
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolysulfone metal-activated carbon magnetic nanocomposites with enhanced CO2 capture
Muhammad Nisar *a,
Pascal S. Thueb,
Myriam B. Maghousa,
Julian Geshevc,
Eder C. Limab and
Sandra Einloft*a
*a,
Pascal S. Thueb,
Myriam B. Maghousa,
Julian Geshevc,
Eder C. Limab and
Sandra Einloft*a
aPrograma de Pós-Graduação em Engenharia e Tecnologia de Materiais (PGETEMA), Pontifical Catholic University of Rio Grande do Sul (PUCRS), Porto Alegre, Brazil. E-mail: Nisarchem1984@gmail.com; Einloft@pucrs.br
bInstitute of Chemistry – Federal University of Rio Grande do Sul (UFRGS), Av. Bento Gonçalves 9500, Porto Alegre, RS, Brazil
cInstitute of Physics, Federal University of Rio Grande do Sul (UFRGS), Av. Bento Gonçalves 9500, Porto Alegre, RS, Brazil
First published on 18th September 2020
Abstract
In the present study, polysulfone (PSF)-activated carbon nanocomposites were synthesized by a melt mixing technique. Here, 2 wt% activated carbon (CA, CA–Ni, and CA–Co) was used as filler, and effects on thermal, mechanical, magnetic, morphological, and carbon dioxide capture properties were studied. The pyrolysis of wood sawdust produced carbon materials activated by Co and/or Ni salt. The thermal degradation and the amount of metal in the carbon materials were investigated by thermogravimetric analysis. The maximum degradation temperature showed an improvement of up to 3 °C, while the initial degradation temperature decreased up to 4 °C with the addition of metal-activated carbons. The values of Tg estimated by differential scanning calorimetry appear to be practically identical for pure PSF and its nanocomposites. The elasticity modulus of the nanocomposite shows an enhancement of 17% concerning the neat PSF. The water contact angle showed a decrease with the incorporation of the fillers, indicating the hydrophilic nature of the composite. The carbon dioxide sorption capacity of the nanocomposite showed an enhancement of almost 10% in contrast to neat PSF. Ferromagnetic behavior of the thermoplastic nanocomposite was observed with the introduction of 2.0 wt% metal-carbonized filler. The exceptional magnetic properties, for a thermoplastic material such as polysulfone, make it promising for various industrial applications.
1. Introduction
In recent times, large-scale emission of CO2 into the atmosphere triggered by fossil-fuel utilization is considered a critical source for climatic changes and global warming.1 As a record, since 1958, the atmospheric CO2 concentration dominantly increased every year with the latest highest recorded value of 414 ppm (https://www.esrl.noaa.gov/gmd/ccgg/trends/). The rise in CO2 level in the atmosphere is predicted to reach a value of 450 ppm in 2100, with an increase of more than 2 °C of the global temperature, due to continuing industrial revolution.2 To limit damage on earth's climate, the effective reduction of CO2 emission to the atmosphere is vital for the economic development and ecological food production.3 Not only is the CO2 capture significant from the environmental point of view, but also the capture of CO2 from the gas stream is obligatory for other essential application areas, including air refining in a confined gas space and treatment of natural gases.4 Synthesis of new materials including inorganic, organic, and composite membranes for gas separation to address the CO2 issue is gaining interest in last few decades. Despite the excellent separation efficiency of inorganic membranes such as zeolite and alumina, compared to polymeric membranes, the high cost, fragility, complicated processing, and low mechanical properties make them less desirable. However, improved permeability, low cost, relaxed processing, and reasonable gas separation of organic membranes make them the material of choice.5 The use of polymeric nanocomposites in the carbon capture technology has well-established advantages such as simplicity, energy efficiency, engineering scalability, and economic cost potentiality, over the conventional liquid and solid sorbents.6,7 Polymeric nanocomposites are multiphase materials in which the matrices are filled with nanosized particles (in the range of 1–100 nm) and can be formed by the introduction of nanofillers into the polymers.8 The formulation of the nanocomposite membranes commonly depends on the choice of materials that competently adsorb CO2, for example, activated carbons, modified silica, zeolites and metal organic frameworks (MOFs).5 Besides, the design of polymer composites can be advantageous in applications such as gas separation membranes to space transportation.The membrane technology is gaining interest in various fields such as medicine, biochemistry, food packaging, water treatment, and separation processes.9,10 For gas and liquid separation, polymers are more favorable to make selective membranes than inorganic materials, forming a thin membrane that selects what permeates through it.10,11 The preparation method depends on the morphology and material of the membrane, and the principal methods used to produce polymer membranes are phase inversion, stretching, sintering, track etching, electrospinning, coating, layer-by-layer assembly (LBL), and graft polymerization.12 The type of filler and the concentration produce different effects on the properties. Thermoplastic polysulfone is commonly used to produce films. It is widely used in engineering, presenting great thermic, mechanic, and biochemical stabilities,13 like oxidation and hydrolysis resistance, excessive functionality at different pH values, and high rigidity.14 An example of the advantages obtained by the addition of nanoparticles is the notable reduction of the bacterial attack on filled polysulfone membranes. Because of their advanced properties, polysulfone nanocomposites are a growing focus of the membrane researchers.14
In recent years, special attention has been paid to nanoparticles possessing magnetic properties (MNPs) due to their promising use in medicine, biotechnology, water treatment, tissue engineering, data storage, and catalysis.15–18 The choice of magnetic particles depends on the application's purpose.19–22 Furthermore, metals are exposed to high toxicity, and oxidation needs proper surface treatment of the magnetic fillers to assure their chemical stability and safety.23 Many research studies have been reported to fix these complications involving the coating method, including the encapsulation process, by non-magnetic surfaces such as silica, carbons, and polymers, effectively preventing the agglomeration and oxidation.24,25 Iron-based magnetic fillers are commonly used to provide magnetism, together with cobalt and nickel.26,27
Among the other methods used for the synthesis of polymer nanocomposites, melt mixing is the most widely adopted technique due to its industrial applicability, easy operation procedure, high yields, fast production rate, and solventless process to avoid any health risk.28 More recently, we have studied how different synthetic talcs affect the polyurethane nanocomposites' properties.29–31 The focus of the present work is to compare the use of different metal-activated carbons obtained from the biomass32 (low-cost source) as fillers to synthesize polysulfone (PSF) nanocomposites with enhanced CO2 capture capacity, good thermal, mechanical and magnetic properties by a melt-mixing technique.
2. Experimental
2.1 Materials
As a polymer matrix, commercial polysulfone (average Mw ∼35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by LS, average Mn ∼16000 by MO, pellets, Sigma-Aldrich), as well as antioxidant agent Irganox 1010, was used for composite preparation.
000 by LS, average Mn ∼16000 by MO, pellets, Sigma-Aldrich), as well as antioxidant agent Irganox 1010, was used for composite preparation.
Metal-carbonized materials containing nickel (CA–Ni) and cobalt (CA–Co) were obtained following the procedure reported in ref. 32.
As activating agents, CoCl2 and NiCl2 metal salts purchased from Neon (São Paulo, Brazil) were utilized without further treatment; CoCl2 and NiCl2 show an oxidation state of +2 in aqueous solutions (pH close to 6).
2.2 Preparation of activated carbon-based cobalt and nickel from biomass
Ayous biomass is a waste from wood processing. Some studies carried out in sawmills in the Adamaoua Region, Cameroon, revealed very high use of Ayous trees. They have low durability, and hence, they are widely used for light carpentry. The transformation of this species generates tons of wastes per year. Therefore, any attempt to reuse this waste would be useful for the country. The Ayous residues were kindly provided by Cameroon's sawmill industries and used without any pre-treatment. The residue contains about 98% cellulose, hemicellulose, and lignin, with the remaining 2% made up of extractables (pectins, tannins, and polymers with a low degree of polymerization) and inorganics such as Ca2+, Na+, Cl− and SO2−. The Ayous residue was used as a carbon source without any pre-treatment for the preparation of activated carbons with two inorganics, namely, chlorides cobalt(II) and nickel(II). The biomass residue was first ground into sawdust (diameter < 300 μm) before use in the preparation of the activated carbon.33The process preparation is as follows: an appropriate amount of chloride salt of cobalt(II) or nickel(II) was completely dissolved in 60.0 mL of treated water, which was followed by the addition of 60.0 g of dried sawdust. The solution was then mixed continuously at approximately 80 °C for 2 hours, and the sample was dried overnight before the carbonization step. The mass impregnation ratio of the inorganic salt to the Ayous sawdust was 1:1 (w/w) for both nickel chloride and cobalt chloride.
The carbonization of the dried powder impregnated with cobalt or nickel salts has been described in our previous study;32 the final carbonization temperature was 700 °C. For comparison with the nanocomposites, the activated carbon material (C-neat) was prepared under the same conditions but without the addition of cobalt(II) chloride or nickel(II) chloride.
2.3 Synthesis of polysulfone nanocomposites
The preparation of nanocomposites was carried out using a Thermo Haake Rheomix-600 (Thermo Electron Corp, Karlsruhe, Germany), operating at 60 rpm and 340 °C. A defined amount of polymer matrix, carbonized metal, and 0.005 g of Irganox1010 were introduced per mixing, which contributes to a total quantity of approximately 60 g. The variation of the filler in the matrix was set from 0 to 2 wt%. In the first step, PSF (∼30 g) and the antioxidant were mixed. After 2 min, different quantities of the filler (0–2 wt%) was gradually added to the melted polymer for 3 min. In the last step, the remaining polymer pellets were added by keeping the mixer speed constant at 60 rpm for 5 min. The whole mixing time was approximately 10 min. Finally, the blend was hot-pressed to form sheets at 325 °C under a pressure of 200 bars.3. Characterization
An elemental analyzer (Perkin-Elmer M CHNS/O model 2400) was used to analyze the CNH/O of the Ayous residue and the activated carbon filler. Briefly, 0.05 g of the oven-dried sample was used to determine the total carbon content (C), nitrogen content (N), and hydrogen content (H). The oxygen-containing (O) mass percentage was found as the ash-containing C, N, and H mass percentages were subtracted from the total mass.The thermal stabilities of the Ayous residue and the activated carbon were checked by thermogravimetric analysis (TGA), exploiting a thermogravimetric analyzer (TA model SDT Q600). The sample was heated up from 20 °C to 800 °C in an inert N2 atmosphere (10 °C min−1) and from 800 °C to 1000 °C in an atmosphere containing oxidants. The oxidant atmosphere allows finding the ash content fraction of the sample.
A transmission electron microscope (TEM, Tecnia G2T20 FEI) was used to perform the morphological analysis to investigate the degree of dispersion on the nanocomposites. Polymeric nanocomposite samples were prepared from ∼50 nm thick ultrathin films cut under cryogenic conditions using a Leica ultra cut UCT microtome placed on a grid. The sample morphology was examined by field-emission scanning electron microscopy (FESEM) using an Inspect F50 equipment (FEI Instruments) in secondary electron mode. The samples were placed on an aluminum stub and coated with gold to make them conducting.
Room-temperature magnetic characterization was performed using an EZ9 MicroSense magnetometer, which is the magnetic field, H, ranging from −18 kOe to +18 kOe.
The values of Tg of pure polysulfone and metal nanocomposites were obtained using a Perkin-Elmer differential calorimeter (TA Instruments model Q20). The heating rate was 10 °C min−1 from −25 to 200 °C in a nitrogen atmosphere. Thermal decomposition of pure PSF and its nanocomposites was performed by thermogravimetric analysis (TGA) using an SDT Q600 thermal analyzer (TA Instruments) at a scanning rate of 20 °C min−1 from 25 °C to 800 °C at a constant nitrogen flow of 100 mL min−1 for polymeric nanocomposites and air for carbon materials.
The pressure-decay technique determined the CO2 sorption capacity. The dual-chamber gas sorption cell was similar to that reported in Koros et al.34 Before measurements, 0.7 g of the sample was weighed and dried at 70 °C (343.15 K) for 1 h. CO2 sorption experiments were carried out at 45 °C (318.15 K) and 4 bars. The CO2 sorption capacity was calculated using eqn (1) and (2):
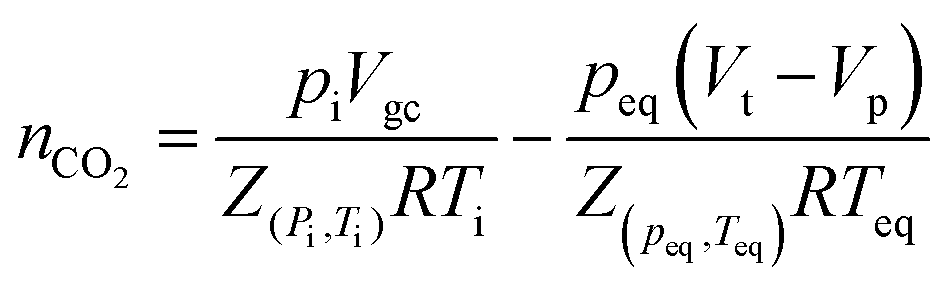 | (1) |
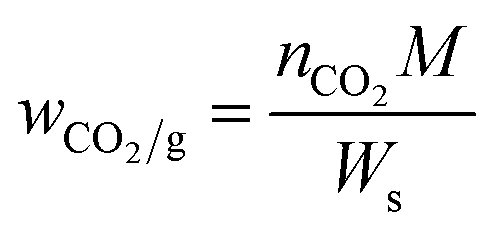 | (2) |
The mechanical properties were analyzed using an HP model D-500 dynamometer according to ASTMD 638-10 at ∼25 °C. For each weight percentage of the nanocomposite, the average of results of five measurements (typical deviation ∼5%) is presented. The bone-shaped samples of overall length 30 mm, width 4.0 mm, and thickness 0.5 mm were tested at a crosshead rate of 50 mm min−1.
A goniometer (Phoenix 300, SEO) was used to measure the static water contact angle by the sessile drop method. Images were obtained using a “Drop Shape Analysis system.” The mean contact angle was calculated by adding five drops of deionized water onto each sample, considering an experimental error of 2 degrees among the measurements.
4. Results and discussion
4.1 Elemental analysis of the Ayous biomass and metal-activated carbons
The elemental composition of the activated carbon material containing nickel and cobalt metals is given in Table 1. When compared with the precursor material, the prepared activated carbons showed a very high content of carbon and a low content of oxygen. The Ayous residue biomass contained initially 46.05% of carbon, 6.26% of hydrogen, and 45.78% of oxygen. Notwithstanding, after pyrolysis and the washing process, the carbon contents in the CA–Ni and CA–Co materials increased to 58.05% and 53.57%, respectively. Indeed, the aromatic structure of the materials was formed during the carbonization process in the presence of nickel or cobalt metals, as indicated by the enhanced carbon content.33| Samples | % C | % H | % N | % Oa | % Ashb | M-content (%) | SBETc (m2 g−1) | Vtotc (cm3 g−1) | Dpc (nm) |
|---|---|---|---|---|---|---|---|---|---|
| a Yield by difference (% O = 100% − % C − % H – % N − % Ash).b Obtained by TGA.c SBET = BET surface area; Vtot = total pore volume; Dp = average pore diameter. | |||||||||
| Biomass | 46.05 | 6.26 | 0.33 | 45.78 | 1.56 | 0.00 | — | — | — |
| CA–Ni | 58.05 | 2.31 | 0.15 | 11.29 | 28.20 | 26.64 | 381 | 0.321 | 3.3 |
| CA–Co | 53.57 | 2.49 | 0.38 | 19.97 | 23.60 | 22.04 | 619 | 0.340 | 2.2 |
The ash content was determined by TGA analysis. The residue remained in the TGA analysis performed in a synthetic air atmosphere is commonly attributed to the ash content. The ash content was used to determine the amount of nickel or cobalt present in the metal–carbon materials after the preparation. One can obtain the carbon–composite (CA–M) metal content knowing the ash content of the Ayous biomass residue and metal–carbon materials. As one can see in Table 1, the CA–Ni composite showed more metal embedded in the structure of the carbon sample; moreover, it also shows the textural parameters of the synthesized activated carbon. The sample CA–Co shows the highest surface area, total pore volume, and the smallest pore diameter.
4.2 XRD analysis of the nanofiller
Fig. 1 presents the XRD patterns of the biomass (CA) and different metal particles embedded in the amorphous carbon. The CA has an amorphous structure,36 where the XRD of Ni–C showed typical sharp peaks at 2θ = 44.5, 51.8, and 76.3° corresponding to planes (111), (200), and (222), respectively, representing the face-centered cubic (fcc) structure of Ni.37 The mean crystallite size of Ni was estimated employing the Scherer38 equation through the broadening of the maximum intense peak (111), and it was 46 nm. Finally, in the XRD of Co–C particles, it can be seen that the pattern of a simple cubic crystalline structure was represented by diffraction peaks at 2θ: 20.1, 31.0, 36.7, 44.4, 47.4, 59.2 and 65.2° corresponding to the (111), (220), (311), (222), (422), (511) and (440) planes.39 The peak at 36.7 (311) was used to calculate the crystal size, and it was 11 nm. In the resume, the XRD of the M–C particles shows that all the metals have crystalline nanometric structures.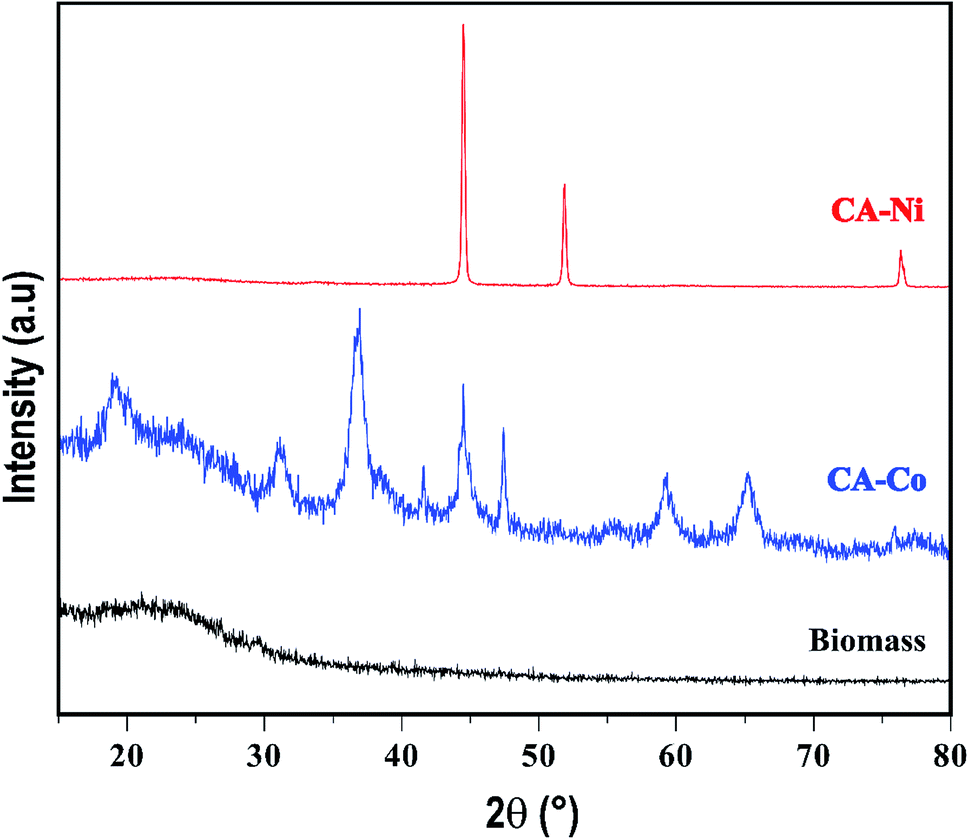 | ||
| Fig. 1 XRD analysis of the biomass and metal-activated carbons. | ||
4.3 Morphological analysis
The dispersion of the filler in the PSF matrix is ascertained by SEM analysis. It is clear from Fig. 2 that, by incorporating the filler in the polymer, the cross-section morphology changed. A better-defined layered morphology appeared with the addition of the fillers, which shows that the polymer is coated around the filler. Furthermore, the nanofiller particles were invisible, indicating a uniform dispersion of the nanofillers in the PSF matrix. Moreover, the presence of metal-activated carbons turns the PSF membrane surface rougher due to the presence of more hydrophilic carbon-based fillers. | ||
| Fig. 2 SEM micrographs of (A) cross-section of PSF-neat, (B) PSF–CA, (C) CA–Ni, and (D) PSF–CA–Co. | ||
Fig. 3 shows the TEM images of the PSF nanocomposites with 2 wt% nanofillers. The filler is very well distributed in the PSF matrix, with no apparent evidence of agglomerates. It can be seen that, in the polymer matrix, the metal particles, cobalt, and nickel, see Fig. 3 (B), (C) and (D) respectively, are surrounded by amorphous carbons. It can be noted that the existence of the black spots in Fig. 3(B), (C) and (D) is due to the presence of the nanoscale metal particles in the PSF matrix. The presence of these black spots is noticeable in the sample containing the nickel particles in the PSF matrix. The existence of the metal as a block spot in the polymer and a similar morphology have been reported in other studies.40–42 The micrographs do not present filler aggregation.
 | ||
| Fig. 3 TEM images of PSF–CA-neat (A), PSF–CA–Co (B), PSF–CA–Ni (C), and (D). | ||
The XRD peaks of PSF and its nanocomposites containing the metal particles are shown in Fig. 4. Diffractogram of PSF and nanocomposites analyzed in the 2θ range from 0 to 80 degrees showed only a peak of 2θ at 17.5° without any change in the peak for its nanocomposites. This is because the small amount of metal particles might be uniformly dispersed into the polymer matrix, resulting in no significant changes in the peaks.
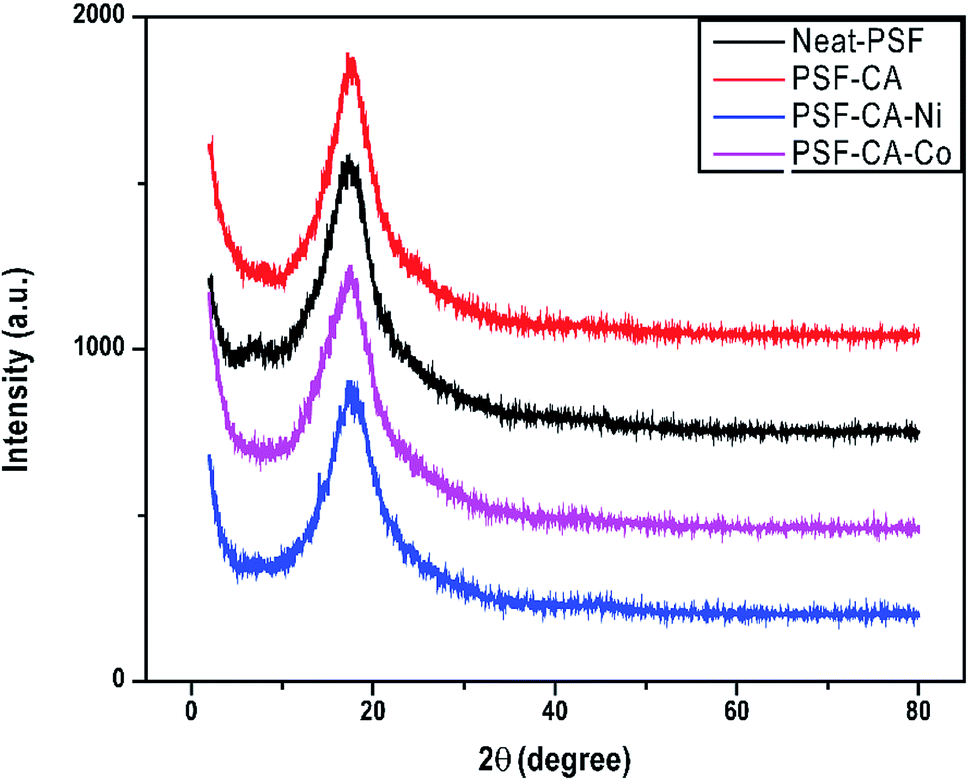 | ||
| Fig. 4 XRD patterns of neat PSF and nanocomposites with different fillers. | ||
4.4 Thermal stability analysis
The thermal degradation properties of all samples shown by TGA analysis are resumed in Table 2 and Fig. 5. The results indicated a slight enhancement in the maximum degradation temperature (Tmax) around 3 °C for the sample with the addition of filler CA–Co, whereas the rest of the samples (PSF–CA and PSF–CA–Ni) showed a similar maximum degradation temperature to that of neat PSF. The thermal stability enhancement in the nanocomposites could be attributed to the fillers serving as a barrier, hindering the oxygen-molecules' transmission into the nanocomposites as well as to the labyrinth effect, which deferred the volatilization.43 The small amount of the filler around 2 wt% was not sufficient to impart a reasonable barrier and labyrinth effect on the thermal enhancement stability of the nanocomposites. Furthermore, the initial degradation temperature (Tonset) of the nanocomposites containing metals (Ni or Co) in the filler was slightly lower around 4 °C than that of neat PSF. Previous studies showed the influence of metals (Ni, Al, Fe, Cu, and Co) on the thermal behavior of plastics, for instance, polypropylene,44 linear low-density polyethylene,45 chitosan,46 or high-density polyethylene.47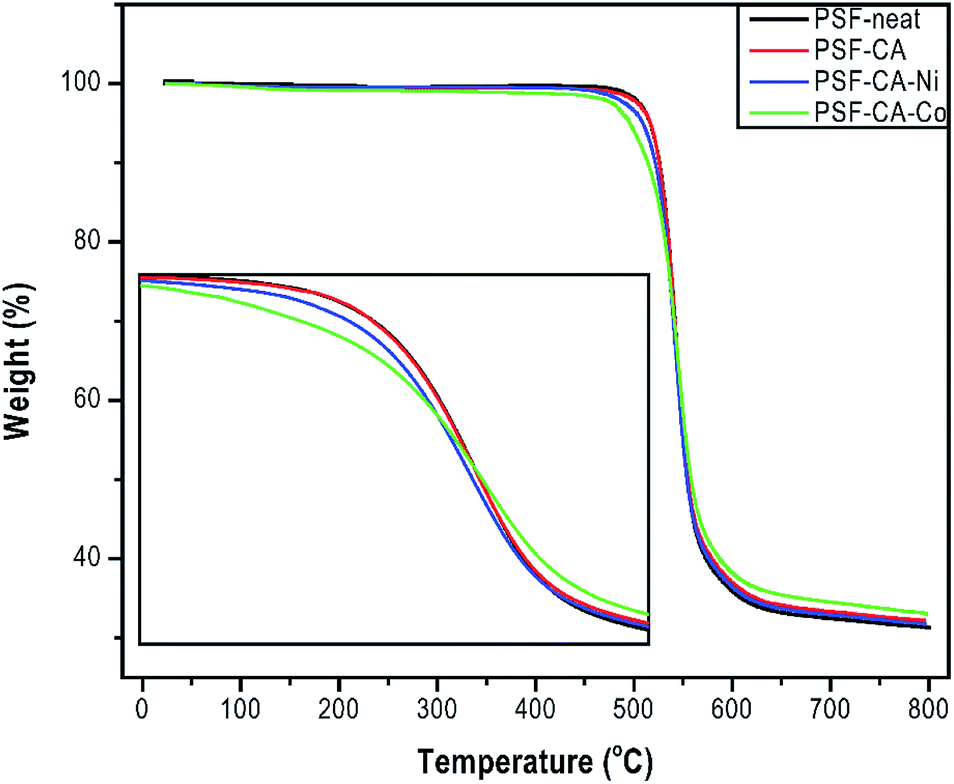 | ||
| Fig. 5 TGA thermogram of PSF-neat and its nanocomposites. | ||
These results indicated that the surface contact of the metals with polymer could stimulate the degradation of the organic part at different rates. Pd nanoparticles accelerated the thermal degradation of PET and PA6.48 Our results support the works as mentioned earlier; Table 2 shows that upon incorporation of CA (activated carbons without metals) into the sample PSF–CA, the Tonset showed an improvement of 2 °C, whereas the addition of the same filler with metals decreased the initial thermal stability. It is thus concluded that the filler containing metals behaves as a catalyst resulting in polymer's degradation, the latter beginning at relatively low temperatures.49 Due to the aromatic structure of the PSF backbone, the residual mass of the sample remains around 32 wt% at 800 °C, which, in turn, forms more thermally stable carbonaceous materials.50
The impacts of the filler on Tg of neat PSF and its nanocomposites are presented in Table 2 and Fig. 6. Tg is typically used to understand the structure of the membrane when an amorphous polymer like polysulfone is employed. Tg of the nanocomposites shows an improvement of around 1 °C, although this effect is not very significant and lies in the error limits of the equipment. Meanwhile, there is a tendency of enhancement of Tg, which can be achieved by the incorporation of higher amounts of the filler. From the above-mentioned results, one can infer the existence of interactions between the polymeric chain and the nanofiller particles. This implies an increase in the rigidity of the polymer chain and, hence, an increase in the amount of energy needed for the polymer chain movement or breakage.51
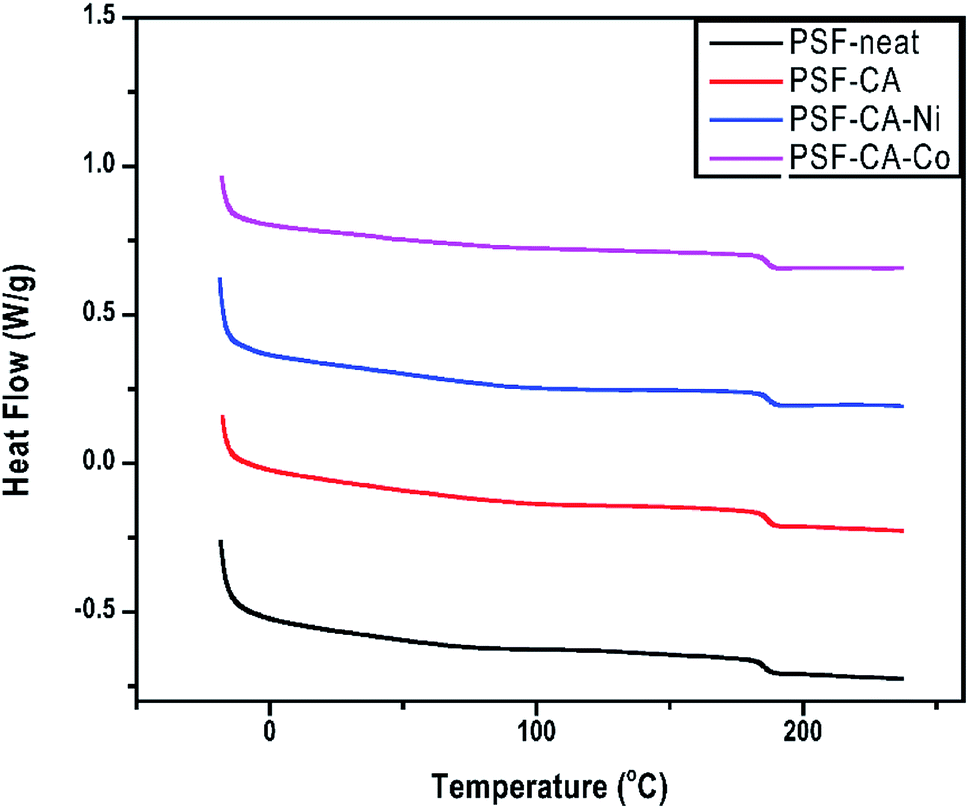 | ||
| Fig. 6 DSC thermograms of PSF-neat and its nanocomposites. | ||
4.5 Carbon dioxide capture capacity
The CO2 sorption capacity of PSF and its nanocomposites was studied at 4 bars and 318.15 °C, as shown in Fig. 7. The interaction between the polar groups and the CO2 molecule52,53 leads, probably, to the adsorptive properties (49.3 mg CO2/g ± 0.31) of PSF. Fig. 7 shows that the CO2 capture capacity of the nanocomposites was slightly improved with the filler's addition. The nanocomposite with 2 wt% of C-neat (activated carbons without metals) and C–Co (activated carbons with cobalt) shows a sorption capacity of 52.32 mg CO2/g ± 0.26 and 54.32 mg CO2/g ± 0.16, respectively, which is almost 10% higher than that of pure PSF. The high uptake of CO2 by the nanocomposites is associated with the existence of activated carbons in the polymer structure with the presence of a large number of ultra-micropores, which helps in the trapping of the CO2 molecules.54 Furthermore, the nanocomposite with the addition of C–Ni (activated carbons with nickel) shows a slight decrease in the capture capacity of CO2. Most probably, this minor decreasing effect of the nanocomposite comes from some agglomeration of filler's in the matrix of PSF. Ko et al.55 immobilized primary, secondary and tertiary amines on mesoporous silica and used it as an absorbent for CO2 capture. The maximum capacity of primary, secondary, and tertiary amine-supported silica was 0.95, 0.75, and 0.17 mmol CO2/g, where our sample shows a high sorption capacity of 54.3 mg CO2/g (∼1.2 mmol CO2/g). Similarly, Huang et al.56 used a polyurea and alkylated graphene oxide IL capsule for CO2 capture, reporting an absorption of only 3.0 mg CO2/g at a pressure of 1.3 bars. More recently, Khdary et al.5 have reported the synthesis of polyvinylidene-fluoride-hexafluoropropylene (PVDF-HFP) amino–silica composites by a phase separation technique and prepared membranes for CO2 capture. Their results indicated CO2 uptake of 33.75 mg CO2/g with the highest loading of 40 wt% of SiO2 in the composite material. Herein, we demonstrate CO2 uptake to be higher than their results with the incorporation of a tiny amount of 2.0 wt% of activated carbon into the polymer matrix. The reason for the highest CO2 capture by the sample PSF–CA–Co could be the higher BET surface area and micropore volume of the activated carbon, as clear from Table 1. The activation of the biomass with the cobalt leads to a high surface area and pore volume, which, in turn, provides the maximum adsorption sites for the petite size CO2 molecule to be captured by the micropores.57 Huang et al.58 reported the preparation of activated carbons from coconut shells and investigated the effect of experimental conditions on the textural properties of the final product. Their results indicated the highest CO2 capture by the sample having the highest BET specific surface area and total pore volume; our results are in close agreement with their findings, thus supporting the results of Hung et al. These results indicated that the presence of an appropriate amount of filler has a positive influence on CO2 capture capacity, as can be seen from Fig. 7.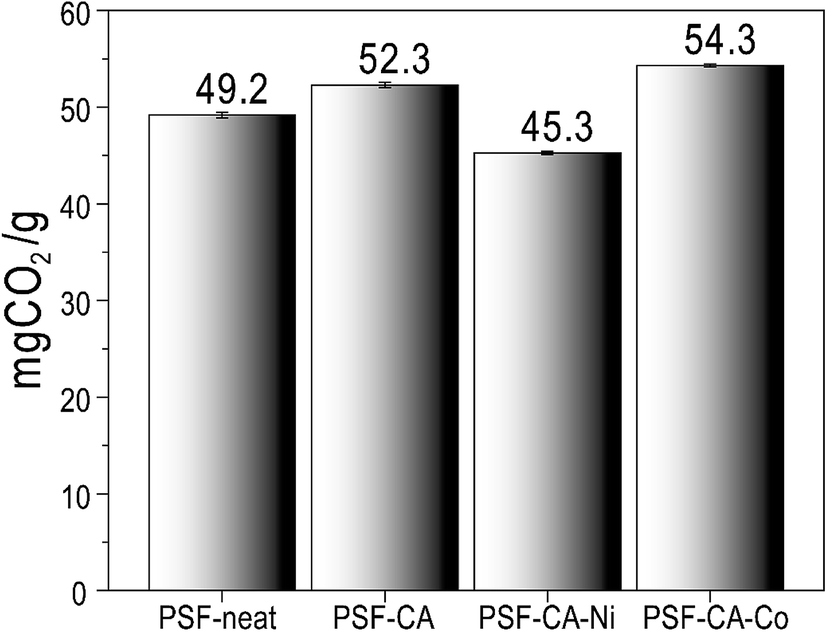 | ||
| Fig. 7 CO2 capture capacity of PSF-neat and its nanocomposites at 45 °C. | ||
4.6 Contact angle measurement
The water contact angle of neat PSF and its nanocomposites was estimated by the sessile drop method to evaluate the wettability of a membrane; the contact of a smooth surface with a water drop is considered the most convenient method. Thus, the hydrophilicity of the materials can be qualitatively obtained using this measurement.59,60 A significant trend of decline in the contact angle with the incorporation of the activated carbon with or without metals is observed in Fig. 8. Dropping of the contact angle leads to the indication that the introduction of hydrophilic activated carbons can be advantageous to increase the membrane hydrophilicity.61 It has already been shown59,61–63 that adding different fillers (i.e., carbon nanotubes, graphene oxide, clay) may result in an upturn in the hydrophilic nature of the PSF-composites.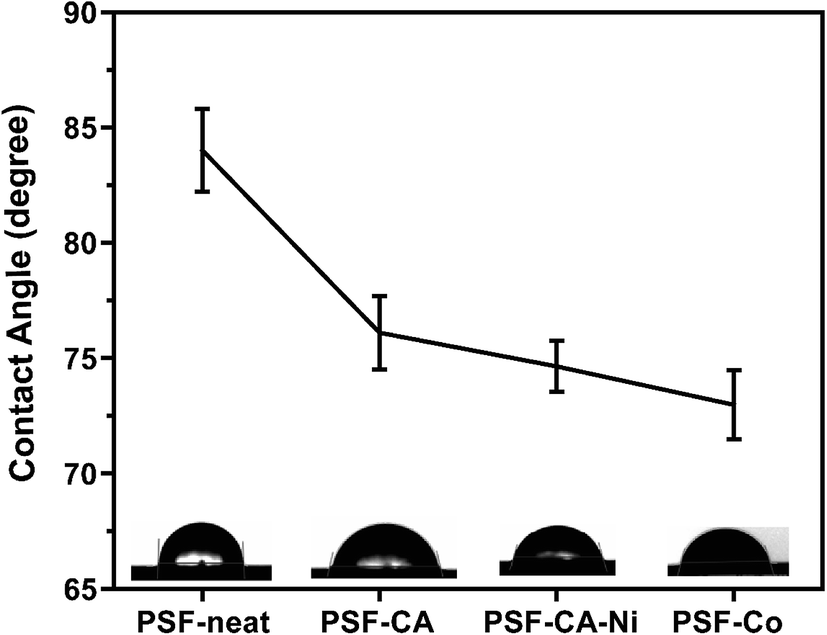 | ||
| Fig. 8 Water contact angle measurement of all samples at 25 °C. | ||
4.7 Mechanical behavior of nanocomposites
Stress–strain curves and elongation at break of the PSF and its nanocomposites with the addition of carbon-based fillers are presented in Table 3 and Fig. 9. The addition of activated carbons to the polymer matrix significantly affects, see Fig. 9 (A), the elastic modulus (enhanced from 1250 to 1464 MPa) despite the meager amount of the filler around 2 wt%. Among the synthesized composite materials, samples PSF–CA and PSF–CA–Co appear to afford maximum reinforcement, i.e., enhancement of almost 17% in the elastic modulus as contrasted with neat PSF. Such an improvement could be attributed to the addition of the stiff-activated carbon with metals, which may prominently affect the mechanical behavior of the composite and by the uniform dispersion, which favors the formation of strong interface PSF/activated carbon nanocomposites.64 However, in comparison with the neat PSF, a decrease in the modulus of the sample PSF–CA–Ni was observed. This is most probably caused by some agglomeration of metal-activated carbons in the PSF matrix, which limits the reinforcement influence of the nanofiller substantially since it restricts the development of significant polymer/filler interfacial adhesion bonds.| Samples | Filler (%) | Tensile modulus (MPa) | Elongation at break (%) |
|---|---|---|---|
| PSF-neat | 0 | 1250 | 7.3 |
| PSF–CA | 2.0 | 1464 | 3.3 |
| PSF–CA–Ni | 2.0 | 1054 | 2.7 |
| PSF–CA–Co | 2.0 | 1451 | 3.4 |
 | ||
| Fig. 9 Characteristic stress–strain curves of PSF and nanocomposites (A). Elongation at break (B). | ||
Elongation at a breakpoint is a measure associated with the material's ductility. Generally, the addition of nanoparticles turns the nanocomposites more fragile.61 It is clear from Fig. 9 (B) and Table 3 that the elongation at break is reduced by almost 70% as compared to the pure PSF as the filler is included. The nanoparticles toughly constrain the movement of the polymer chain during the stress.65 The superior intrinsic mechanical behavior of the filler and an interfacial interaction of the filler/PSF matrix is the crucial motivation for the enhancement of mechanical properties.66
4.8 Magnetic characterization
Fig. 10 presents the major magnetization hysteresis loops of PSF–CA–Ni and PSF–CA–Co nanocomposites. The nanocomposite with Co filler saturates in magnetic fields much higher than the others (see the insets in this figure), which should be attributed to the higher magnetic anisotropy energy of Co as compared to that of Ni. The amplitude of H used here was sufficiently significant to circumvent minor loop effects.67–69 This difference in anisotropy could also explain the preferably higher value of the coercive field HC (≈240 Oe) of the nanocomposite with the Co filler as compared to that with Ni (≈100 Oe). However, the value of the normalized remnant magnetization of 0.10 of the Co-filler sample is smaller than that of the Ni-filler nanocomposite equal to 0.14. This result could be attributed to the stronger dipolar interactions present in the Ni-filler nanocomposite.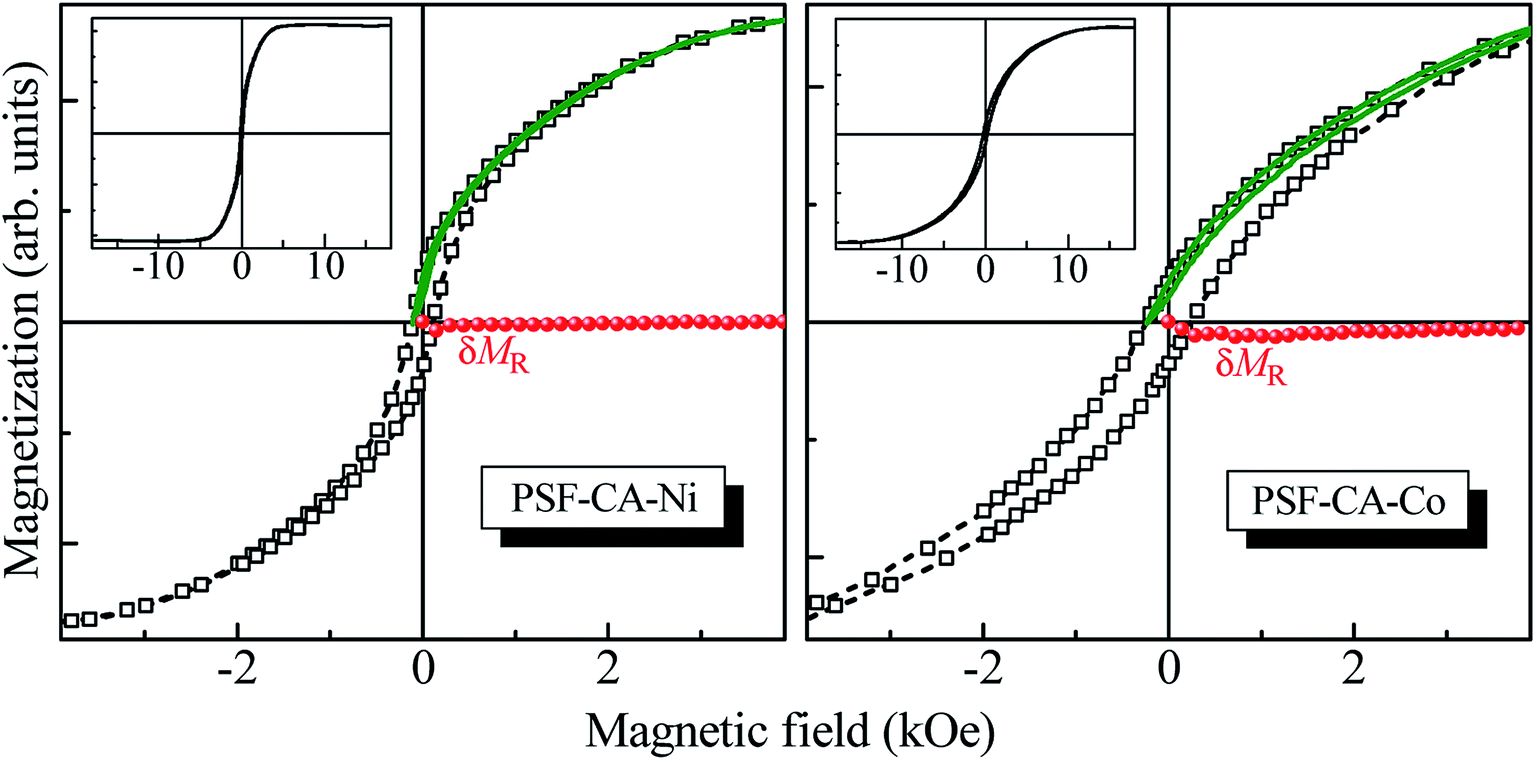 | ||
| Fig. 10 Major magnetization hysteresis loops (empty squares) and the respective interaction δMR plots (full circles), constructed from recoil curves (solid lines) with the recoil field equal to HC for the PSF–CA–Ni (left panel) and PSF–CA–Co (right panel) nanocomposites. The insets show the major loops in the whole field range. | ||
The effects of magnetic interactions of the nanocomposites with magnetic fillers were estimated herein through the recently introduced recoil-loop δMR plots.70,71 Any nonzero deviation of δMR(H) should be ascribed to the magnetic interactions if the magnetic anisotropy is uniaxial. For the present study, we used recoil loops (also shown in Fig. 10) with a recoil field equal to HC. The δMR plot for the Ni filler sample indicates no noticeable magnetic interactions, given that δMR ≈ 0 for all H values. In contrast, the δMR plot obtained for the Co-filler nanocomposite is weak though noticeably negative, stabilizing the demagnetized state interactions. The latter leads to a decrease in the remnant magnetization, explaining the experimental observation.
5. Conclusions
The use of cost-effective materials as fillers for the synthesis of polymeric nanocomposites is an exciting area of research. We demonstrated the use of a new cost-effective carbon-based magnetic filler for the synthesis of polysulfone magnetic nanocomposites. The activation of the carbon with metals, here either Ni or Co, leads to the encapsulation of these metals by amorphous carbons, protecting them from natural oxidation and improving their dispersion within the polymer matrix. Morphological SEM and TEM analyses show that the filler was uniformly dispersed in the polymer matrix. The maximum degradation temperature of the nanocomposites was enhanced by the addition of the fillers, indicating an improvement of 3 °C. The initial degradation temperature of the nanocomposites shows a decrease of 4 °C as compared to neat PSF, which can be explained by the catalytic action of the metals in the filler. The glass transition temperature did not show any effective change with the filler loading.Moreover, the contact angle of the nanocomposites was reduced, indicating the hydrophilic nature of the nanocomposites. The elastic modulus of the nanocomposites indicated a significant improvement of around 17% at such low loading of fillers of 2 wt%. A critical usage of a polymeric nanocomposite is as a membrane for CO2 capture. Our results indicated that, when contrasted with the pure PSF matrix, the absorption capacity of the nanocomposites was enhanced by almost 10%. The nanocomposites showed ferromagnetic behavior with the addition of the Ni- and Co-activated carbon. These results are significant, given that the majority of the studies on such systems reported magnetic properties at low temperatures, whereas we obtained a high coercive field of ≈240 Oe at ambient temperature. Thus, we synthesized multifunctional PSF nanocomposites using a cost-effective filler, which can be beneficial in various industrial applications. This material has the potential to be used in a variety of applications, where a flexible magnetic material with good processability is required, for example, electromagnetic device applications such as electromagnetic interference suppression, aircraft industries, and microelectronics.
Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
CNPq has financed this work − grants 305796/2016-0, and 303.622/2017-2 and 422740/2018-7, and CAPES – Finance Code 001.References
- R. Selyanchyn and S. Fujikawa, Sci. Technol. Adv. Mater., 2017, 18, 816–827 CrossRef CAS.
- M. Vinoba, M. Bhagiyalakshmi, S. K. Jeong, S. C. Nam and Y. Yoon, Chem.–Eur. J., 2012, 18, 12028–12034 CrossRef CAS.
- J. Rogelj, M. Den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS.
- H. Cheng, P. Wang, J. Luo, J. Fransaer, D. E. De Vos and Z. Luo, Ind. Eng. Chem. Res., 2015, 54, 3107–3115 CrossRef CAS.
- N. H. Khdary and M. E. Abdelsalam, Arab. J. Chem., 2020, 13, 557–567 CrossRef CAS.
- T. C. Merkel, H. Lin, X. Wei and R. Baker, J. Memb. Sci., 2010, 359, 126–139 CrossRef CAS.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, 1138–1148 CrossRef.
- P. H. C. Camargo, K. G. Satyanarayana and F. Wypych, Mater. Res., 2009, 12, 1–39 CrossRef CAS.
- L. Phelane, F. N. Muya, H. L. Richards, P. G. L. Baker and E. I. Iwuoha, Electrochim. Acta, 2014, 128, 326–335 CrossRef CAS.
- Z. Pientka, J. Peter, J. Zitka and P. Bakonyi, Curr. Biochem. Eng., 2014, 1, 99–105 CrossRef CAS.
- S. M. Momeni and M. Pakizeh, Brazilian J. Chem. Eng., 2013, 30, 589–597 CrossRef CAS.
- X. Tan and D. Rodrigue, Polymers, 2019, 11, 1160 CrossRef.
- S. T. Muntha, M. Ajmal, H. Naeem, A. Kausar, M. A. Zia and M. Siddiq, Polym. Compos., 2019, 40, 1897–1910 CrossRef CAS.
- A. M. Alosaimi, M. A. Hussein, M. Y. Abdelaal, M. A. Elfaky, T. R. Sobahi and A. M. Abdel-Daiem, Cogent Chem., 2017, 3, 1–17 Search PubMed.
- S. C. N. Tang and I. M. C. Lo, Water Res., 2013, 47, 2613–2632 CrossRef CAS.
- M. Neamtu, C. Nadejde, V. D. Hodoroaba, R. J. Schneider, L. Verestiuc and U. Panne, Sci. Rep., 2018, 8, 1–11 CrossRef CAS.
- A. Hasan, M. Morshed, A. Memic, S. Hassan, T. J. Webster and H. E. S. Marei, Int. J. Nanomedicine, 2018, 13, 5637–5655 CrossRef CAS.
- A. Ito, M. Shinkai, H. Honda and T. Kobayashi, J. Biosci. Bioeng., 2005, 100, 1–11 CrossRef CAS.
- Q. Huang, Y. Guo, X. Wang, L. Chai, J. Ding, L. Zhong, T. T. Li, Y. Hu, J. Qian and S. Huang, Nanoscale, 2020, 12, 10019–10025 RSC.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T. T. Li, Y. Hu, J. Qian and S. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS.
- X. Wang, L. Chai, J. Ding, L. Zhong, Y. Du, T. T. Li, Y. Hu, J. Qian and S. Huang, Nano Energy, 2019, 62, 745–753 CrossRef CAS.
- X. Wang, Z. Zhu, L. Chai, J. Ding, L. Zhong, A. Dong, T. T. Li, Y. Hu, J. Qian and S. Huang, J. Power Sources, 2019, 440, 227158 CrossRef CAS.
- N. Tran and T. J. Webster, J. Mater. Chem., 2010, 20, 8760–8767 RSC.
- K. Saleh and P. Guigon, Handb. Powder Technol., 2007, 11, 323–375 Search PubMed.
- N. Zhu, H. Ji, P. Yu, J. Niu, M. U. Farooq, M. W. Akram, I. O. Udego, H. Li and X. Niu, Nanomaterials, 2018, 8, 1–27 Search PubMed.
- T. Guo, M. Lin, J. Huang, C. Zhou, W. Tian, H. Yu, X. Jiang, J. Ye, Y. Shi, Y. Xiao, X. Bian and X. Feng, J. Nanomater., 2018, 1–8 Search PubMed.
- J. Neamtu, I. Jitaru, T. Malaeru, G. Georgescu, W. Kappel and V. Alecu, Nanotechnology, 2005, 1, 222–224 CAS.
- S. P. Bao and S. C. Tjong, Mater. Sci. Eng. A, 2008, 485, 508–516 CrossRef.
- M. A. Prado, G. Dias, C. Carone, R. Ligabue, A. Dumas, C. Le Roux, P. Micoud, F. Martin and S. Einloft, J. Appl. Polym. Sci., 2015, 132, 1–8 CrossRef.
- G. Dias, M. Prado, R. Ligabue, M. Poirier, C. Le Roux, F. Martin, S. Fery-Forgues and S. Einloft, Appl. Clay Sci., 2018, 158, 37–45 CrossRef CAS.
- G. Dias, M. Prado, C. Carone, R. Ligabue, A. Dumas, C. Le Roux, P. Micoud, F. Martin and S. Einloft, Macromol. Symp., 2016, 367, 136–142 CrossRef CAS.
- P. S. Thue, E. C. Lima, J. M. Sieliechi, C. Saucier, S. L. P. Dias, J. C. P. Vaghetti, F. S. Rodembusch and F. A. Pavan, J. Colloid Interface Sci., 2017, 486, 163–175 CrossRef CAS.
- J. M. Sieliechi and P. S. Thue, Desalin. Water Treat., 2015, 55, 986–998 CrossRef CAS.
- W. J. Koros and D. R. Paul, J. Polym. Sci. Polym. Phys. Ed., 1976, 14, 1903–1907 CrossRef CAS.
- W. Span and R. Wagner, J. Phys. Chem. Ref. Data, 1996, 25, 1509–1596 CrossRef.
- L. Li, Z. H. Zhu, Z. F. Yan, G. Q. Lu and L. Rintoul, Appl. Catal. A Gen., 2007, 320, 166–172 CrossRef CAS.
- D. Dutta, B. J. Borah, L. Saikia, M. G. Pathak, P. Sengupta and D. K. Dutta, Appl. Clay Sci., 2011, 53, 650–656 CrossRef CAS.
- P. Scherrer, Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Math. Klasse., 1918, 1–2 Search PubMed.
- Z. Kanwal, M. A. Raza, F. Manzoor, S. Riaz, G. Jabeen, S. Fatima and S. Naseem, Nanomaterials, 2019, 9(2), 309 CrossRef CAS.
- Q. He, T. Yuan, J. Zhu, Z. Luo, N. Haldolaarachchige, L. Sun, A. Khasanov, Y. Li, D. P. Young, S. Wei and Z. Guo, Polymer (Guildf)., 2012, 53, 3642–3652 CrossRef CAS.
- M. Nisar, P. S. Thue, C. A. Heck, J. L. Salazar Cuaila, J. Geshev, E. C. Lima, M. M. Jacobi and G. B. Galland, Eur. Polym. J., 2018, 99, 378–383 CrossRef CAS.
- M. Nisar, P. S. Thue, C. A. Heck, J. Geshev, E. C. Lima and G. B. Galland, Polym. Eng. Sci., 2020, 1–8 Search PubMed.
- H. Lu, Y. Hu, M. Li, Z. Chen and W. Fan, Compos. Sci. Technol., 2006, 66, 3035–3039 CrossRef CAS.
- M. Day, J. D. Cooney and M. MacKinnon, Polym. Degrad. Stab., 1995, 48, 341–349 CrossRef CAS.
- P. K. Roy, P. Surekha, R. Raman and C. Rajagopal, Polym. Degrad. Stab., 2009, 94, 1033–1039 CrossRef CAS.
- C. Ou, S. Li, J. Shao, T. Fu, Y. Liu, W. Fan, X. Yang and X. Bi, Cogent Chem, 2016, 2, 1–8 Search PubMed.
- L. M. Gorghiu, S. Jipa, T. Zaharescu, R. Setnescu and I. Mihalcea, Polym. Degrad. Stab., 2004, 84, 7–11 CrossRef CAS.
- J. Y. Lee, Y. Liao, R. Nagahata and S. Horiuchi, Polymer, 2006, 47, 7970–7979 CrossRef CAS.
- M. Nisar, C. Bergmann, J. Geshev, R. Quijada and G. B. Galland, Polymer, 2016, 97, 131–137 CrossRef CAS.
- H. Li, Q. Wang, H. Wang, Y. Cui, Y. Zhu and B. Wang, Macromol. Mater. Eng., 2016, 301, 1473–1481 CrossRef CAS.
- Y. Yang, P. Wang and Q. Zheng, J. Polym. Sci. Part B Polym. Phys., 2006, 44, 879–887 CrossRef CAS.
- A. A. Gabrienko, A. V. Ewing, A. M. Chibiryaev, A. M. Agafontsev, K. A. Dubkov and S. G. Kazarian, Phys. Chem. Chem. Phys., 2016, 18, 6465–6475 RSC.
- D. L. Tomasko, H. Li, D. Liu, X. Han, M. J. Wingert, L. J. Lee and K. W. Koelling, Ind. Eng. Chem. Res., 2003, 42, 6431–6456 CrossRef CAS.
- G. Sethia and A. Sayari, Carbon, 2015, 93, 68–80 CrossRef CAS.
- Y. G. Ko, S. S. Shin and U. S. Choi, J. Colloid Interface Sci., 2011, 361, 594–602 CrossRef CAS.
- Q. Huang, Q. Luo, Y. Wang, E. Pentzer and B. Gurkan, Ind. Eng. Chem. Res., 2019, 58, 10503–10509 CrossRef CAS.
- A. Serge, L. K. C. De Souza, A. Trokourey and M. Jaroniec, Microporous Mesoporous Mater, 2013, 180, 280–283 CrossRef.
- P. Huang, H. Cheng and S. Lin, J. Chem., 2015, 1–10 Search PubMed.
- Z. Yang, H. Peng, W. Wang and T. Liu, J. Appl. Polym. Sci., 2010, 116, 2658–2667 CAS.
- B. M. Ganesh, A. M. Isloor and A. F. Ismail, Desalination, 2013, 313, 199–207 CrossRef CAS.
- P. Anadão, L. F. Sato, H. Wiebeck and F. R. Valenzuela-Díaz, Appl. Clay Sci., 2010, 48, 127–132 CrossRef.
- J. H. Choi, J. Jegal and W. N. Kim, J. Memb. Sci., 2006, 284, 406–415 CrossRef CAS.
- B. M. Ganesh, A. M. Isloor and A. F. Ismail, Desalination, 2013, 313, 199–207 CrossRef CAS.
- M. Ionita, A. M. Pandele, L. Crica and L. Pilan, Compos. Part B Eng., 2014, 59, 133–139 CrossRef CAS.
- S. Mohanty and S. K. Nayak, Polym. Compos., 2007, 28, 153–162 CrossRef CAS.
- M. Nisar, P. S. Thue, C. A. Heck, J. L. Salazar Cuaila, J. Geshev, E. C. Lima, M. M. Jacobi and G. B. Galland, Eur. Polym. J., 2018, 99, 378–383 CrossRef CAS.
- J. Geshev, Appl. Phys. Lett., 2008, 93, 176101 CrossRef.
- J. Geshev, J. Appl. Phys., 2009, 105, 066108 CrossRef.
- A. Harres, M. Mikhov, V. Skumryev, A. M. H. De Andrade, J. E. Schmidt and J. Geshev, J. Magn. Magn. Mater., 2016, 402, 76–82 CrossRef CAS.
- J. Geshev, J. Magn. Magn. Mater., 2018, 467, 135–138 CrossRef CAS.
- J. Geshev, L. L. Bianchi, R. F. Lopes, J. L. Salazar Cuaila and A. Harres, J. Magn. Magn. Mater., 2019, 497, 166061 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |