
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the solubility of 1,4-diaminoanthraquinones in electrolytes for organic redox flow batteries through molecular modification†
Pieter Geysens a,
Jorik Eversa,
Wim Dehaena,
Jan Fransaerb and
Koen Binnemans*a
a,
Jorik Eversa,
Wim Dehaena,
Jan Fransaerb and
Koen Binnemans*a
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: Koen.Binnemans@kuleuven.be
bKU Leuven, Department of Materials Engineering, Kasteelpark 44, P.O. box 2450, B-3001 Leuven, Belgium
First published on 29th October 2020
Abstract
1,4-Diaminoanthraquinones (DAAQs) are a promising class of redox-active molecules for use in nonaqueous redox flow batteries (RFBs) because they can have up to five electrochemically accessible and reversible oxidation states. However, most of the commercially available DAAQs have a low solubility in the polar organic solvents that are typically used in RFBs, in particular when supporting electrolyte salts are present. This significantly limits the energy densities that can be achieved. We have functionalized the amino groups in the DAAQ structure with three types of chains, namely alkyl chains, cationic alkyl chains and oligoethylene glycol ether chains, and measured the solubility of these derivatives in various organic solvents by quantitative UV-Vis absorption spectroscopy. The DAAQ derivatives with higher polarity exhibit a significantly higher solubility in commonly used organic electrolytes in comparison to apolar derivatives. Cyclic voltammetry was used to assess the viability of the DAAQs as redox-active species for RFBs. Although the cationic DAAQ derivatives have an enhanced solubility in the electrolytes, the cathodic redox reactions have a poor reversibility, most likely due to an internal decomposition reaction of their reduced forms. The oligoethylene-glycol-ether-functionalized DAAQs are the most promising compounds for use in organic RFB electrolytes because they have the optimal combination of high solubility and a high reversibility of the redox couples.
Introduction
Redox flow batteries (RFBs) are regarded as a promising electrochemical energy storage (EES) technology that can facilitate the integration of intermittent renewable energy sources, such as solar and wind, in the electrical grid.1–3 Because the energy is stored in redox couples that are dissolved in the electrolyte, as opposed to storage in solid electrodes (e.g. lithium-ion, lead-acid), the power and capacity of RFBs scale independently from each other. This makes RFBs highly versatile and conveniently scalable EES systems, capable of storing power and energy on a multi-MW and multi-MW h scale, respectively.4–6 Moreover, they have a good tolerance against over(dis)charging, a high stability against power fluctuations, a deep discharge ability and fast response times.6–8Many aqueous RFB chemistries such as all-vanadium,9,10 iron–vanadium,11 iron-chrome,12 zinc–bromine13 etc. have been widely studied in the literature and new developments are still being reported frequently. However, the achievable cell voltage, and therefore the energy density of aqueous RFB systems is limited by the narrow electrochemical stability window of water. This has caused a shift in RFB research towards non-aqueous electrolytes such as ionic liquids, deep eutectic solvents, and molecular organic solvents that typically have a wider electrochemical window, thus enabling higher cell voltages.14–18 Additionally, using non-aqueous solvents opens the door to a wide range of organic redox-active compounds that are insoluble in aqueous electrolytes. An all-organic redox flow battery (AORFB) based on organic redox couples with a high cell potential and a high solubility in organic electrolytes can reach much higher energy densities than any aqueous RFB. From an economical and environmental point-of-view, organic redox-active species also have an advantage over transition metal-based species such as vanadium,9,10,19 chromium20,21 and cobalt,22 that are often scarce and/or toxic.
An ideal organic redox-active species for RFBs should have the following properties: (1) it should either be commercially available at low cost or require few steps to synthesize from inexpensive precursors; (2) it should have a high solubility in polar organic solvents, also when high concentrations of supporting electrolyte salts are present; (3) the electron-transfer reactions between its different oxidation states should be highly reversible and have fast kinetics; (4) it should be chemically stable over time in all its oxidation states to prevent capacity losses through side reactions; (5) it should have at least two electrochemically accessible redox reactions at well separated potentials, as this would allow to construct a symmetric redox flow battery (SRFB), similarly to the aqueous all-vanadium redox flow battery (VRB). Because they are based on one single active species, SRFBs do not suffer from electrolyte contamination through crossover which simplifies the choice of a separator. Many commercially available organic compounds and their derivatives have shown promising reversible electrochemical behavior for use as redox-active species, most notably 2,2,6,6-tetramethylpiperidinooxyl (TEMPO)23–25 and phenothiazines.26,27 However, these compounds exhibit only one reversible redox reaction, so they can only be used in one half-cell. For a symmetric RFB cell setup, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO) is more interesting because it is highly soluble in organic solvents, and has two reversible redox reactions enabling a cell voltage of 1.7 V.28 Unfortunately, the high cost and fast capacity fading during cycling limits the usefulness of this compound.
Recently, 1,4-diaminoanthraquinones (DAAQs) have been proposed as an exceptionally promising compound class that can be used as active species in a SRFB electrolyte.29 Anthraquinones (AQs) in general exhibit two subsequent cathodic one-electron transitions in non-aqueous conditions, corresponding to the well-known reduction of the quinone functional unit.30,31 The amino functional groups on the 1- and 4-positions of DAAQs are sensitive to oxidation, resulting in two additional subsequent anodic one-electron reactions that correspond to the formation of a divalent diimine cation.32 The aromatic base structure serves to stabilize the reduced and oxidized DAAQ species via electron delocalization. Potash et al. studied the electrochemical behavior of several commercially available DAAQs and observed that the dye Disperse Blue 134 (DB-134, 1,4-bis(isopropylamino)anthraquinone) had the highest reversibility for all four redox couples.29 A SRFB using DB-134 as redox-active component can store two electrons per molecule of active species in both the anolyte and catholyte, resulting in an impressive theoretical molar energy density of 120 W h mol−1 at a cell voltage of 2.7 V. This is much higher than the theoretical molar energy density of VRB electrolytes (34 W h mol−1). However, the achievable energy density is limited by the poor solubility of DB-134 in organic solvents (approx. 20 mmol L−1 in acetonitrile and approx. 200 mmol L−1 in toluene).29
We have recently reported a highly soluble DAAQ derivative, 1,4-bis((2-(2-(2-methoxyethoxy)ethoxy)ethyl)amino)anthracene-9,10-dione (Me-TEG-DAAQ) that is miscible in any ratio with many polar organic solvents, while also retaining the reversible electrochemical behavior of DAAQs.33 In the present paper, we report the general strategy that was used to discover this compound. The DAAQ structure was functionalized with various chains on the amino positions (Fig. 1). The influence of solvent polarity and supporting electrolyte concentration on the solubility of these derivatives was studied by quantitative UV-Vis absorption spectroscopy. The electrochemical behavior of the DAAQ derivatives was characterized by cyclic voltammetry to evaluate their suitability for SRFB applications.
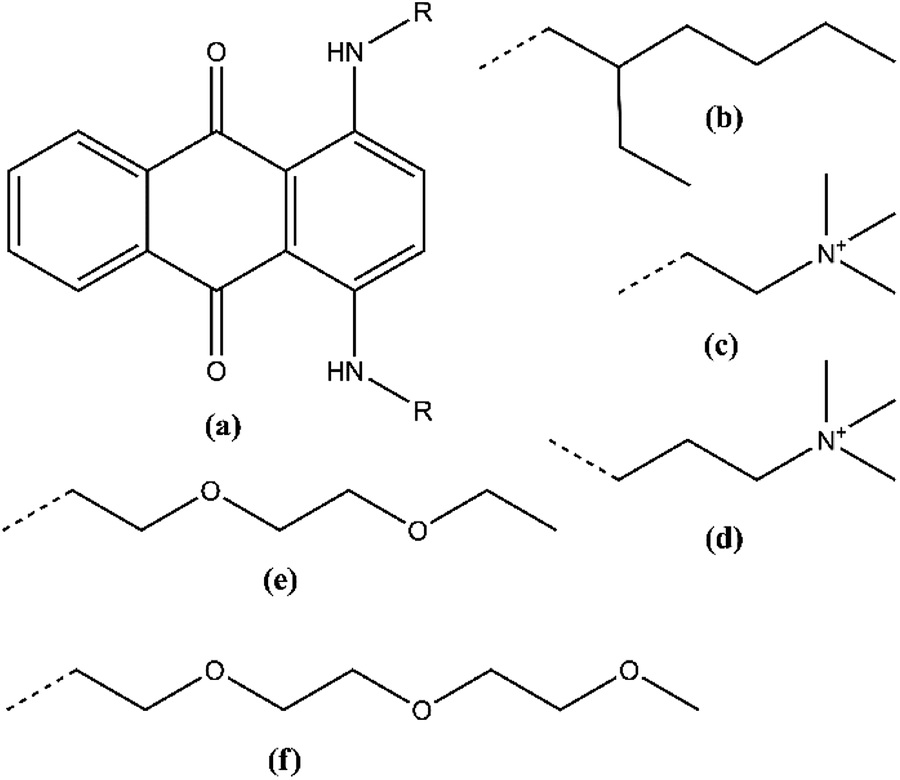 | ||
| Fig. 1 Chemical structures of (a) 1,4-diaminoanthraquinone (DAAQ) and the side chains (R) on the amino positions: (b) 2-ethylhexyl (Et-He), (c) N,N,N-trimethylethanaminium (Me3N-Et), (d) N,N,N-trimethylpropan-1-aminium (Me3N-Pr), (e) 2-(2-ethoxyethoxy)ethyl (Et-DEG), and (f) 2-(2-(2-methoxyethoxy)ethoxy)ethyl (Me-TEG). The dashed bonds represent the connection point of the side chains on the amino groups. | ||
Experimental
Chemicals
1,4-Dihydroxyanthraquinone (quinizarin, 96%), 2-ethylhexylamine (99%), 2(2-ethoxyethoxy)ethanol (diethylene glycol monoethyl ether, Et-DEG-OH, ≥98%), heptane (for HPLC, 99%), methanesulfonyl chloride (MsCl, 99.5%), 2-butanone (methyl ethyl ketone, MEK, ≥99%, extra pure), acetonitrile (MeCN, 99.9%, extra dry, over molecular sieves) and dichloromethane (DCM, 99.8%, extra dry, over molecular sieves) were purchased from Acros Organics (Geel, Belgium). Sodium dithionite (Na2S2O4, purified, ≥86%) and ethyl acetate (EtOAc, ≥99.7%, for HPLC) were purchased from Honeywell Riedel-deHaën (Seelze, Germany). Ammonia (NH3) 25 wt% aqueous solution (analytical reagent grade) was purchased from VWR (Haasrode, Belgium). Toluene (analytical reagent grade), sodium hydroxide (NaOH, analytical reagent grade) and methanol (MeOH, ≥99.9%, synthesis grade) were purchased from Fisher Scientific (Asse, Belgium). Triethylamine (TEA, ≥99.5%, for synthesis) was purchased from Carl Roth (Karlsruhe, Germany). Potassium carbonate (K2CO3, analytical reagent grade) and ammonium chloride (NH4Cl, analytical reagent grade) were purchased from Chem-Lab (Zedelgem, Belgium). Lithium bis(trifluoromethylsulfonyl)imide (LiTf2N, 99%) was purchased from Iolitec (Heilbronn, Germany). Silver nitrate (AgNO3, ≥99%) was purchased from Alfa Aesar (Karlsruhe, Germany) and was dried for 12 hours on a Schlenk line (0.6–1.6 mbar) at room temperature prior to use. The synthesized DAAQ derivatives and tetraethylammonium bis(trifluoromethylsulfonyl)imide (TEATf2N) were dried on a Schlenk line (0.6–1.6 mbar) at 70 °C for 12 hours prior to use and were stored in an argon-filled glovebox with water and oxygen concentrations below 1 ppm. All chemicals were used as received, unless stated otherwise.Characterization
Melting points were measured on a Mettler-Toledo DSC-1 differential scanning calorimeter at a heating rate of 10 °C min−1 under a helium atmosphere. Samples were weighed (3 to 12 mg) inside aluminum crucibles and subsequently sealed with an aluminum cap. The samples were cycled twice (heating first) between −70 °C and an appropriate upper temperature limit to achieve complete melting. The melting point value determined in the second cycle is reported. Infrared spectra were recorded on a Bruker Vertex 70 FT-IR spectrometer in attenuated total reflectance (ATR) mode, equipped with the Bruker platinum ATR module and a diamond ATR crystal. Each measurement consisted of 32 scans at a resolution of 4 cm−1. FTIR spectra were analyzed with OPUS 7.0 software. NMR spectra were recorded on either a Bruker 400 MHz Avance III HD spectrometer, operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR or a Bruker 300 MHz Avance spectrometer, operating at 300 MHz for 1H NMR and 75 MHz for 13C NMR. Tetramethylsilane (TMS) was used as internal reference (0.00 ppm for 1H and 13C). NMR spectra were analyzed with TopSpin 4.0.2 software. UV-Vis absorption spectra were recorded on a Varian Cary 6000i UV-Vis-NIR spectrophotometer at a scan rate of 10 nm s−1 with 1 nm intervals. The instrument was used in double-beam mode. Samples were diluted to fall within the absorbance range from 0.1 to 1.0 and were measured in quartz glass cuvettes with a path length of 1 cm. Blanks consisted of the supporting electrolyte (TEATf2N) dissolved in the same solvent system and at the same concentration as the samples, and were measured in a matched pair of cuvettes. The saturated samples were diluted in threefold and the determined concentrations are given as an average of three separate measurements.Electrochemistry
All the electrochemical measurements were performed inside an argon-filled glovebox with water and oxygen concentrations below 1 ppm. The electrolyte solutions were purged for 10 min with argon gas prior to the measurements in order to remove traces of dissolved oxygen. Cyclic voltammograms (CVs) were measured using an Autolab PGSTAT302N potentiostat and Nova 2.1 software. The working electrode for CV was a polished platinum disk electrode (∅ = 0.5 mm), the counter electrode was a piece of platinum-coated silicon wafer (approx. surface area = 0.0025 dm2) and the scan rate was 50 mV s−1, unless stated otherwise. The reference electrode consisted of a silver wire, submerged in a solution of silver nitrate (0.01 mol L−1) and TEATf2N (0.1 mol L−1) in anhydrous acetonitrile, contained inside a fritted glass tube. The CVs were started at open circuit potential (OCP). For the CV measurements, IR drop compensation was applied by measuring the impedance of the setup by electrochemical impedance spectroscopy (EIS) and manually correcting the potential. EIS spectra were recorded in threefold at OCP in a frequency range of 1 MHz–1000 MHz, using an Autolab PGSTAT302N potentiostat and Nova 2.1 software.Synthesis procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 heptane/ethyl acetate. The dark blue band (Rf = 0.60) was isolated to give to give 1,4-bis[(2-ethylhexyl)amino]-9,10-anthracenedione (2.37 g, 5.12 mmol, 20% yield) as a dark blue solid. 1H NMR (400 MHz, CD2Cl2, δ/ppm): 0.93–0.97 (m, 12H), 1.36 (m, 8H), 1.47 (m, 4H), 1.53 (m, 4H), 1.72 (m, 2H), 3.35 (m, 4H), 7.30 (s, 2H), 7.69–7.70 (m, 2H) 8.33 (m, 2H), 10.97 (s, 2H). 13C NMR (400 MHz, CDCl3, δ/ppm): 10.97, 14.11, 23.02, 24.52, 29.03, 31.27, 39.47, 45.98, 109.66, 123.62, 126.02, 131.82, 134.56, 146.52, 182.09. IR (ATR, νmax/cm−1): 3080, 2958, 2922, 2871, 2859, 1645, 1605, 1578, 1554, 1520, 1457, 1416, 1377, 1349, 1281, 1255 (ν C–N), 1163, 1141, 1076, 1041, 1010, 967, 920, 891, 814, 799, 768, 723, 668, 562, 518, 497, 458, 432. CHN found (calculated) for C30H42N2O2: C 77.97 (77.88) %, H 8.63 (9.15) %, N 5.89 (6.05) %. Mp: 84 °C.:3 and the crystals were dried overnight on a Schlenk line at 80 °C to give 2,2′-((9,10-dioxo-9,10-dihydroanthracene-1,4-diyl)bis(azanediyl))bis(N,N,N-trimethylethanaminium)bis(trifluoromethylsulfonyl)imide (1.06 g, 1.092 mmol, 77% yield). 1H NMR (400 MHz, DMSO-d6, δ/ppm): 3.32 (s, 18H), 3.64 (t, J = 6.5 Hz, 4H), 3.99 (q, J = 6.2 Hz, 4H), 7.57 (s, 2H), 7.83–7.87 (m, 2H), 8.23–8.26 (m, 2H), 10.64 (t, J = 5.99 Hz, 2H). 13C NMR (300 MHz, CDCl3, δ/ppm): 36.62, 53.25, 64.29, 110.27, 124.65, 126.30, 133.40, 134.07, 145.44, 182.39. IR (ATR, νmax/cm−1): 3044, 2959, 2883, 2825, 1639, 1613, 1598, 1583, 1564, 120, 1480, 1423, 1394, 1346, 1328, 1299, 1263, 1227, 1174, 1131, 1051, 1022, 967, 953, 916, 865, 811, 788, 763, 739, 730, 652, 613, 598, 569, 533, 509, 471, 456, 450, 432, 405. Mp: 183 °C.:1 DCM/methanol + 20 vol% triethylamine. The dark blue band was isolated to give to give 1,4-bis[[3-(dimethylamino)propyl]amino]-9,10-anthracenedione (3.54 g, 8.665 mmol, 52% yield) as a dark blue solid. 1H NMR (300 MHz, CDCl3, δ/ppm): 1.86–1.95 (m, 4H), 2.26 (s, 12H), 2.43 (t, J = 7.0 Hz, 4H), 3.41–3.48 (m, 4H), 7.25 (s, 2H), 7.66–7.69 (m, 2H), 8.32–8.35 (m, 2H), 10.81 (t, J = 5.3 Hz, 2H).:3 mixture of 1 mol L−1 aqueous HCl and brine (2 × 50 mL), followed by pure brine (2 × 50 mL). The dried organic phases were dried over MgSO4 and concentrated on a rotary evaporator to give 1,4-bis((2-(2-(2-methoxyethoxy)ethoxy)ethyl)amino)anthracene-9,10-dione as a dark blue solid (1.86 g, 3.95 mmol, 53% crude yield). The crude product was purified by column chromatography on silica gel with eluent EtOAc. The bright blue band (Rf = 0.58 with EtOAc) was isolated (1.42 g, 3.02 mmol, 41% final yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 1.22 (t, J = 7.0 Hz, 6H), 3.55 (q, J = 7.0 Hz, 4H), 3.60–3.65 (m, 8H), 3.69–3.72 (m, 4H), 3.81 (t, J = 5.8 Hz, 4H), 7.29 (s, 2H), 7.66–7.70 (m, 2H), 8.31–8.36 (m, 2H), 10.82 (t, J = 5.2 Hz, 2H). 13C NMR (400 MHz, CDCl3, δ/ppm): 15.20, 42.73, 66.74, 69.88, 70.19, 70.91, 110.21, 123.49, 126.08, 132.05, 134.51, 146.14, 182.69. IR (ATR, νmax/cm−1): 3388, 3220, 3068 (ν N–H), 2972, 2866 (ν C–H CH2), 1611 (ν C
:1 heptane/ethyl acetate. The dark blue band (Rf = 0.60) was isolated to give to give 1,4-bis[(2-ethylhexyl)amino]-9,10-anthracenedione (2.37 g, 5.12 mmol, 20% yield) as a dark blue solid. 1H NMR (400 MHz, CD2Cl2, δ/ppm): 0.93–0.97 (m, 12H), 1.36 (m, 8H), 1.47 (m, 4H), 1.53 (m, 4H), 1.72 (m, 2H), 3.35 (m, 4H), 7.30 (s, 2H), 7.69–7.70 (m, 2H) 8.33 (m, 2H), 10.97 (s, 2H). 13C NMR (400 MHz, CDCl3, δ/ppm): 10.97, 14.11, 23.02, 24.52, 29.03, 31.27, 39.47, 45.98, 109.66, 123.62, 126.02, 131.82, 134.56, 146.52, 182.09. IR (ATR, νmax/cm−1): 3080, 2958, 2922, 2871, 2859, 1645, 1605, 1578, 1554, 1520, 1457, 1416, 1377, 1349, 1281, 1255 (ν C–N), 1163, 1141, 1076, 1041, 1010, 967, 920, 891, 814, 799, 768, 723, 668, 562, 518, 497, 458, 432. CHN found (calculated) for C30H42N2O2: C 77.97 (77.88) %, H 8.63 (9.15) %, N 5.89 (6.05) %. Mp: 84 °C.:3 and the crystals were dried overnight on a Schlenk line at 80 °C to give 2,2′-((9,10-dioxo-9,10-dihydroanthracene-1,4-diyl)bis(azanediyl))bis(N,N,N-trimethylethanaminium)bis(trifluoromethylsulfonyl)imide (1.06 g, 1.092 mmol, 77% yield). 1H NMR (400 MHz, DMSO-d6, δ/ppm): 3.32 (s, 18H), 3.64 (t, J = 6.5 Hz, 4H), 3.99 (q, J = 6.2 Hz, 4H), 7.57 (s, 2H), 7.83–7.87 (m, 2H), 8.23–8.26 (m, 2H), 10.64 (t, J = 5.99 Hz, 2H). 13C NMR (300 MHz, CDCl3, δ/ppm): 36.62, 53.25, 64.29, 110.27, 124.65, 126.30, 133.40, 134.07, 145.44, 182.39. IR (ATR, νmax/cm−1): 3044, 2959, 2883, 2825, 1639, 1613, 1598, 1583, 1564, 120, 1480, 1423, 1394, 1346, 1328, 1299, 1263, 1227, 1174, 1131, 1051, 1022, 967, 953, 916, 865, 811, 788, 763, 739, 730, 652, 613, 598, 569, 533, 509, 471, 456, 450, 432, 405. Mp: 183 °C.:1 DCM/methanol + 20 vol% triethylamine. The dark blue band was isolated to give to give 1,4-bis[[3-(dimethylamino)propyl]amino]-9,10-anthracenedione (3.54 g, 8.665 mmol, 52% yield) as a dark blue solid. 1H NMR (300 MHz, CDCl3, δ/ppm): 1.86–1.95 (m, 4H), 2.26 (s, 12H), 2.43 (t, J = 7.0 Hz, 4H), 3.41–3.48 (m, 4H), 7.25 (s, 2H), 7.66–7.69 (m, 2H), 8.32–8.35 (m, 2H), 10.81 (t, J = 5.3 Hz, 2H).:3 mixture of 1 mol L−1 aqueous HCl and brine (2 × 50 mL), followed by pure brine (2 × 50 mL). The dried organic phases were dried over MgSO4 and concentrated on a rotary evaporator to give 1,4-bis((2-(2-(2-methoxyethoxy)ethoxy)ethyl)amino)anthracene-9,10-dione as a dark blue solid (1.86 g, 3.95 mmol, 53% crude yield). The crude product was purified by column chromatography on silica gel with eluent EtOAc. The bright blue band (Rf = 0.58 with EtOAc) was isolated (1.42 g, 3.02 mmol, 41% final yield). 1H NMR (400 MHz, CDCl3, δ/ppm): 1.22 (t, J = 7.0 Hz, 6H), 3.55 (q, J = 7.0 Hz, 4H), 3.60–3.65 (m, 8H), 3.69–3.72 (m, 4H), 3.81 (t, J = 5.8 Hz, 4H), 7.29 (s, 2H), 7.66–7.70 (m, 2H), 8.31–8.36 (m, 2H), 10.82 (t, J = 5.2 Hz, 2H). 13C NMR (400 MHz, CDCl3, δ/ppm): 15.20, 42.73, 66.74, 69.88, 70.19, 70.91, 110.21, 123.49, 126.08, 132.05, 134.51, 146.14, 182.69. IR (ATR, νmax/cm−1): 3388, 3220, 3068 (ν N–H), 2972, 2866 (ν C–H CH2), 1611 (ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1582, 1554 (ν C–C aromatic ring), 1522 1447, 1350, 1231, 1196, 1162, 1112, 1050, 1022, 980, 940, 920, 841, 816, 797, 725, 656, 613, 565, 510, 458, 432. CHN found (calculated) for C26H34N2O6: C 66.38 (66.36) %, H 7.23 (7.28) %, N 5.58 (5.95) %. Mp: 50 °C.
O), 1582, 1554 (ν C–C aromatic ring), 1522 1447, 1350, 1231, 1196, 1162, 1112, 1050, 1022, 980, 940, 920, 841, 816, 797, 725, 656, 613, 565, 510, 458, 432. CHN found (calculated) for C26H34N2O6: C 66.38 (66.36) %, H 7.23 (7.28) %, N 5.58 (5.95) %. Mp: 50 °C.Results and discussion
Synthesis and physicochemical properties
Many 1,4-diaminoanthraquinones are commercially available as dyes that are used extensively in the textile industry: e.g. Disperse Blue 14 (1,4-bis(methylamino)anthraquinone), Disperse Blue 134 (1,4-bis(isopropylamino)anthraquinone), Solvent Blue 59 (1,4-bis(ethylamino)anthraquinone) etc.34,35 Furthermore, because they have been shown to have biological activity as anti-cancer agents, the synthesis procedures of many DAAQ derivatives have been reported in medicinal chemistry journals.36–38 The reaction of the commercially available orange dye 1,4-dihydroxyanthraquinone (quinizarin, Solvent Orange 86) or its reduced form leucoquinizarin with primary amines will generally give the corresponding DAAQs in good yield.37,38 This offers a convenient strategy to functionalize the DAAQ structure by using differently substituted amines with the goal of increasing the solubility. For this study, three types of amino substituents were investigated: firstly, the branched alkyl chain 2-ethylhexyl (Et-He) that has been used before to increase the solubility of organic compounds because of its high degree of conformational freedom, thereby increasing the entropy of dissolution;39 secondly, alkyl chains with terminal quaternary ammonium groups, N,N,N-trimethylethanammonium (Me3N+-Et) and N,N,N-trimethylpropan-1-aminium (Me3N+-Pr). Because these chains contain charged groups, the derived DAAQs are highly polar, increasing their solubility in polar solvents. Thirdly oligoethylene glycol ether chains were considered: diethylene glycol monoethyl ether (Et-DEG), and triethylene glycol monomethyl ether (Me-TEG). These chains are significantly more polar than alkyl chains, while also having a high degree of conformational freedom.Fig. 2 gives an overview of the synthesis routes that were used to prepare these derivatives. The alkyl (A) and cationic (B) derivatives were prepared by the reaction of quinizarin and the corresponding alkyl- or N,N-dimethylaminoalkylamines. For the cationic derivatives, the terminal ternary amino groups were quaternized with iodomethane, resulting in a cationic DAAQ iodide salt. Subsequently the iodide salt was converted to a bis(trifluoromethylsulfonyl)imide (Tf2N−) salt via ion-exchange with lithium bis(trifluoromethylsulfonyl)imide. This anion is electrochemically more stable than iodide and it is more hydrophobic, resulting in higher solubility in organic solvents. The oligoethylene glycol ether derivatives were prepared via the leucoquinizarin route (C1). In this case, the required primary amines are not commercially available and were synthesized via a two-step procedure (C2).40 The hydroxyl group of the oligoethylene glycol monoalkyl ethers was first converted to methanesulfonate by reacting with methanesulfonyl chloride. Subsequently, the methanesulfonate groups were converted to primary amine groups by reaction with an excess of aqueous ammonia solution. The detailed reaction conditions for the DAAQs and precursors can be found in the Experimental section. The synthesis of Me-TEG-DAAQ and its precursors was reported in our previous work.33
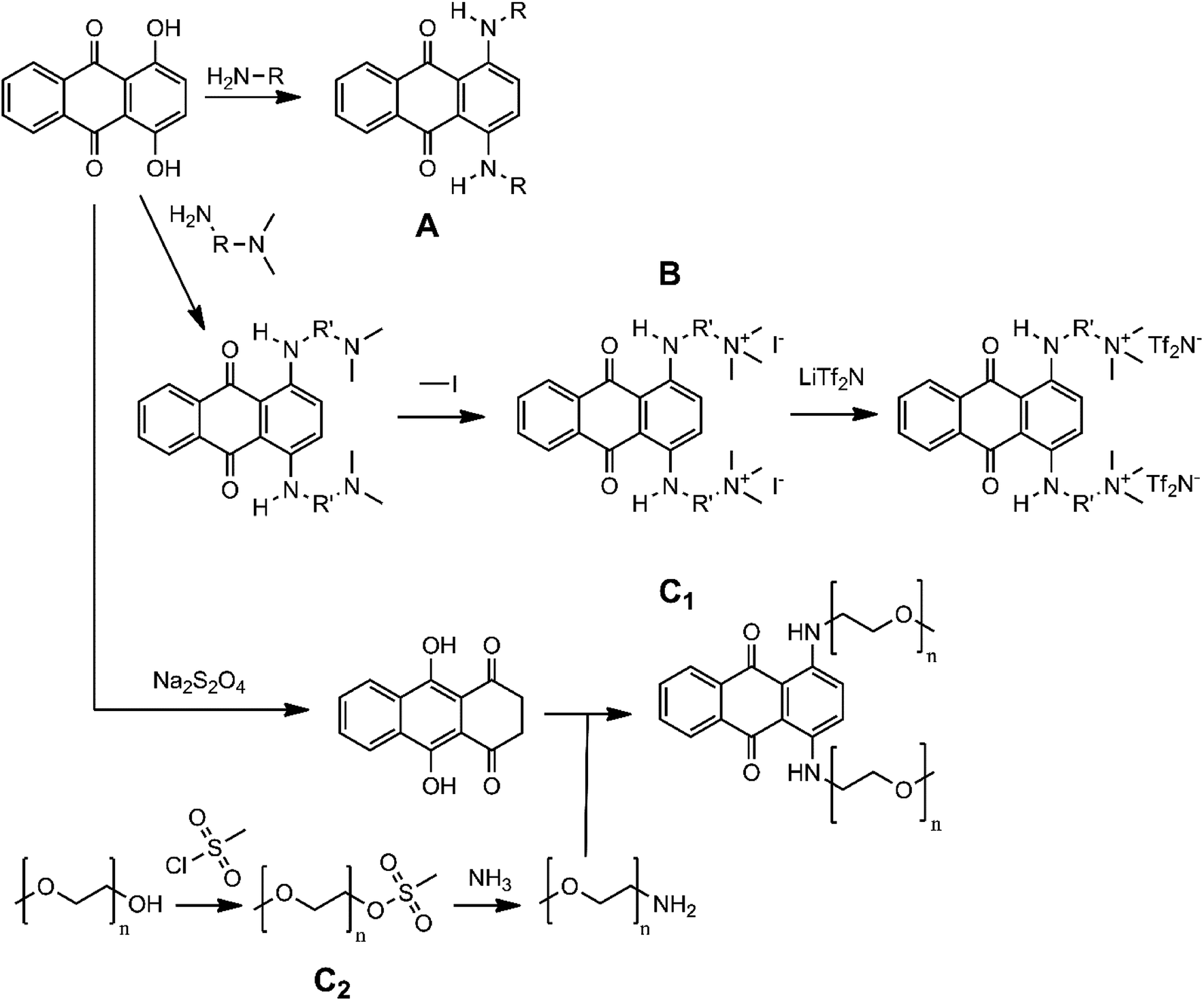 | ||
| Fig. 2 Procedures for the synthesis of the DAAQ derivatives: (A) alkyl DAAQs, (B) cationic DAAQs, (C) oligoethylene glycol ether DAAQs. | ||
All the DAAQ derivatives are dark blue solids at room temperature. The melting points of the alkyl and oligoethylene glycol ether derivatives are significantly lower than the melting point of DB-134 (170 °C) due to the high degree of conformational freedom associated with these chains. This can also be advantageous for achieving a high solubility. Another important parameter to consider for solubility is the polarity of the solute compared to the solvent. For organic compounds, the polarity can be conveniently compared by thin layer chromatography (TLC) on silica gel plates (Fig. S1†). The aromatic core of the DAAQ structure is rather apolar due to an absence of polar groups. For the alkyl DAAQ derivatives, this apolar character is further increased by the alkyl chains on the amino groups. This is demonstrated by the increase in Rf values (eluens = EtOAc/heptane 1/1) from DB-134 (Rf = 0.74) to Et-He-DAAQ (Rf = 0.87). Contrarily, the oligoethylene glycol ether derivatives are significantly more polar due to the many ether functionalities in the side chains, as shown by the small Rf values: Rf = 0.16 for Et-DEG-DAAQ and Rf = 0.02 for Me-TEG-DAAQ. Like all ionic compounds, Me3N-Et-DAAQ and Me3N-Pr-DAAQ exhibit very slow migration over the silica TLC plates due to their strong interaction with the polar stationary phase.
Solubility studied by UV-Vis absorption spectroscopy
All the DAAQs that are included in this study strongly absorb electromagnetic radiation in the visible spectral region, resulting in an intense blue color. Typically, two maxima are observed in the optical absorption spectra in the wavelength ranges 580–605 nm and 620–650 nm, depending on the solvent (Fig. S2†). With the exception of Me3N-Et-DAAQ, the latter peak is the most intense with a molar absorption coefficient in the 104 L mol−1 cm−1 order of magnitude.33 Therefore, quantitative UV-Vis absorption spectroscopy was used as an inexpensive and fast technique to determine the solubility of DAAQs with good precision. Several standards were prepared, typically between 10 μmol L−1 and 70 μmol L−1, in order to construct calibration curves. The solubility of the DAAQs was determined by precisely diluting saturated samples down to fall within the absorbance range of the calibration curves. The influence of solvent polarity on the solubility was determined by using mixtures of the polar solvent acetonitrile (MeCN), and the apolar solvent toluene in different volume ratios. Another parameter that was studied was the influence of a supporting electrolyte salt on the solubility of the active species, which is often overlooked in the literature on non-aqueous RFBs. This was achieved by measuring the solubility in a 1 mol L−1 solution of TEATf2N in acetonitrile, which is a realistic supporting electrolyte used in non-aqueous RFBs. The results of the solubility experiments are summarized in Fig. 3. A logarithmic scale was used because the solubility of the different DAAQs varied over several orders of magnitude (10−3 to 100 mol L−1). The numerical solubility values can be found in the ESI, Tables S1–S6.†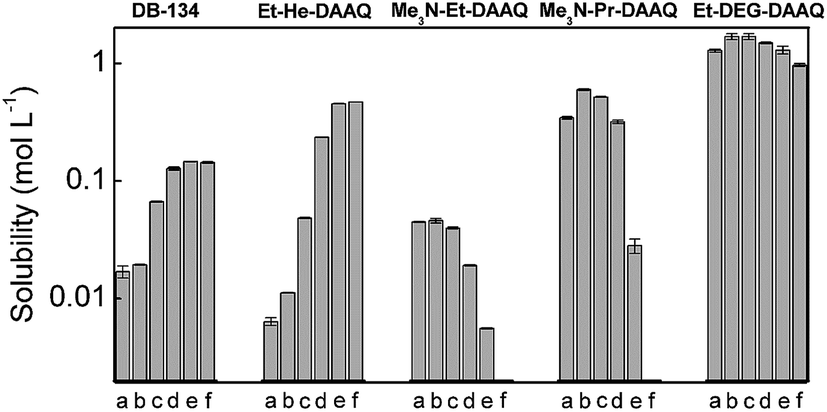 | ||
| Fig. 3 Plot of the solubility (logarithmic scale) of the DAAQs in different solvent systems: (a) acetonitrile + 1 mol L−1 TEATf2N, (b) acetonitrile/toluene 1/0, (c) acetonitrile/toluene 3/1, (d) acetonitrile/toluene 1/1, (e) acetonitrile/toluene 1/3, and (f) acetonitrile/toluene 0/1. Error bars are indicated at the top of the columns. | ||
The solubility of DB-134 (1,4-bis(isopropylamino)anthraquinone) in pure acetonitrile is 19 mmol L−1, as determined in our previous work.33 When toluene is added, a gradual increase in solubility is observed as the solvent mixture becomes increasingly apolar. In pure toluene, DB-134 has a solubility of 144 mmol L−1, an eightfold increase compared to pure acetonitrile. The addition of 1 mol L−1 TEATf2N to acetonitrile results in a 20% decrease in solubility due to the increase in polarity associated with the high salt concentration. Because of the 2-ethylhexyl chains in Et-He-DAAQ, this DAAQ is more apolar than DB-134. This results in a lower solubility in pure acetonitrile (12 mmol L−1), but the increasing trend as toluene is added is much more drastic, with a solubility of 471 mmol L−1 in pure toluene. Additionally, the presence of 1 mol L−1 TEATf2N also results in a stronger decrease in solubility of 54%. For Me3N-Et-DAAQ and Me3N-Pr-DAAQ, a decrease in solubility is observed as the fraction of toluene in the solvent mixtures is increased. In pure toluene, the solubility of both DAAQs is below the detection limit of the spectrophotometer. This behavior is expected because ionic compounds dissolve better in polar solvents. Interestingly, the solubility of Me3N-Et-DAAQ in pure acetonitrile is only 46 mmol L−1, which is a significant improvement compared to DB-134, but still insufficient for a practical RFB electrolyte. Remarkably, the slightly longer ionic side chains in Me3N-Pr-DAAQ result in a solubility of 601 mmol L−1 in pure acetonitrile, a thirteenfold increase compared to Me3N-Et-DAAQ. In acetonitrile + 1 mol L−1 TEATf2N the solubility of both ionic DAAQs is decreased compared to pure acetonitrile, most likely due to the common anion effect. In our previous work, we reported on the complete miscibility of Me-TEG-DAAQ with acetonitrile and 1,4-dimethoxyethane (DME), with and without 1 mol L−1 TEATf2N added in both solvents. The same behavior was observed with all the acetonitrile/toluene mixtures. Because this compound is liquid close to room temperature (mp = 25 °C), it readily forms homogeneous mixtures with most organic solvents. The slightly shorter side chain in Et-DEG-DAAQ results in a higher melting point (mp = 50 °C), and this allowed for the determination of its maximum solubility in the solvent mixtures. In pure acetonitrile, an impressive solubility of 1.7 mol L−1 is observed for this compound. Due to its high polarity, the solubility decreases as toluene is added to 0.97 mol L−1 in pure toluene. When 1 mol L−1 TEATf2N is added, the solubility of Et-DEG-DAAQ is decreased to 1.3 mol L−1. Nevertheless, this is still a very promising value for a redox-active compound for all-organic RFBs. To summarize: the modification of the DAAQ structure with a branched alkyl chain such as 2-ethylhexyl was successful in increasing the solubility in apolar solvents. However, the use of apolar solvents in RFB electrolytes is not recommended because herein, supporting electrolyte salts are poorly soluble, resulting in low electrical conductivity of the electrolyte. The modification with polar side chains did result in an increased solubility in polar solvents, in particular for the oligo ethyleneglycol-ether-functionalized DAAQs. Other than matching the polarity of the DAAQ with the solvent/electrolyte, a long chain length is also advantageous to achieve a high solubility. This is most likely related to the high degree of conformational freedom associated with these chains which increases the entropy of dissolution.
Cyclic voltammetry
A high solubility in organic supporting electrolyte solutions is only one of the desirable properties of redox-active compounds for RFBs. The different amino substituents that are attached to the DAAQ structure can also have a significant influence on the reversibility of the electron-transfer reactions.29 Therefore, the electrochemical behavior of the DAAQ derivatives was evaluated by cyclic voltammetry (Fig. 4).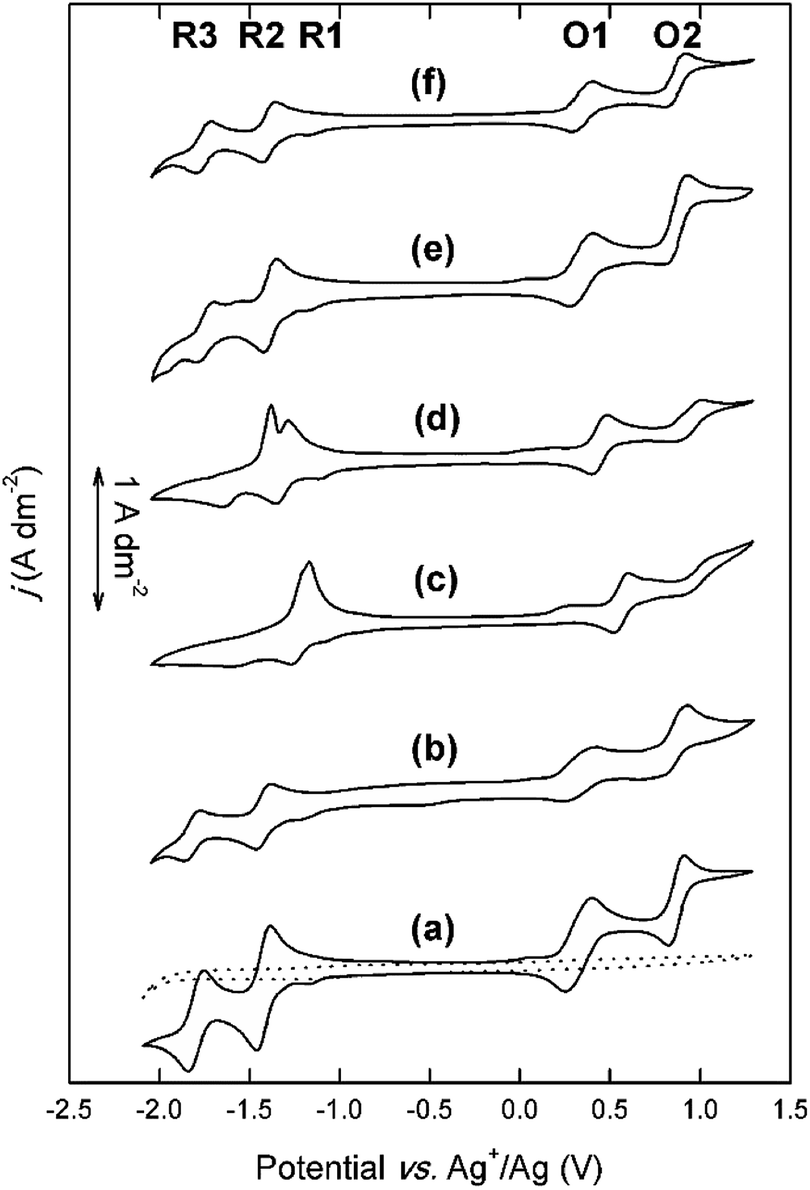 | ||
| Fig. 4 Cyclic voltammograms of 0.01 mol L−1 DB-134 (a), Et-He-DAAQ (b), Me3N-Et-DAAQ (c), Me3N-Pr-DAAQ (d), Et-DEG-DAAQ (e), and Me-TEG-DAAQ (f) in 0.1 mol L−1 TEATf2N in acetonitrile, recorded at ambient temperature on a platinum disk working electrode (∅ = 0.5 mm) with a scan rate of 50 mV s−1. The reference electrode was a silver wire immersed in 0.01 mol L−1 AgNO3 in acetonitrile. The counter electrode was a piece of platinum-coated silicon wafer with a surface area of approx. 25 mm2. The dotted curve is a blank CV of 0.1 mol L−1 TEATf2N in acetonitrile, recorded under identical conditions. | ||
All CVs were compensated for solution resistance. An example of the IR potential shift can be found in the ESI (Fig. S3†). In every CV, a small cathodic wave can be observed at approx. −1.2 V vs. Ag+/Ag (R1). This artefact was found to originate from the supporting electrolyte solution, as indicated in the blank measurement (Fig. S4†). In the CV of the model compound DB-134, the four expected highly reversible one-electron transfer reactions are observed. The two subsequent cathodic waves at −1.42 V vs. Ag+/Ag (R2) and −1.80 V vs. Ag+/Ag (R3) correspond to the reduction of the quinone functionality, and the two subsequent anodic waves at +0.33 V vs. Ag+/Ag (O1) and +0.87 V vs. Ag+/Ag (O2) correspond to the oxidation of the two amino functionalities. In the CV of Et-He-DAAQ, two reversible cathodic waves can be observed at −1.43 V vs. Ag+/Ag (R1) and −1.82 V vs. Ag+/Ag (R2). Thus, the 2-ethylhexyl substituents do not have a significant influence on the cathodic behavior, as expected from their chemically inert nature. The two anodic waves are present as well at +0.33 V vs. Ag+/Ag (O1) and +0.80 V vs. Ag+/Ag (O2). However, judging from the relative anodic and cathodic peak current densities, these electron-transfer reactions are less reversible, in particular O2.
The overall electrochemical behavior of Me-TEG-DAAQ and Et-DEG-DAAQ is very similar to that of DB-134, with four accessible and reversible redox couples at comparable potentials. For Me-TEG-DAAQ, the redox couples are located at −1.40 V (R2), −1.76 V (R3), +0.35 V (O1), and +0.84 V vs. Ag+/Ag (O2), and for Et-DEG-DAAQ at −1.39 V (R2), −1.75 V (R3), +0.34 V (O1), and +0.86 V vs. Ag+/Ag (O2). As discussed in our previous work, O2 is less reversible than the other redox couples, and this is also the case for Et-DEG-DAAQ.33
The overall electrochemical behavior of the cationic DAAQ derivatives, and in particular the cathodic electron-transfer reactions, is significantly altered by the presence of the quaternary ammonium groups in the side chains. The two anodic redox couples are accessible for both cationic DAAQs, but O2 is significantly less reversible than O1. For Me3N-Et-DAAQ, the expected two cathodic peaks are observed at −1.27 V vs. Ag+/Ag and −1.61 V vs. Ag+/Ag. These potentials are significantly more positive compared to those of the alkyl DAAQs, which is most likely a consequence of the electron-withdrawing quaternary ammonium groups that are located relatively close to the aromatic core of the molecule. However, in the backwards scan, one single sharp anodic peak is observed at −1.17 V vs. Ag+/Ag. The cathodic behavior of Me3N-Pr-DAAQ is similar, with two cathodic peaks at −1.35 V vs. Ag+/Ag and −1.65 V vs. Ag+/Ag but here, a sharp double peak at −1.29 V vs. Ag+/Ag and −1.40 V vs. Ag+/Ag is observed in the backwards scan. The cathodic behavior of the cationic DAAQs was further studied by varying the scan rate (Fig. 5).
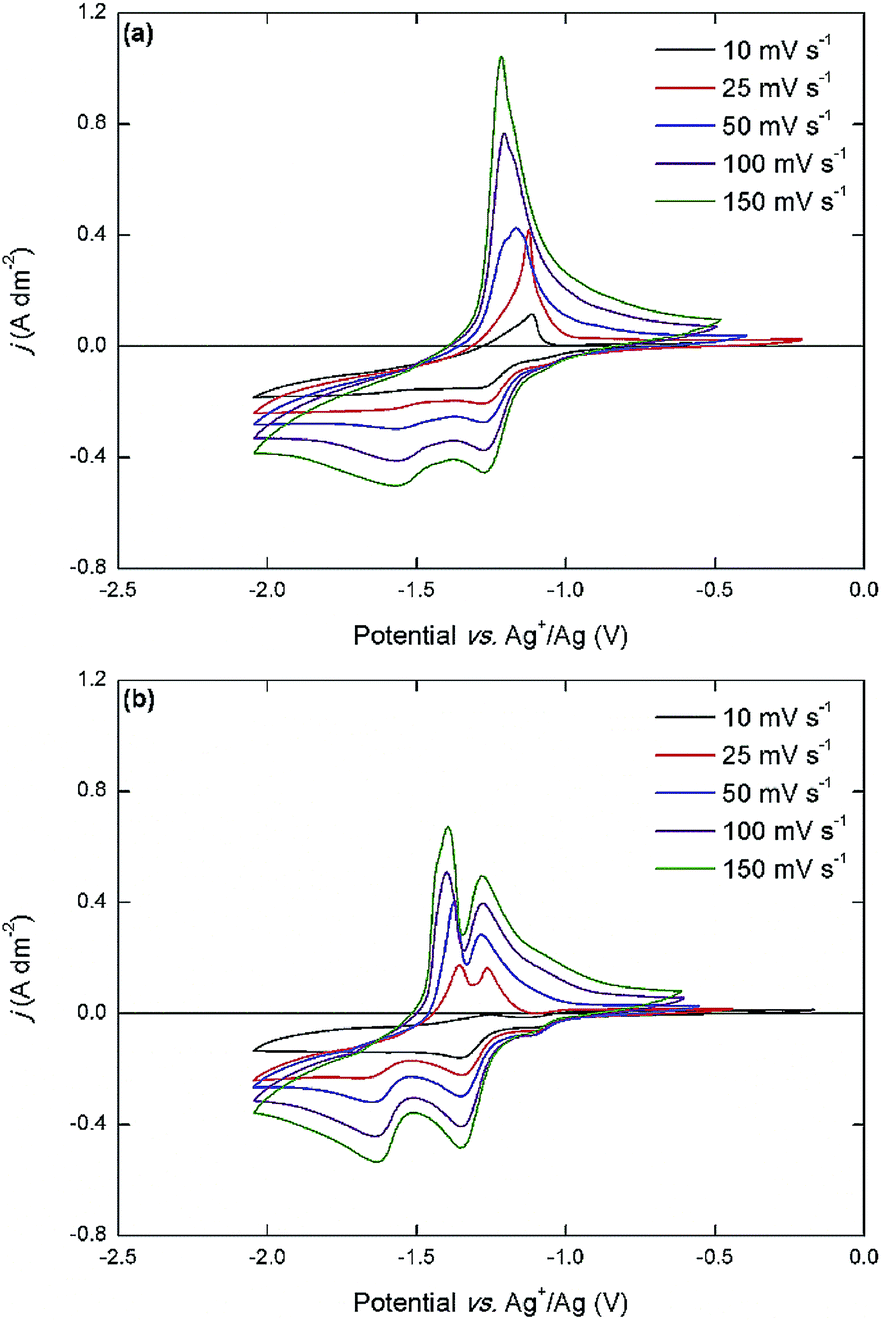 | ||
| Fig. 5 Cyclic voltammograms of 0.01 mol L−1 Me3N-Et-DAAQ (a) and Me3N-Pr-DAAQ (b) in 0.1 mol L−1 TEATf2N in acetonitrile, recorded at ambient temperature on a platinum disk working electrode (ϕ = 0.5 mm) with scan rates varying from 10 mV s−1 to 150 mV s−1. The reference electrode was a silver wire immersed in 0.01 mol L−1 AgNO3 in acetonitrile. The counter electrode was a piece of platinum-coated silicon wafer with a surface area of approx. 25 mm2. | ||
At a low scan rate (10 mV s−1), the reversibility of the cathodic electron-transfer reactions is very poor, judging from the low current density of the anodic peaks. As the scan rate is increased, the anodic peak current density is drastically increased, indicating a higher overall reversibility. It also becomes apparent that the anodic peak in the CV of Me3N-Et-DAAQ actually consists of two separate closely adjacent peaks, and that the more negative peak becomes more intense as the scan rate increases. In the CV of Me3N-Pr-DAAQ, the anodic peaks are more separated, but the same scan-rate-dependent trend is observed. These results are a strong indication that the reduced forms of the cationic DAAQs are chemically unstable and decompose over time. Considering that the reduced anionic quinone functionality is closely located to the cationic quaternary ammonium groups, the possibility of an internal decomposition reaction occurring seems plausible. This makes the ionic DAAQs much less suitable for use in RFB electrolytes, because considerable capacity losses would occur. In general, the oligoethylene-glycol-functionalized DAAQs are the most promising compounds for use in all-organic RFB electrolytes, because they combine a high solubility in polar supporting electrolyte solutions with a high reversibility of their inherent electron-transfer events. In previously reported cycling experiments, we demonstrated fast capacity fading when both sets of redox couples are utilized in a symmetric RFB setup, but when only the inner set of redox couples (R2 and O1) is used, good capacity retention can be achieved.33 Even with this limitation, a saturated electrolyte (1.29 mol L−1) consisting of Et-DEG-DAAQ in 1 mol L−1 TEATf2N in acetonitrile, provides a theoretical energy density of 60 W h L−1 and a cell potential of 1.73 V, which is much higher than any aqueous RFB.
Conclusions
1,4-Diaminoanthraquinones are a promising class of redox-active molecules for use in symmetric nonaqueous RFBs. Several types of amino substituents, namely alkyl groups, cationic groups and oligoethylene glycol ether groups, were successfully introduced in the DAAQ structure via a convenient one-step synthesis procedure starting from inexpensively available precursors. The DAAQs that were functionalized with polar cationic and oligoethylene glycol ether groups had a significantly increased solubility in polar supporting electrolyte solutions, in contrast with the apolar alkyl DAAQs, which had a decreased solubility. It was also observed that the amino substituents had a strong influence on the redox behavior of the molecules. The reversibility of the quinone reduction reaction was severely compromised by the cationic amino substituents, most likely due to an internal decomposition reaction. The oligoethylene glycol ether functionalized DAAQs were found to have the optimal combination of a high solubility in supporting electrolyte solutions, and good retention of the reversibility of the electron-transfer reactions. This enables to design a symmetric nonaqueous RFB with a high cell potential and a high energy density.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the KU Leuven for financial support (project KP/14/005).References
- P. Alotto, M. Guarnieri and F. Moro, Redox flow batteries for the storage of renewable energy: a review, Renew. Sust. Energ. Rev., 2014, 29, 325–335, DOI:10.1016/j.rser.2013.08.001.
- B. Dunn, H. Kamath and J.-M. Tarascon, Electrical energy storage for the grid: a battery of choices, Science, 2011, 334, 928–935, DOI:10.1126/science.1212741.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Electrochemical energy storage for green grid, Chem. Rev., 2011, 111, 3577–3613, DOI:10.1021/cr100290v.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Recent progress in redox flow battery research and development, Adv. Funct. Mater., 2013, 23, 970–986, DOI:10.1002/adfm.201200694.
- P. Leung, X. Li, C. Ponce de León, L. Berlouis, C. T. J. Low and F. C. Walsh, Progress in redox flow batteries, remaining challenges and their applications in energy storage, RSC Adv., 2012, 2, 10125–10156, 10.1039/c2ra21342g.
- C. Ponce de Leòn, A. Frías-Ferrer, J. González-García, D. A. Szánto and F. C. Walsh, Redox flow cells for energy conversion, J. Power Sources, 2006, 160, 716–732, DOI:10.1016/j.jpowsour.2006.02.095.
- M. Bartolozzi, Development of redox flow batteries: a historical bibliography, J. Power Sources, 1989, 27, 219–234, DOI:10.1016/0378-7753(89)80037-0.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, Redox flow batteries: a review, J. Appl. Electrochem., 2011, 41, 1137–1164, DOI:10.1007/s10800-011-0348-2.
- M. Skyllas-Kazacos, M. Rychcik, R. G. Robins, A. G. Fane and M. A. Green, New all-vanadium redox flow cell, J. Electrochem. Soc., 1986, 133, 1057–1058, DOI:10.1149/1.2108706.
- S. Roe, C. Menictas and M. Skyllas-Kazacos, A high energy density vanadium redox flow battery with 3 M vanadium electrolyte, J. Electrochem. Soc., 2016, 163, A5023–A5028, DOI:10.1149/2.0041601jes.
- W. Wang, S. Kim, B. Chen, Z. Nie, J. Zhang, G. G. Xia and L. Li, A new redox flow battery using Fe/V redox couples in chloride supporting electrolyte, Energy Environ. Sci., 2011, 4, 4068–4073, 10.1039/c0ee00765j.
- L. Swette and V. Jalan, Development of electrodes for the NASA iron/chromium redox system and factors affecting their performance, NASA CR-174724, DOE/NASA/0262-1, 1984 Search PubMed.
- M. C. Wu, T. S. Zhao, H. R. Jiang, Y. K. Zeng and Y. X. Ren, High-performance zinc bromine flow battery via improved design of electrolyte and electrode, J. Power Sources, 2017, 355, 62–68, DOI:10.1016/j.jpowsour.2017.04.058.
- W. Wang and V. Sprenkle, Redox flow batteries go organic, Nat. Chem., 2016, 8, 204–206, DOI:10.1038/nchem.2466.
- M. L. Perry and A. Z. Weber, Advanced redox-flow batteries: a perspective, J. Electrochem. Soc., 2016, 163, A5064–A5067, DOI:10.1149/2.0101601jes.
- C. Zhang, Y. Qian, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Biredox eutectic electrolytes derived from organic redox-active molecules: high-energy storage systems, Angew. Chem., Int. Ed., 2019, 58, 7045–7050, DOI:10.1002/anie.201902433.
- Y. V. Pleskov, Nonaqueous Electrochemistry Edited by D. Aurbach, Russ. J. Electrochem., 2001, 37, 871–873, DOI:10.1023/A:1016707724482.
- M. C. Buzzeo, C. Hardacre and R. G. Compton, Extended electrochemical windows made accessible by room temperature ionic liquid-organic solvent electrolyte systems, ChemPhysChem, 2006, 7, 176–180, DOI:10.1002/cphc.200500361.
- I. L. Escalante-García, J. S. Wainright, L. T. Thompson and R. F. Savinell, Performance of a non-aqueous vanadium acetylacetonate prototype redox flow battery: examination of separators and capacity decay, J. Electrochem. Soc., 2015, 162, A363–A372, DOI:10.1149/2.0471503jes.
- P. J. Cabrera, X. Yang, J. A. Suttil, R. E. M. Brooner, L. T. Thompson and M. S. Sanford, Evaluation of tris-bipyridine chromium complexes for flow battery applications: impact of bipyridine ligand structure on solubility and electrochemistry, Inorg. Chem., 2015, 54, 10214–10223, DOI:10.1021/acs.inorgchem.5b01328.
- Q. Liu, A. A. Shinkle, Y. Li, C. W. Monroe, L. T. Thompson and A. E. S. Sleightholme, Nonaqueous chromium acetylacetonate electrolyte for redox flow batteries, Electrochem. Commun., 2010, 12, 1634–1637, DOI:10.1016/j.elecom.2010.09.013.
- X. Xing, D. Zhang and Y. Li, A non-aqueous all-cobalt redox flow battery using 1,10-phenanthrolinecobalt(II) hexafluorophosphate as active species, J. Power Sources, 2015, 279, 205–209, DOI:10.1016/j.jpowsour.2015.01.011.
- Z. Li, S. Li, S. Liu, K. Huang, D. Fang, F. Wang and S. Peng, Electrochemical properties of an all-organic redox flow battery using 2,2,6,6-tetramethyl-1-piperidinyloxy and N-methylphtalimide, Electrochem. Solid-State Lett., 2011, 14, A171–A173, DOI:10.1149/2.012112esl.
- X. Wei, W. Xu, M. Vijayakumar, L. Cosimbescu, T. Liu, V. Sprenkle and W. Wang, TEMPO-based catholyte for high-energy density nonaqueous redox flow batteries, Adv. Mater., 2014, 26, 7649–7653, DOI:10.1002/adma.201403746.
- Y. Liu, M.-A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, A Long Lifetime All-Organic Aqueous Flow Battery Utilizing TMAP-TEMPO Radical, Chem, 2019, 5, 1861–1870, DOI:10.1016/j.chempr.2019.04.021.
- A. P. Kaur, N. E. Holubowitch, S. Ergun, C. F. Elliott and S. A. Odom, A highly soluble organic catholyte for non-aqueous redox flow batteries, Energy Technol, 2015, 3, 476–480, DOI:10.1002/ente.201500020.
- J. D. Milshtein, A. P. Kaur, M. D. Casselman, J. A. Kowalski, S. Modekrutti, P. L. Zhang, N. H. Attanayake, C. F. Elliott, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, High current density, long duration cycling of soluble organic active species for non-aqueous redox flow batteries, Energy Environ. Sci., 2016, 9, 3531–3543, 10.1039/c6ee02027e.
- W. Duan, R. S. Vemuri, J. D. Milshtein, S. Laramie, R. D. Dmello, J. Huang, L. Zhang, D. Hu, V. Murugesan, W. Wang, J. Liu, R. Darling, L. Thompson, K. C. Smith, J. Moore, F. R. Brushett and X. Wei, A symmetric organic-based nonaqueous redox flow battery and its state of charge diagnostics by FTIR, J. Mater. Chem. A, 2016, 4, 5448–5456, 10.1039/c6ta01177b.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, On the benefits of a symmetric redox flow battery, J. Electrochem. Soc., 2016, 163, A338–A344, DOI:10.1149/2.0971602jes.
- W. Wang, W. Xu, L. Cosimbescu, D. Choi, L. Li and Z. Yang, Anthraquinone with tailored structure for a nonaqueous metal–organic redox flow battery, Chem. Commun., 2012, 48, 6669–6671, 10.1039/c2cc32466k.
- J. Huang, Z. Yang, M. Vijayakumar, W. Duan, A. Hollas, B. Pan, W. Wang, X. Wei and L. Zhang, A two-electron storage nonaqueous organic redox flow battery, Adv. Sustain. Syst., 2018, 2, 1700131–1700137, DOI:10.1002/adsu.201700131.
- V. Vatanen and J. A. Pedersen, Electron paramagnetic resonance and cyclic voltammetry studies of the cations and anions of α-aminoanthraquinones obtained by electrochemical oxidation/reduction, J. Chem. Soc., Perkin Trans. 2, 1996, 2, 2207–2212, 10.1039/P29960002207.
- P. Geysens, Y. Li, I. Vankelecom, J. Fransaer and K. Binnemans, Highly soluble 1,4-diaminoanthraquinone derivative for nonaqueous symmetric redox flow batteries, ACS Sustain. Chem. Eng., 2020, 8, 3832–3843, DOI:10.1021/acssuschemeng.9b07244.
- M. Gordillo, C. Pereyra and E. J. Martinez de la Ossa, Solubility estimations for Disperse Blue 14 in supercritical carbon dioxide, Dyes Pigm., 2005, 67, 167–173, DOI:10.1016/j.dyepig.2004.12.013.
- R. S. Alwi, T. Tanaka and K. Tamura, Measurement and correlation of solubility of anthraquinone dyestuffs in supercritical carbon dioxide, J. Chem. Thermodynamics, 2014, 74, 119–125, DOI:10.1016/j.jct.2014.01.015.
- A. P. Krapcho, Z. Getahun, K. L. Avery, Jr, K. J. Vargas and M. P. Hacker, Synthesis and antitumor evaluations of symmetrically and unsymmetrically substituted 1,4-bis[(aminoalkyl)amino]anthracene-9,10-diones and 1,4-bis[(aminoalkyl)amino]-5,8-dihydroxyanthracene-9,10-diones, J. Med. Chem., 1991, 34, 2373–2380, DOI:10.1021/jm00112a009.
- K. C. Murdock, R. G. Child, P. F. Fabio and R. B. Angier, Antitumor agents. 1. 1,4-bis[(aminoalkyl)amino]-9,l0-anthracenediones, J. Med. Chem., 1979, 22, 1024–1030, DOI:10.1021/jm00195a002.
- A. Gianoncelli, S. Basili, M. Scalabrin, A. Sosic, S. Moro, G. Zagotto, M. Palumbo, N. Gresh and B. Gatto, Rational design, synthesis, and DNA binding properties of novel sequence-selective peptidyl congeners of ametantrone, ChemMedChem, 2010, 5, 1080–1091, DOI:10.1002/cmdc.201000106.
- J. Mei and Z. Bao, Side chain engineering in solution-processable conjugated polymers, Chem. Mater., 2014, 26, 604–615, DOI:10.1021/cm4020805.
- D. Komáromy, M. C. A. Stuart, G. M. Santiago, M. Tezcan, V. V. Krasnikov and S. Otto, Self-assembly can direct covalent bond formation towards diversity or specificity, J. Am. Chem. Soc., 2017, 139, 6234–6241, DOI:10.1021/jacs.7b01814.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with detailed solubility data of DAAQs. TLC experiment comparing the DAAQs. Optical absorption spectra of DAAQs in different solvent systems. Comparative cyclic voltammograms of 0.01 mol L−1 DB-134 in 0.1 mol L−1 TEATf2N in acetonitrile with and without IR drop correction. Blank cyclic voltammogram of 0.1 mol L−1 TEATf2N in acetonitrile. See DOI: 10.1039/d0ra06851a |
| This journal is © The Royal Society of Chemistry 2020 |