNA and pEGFP-C1 with the N/P ratio of 5:1, which was also suitable for the control groups, including pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, and Ang-TAT-pDNA-(ss)373 LPNPs.
The mean diameters and zeta potential of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs and control groups were measured using Zetasizer (Nano-ZS, Malvern instruments Ltd., Malvern, UK). The storage stability of the different modified LPNPs was determined by measuring the size change at 4 °C for 7 days. The work stability of the different modified LPNPs was determined by measuring the size change at 37 °C with the addition of ECM (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS (Gibco, Carlsbad, CA, USA) for 48 h. The particle morphology was detected by AFM (Multimode 8, Veeco Instruments, Santa Barbara, CA, USA). A CLSM (LSM 710, Carl Zeiss Meditec AG, Gena, Germany) was used to observe the location of the Cy5-labled pDNA in the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs.
Cell culture
Human brain microvascular endothelial (hCMEC/D3) cells (Thermo Fisher Scientific Company, Waltham, MA, USA) were cultured in ECM supplemented with 10% FBS. C6 Glioma (C6) cells (ACTT, Zhongyuan Ltd, Beijing, China) were cultured in DMEM/F12 medium (Gibco, Carlsbad, CA, USA) containing 10% FBS. All cells were cultured at 37 °C.The cytotoxicity was evaluated by MTS assay.37 Briefly, hCMEC/D3 and C6 cells were separately seeded in two 96-well plates at a density of 5 × 103 cells per well, and incubated at 37 °C for 24 h. Then, Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs were diluted with PBS (pH 7.4) to 50 μg mL−1, 100 μg mL−1, 150 μg mL−1, 200 μg mL−1, 300 μg mL−1, and 400 μg mL−1. Each concentration of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was added to six wells of each 96-well plate. Two additional groups were set as the control. One was a blank control group that only had the ECM and incubated cells without LPNPs; the other was the negative control group that only had the ECM and the LPNPs without incubated cells. After processing, the two 96-well plates were incubated at 37 °C for 24 h. Then, the MTS reagent (20 μL) was added to each well and incubated at 37 °C for 2 h in the dark. The plate was read at 490 nm using a microplate reader (Bio-Rad, Hercules, CA, USA) to identify the cytotoxicity of the different modified LPNPs. In the pre-experiment, we determined that the appropriate concentration of pDNA was 0.4 μg mL−1 (12-well plate). The corresponding concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was calculated as approximately 150 μg mL−1, and this concentration was used for the entire study.
Preparation of monolayer cell BBB model in vitro and measurement of penetration rate
A Transwell plate and hCMEC/D3 cells were used to establish the monolayer cell BBB model in vitro. The second or third generation passage hCMEC/D3 cells were placed at a density of 4 × 105 per well in the upper chambers of a Transwell 3460 (Corning, USA) plate. ECM medium was added to the lower chambers of Transwell plate. The medium was changed every day, and the Transwell plate was placed in an incubator at 37 °C. The transendothelial electric resistance (TEER) value of the BBB model was detected using Millicell ERS (MERS00001, Millipore, MA, USA). When the TEER value reached above 100 Ω cm2 and was stabilized, the actin-tracker Green and DAPI were used for staining in the well. The formation of tight connections between the hCMEC/D3 cells was observed by CLSM to determine whether the BBB model was successfully constructed. We also detected the TEER value of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs at the concentration of 150 μg mL−1, and the TEER value of the other different modified LPNPs in the control groups before and after penetrating the BBB under a magnetic field. The aim was to identify whether the carriers themselves would destroy the BBB structure. With the concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs at 150 μg mL−1, the BBB penetration rates of the different modified LPNPs were determined by observing the fluorescence intensity of the Cy5-labled pDNA using a fluorescence spectrophotometer (FL 6500, PerkinElmer, MA, USA).Preparation of the glioma model in vitro and measurement of the transfection rate of C6 cells after BBB penetration
C6 cells were seeded at a density of 4 × 105 cells per well in the lower chamber of a Transwell plate (70–80% confluence). The hCMEC/D3 cells, which had reached the criteria of the BBB model mentioned above, were placed in the upper chamber. To the upper chamber of the Transwell plate, a 2 μg per well mixture was added that contained 150 μg mL−1 Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs and DMEM/F12 medium. The magnet was placed at the bottom of the plate, and the cells were cultivated for 24 h. After processing, the expression of GFP was observed in the different groups, which had penetrated the BBB model to transfect the C6 cells using CLSM, and the transfection rates of the different modified LPNPs were analyzed using flow cytometry (BD FACS Calibur, BD Biosciences, CA, USA).Statistical analysis
All data are presented as the mean ± standard deviation (SD). The differences among the different modified LPNPs were analysed by one-way analysis of variance (ANOVA) using SPSS 23.0. P-value <0.05 was considered to be significant, and P-value <0.01 was considered as highly significant.Results
The preparation and characterization of PCL3750-ss-PEG7500-ss-PCL3750 [(ss)373]
As shown in Fig. 1A, the reduction-sensitive material disulfide-bridged triblock copolymer poly(ε-caprolactone)3750-ss-poly(ethylene glycol)7500-ss-poly(ε-caprolactone)3750(PCL3750-ss-PEG7500-ss-PCL3750) was synthesized by ring-opening polymerization of ε-CL initiated by HO-ss-PEG7500-ss-OH using Sn(Oct)2 as a catalyst. The product was characterized by 1H NMR. The 1H NMR spectrum was recorded, and is shown in Fig. 1B. The peak at 3.63 ppm was assigned to the methylene protons of –OCH2CH2– in the PEG units. Furthermore, the peaks at 1.40, 1.64, 2.30 and 4.04 ppm were assigned to the methylene protons of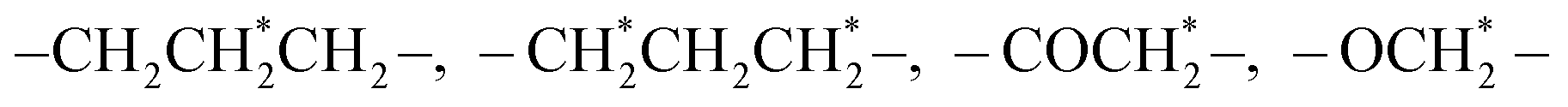 in the PCL units, respectively. In addition, the number average molecular weight (Mn) based on 1H NMR was 13442. Besides, the Gel Permeation Chromatography (GPC) spectrum reported that the Mn was 19844, and the weight average molecular weight (Mw) was 30116. These results indicated that the copolymer was synthesized successfully.
in the PCL units, respectively. In addition, the number average molecular weight (Mn) based on 1H NMR was 13442. Besides, the Gel Permeation Chromatography (GPC) spectrum reported that the Mn was 19844, and the weight average molecular weight (Mw) was 30116. These results indicated that the copolymer was synthesized successfully.
 | ||
| Fig. 1 (A) Synthesis of PCL3750-ss-PEG7500-ss-PCL3750; (B) 1H NMR spectrum of PCL3750-ss-PEG7500-ss-PCL3750. | ||
Structure and characterization of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs
A schematic diagram of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs is shown in Fig. 2A. PCL3750-ss-PEG7500-ss-PCL3750 was successfully synthesized, and is named (ss)373 NPs herein. DOTAP, DSPE-PEG2000-MAL, and (ss)373 NPs formed the basic structure of LPNPs by self-assembly, which had a hydrophobic core and hydrophilic shell. The PCL groups of (ss)373 NPs constructed the hydrophobic core, and the Fe3O4 MNPs were wrapped in the hydrophobic core to achieve the effect for magnetic targeting. The PEG groups of the (ss)373 NPs formed the hydrophilic shell, which was separated from the hydrophobic core by disulfide bonds. Then, TAT-cys and Ang-cys were coupled with the MAL molecule group of DSPE-PEG2000-MAL. Finally, pDNA was compressed on the positively charged DOTAP to form a complete gene delivery system. | ||
| Fig. 2 (A) Schematic diagram of the preparation of Ang-TAT-Fe3O4-(ss)373 lipid-polymer hybrid nanoparticles (LPNPs). (B) Atomic force microscope image of Ang-TAT-Fe3O4-(ss)373 LPNPs. (C) Stability of LPNPs with different modifications stored at 4 °C dissolved in phosphate-buffered saline. (D) Stability of LPNPs with different modifications stored at 37 °C dissolved in endothelial cell medium with 10% fetal bovine serum. | ||
The morphology of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was characterized using atomic force microscopy (AFM). Fig. 2B indicates that the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs showed a well-formed spherical shape. As shown in Table 1, the average particle size of the (ss)373 NPs, pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, and Ang-TAT-pDNA-(ss)373 LPNPs was 84.55 nm, 181.00 nm, 193.00 nm, and 199.33 nm, respectively. The PDI values of those formulations all demonstrated a fairly homogeneous distribution. The average particle size of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs increased to 302.33 nm, demonstrating the successful encapsulation of the Fe3O4 MNPs. The average zeta potential of (ss)373 NPs, pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, and Ang-TAT-pDNA-(ss)373 LPNPs was −11.73 mV, 6.00 mV, 8.36 mV, and 9.07 mV, respectively. Owing to the influence of Fe3O4, the average zeta potential of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was decreased to 4.66 mV. Fig. 2C shows that the different modified LPNPs were able to maintain the size for up to 7 days at almost a constant level at 4 °C in phosphate-buffered saline (PBS). Fig. 2D shows that the different modified LPNPs were able to maintain stability for at least 2 days at 37 °C in endothelial cell medium (ECM) supplemented with 10% fetal bovine serum (FBS).
| Formulation | Size (nm) | Zeta potential (mV) | Polydispersity index (PDI) |
|---|---|---|---|
| (ss)373 | 84.55 ± 0.27 | −11.73 ± 0.37 | 0.28 ± 0.03 |
| pDNA-(ss)373 | 181.00 ± 3.74 | 6.00 ± 0.10 | 0.28 ± 0.02 |
| Ang-pDNA-(ss)373 | 193.00 ± 0.82 | 8.36 ± 0.22 | 0.27 ± 0.03 |
| Ang-TAT-pDNA-(ss)373 | 199.33 ± 1.70 | 9.07 ± 0.24 | 0.23 ± 0.02 |
| Ang-TAT-Fe3O4-pDNA-(ss)373 | 302.33 ± 3.68 | 4.66 ± 0.15 | 0.13 ± 0.02 |
Determination of the optimal nanoparticles/pDNA (N/P) ratio
Based on the results of the gel retardation assay, when the N/P ratio is >5:1, the entrapped pDNA can remain in the wells (Fig. 3A). Considering the improvement in the encapsulation rate, the N/P ratio = 5:1 was chosen for application in the entire study. As shown in Fig. 3B, as observed by laser scanning confocal microscopy (CLSM), the Cy5-labled pDNA was connected tightly with the carrier, and still maintained a stable spherical shape.
 | ||
| Fig. 3 (A) Gel retardation assay of the Ang-TAT-Fe3O4-pDNA-(ss)373 lipid-polymer hybrid nanoparticles (LPNPs) at different N/P ratios. (B) Laser scanning confocal microscopy image of Ang-TAT-Fe3O4-(Cy5) pDNA-(ss)373 LPNPs. | ||
Cytotoxicity of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs in vitro
The cytotoxicity of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs at a series of concentrations was evaluated in hCMEC/D3 and C6 cells using MTS reagent kits. As shown in Fig. 4, although the concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs reached 400 μg mL−1, the cell viability of both hCMEC/D3 and C6 cells was more than 95% at 2 h and 24 h, and there was no significant difference between the two time periods. According to the results of the pre-experiment, the appropriate concentration of pDNA was 0.4 μg mL−1 (12-well plate) and the corresponding concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was calculated as approximately 150 μg mL−1, which indicated that there is no apparent cytotoxicity of the gene delivery system toward C6 and hCMEC/D3 cells. The Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs at the concentration of 150 μg mL−1 were used in all experiments in this study.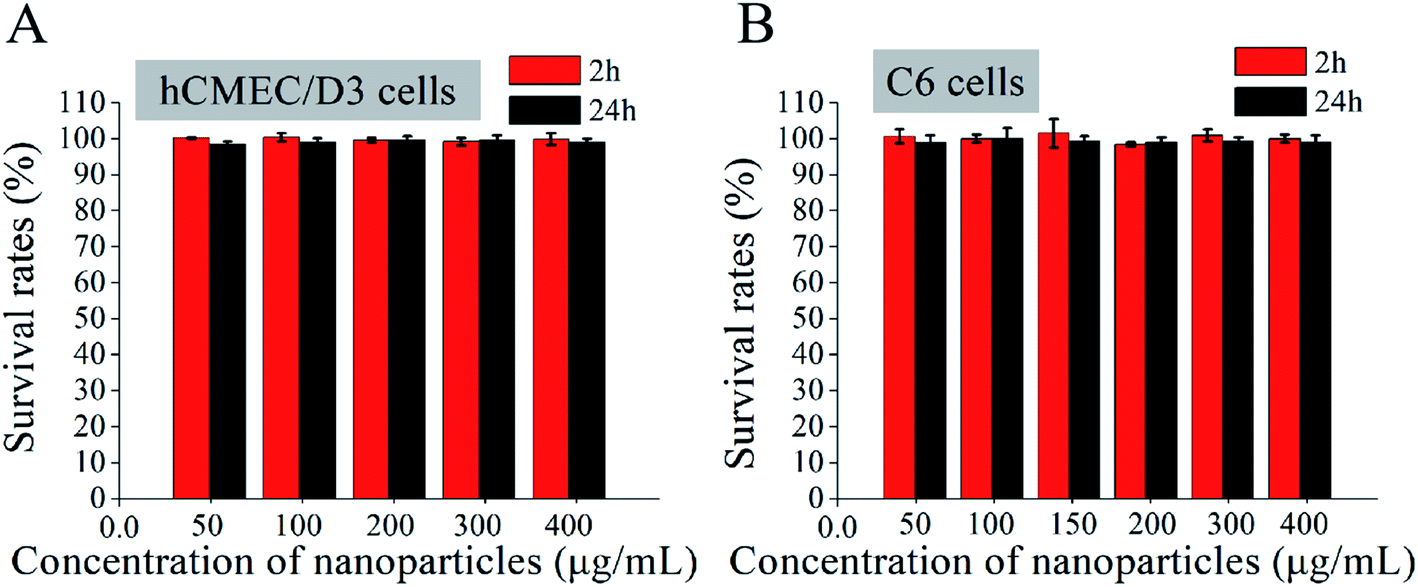 | ||
| Fig. 4 (A) Survival rates of hCMEC/D3 cells cultured with different concentrations of Ang-TAT-Fe3O4-pDNA-(ss)373 lipid-polymer hybrid nanoparticles (LPNPs) at 2 h/24 h. (B) Survival rates of C6 cells cultured with different concentrations of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs at 2 h/24 h. All data are represented as the mean ± SD (n = 5). | ||
BBB penetration rate of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs
Fig. 5A shows the in vitro monolayer BBB model formed by hCMEC/D3 cells to identify the BBB penetration efficiency of different modified LPNPs. The trans-endothelial electrical resistance (TEER) value of the BBB model was detected to ascertain whether the hCMEC/D3 cells had formed the BBB structure successfully. As shown in Fig. 5B, the TEER value in this study tended to be stable on the fourth day and exceeded 130 Ω cm2. Moreover, as shown in Fig. 5C, the CLSM images of hCMEC/D3 cells stained with actin-tracker Green and DAPI further confirmed the formation of tight junctions. After different modified LPNPs had penetrated the BBB model, its TEER value was also detected. As shown in Fig. 5D, there was no significant change, which indicates that the different modified LPNPs had almost no damage on the integrity of the BBB model. Fig. 5E shows the BBB penetration rate of different modified LPNPs analyzed semi-quantitatively at 2 h, 6 h, 12 h, and 24 h with the magnet under the Transwell plate. The concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was 150 ug mL−1, and the pDNA was labeled by the Cy5 molecule in this experiment. The BBB penetration rate of pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, Ang-TAT-pDNA-(ss)373 LPNPs, and Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was 0.44%, 0.63%, 1.02%, and 1.55%, respectively, at 2 h. The accumulated BBB penetration rate of pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, Ang-TAT-pDNA-(ss)373 LPNPs, and Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was 22.00%, 23.35%, 27.22%, and 29.69% respectively, in a total of 24 h. Compared with the others, the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs showed a significant difference in penetrating the BBB (P < 0.05), and the penetration rate increased with prolonged incubation.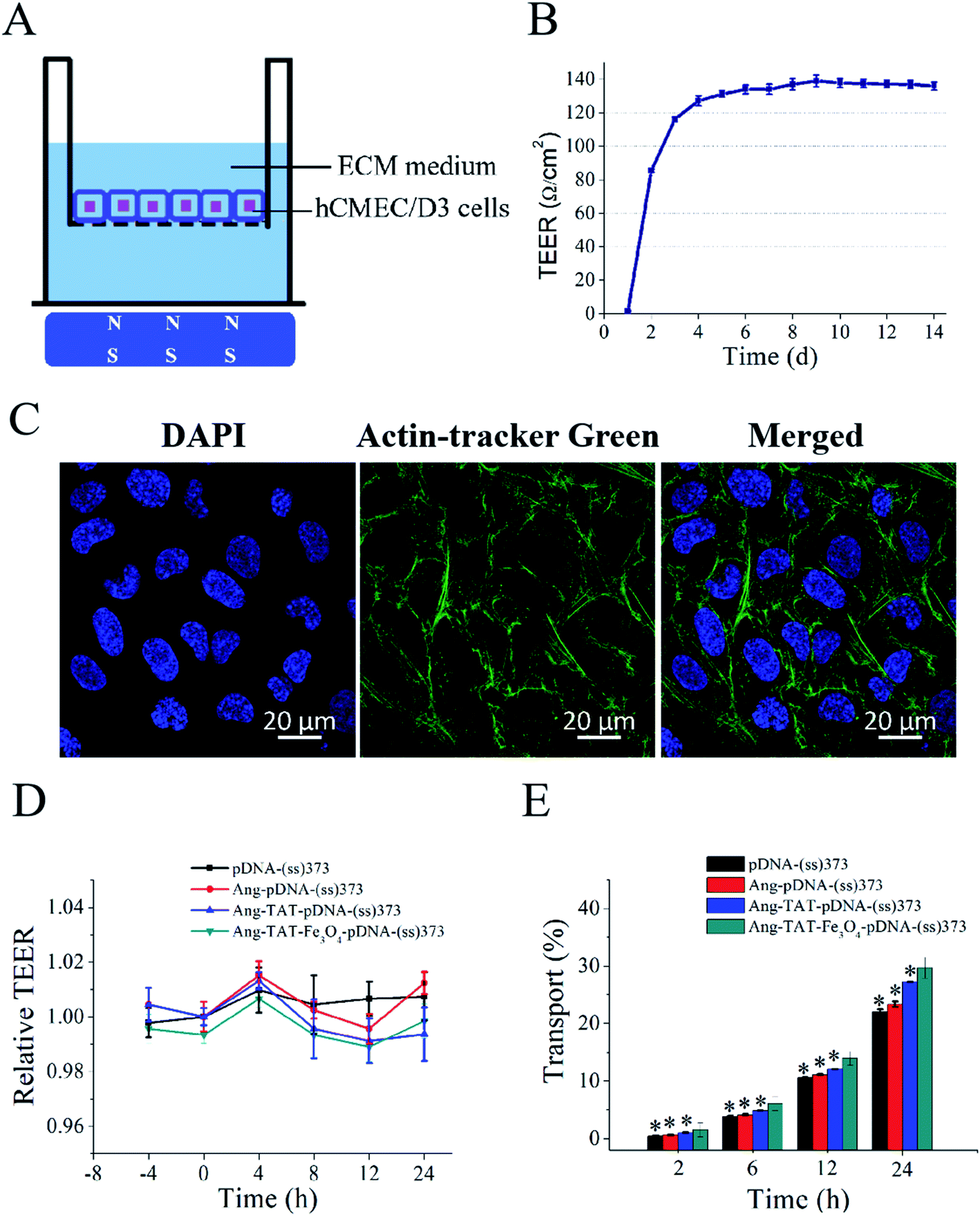 | ||
| Fig. 5 (A) Schematic diagram of the BBB model constructed by the monolayer hCMEC/D3 cells in vitro [the model was placed on a magnetic field]. (B) Trend of trans-endothelial electrical resistance (TEER) values of the constructed BBB model with time. (C) Laser scanning confocal microscopy images of the actin-tracker Green- and DAPI-stained BBB model (scale bar = 20 μm). (D) TEER value changes in the BBB model after treatment with lipid-polymer hybrid nanoparticles (LPNPs) with different modifications. (E) Mean relative transport rate of LPNPs with different modifications in the BBB model. [Magnet placed under the BBB model.] All data are represented as mean ± SD (n = 3); *P < 0.05 versus Ang-TAT-Fe3O4-pDNA-(ss)373. | ||
In vitro experiment for transfection of C6 cells through BBB
Fig. 6A shows the multiple targeting model constructed using hCMEC/D3 and C6 cells, which was designed to detect the transfection of C6 cells through the BBB in vitro. The C6 cells were seeded in the lower chamber of Transwell plates. The hCMEC/D3 cells, which had reached the criteria of the BBB model mentioned above, were placed in the upper chamber. The magnet was placed under the Transwell plate to exert a magnetic targeting effect. Using pEGFP-C1 as the reporter gene, the green fluorescent protein (GFP) was expressed after successfully transfecting the C6 cells. GFP was used as a tracer to detect the transfection of C6 cells. The CLSM images in Fig. 6B show the transfection of C6 cells with the different modified LPNPs after 24 h. The concentration of Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was 150 μg mL−1. There was almost no GFP expression in the free pEGFP-C1 group. With the addition of target molecules, the GFP expression increased gradually. The maximum GFP expression was achieved in the pEGFP-C1 loaded with the Ang-TAT-Fe3O4-(ss)373 LPNPs group. The flow cytometry results are shown in Fig. 6C. The transfection rate of C6 cells with free pDNA, pDNA-(ss)373 LPNPs, Ang-pDNA-(ss)373 LPNPs, Ang-TAT-pDNA-(ss)373 LPNPs, and Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was 0.143%, 1.51%, 5.20%, 8.23%, and 14.6% at 24 h, respectively. Fig. 6D shows the processed data, which illustrates that compared with free pEGFP-C1, the transfection rate of pEGFP-C1 loaded with the different modified LPNPs was improved gradually. The transfection efficiency of the C6 cells with Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was the highest, reaching 14.6% with significant differences from that of other LPNPs at all detected time points (P < 0.001). With the addition of target molecules, the transfection efficiency of C6 treated with different modified LPNPs was significantly different (P < 0.001).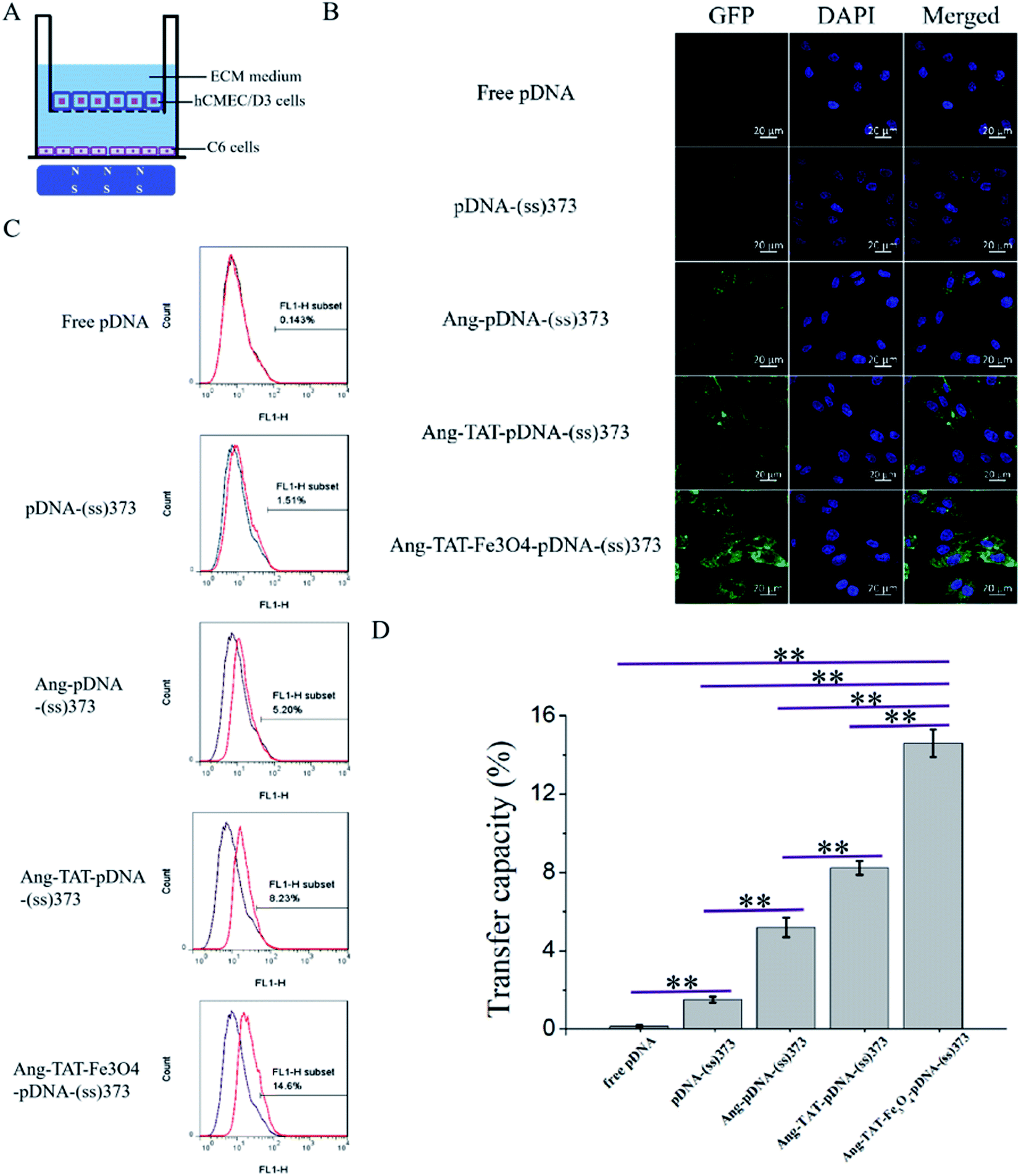 | ||
| Fig. 6 (A) Schematic diagram of the multiple target model constructed using monolayer hCMEC/D3 and C6 cells [the model was placed on the magnetic field]. (B) Fluorescent images of free pDNA and lipid-polymer hybrid nanoparticles (LPNPs) with different modifications to transfect the C6 cells through the multiple target model under laser scanning confocal microscopy after 24 h (scale bar = 20 μm). (C) Ability of free pEGFP-C1 and LPNPs with different modifications to transfect C6 cells through the BBB model measured by flow cytometer after 24 h. (D) Transfer capacity of free pDNA and LPNPs with different modifications to transfect C6 cells through the BBB model. **P < 0.001. a: Free pDNA; b: pDNA-(ss)373; c: Ang-pDNA-(ss)373; d: Ang-TAT-pDNA-(ss)373; e: Ang-TAT-Fe3O4-pDNA-(ss)373. | ||
Discussion
Currently, it is urgent to improve the therapeutic effect of glioma, and gene therapy may be a potential and promising therapeutic method. Two key points to achieve glioma gene therapy are to increase the BBB penetration efficiency and the transfection efficiency of glioma cells. We designed and synthesized Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs in this study to partially solve the two issues. The Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs showed greater efficiency of BBB penetration and glioma cells transfection in vitro. In this study, three methods were used to improve the targeting of the gene to the brain, namely, the introduction of Ang, TAT, and Fe3O4. Previous studies have introduced either or both Ang and TAT, which achieved good effects in BBB penetration.19,29,38 When the ratio of Ang:TAT is 1–2:1, relatively high targeting and low cytotoxicity can be obtained.28 Magnetic targeting is also a popular brain targeting method.39–41 Our research integrated the above three targeting methods to greatly improve the penetration efficiency of the BBB under an external magnetic field.
However, with the introduction of Fe3O4, the size of the LPNPs also expanded. The mean diameter of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs increased to 302.33 nm. In this study, the result of the cytotoxicity assay showed that this particle size did not cause any apparent damage to the hCMEC/D3 and C6 cells. Moreover, excellent stability plays an important role in the long-term storage and normal exertion of functions. The Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs with this size were able to sustain at least 48 h without aggregation and decomposition under working conditions, as well as under 4 °C storage conditions. This result was similar to that reported by the other studies with a particle size greater than 300 nm, which was still within a safe range and did not cause damage to the brain cells.42,43 Therefore, Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs with a diameter of approximately 302.33 nm might safely and effectively penetrate the BBB, and enter glioma cells to achieve gene transfection in vitro.
The average zeta potential of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs was around 4.66 mV, which made them easier to approach the negatively charged membrane of the brain capillary endothelial cells.44,45 The reason the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs possessed a positive zeta potential was that DOTAP and DSPE-PEG2000-MAL are cationic liposomes, especially DOTAP, which can contribute to the whole lipid formulation to present a positive zeta potential.46,47 The positively charged DOTAP also allows it to connect tightly with the negative charge of pDNA.48 Both TAT and Ang-2 have a positive charge. When they were used as a modification, the zeta potential increased positively.49,50 The zeta potential of Fe3O4 MNPs will change with different pH values. In this study, the pH value of the PBS buffer was 7.45, which was used to measure the zeta potential. At this pH value, the Fe3O4 MNPs show negative zeta potential and can neutralize parts of the positive charges.51
Preparing the BBB model successfully is essential for identifying the BBB penetration rate. In this study, the BBB model was established using a monolayer of hCMEC/D3 cells. The formation of the BBB structure was determined by two methods: by measuring the TEER value of the BBB model and observing the stained hCMEC/D3 cells with CLSM. It is generally considered that BBB in mammalian systems show high TEER values, well above 1000 Ω cm2. However, it is difficult to achieve the TEER value above 1000 Ω cm2 in vitro, especially in cell lines compared with that in primary culture. The monolayers that are formed by hCMEC/D3 cells develop only a low to medium level TEER (around 30–50 Ω cm2) under static culture conditions, according to various reports.52 The TEER value in this study exceeded 130 Ω cm2 and maintained stability. Previous studies have shown that the BBB structure is formed under this TEER value.53–55 It has also been reported that a higher TEER value can be obtained by co-culturing with other cells, such as astrocytes, which can increase the TEER value to approximately 40 Ω cm2.56 The successful release of pDNA by the gene delivery system in glioma cells is very important to improve gene transfection. In order to improve the gene transfection efficiency, the reduction-sensitive material (ss)373 NPs with disulfide bonds was selected. Previous studies have proven that the dissociation of carriers containing disulfide bonds increases in glioma cells.31,57 Disulfide bonds were introduced based on multi-targeting, so that the gene delivery system can decompose relatively specifically in C6 cells with a high concentration of glutathione. Through analyzing the flow cytometry results and CLSM images with GFP as the tracer, the introduction of disulfide bonds that can improve the transfection efficiency of C6 cells was confirmed. According to the results of this study, the mechanism of LPNPs penetrating the BBB should be receptor-mediated endocytosis. However, we did not perform the relevant experiments for further confirmation. In this experiment, the types of cells were relatively simple; only the most typically used hCMEC/D3 cells and C6 glioma cells were selected, and the results were not compared with those of other cell lines. In the follow-up studies, we will verify the effectiveness of the Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs-loaded therapeutic gene for glioma treatment through animal experiments, and further clarify the mechanism of BBB penetration.
Conclusions
To summarize, gene therapy is currently considered a promising approach for glioma treatment. The Ang-TAT-Fe3O4-pDNA-(ss)373 LPNPs designed and synthesized in this study are a multi-targeting gene delivery system with several advantages, such as high BBB penetration efficiency, low toxicity, stability, and high transfection efficiency. These advantages may provide new opportunities for the therapeutic gene to penetrate the BBB to treat brain diseases.Author contributions
X. J. and L. Z. conceived and designed the experiments. L. Q., Y. Q., and D. Z. performed the experiments. L. Q. contributed to the data analysis. Writing, editing, and reviewing were performed by Y. W., L. L., W. X., L. L., and D. B. All authors have read and agreed to the published version of the manuscript.Funding
This work was supported by the National Natural Science Foundation of Beijing (No. 7172072); National Natural Science Foundation of China (No. 81671806 and 81571793); CAMS Initiative for Innovative Medicine (No. 2017-I2M-4-001 and 2017-I2M-3-020); Beijing Dongcheng District Outstanding Talent Funding Project (No. 2019DCT-M-17); Scientific Research and Cultivation Fund of Capital Medical University (No. PYZ19097); Scientific Research and Cultivation Fund of Beijing Tiantan Hosipital (No. YZR-2020-13); and Tianjin Municipal Natural Science Foundation (No. 15JCZDJC38300).List of abbreviations
| AFM | Atomic force microscopy |
| Ang | Angiopep-2 peptide |
| BBB | Blood–brain barrier |
| CLSM | Laser scanning confocal microscopy |
| CPPs | Cell-penetrating peptides |
| DOTAP | 1,2-Dioleoyl-3-trimethylammonium-propane 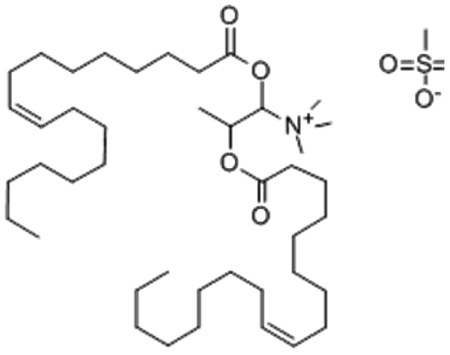 |
| DSPE-PEG2000-MAL | 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N [maleimide (poly(ethylene glycol)-2000)]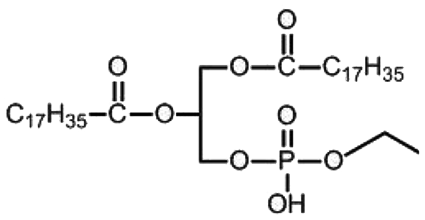 |
| ECM | Supplemented with 10% fetal bovine serum |
| FBS | Endothelial cell medium |
| GFP | Green fluorescent protein |
| GPC | Gel permeation chromatography |
| LPNPs | Lipid-polymer hybrid nanoparticles |
| MNPs | Magnetic nanoparticles |
| PBS | Phosphate-buffered saline |
| PCL | Polycaprolactone 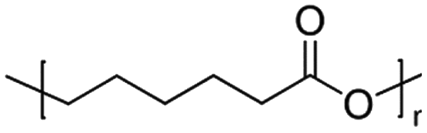 |
| PCL3750-ss-PEG7500-ss-PCL3750 | Poly(ε-caprolactone)3750-ss-poly(ethylene glycol)7500-ss-poly(ε-caprolactone)3750 |
| PEG | Polyethylene glycol  |
| (ss)373 | PCL3750-ss-PEG7500-ss-PCL3750 |
| TAT | Transactivator of transcription |
| TEER | Trans-endothelial electrical resistance |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Hailing Zhang, a professor from the Institute of Medical Biology Chinese Academy of Medical Sciences, for her support with plasmid DNA.References
- L. Zhang, L. Pang, S. Zhu, J. Ma, R. Li, Y. Liu, L. Zhu, X. Zhuang, W. Zhi, X. Yu, L. Du, H. Zuo and Y. Jin, Int. J. Pharm., 2020, 583, 119384 CrossRef CAS.
- S. L. Hervey-Jumper and M. S. Berger, Curr. Treat. Options Neurol., 2014, 16, 284 CrossRef.
- H. Yao, K. Wang, Y. Wang, S. Wang, J. Li, J. Lou, L. Ye, X. Yan, W. Lu and R. Huang, Biomaterials, 2015, 37, 345–352 CrossRef CAS.
- S. A. Pena, R. Iyengar, R. S. Eshraghi, N. Bencie, J. Mittal, A. Aljohani, R. Mittal and A. A. Eshraghi, J. Drug Target., 2020, 28, 111–128 CrossRef CAS.
- B. Caffery, J. S. Lee and A. A. Alexander-Bryant, Nanomaterials, 2019, 9, 105 CrossRef.
- A. Wahane, A. Waghmode, A. Kapphahn, K. Dhuri, A. Gupta and R. Bahal, Molecules, 2020, 25, 2886 CrossRef.
- D. L. Puhl, A. R. D'Amato and R. J. Gilbert, Brain Res. Bull., 2019, 150, 216–230 CrossRef CAS.
- E. Neuwelt, N. J. Abbott, L. Abrey, W. A. Banks, B. Blakley, T. Davis, B. Engelhardt, P. Grammas, M. Nedergaard, J. Nutt, W. Pardridge, G. A. Rosenberg, Q. Smith and L. R. Drewes, Lancet Neurol., 2008, 7, 84–96 CrossRef CAS.
- A. Alexander, M. Agrawal, A. Uddin, S. Siddique, A. M. Shehata, M. A. Shaker, S. Ata Ur Rahman, M. I. M. Abdul and M. A. Shaker, Int. J. Nanomed., 2019, 14, 5895–5909 CrossRef CAS.
- N. J. Abbott, A. A. Patabendige, D. E. Dolman, S. R. Yusof and D. J. Begley, Neurobiol. Dis., 2010, 37, 13–25 CrossRef CAS.
- M. M. Patel and B. M. Patel, CNS Drugs, 2017, 31, 109–133 CrossRef CAS.
- K. Nagpal, S. K. Singh and D. N. Mishra, Expet Opin. Drug Deliv., 2013, 10, 927–955 CrossRef CAS.
- Y. Song, D. Du, L. Li, J. Xu, P. Dutta and Y. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 20410–20416 CrossRef CAS.
- L. I. Goulatis and E. V. Shusta, Curr. Opin. Struct. Biol., 2017, 45, 109–115 CrossRef CAS.
- Y. Cui, Q. Xu, P. K. Chow, D. Wang and C. H. Wang, Biomaterials, 2013, 34, 8511–8520 CrossRef CAS.
- S. Ohtsuki, C. Ikeda, Y. Uchida, Y. Sakamoto, F. Miller, F. Glacial, X. Decleves, J. M. Scherrmann, P. O. Couraud, Y. Kubo, M. Tachikawa and T. Terasaki, Mol. Pharm., 2013, 10, 289–296 CrossRef CAS.
- T. Tian, J. Li, C. Xie, Y. Sun, H. Lei, X. Liu, J. Xia, J. Shi, L. Wang, W. Lu and C. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 3414–3420 CrossRef CAS.
- Y. Bertrand, J. C. Currie, M. Demeule, A. Regina, C. Che, A. Abulrob, D. Fatehi, H. Sartelet, R. Gabathuler, J. P. Castaigne, D. Stanimirovic and R. Beliveau, J. Cell Mol. Med., 2010, 14, 2827–2839 CrossRef CAS.
- H. Xin, X. Sha, X. Jiang, L. Chen, K. Law, J. Gu, Y. Chen, X. Wang and X. Fang, Biomaterials, 2012, 33, 1673–1681 CrossRef CAS.
- A. Ur Rahman, S. Khan and M. Khan, J. Pharm. Pharmacol., 2020, 72, 519–530 CrossRef CAS.
- M. Kristensen, D. Birch and H. Morck Nielsen, Int. J. Mol. Sci., 2016, 17, 185 CrossRef.
- D. Zhang, J. Wang and D. Xu, J. Controlled Release, 2016, 229, 130–139 CrossRef CAS.
- N. Todorova, C. Chiappini, M. Mager, B. Simona, I. I. Patel, M. M. Stevens and I. Yarovsky, Nano Lett., 2014, 14, 5229–5237 CrossRef CAS.
- J. Cai, Y. Q. Miao, B. Z. Yu, P. Ma, L. Li and H. M. Fan, Langmuir, 2017, 33, 1662–1669 CrossRef CAS.
- L. Shen, B. Li and Y. Qiao, Materials, 2018, 11, 324 CrossRef.
- C. H. Su, C. Y. Tsai, B. Tomanek, W. Y. Chen and F. Y. Cheng, Nanoscale, 2016, 8, 7866–7870 RSC.
- N. Zhang, Y. Wang, C. Zhang, Y. Fan, D. Li, X. Cao, J. Xia, X. Shi and R. Guo, Theranostics, 2020, 10, 2791–2802 CrossRef CAS.
- Y. Zhu, Y. Jiang, F. Meng, C. Deng, R. Cheng, J. Zhang, J. Feijen and Z. Zhong, J. Controlled Release, 2018, 278, 1–8 CrossRef CAS.
- W. Han, G. Yin, X. Pu, X. Chen, X. Liao and Z. Huang, J. Biomater. Sci., Polym. Ed., 2017, 28, 1695–1712 CrossRef CAS.
- B. B. M. Garcia, O. Mertins, E. R. D. Silva, P. D. Mathews and S. W. Han, Colloids Surf. B Biointerfaces, 2020, 193, 111131 CrossRef CAS.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS.
- Y. Su, Y. Hu, Y. Du, X. Huang, J. He, J. You, H. Yuan and F. Hu, Mol. Pharm., 2015, 12, 1193–1202 CrossRef CAS.
- H. Y. Wen, H. Q. Dong, W. J. Xie, Y. Y. Li, K. Wang, G. M. Pauletti and D. L. Shi, Chem. Commun., 2011, 47, 3550–3552 RSC.
- L. H. Zhang, C. Y. Hu, S. J. Wu, Z. Chen, F. Fan, Y. Qin and D. W. Zhu, J. Controlled Release, 2017, 259, E84–E85 CrossRef.
- Q. X. Chen, K. Osada, Z. S. Ge, S. Uchida, T. A. Tockary, A. Dirisala, A. Matsui, K. Toh, K. M. Takeda, X. Y. Liu, T. Nomoto, T. Ishii, M. Oba, Y. Matsumoto and K. Kataoka, Biomaterials, 2017, 113, 253–265 CrossRef CAS.
- M. Z. Cao, Y. Gao, N. S. Qiu, Y. Q. Shen and P. H. Shen, J. Drug Deliv. Sci. Technol., 2020, 56, 1773–2247 Search PubMed.
- A. Rizzo, S. Donzelli, V. Girgenti, A. Sacconi, C. Vasco, A. Salmaggi, G. Blandino, M. Maschio and E. Ciusani, J. Exp. Clin. Canc. Res., 2017, 36, 76 CrossRef.
- S. Huang, J. Li, L. Han, S. Liu, H. Ma, R. Huang and C. Jiang, Biomaterials, 2011, 32, 6832–6838 CrossRef CAS.
- L. L. Israel, A. Galstyan, E. Holler and J. Y. Ljubimova, J. Controlled Release, 2020, 320, 45–62 CrossRef CAS.
- A. Skouras, K. Papadia, S. Mourtas, P. Klepetsanis and S. G. Antimisiaris, Eur. J. Pharm. Sci., 2018, 123, 162–172 CrossRef CAS.
- F. D'Agata, F. A. Ruffinatti, S. Boschi, I. Stura, I. Rainero, O. Abollino, R. Cavalli and C. Guiot, Molecules, 2017, 23, 9 CrossRef.
- B. Ramaswamy, S. D. Kulkarni, P. S. Villar, R. S. Smith, C. Eberly, R. C. Araneda, D. A. Depireux and B. Shapiro, Nanomedicine, 2015, 11, 1821–1829 CrossRef CAS.
- H. Ding, V. Sagar, M. Agudelo, S. Pilakka-Kanthikeel, V. S. Atluri, A. Raymond, T. Samikkannu and M. P. Nair, Nanotechnology, 2014, 25, 055101 CrossRef.
- X. Wang, Y. Zhao, S. Dong, R. J. Lee, D. Yang, H. Zhang and L. Teng, Molecules, 2019, 24, 3540 CrossRef CAS.
- Y. C. Kuo and I. C. Chen, J. Phys. Chem. B, 2007, 111, 11228–11236 CrossRef CAS.
- B. Dos Santos Rodrigues, S. Lakkadwala, T. Kanekiyo and J. Singh, Int. J. Nanomed., 2019, 14, 6497–6517 CrossRef.
- B. D. S. Rodrigues, T. Kanekiyo and J. Singh, Mol. Pharm., 2020, 17, 2054–2063 CrossRef CAS.
- G. Caracciolo and H. Amenitsch, Eur. Biophys. J., 2012, 41, 815–829 CrossRef CAS.
- A. Borrelli, A. L. Tornesello, M. L. Tornesello and F. M. Buonaguro, Molecules, 2018, 23, 295 CrossRef.
- M. Demeule, A. Regina, C. Che, J. Poirier, T. Nguyen, R. Gabathuler, J. P. Castaigne and R. Beliveau, J. Pharmacol. Exp. Ther., 2008, 324, 1064–1072 CrossRef CAS.
- I. Antal, O. Strbak, I. Khmara, M. Koneracka, M. Kubovcikova, V. Zavisova, M. Kmetova, E. Baranovicova and D. Dobrota, Nanomaterials, 2020, 10, 394 CrossRef CAS.
- B. Weksler, I. A. Romero and P. O. Couraud, Fluids Barriers CNS, 2013, 10, 16 CrossRef.
- Y. Mi, Y. Mao, H. Cheng, G. Ke, M. Liu, C. Fang and Q. Wang, Fitoterapia, 2020, 140, 104447 CrossRef CAS.
- L. Chen, W. Liu, P. Wang, Y. Xue, Q. Su, C. Zeng and X. Shang, Brain Res., 2015, 1606, 44–53 CrossRef CAS.
- W. Liu, P. Wang, C. Shang, L. Chen, H. Cai, J. Ma, Y. Yao, X. Shang and Y. Xue, Brain Res., 2014, 1573, 17–26 CrossRef CAS.
- B. P. Daniels, L. Cruz-Orengo, T. J. Pasieka, P. O. Couraud, I. A. Romero, B. Weksler, J. A. Cooper, T. L. Doering and R. S. Klein, J. Neurosci. Methods, 2013, 212, 173–179 CrossRef CAS.
- L. Lu, X. Zhao, T. Fu, K. Li, Y. He, Z. Luo, L. Dai, R. Zeng and K. Cai, Biomaterials, 2020, 230, 119666 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |