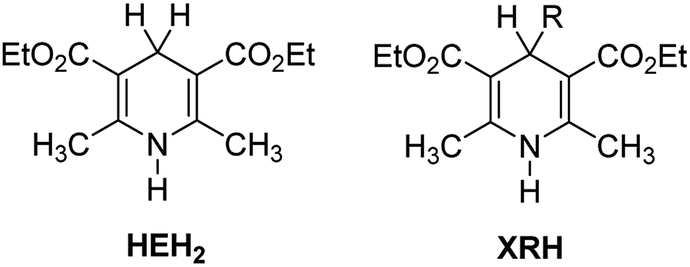DOI:
10.1039/D0RA06745H
(Paper)
RSC Adv., 2020,
10, 31425-31434
Theoretical investigation on the nature of 4-substituted Hantzsch esters as alkylation agents
Received
5th August 2020
, Accepted 20th August 2020
First published on 25th August 2020
Abstract
Recently, a variety of 4-substituted Hantzsch esters (XRH) with different structures have been widely researched as alkylation reagents in chemical reactions, and the key step of the chemical process is the elementary step of XRH˙+ releasing R˙. The purpose of this work is to investigate the essential factors which determine whether or not an XRH is a great alkylation reagent using density functional theory (DFT). This study shows that the ability of an XRH acting as an alkylation reagent can be reasonably estimated by its ΔG≠RD(XRH˙+) value, which can be conveniently obtained through DFT computations. Moreover, the data also show that ΔG≠RD(XRH˙+) has no simple correlation with the structural features of XRH, including the electronegativity of the R substituent group and the magnitude of steric resistance; therefore, it is difficult to judge whether an XRH can provide R˙ solely by experience. Thus, these results are helpful for chemists to design 4-substituted Hantzsch esters (XRH) with novel structures and to guide the application of XRH as a free radical precursor in organic synthesis.
1 Introduction
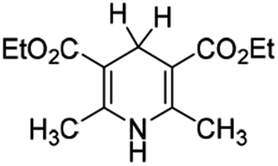
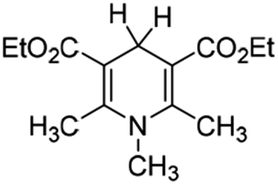
It is well known that the Hantzsch ester (HEH2, Scheme 1) was first synthesized by Hantzsch, a German chemist, in 1881.1 Due to the simple synthesis method and excellent reduction performance, HEH2 has long been used as an organic hydrogen molecular reductant to hydrogenate various types of unsaturated compounds.2–4 HEH2 releases two hydrogen atoms or hydrogen ions to unsaturated substrates while itself aromatizes into pyridine compounds, meanwhile the unsaturated bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, CO, etc.) in the substrates are reduced. Organic synthetic chemists have done excellent research work in designing novel catalysts, expanding new reaction substrate and improving product chirality and yield using HEH2 as reducer. Since 2013, chemists found that 4-substituted Hantzsch esters (XRH, Scheme 1) can break C–C bond and release alkyl radical (R˙) under the condition of photocatalysis.5 Then R˙ reacts with electron-activated substrate to produce alkyl substitution or alkyl addition products, and hundreds of literatures have reported the alkylation reaction between XRH with various substrates.6–18
C, CN, CO, etc.) in the substrates are reduced. Organic synthetic chemists have done excellent research work in designing novel catalysts, expanding new reaction substrate and improving product chirality and yield using HEH2 as reducer. Since 2013, chemists found that 4-substituted Hantzsch esters (XRH, Scheme 1) can break C–C bond and release alkyl radical (R˙) under the condition of photocatalysis.5 Then R˙ reacts with electron-activated substrate to produce alkyl substitution or alkyl addition products, and hundreds of literatures have reported the alkylation reaction between XRH with various substrates.6–18
 |
| | Scheme 1 Structures of HEH2 and XRH investigated in this work. | |
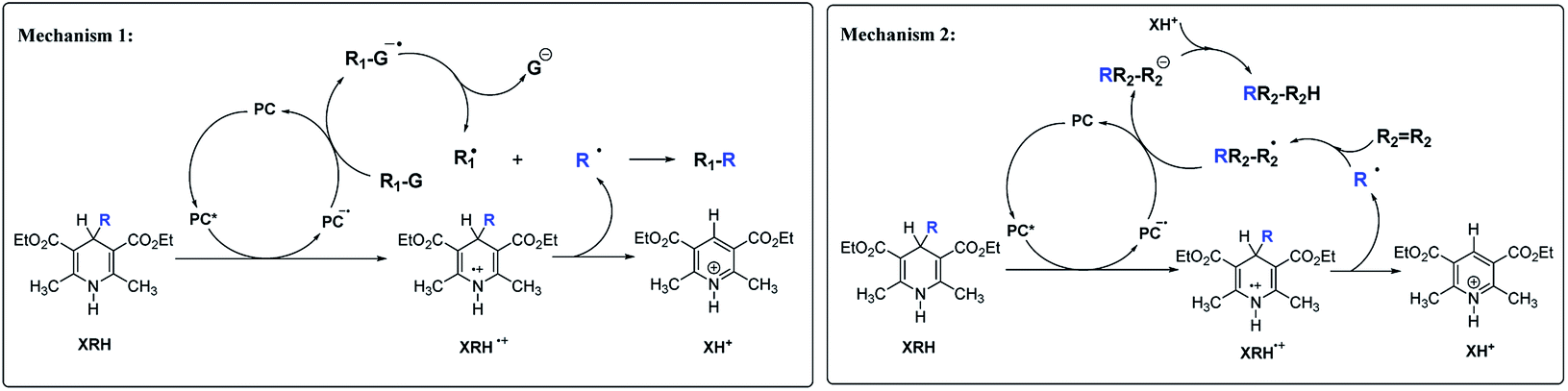
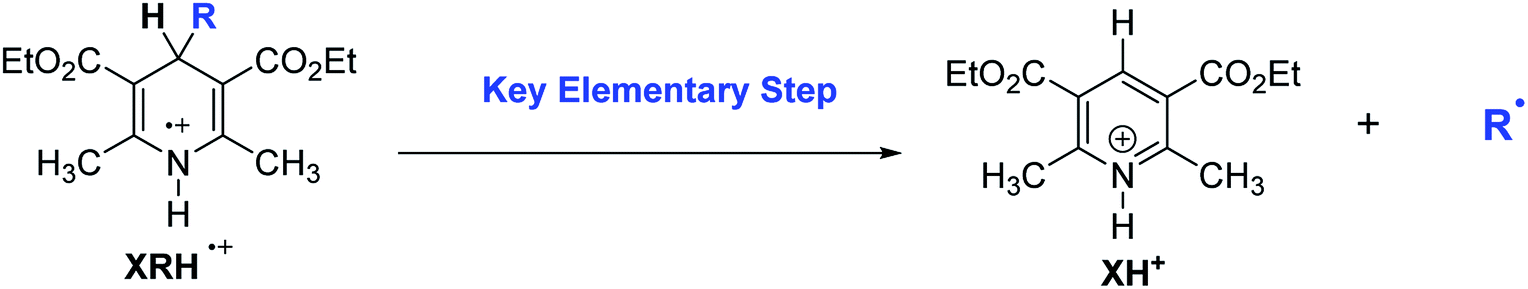
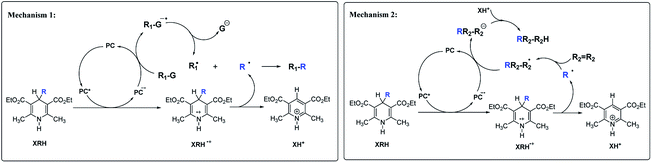
There are two main mechanisms (Scheme 2) for the reactions of XRH as alkylating agents reacting with substrates. Under certain specific wavelength, the photocatalyst (PC) is activated into an excited state photocatalyst (PC*) with strong single electron oxidation ability. PC* oxidizes XRH to XRH˙+ and transforms itself into PC˙−, a strong single electron reducer. XRH˙+ is an unstable and active key intermediate, which will release R˙ to substrate. The first mechanism7–12 is that PC˙− reduces the reaction substrate (R1–G) to R1–G˙−, subsequently, R1–G˙− releases stable G− group to generate R1˙. Recombination of R˙ and R1˙ radicals generates the desired product R1–R. Since the concentrations of R˙ and R1˙ are very low in solution, hydrogen-abstraction by the radicals may possibly occur as a side-reaction (Mechanism 1 in Scheme 2). Alkylation of haloalkanes is a typical reaction of this mechanism. The second mechanism13–18 is that R˙ reacts with unsaturated substrate (such as R2R2) to form active free radical intermediate RR2–R2˙ first, then RR2–R2˙ obtains electrons from PC˙− and protons from XH+ to form alkylation product RR2–R2H (Mechanism 2 in Scheme 2). After carefully analyzing the reaction mechanism of XRH acting as alkylating reagents, it is clear that the key step is the elementary step of XRH˙+ releasing R˙ (Scheme 3), but not all XRH with various R substituent groups in 4-position can give the alkyl radicals, for example, 4-Ph-HEH cannot provide Ph˙ in alkylation reaction.
 |
| | Scheme 2 Two mechanisms of XRH being used as alkylation reagents in chemical reactions. | |
 |
| | Scheme 3 Key elementary step of XRH being used as alkylation reagents in chemical reactions. | |
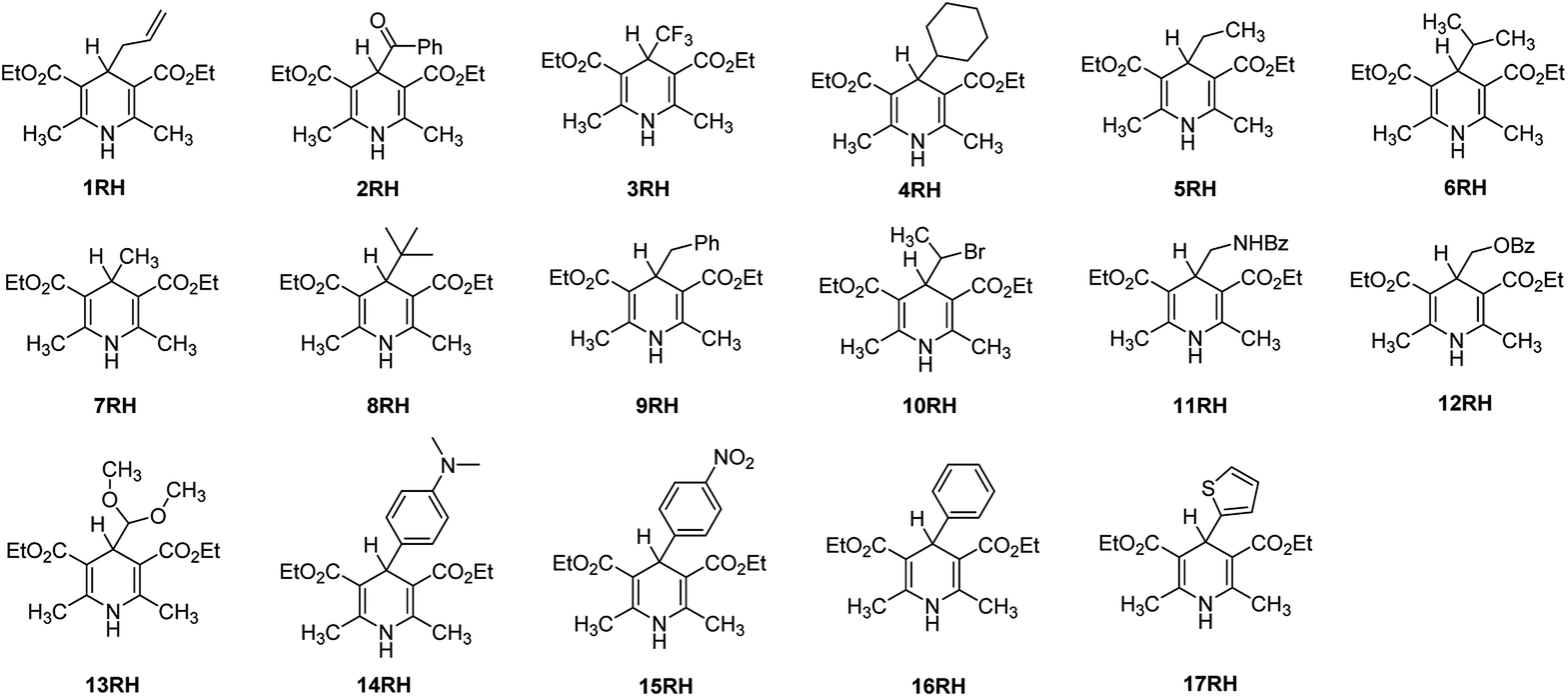
Then we begin to wonder: why some structures of XRH (R = Bn, tert-Bu, cyclohexyl, allyl, etc.) are excellent alkylation reagents, while some structures of XRH (R = Ph, thienyl, etc.) cannot be used as alkylation reagents, and what determines whether an XRH can be a good alkylation reagent? At present, the above question have not been well answered, and it is difficult to determine these important thermodynamic and kinetic data by experimental methods. Therefore, there is an urgent need for an accurate and reliable theoretical method to guide the experimental design in the field of organic synthesis. Further study on the thermodynamic and kinetic properties of the key intermediate XRH˙+ releasing R˙ can guide chemists to better design 4-substituted Hantzsch esters (XRH) as novel alkylation reagents and understand the alkylation mechanism in organic synthesis.
Owing to the rapid development of density functional theory (DFT)19–23 in recent years and the large enhancement of computational capability, the thermodynamic driving forces and activation energies of chemical reactions could be accurately computed by employing suitable density functionals. Based on the important structure of 4-substituted Hantzsch esters (XRH) reported in previous literatures, 17 XRH have been well-designed in this work (Scheme 4). The thermodynamic driving forces [free energy change, ΔGoRD(XRH˙+)] and activation Gibbs free energies [ΔG≠RD(XRH˙+)] of different structures of XRH˙+ releasing R˙ were computed using DFT, and the essential factors determining whether or not an XRH is alkylation reagent have been revealed in detail in this work.
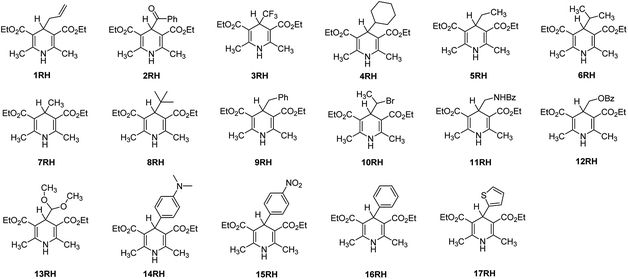 |
| | Scheme 4 Chemical structures of 17 XRH examined in this work. | |
2 Computational section
Molar free energy change of XRH˙+ releasing R˙ [ΔGoRD(XRH˙+)], molar enthalpy changes of XRH˙+ releasing R˙ [ΔHoRD(XRH˙+)], and activation free energy of XRH˙+ releasing R˙ [ΔG≠RD(XRH˙+)] were computed in this work.
All quantum chemical calculations in this work were performed using Gaussian 09,19a and wave function analysis was performed using Multiwfn 3.7.19b First, find out the transition state of XRH˙+ releasing R˙, and calculate the Gibbs free energy and enthalpy of reactant, transition state and product of each elemental step. Through analyzing the difference between reactant and product, free energy changes and enthalpy changes of XRH˙+ releasing R˙ were obtained. The activation free energy and activation enthalpy could be obtained by comparing the reactant and the transition state in similar method.
First, the reactants, transition states and products of elemental step were geometrically optimized to convergence using the functional B3LYP20 and the basis set 6-31G(d,p).21 DFT-D3 empirical dispersion correction (BJ damping)22 was applied, and SMD solvent model23 (acetonitrile as the solvent) was used during the computation. After geometrical optimization, the vibration analysis was carried out using the same parameters. The results of vibration analysis showed that there is no imaginary frequency for all reactants and products; meanwhile, only one imaginary frequency was found for each transition state, and the direction of imaginary frequency was the same as that of C–C bond stretch, indicating that the transition states were correct. Intrinsic reaction coordinate (IRC) was then calculated for each transition state, and electron spin density analysis of IRC-derived products showed that the cleavage products were alkyl radicals, not alkyl cations. Then, the thermodynamic correction was applied for each reactant, transition state and product at 298.15 K. The free energy was obtained by adding the single point energy to the Gibbs free energy correction value, and the enthalpy was obtained by adding the single point energy to the enthalpy correction value. Reaction rate constant (k2) was obtained by Eyring equation [k2 = (kBT/h)exp(−ΔG≠/RT)].
In order to verify the applicability and accuracy of the calculation method in this work, first we calculated the molar enthalpy changes of four XRH˙+ releasing H˙ using the above method (termed “calculation values” in this work). The molar enthalpy changes of XRH˙+ releasing H˙ determined by the calorimetric titration method in previous literature were termed “experimental values”.24 The calculated value and the experimental value are listed in Table 1, and the data shows that the deviation between calculated values and the experimental values is less than 2.0 kcal mol−1. Therefore, considering the experimental measurement error, it is appropriate to use the above calculation method to study the energy changes of XRH˙+ releasing R˙, and the results obtained are accurate and reliable.
Table 1 Comparison between DFT calculation data and experimental data (kcal mol−1)
| Compounds |
ΔHoHD(XRH˙+) (DFT)a |
ΔHoHD(XRH˙+) (exp)a |
ΔΔHoHDb |
| The unit is kcal mol−1. ΔΔHoHD = ΔHoHD(XRH˙+) (DFT) − ΔHoHD(XRH˙+) (exp), the unit is kcal mol−1. |
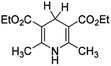 |
30.27 |
31.40 (ref. 24a) |
−1.13 |
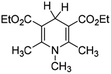 |
30.72 |
32.20 (ref. 24a) |
−1.48 |
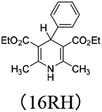 |
26.85 |
27.80 (ref. 24b) |
−0.95 |
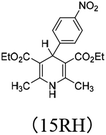 |
27.85 |
29.80 (ref. 24b) |
−1.95 |
3 Results and discussions
The calculation results, ΔHoRD(XRH˙+), ΔGoRD(XRH˙+) and ΔH≠RD(XRH˙+), ΔG≠RD(XRH˙+) are presented in Table 2.
Table 2 Thermodynamic data and activation free energy data of XRH˙+ releasing R˙
| XRH |
ΔHoRD(XRH˙+)a |
ΔGoRD(XRH˙+)a |
ΔH≠RD(XRH˙+)a |
ΔG≠RD(XRH˙+)a |
| The unit is kcal mol−1. |
| 1RH |
0.48 |
−12.15 |
3.29 |
3.47 |
| 2RH |
9.43 |
−3.13 |
3.24 |
3.44 |
| 3RH |
17.76 |
4.75 |
13.92 |
12.95 |
| 4RH |
13.16 |
−0.02 |
5.68 |
5.38 |
| 5RH |
13.71 |
0.58 |
9.64 |
8.66 |
| 6RH |
11.49 |
−2.42 |
4.76 |
4.59 |
| 7RH |
17.15 |
5.60 |
15.32 |
13.62 |
| 8RH |
7.73 |
−7.33 |
1.44 |
1.17 |
| 9RH |
5.37 |
−7.73 |
3.22 |
3.11 |
| 10RH |
12.79 |
−1.28 |
7.77 |
7.45 |
| 11RH |
13.47 |
−0.11 |
2.05 |
3.37 |
| 12RH |
17.16 |
1.88 |
6.65 |
6.39 |
| 13RH |
15.64 |
2.27 |
0.85 |
0.83 |
| 14RH |
38.83 |
26.85 |
32.81 |
31.80 |
| 15RH |
27.12 |
15.17 |
22.49 |
22.28 |
| 16RH |
27.29 |
1.60 |
21.93 |
21.48 |
| 17RH |
32.77 |
21.72 |
26.95 |
26.60 |
3.1 ΔGoRD(XRH˙+)
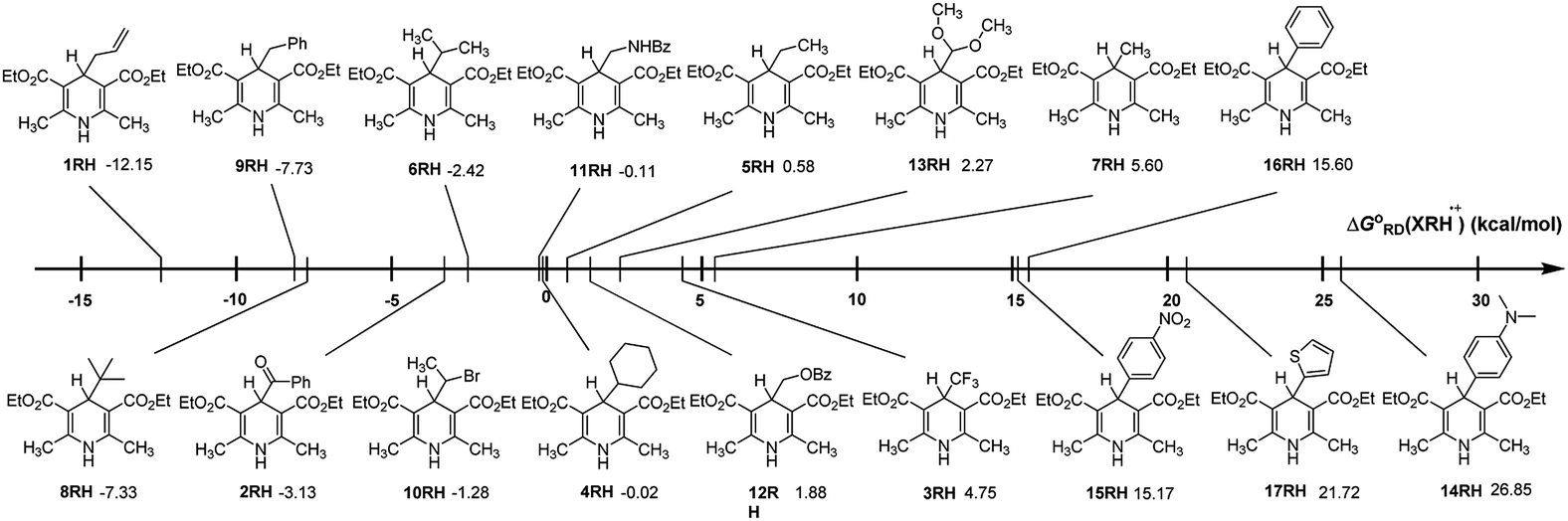
In order to discover the influence of structural factors on ΔGoRD(XRH˙+) values, the free energies changes of 17 XRH˙+ releasing R˙ are listed in Fig. 1 for comparison. From Fig. 1, it is evident that the free energy changes of breaking C–C bond for XRH˙+ releasing R˙ varies greatly with the substituents, which ranges from −12.15 to 26.85 kcal mol−1. Among 4-substituted Hantzsch ester structures investigated in this work, the Gibbs free energy of XRH˙+ releasing R˙ increased from ΔGo ≪ 0 (−12.15 kcal mol−1) to ΔGo ≫ 0 (26.85 kcal mol−1). 1RH˙+ (4-allyl-substituted Hantzsch ester) has the lowest ΔGoRD(XRH˙+) value as −12.15 kcal mol−1, and 14RH˙+ (4-dimethylaminophenyl substituted Hantzsch ester) has the highest value of ΔGoRD(XRH˙+) as 26.85 kcal mol−1, and the former is 39 kcal mol−1 lower than the latter. The thermodynamic data show that 1RH is the best alkylation reagents which spontaneously offers allyl radical in acetonitrile under the reaction condition of 298.15 K; in contrast, 14RH cannot give 4-dimethylaminophenyl group in chemical reactions. Such a large range of ΔGoRD(XRH˙+) values (−12.15–26.85 kcal mol−1) indicates that the R group in the parent structure of XRH has a great influence on the value of ΔGoRD(XRH˙+), and the strength of C–C bond varies greatly in different structures of XRH˙+.
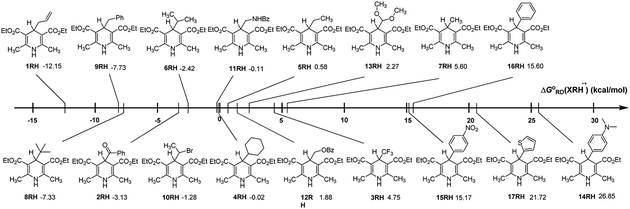 |
| | Fig. 1 Visual comparison of ΔGoRD(XRH˙+) among the 17 XRH in acetonitrile at 298.15 K. | |
Through comprehensive analysis and evaluation of ΔGoRD(XRH˙+), the results show that XRH˙+ is possible (but not necessarily) to be an excellent R˙ donors which are determined by the R substituents in 4-position. If R substituents in XRH˙+ are not aromatic substituents, XRH˙+ would be used as alkylation reagent; in contrast, if R substituents in XRH˙+ are aromatic substituents, XRH˙+ would not be used as R˙ donors.
3.2 ΔG≠RD(XRH˙+)
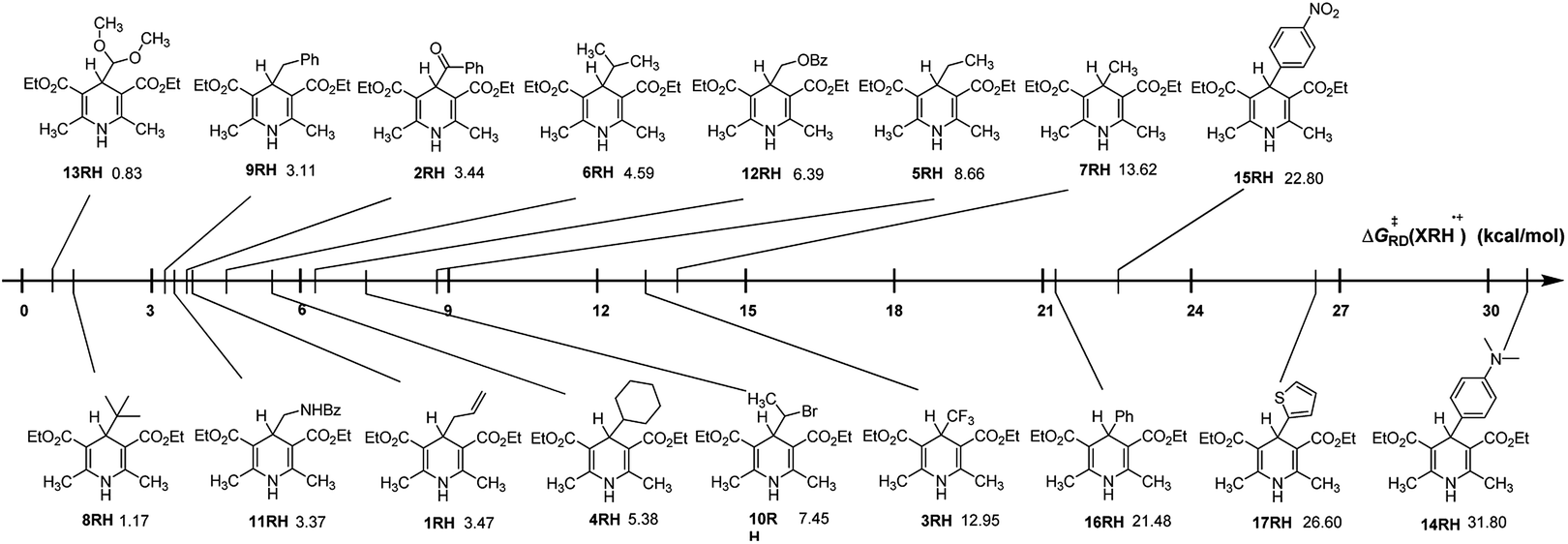
From columns 5 in Table 2, it is clear that the ΔG≠RD(XRH˙+) values of XRH˙+ releasing R˙ range from 0.83 to 31.80 kcal mol−1. As shown in Fig. 2, ΔG≠RD(XRH˙+) value of 13RH˙+ releasing R˙ is the smallest (0.83 kcal mol−1); hence, the rate constant of 13RH˙+ releasing R˙ (1.53 × 1012 M−1 s−1) is greater than diffusion speed (1010 M−1 s−1); while ΔG≠RD(XRH˙+) value of 14RH˙+ releasing R˙ is the largest (31.80 kcal mol−1), so the rate constant of 14RH˙+ releasing R˙ is lowest with 2.87 × 10−12 M−1 s−1 at 298.15 K in acetonitrile, which indicates that the reaction of 14RH˙+ releasing R˙ is extremely slow, and therefore 14RH cannot be used as R˙ donor in chemical reaction. The predicted rate constants of XRH˙+ releasing R˙ in this work range from 2.87 × 10−12 to 1.53 × 1012 (M−1 s−1). Thus, it can be seen that there are many important chemical reactions in vivo or in vitro whose rates cannot be directly measured using experimental methods up to now. The reason is that some reactions are too fast, some are too slow and some have no detectable signals, etc. Because the accuracy of the calculation results is verified by experimental measured values, the rate constants of XRH˙+ releasing R˙ that cannot be measured experimentally can be estimated by the activation energy calculation method used in this work. Among the 17 XRH structures examined, ΔG≠RD(XRH˙+) values of 13 XRH (1RH–13RH) are less than 16 kcal mol−1 (k2 = 6.29 × 102 M−1 s−1 at 298.15 K), ΔG≠RD(XRH˙+) values of 4 XRH (14RH–17RH) span from 21.48 to 31.80 kcal mol−1 (1.07 × 10−3 to 1.07 × 10−11 M−1 s−1 at 298.15 K). The ΔG≠RD(XRH˙+) values indicate that 1RH–13RH could be selected as R˙ donor in chemical reactions, while 14RH–17RH cannot provide R˙ as alkylation reagents. Further taking their structures into consideration, we find that R substituents in 14RH–17RH are aromatic substituents and the ΔG≠RD(XRH˙+) of XRH˙+ releasing R˙ all larger than 21.4 kcal mol−1 which means 4-aromatic-group substituted Hantzsch esters cannot be used as R˙ donors. If R substituents in XRH˙+ are not aromatic substituents, XRH˙+ (1RH–13RH) would be good R˙ donors in radical reactions.
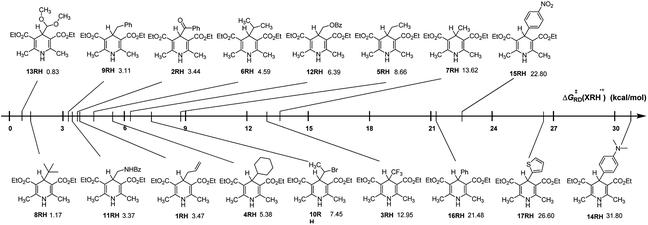 |
| | Fig. 2 Visual comparison of ΔG≠RD(XRH˙+) among the 17 XRH in acetonitrile at 298.15 K. | |
3.3 Entropy change and activation entropy change
Since Gibbs free energies [ΔGoRD(XRH˙+)], enthalpy changes [ΔHoRD(XRH˙+)], activation energies [ΔG≠RD(XRH˙+)] and activation enthalpy changes [ΔH≠RD(XRH˙+)] were obtained now, we calculated the entropy changes (ΔSoRD) and activation entropy changes (ΔS≠RD) of XRH˙+ releasing R˙ which are listed in Table 3.
Table 3 Entropy changes and activation entropy changes of XRH˙+ releasing R˙
| XRH |
ΔSoRDa |
ΔS≠RDb |
| From the equation, ΔS = (ΔH − ΔG)/T, the unit is cal mol−1 K−1. From the equation, ΔS≠ = (ΔH≠ − ΔG≠)/T, the unit is cal mol−1 K−1. |
| 1RH |
42.39 |
−0.60 |
| 2RH |
42.13 |
−0.66 |
| 3RH |
43.65 |
3.26 |
| 4RH |
44.22 |
0.99 |
| 5RH |
44.07 |
3.28 |
| 6RH |
46.69 |
0.58 |
| 7RH |
38.75 |
5.69 |
| 8RH |
50.53 |
0.91 |
| 9RH |
43.98 |
0.36 |
| 10RH |
47.24 |
1.09 |
| 11RH |
45.57 |
−4.42 |
| 12RH |
51.26 |
0.87 |
| 13RH |
44.85 |
0.08 |
| 14RH |
40.21 |
3.37 |
| 15RH |
40.12 |
0.72 |
| 16RH |
39.24 |
1.50 |
| 17RH |
37.09 |
1.18 |
From Table 3, it is found that the entropy changes of XRH˙+ releasing R˙, ΔSoRD, range from 38.75 to 51.26 cal mol−1 K−1, and the activation entropy changes of XRH˙+ releasing R˙, ΔS≠RD, range from −4.42 to 5.69 cal mol−1 K−1. ΔSoRD are positive values (cal mol−1 K−1), which indicates that the chemical process of XRH˙+ releasing R˙ is the large entropy increase process. While the span of activation entropy changes of XRH˙+ releasing R˙, ΔS≠RD, is less than ±5 cal mol−1 K−1, which indicates that the chemical structures of XRH˙+ do not change greatly in the transition state.
3.4 Main factors affecting free energy changes and activation free energy changes of XRH˙+ releasing R˙
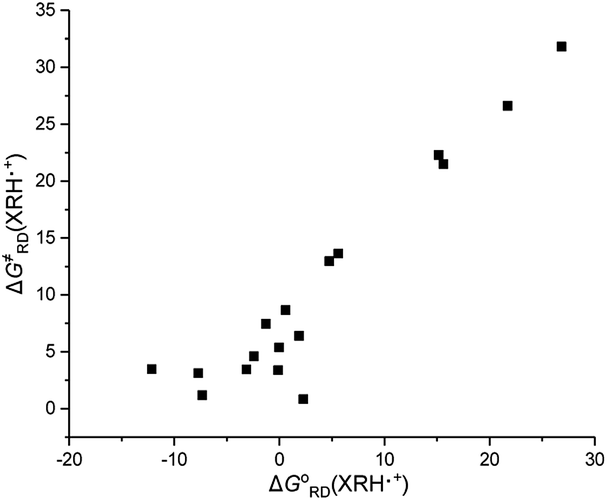
3.4.1 Thermodynamic driving force and activation free energy. Because the XRH structure investigated in this work is 1,4-dihydropyridine compounds, only the R substituents in 4-position are different. In order to further explore the intrinsic relationship between the thermodynamic driving forces [ΔGoRD(XRH˙+)] and the activation energy [ΔG≠RD(XRH˙+)], relationships between them are shown in Fig. 3. It can be seen from Fig. 3 that there is a certain correlation between the thermodynamic driving force and activation free energy changes of XRH˙+ releasing R˙, that is, ΔG≠RD(XRH˙+) increases along with the increase of ΔGoRD(XRH˙+). But for all chemical reactions, there is no good linear correlation between ΔG≠ and ΔGo. This phenomenon shows that the thermodynamic driving force has an important influence on the activation energy, but it is not the only decisive factor.25
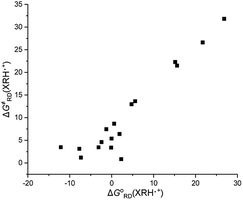 |
| | Fig. 3 Relationship between the activation free energies of 17 XRH˙+ releasing R˙ and the corresponding free energy changes of XRH˙+ releasing R˙ in acetonitrile at 298.15 K. | |
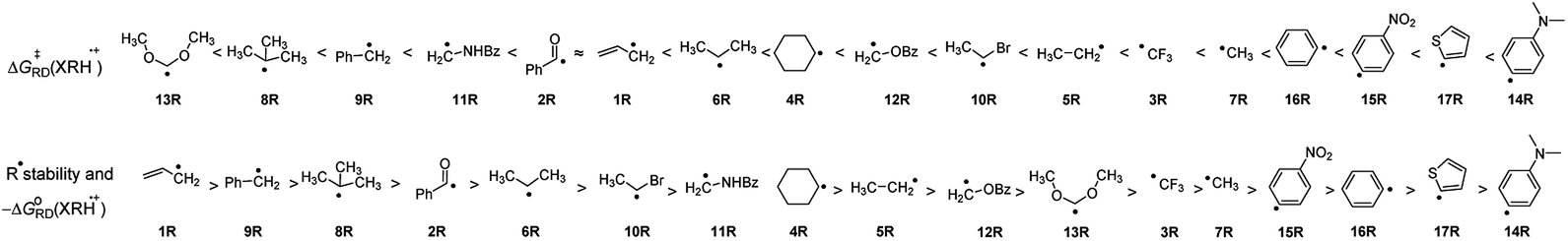
3.4.2 The stability of R˙ and ΔG≠RD(XRH˙+). It can be seen from the above discussion that many XRH with different structures can be used as R˙ donor. ΔG≠RD(XRH˙+) values range from 0.83 to 13.62 kcal mol−1, and the difference between the fastest and slowest rate constant is 1010. Through the previous discussion, we have known that the thermodynamic driving force is an important factor affecting the activation energy of the reaction. Then, does it exist an internal relationship between the stability of R˙ and the reaction activity of XRH˙+ releasing R˙ ΔG≠RD(XRH˙+)? Considering the chemical process of XRH˙+ releasing R˙ with different structure, XRH˙+ → XH+ + R˙, it is found that the values of molar free energy of XRH˙+ releasing R˙ [ΔGoRD(XRH˙+)] do reflect the relative stabilities of R˙. The smaller ΔGoRD(XRH˙+) is, the more stability R˙ is. On the basis of ΔGoRD(XRH˙+) values computed in this work, the R˙ stability order in 1RH to 17RH molecules investigated in this work is 1R > 9R > 8R > 2R > 6R > 10R > 11R > 4R > 5R > 16R > 12R > 13R > 3R > 7R > 15R > 17R > 4R (Fig. 4). But according to the ΔG≠RD(XRH˙+) values, the order of reactivity is 13R > 8R > 9R > 11R > 2R > 6R > 4R > 12R > 10R > 5R > 3R > 7R > 16R > 15R > 17R > 14R (Fig. 4). It is clear that there are significant differences between the order of stability of R˙ and the order of ΔG≠RD(XRH˙+), and the stability of R˙ is not the decisive factor of ΔG≠RD(XRH˙+). The results also show that the influence of structural factors on ΔG≠RD(XRH˙+) is multifaceted and complicated.
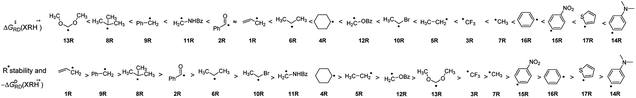 |
| | Fig. 4 The order of R˙ stability and activation free energies of 17 XRH˙+ releasing R˙. | |
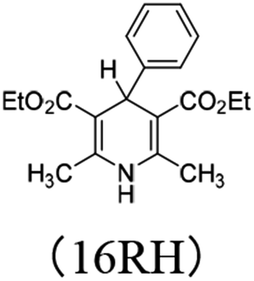
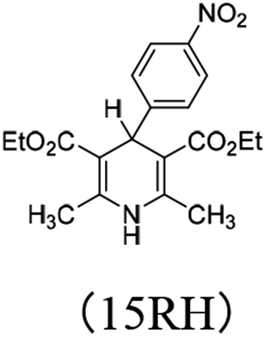
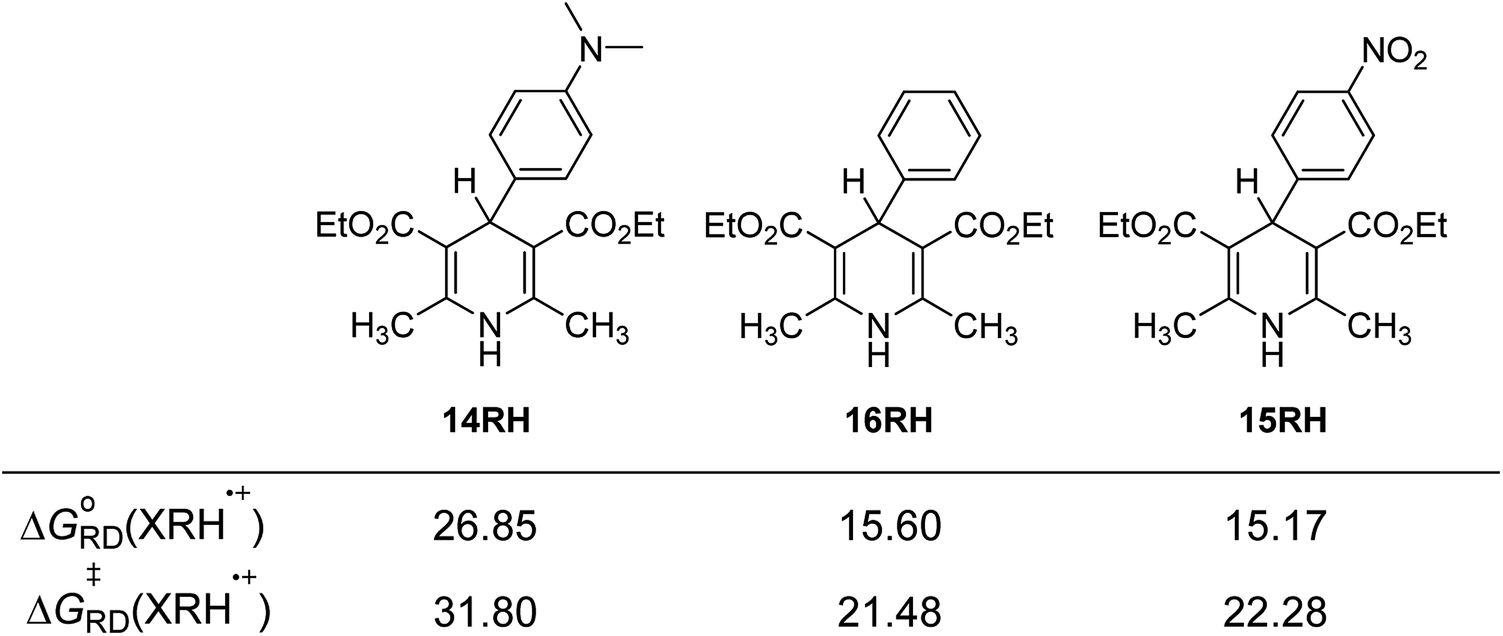
3.4.3 Substituent effect. For 14RH, 16RH and 15RH, the R substituents at the 4-positions are p-dimethylaminophenyl, phenyl and p-nitrophenyl respectively, all of which are benzene ring structures with different p-substituents. Dimethylamino group (σp = −0.83) and nitro group (σp = −0.78) are relatively strong electron-donating and electron-withdrawing groups respectively. We designed these two groups in order to change the ability of XRH˙+ releasing R˙ to provide aromatic free radicals by tremendously changing the electronegativity of substituents. The relevant data are compared in Fig. 5. According to Fig. 5, the following conclusions can be made: (1) when the 4-position is a strong electron-donating group (dimethylamino group), free energy changes and activation energy changes of XRH˙+ releasing R˙ increased significantly (about 5–10 kcal mol−1). (2) When the 4-position is a strong electron-withdrawing group (nitro group), free energy changes and activation energy changes of XRH˙+ releasing R˙ changed slightly (<1.5 kcal mol−1). The above calculation results show that the change of electronegativity of benzene ring does not make XHR the donor of aromatic benzene free radicals. No matter it is electron-donating group or electron-withdrawing group, the abilities of XHR˙+ releasing R˙ are decreased, meanwhile the effect of electron-donating group on ability of XHR˙+ releasing R˙ is greater than that of electron-withdrawing group.26
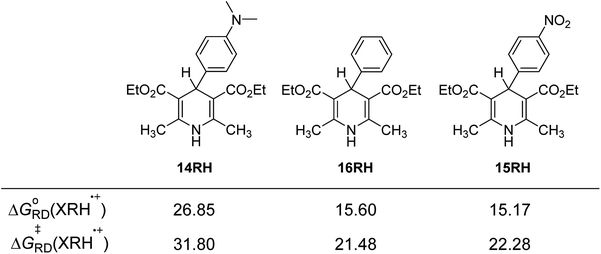 |
| | Fig. 5 Effects of substituent groups on the activation free energy and free energy changes of XRH˙+ releasing R˙ (energy unit: kcal mol−1). | |
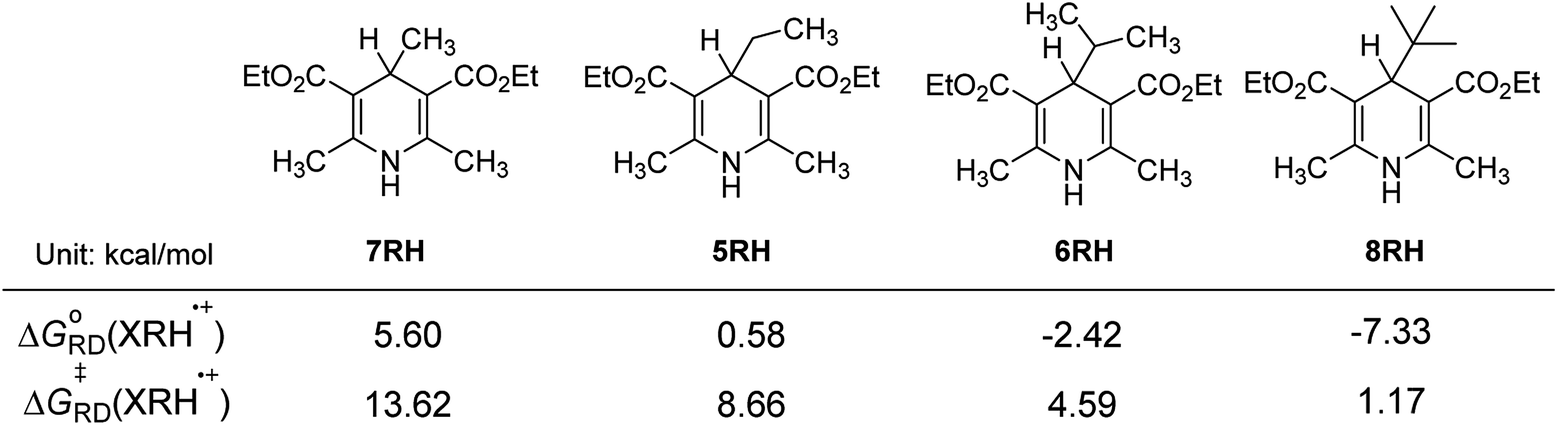
3.4.4 Influence of steric hindrance and stability of R˙. For 4-substituted Hantzsch esters 7RH, 5RH, 6RH and 8RH, R substituent groups are methyl, ethyl, isopropyl and tert-butyl (Fig. 6), respectively. The steric resistance of the substituents increases gradually with increasing of the stability of R˙. From Fig. 6, it is found that ΔGoRD(XRH˙+) of XRH˙+ releasing R˙ decrease from 5.60 to −7.33 kcal mol−1, ΔG≠RD(XRH˙+) of XRH˙+ releasing R˙ decrease from 13.62 to 1.17 kcal mol−1, as gradually increasing of steric hindrance and stability of R˙ from 7RH˙+ to 8RH˙+ (Fig. 6).
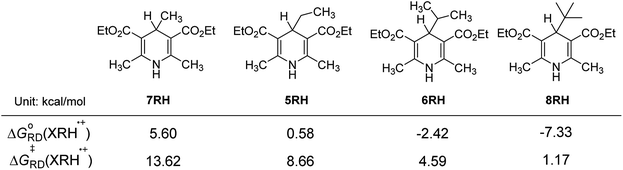 |
| | Fig. 6 Effects of size and stability of substituent groups on the activation free energy and free energy changes of XRH˙+ releasing R˙ (energy unit: kcal mol−1). | |
3.5 Judgment criteria of whether an XRH is a great alkylation reagent
Considering activation free energy of XRH˙+ releasing R˙ [ΔG≠RD(XRH˙+)], the judgment criteria of XRH being a good R˙ donor can be concluded reasonably (Table 4). Generally, in practice, an activation free energy (ΔG≠) of 21 kcal mol−1 is usually used as a criteria in determining whether a reaction could easily occur at room temperature, according to the Transition State Theory (TST);27 an activation free energy of 21 kcal mol−1 corresponds to a half-life of ∼4.5 min and a 97% completion time of 22.3 min at room temperature, which indicate that the reaction can be completed within an acceptable time. Accordingly, the following predictions can be made: (1) if the value of ΔG≠RD(XRH˙+) is larger than 21.0 kcal mol−1 (k2 < 2.42 × 10−3 M−1 s−1), XRH could not be used as alkylation reagent, like 14RH–17RH. (2) If the value of ΔG≠RD(XRH˙+) is larger than 17.5 kcal mol−1 (k2 ≤ 1 M−1 s−1), but smaller than 21.0 kcal mol−1 (k2 ≥ 2.42 × 10−3 M−1 s−1) XRH is a weak alkylation reagent. (3) If the value of ΔG≠RD(XRH˙+) is larger than 12.0 kcal mol−1 (k2 ≤ 9.71 × 103 M−1 s−1), but smaller than 17.5 kcal mol−1 (k2 ≥ 1 M−1 s−1) XRH is a medium alkylation reagent, like 3RH and 7RH. (4) If the value of ΔG≠RD(XRH˙+) is smaller than 12.0 kcal mol−1 (k2 > 9.71 × 103 M−1 s−1), XRH is an excellent alkylation reagent, like 8RH–13RH. Since ΔG≠RD(XRH˙+) has no simple correlation with the XRH′ structure, the electronegativity and steric resistance, it is difficult to judge whether XRH can provide R˙ in chemical reactions solely by experience. The calculation method and results provided in this work are helpful to guide the application of XRH as a free radical donor in organic synthesis.
Table 4 Diagnoses of reactivity of XRH using as radical agent according to ΔG≠RD(XRH˙+)
| XRH |
ΔGoRD(XRH˙+)a |
ΔG≠RD(XRH˙+)a |
k2b |
Diagnose |
| The energy unit is kcal mol−1. The unit is M−1 s−1. |
| 1RH |
−12.15 |
3.47 |
1.76 × 1010 |
Excellent R˙ donor |
| 2RH |
−3.13 |
3.44 |
1.85 × 1010 |
Excellent R˙ donor |
| 3RH |
4.75 |
12.95 |
1.95 × 103 |
Medium R˙ donor |
| 4RH |
−0.02 |
5.38 |
6.99 × 108 |
Excellent R˙ donor |
| 5RH |
0.58 |
8.66 |
2.74 × 106 |
Excellent R˙ donor |
| 6RH |
−2.42 |
4.59 |
2.66 × 109 |
Excellent R˙ donor |
| 7RH |
5.60 |
13.62 |
6.29 × 102 |
Medium R˙ donor |
| 8RH |
−7.33 |
1.17 |
8.59 × 1011 |
Excellent R˙ donor |
| 9RH |
−7.73 |
3.11 |
3.24 × 1010 |
Excellent R˙ donor |
| 10RH |
−1.28 |
7.45 |
2.12 × 107 |
Excellent R˙ donor |
| 11RH |
−0.11 |
3.37 |
2.09 × 1010 |
Excellent R˙ donor |
| 12RH |
1.88 |
6.39 |
1.27 × 108 |
Excellent R˙ donor |
| 13RH |
2.27 |
0.83 |
1.53 × 1012 |
Excellent R˙ donor |
| 14RH |
26.85 |
31.80 |
2.87 × 10−11 |
Not R˙ donor |
| 15RH |
15.17 |
22.28 |
2.78 × 10−4 |
Not R˙ donor |
| 16RH |
15.60 |
21.48 |
1.07 × 10−3 |
Not R˙ donor |
| 17RH |
21.72 |
26.60 |
1.88 × 10−7 |
Not R˙ donor |
4 Conclusions
In this work, 4 characteristic physical parameters, ΔHoRD(XRH˙+), ΔGoRD(XRH˙+), ΔH≠RD(XRH˙+), ΔG≠RD(XRH˙+) of 17 important XRH releasing R˙ were calculated using DFT method. After careful discussion of the relevant thermodynamic and kinetic data, the following conclusions can be made: (1) through comprehensive analysis and evaluation of ΔGo and ΔG≠, it is found that XRH˙+ is possible (but not necessarily) to be an excellent R˙ donors which are determined by the R substituents in 4-position. (2) If R substituents in XRH are not aromatic groups, XRH˙+ can release R˙ as key intermediates, therefore XRH could be used as alkylation reagents. In contrast, if R substituents in XRH are aromatic substituents, the key intermediates XRH˙+ could not release aromatic radical in chemical reactions, so XRH with aromatic substituents could not be used as alkylation reagents. (3) The thermodynamic driving force (Gibbs free energy) has an important influence on the activation energy, but it is not the only decisive factor. (4) There are significant differences between the order of R˙ stability and the order of ΔG≠RD(XRH˙+), and the stability of R˙ is not the decisive factor of ΔG≠RD(XRH˙+). In addition, the influence of structural factors on ΔG≠RD(XRH˙+) is multifaceted and complicated. (5) Change of electronegativity of benzene ring does not make XHR be an aromatic free radical donor. (6) According to ΔG≠RD(XRH˙+) values, the kinetic ability of XRH˙+ releasing R˙ can be predicted reasonably. ① If the value of ΔG≠RD(XRH˙+) is larger than 21.0 kcal mol−1, XRH could not be used as an alkylation reagent. ② If the value of ΔG≠RD(XRH˙+) is larger than 17.5 kcal mol−1, but smaller than 21.0 kcal mol−1, XRH is a weak alkylation reagent. ③ If the value of ΔG≠RD(XRH˙+) is larger than 12.0 kcal mol−1, but smaller than 21.0 kcal mol−1, XRH is a medium alkylation reagent. ④ If the value of ΔG≠RD(XRH˙+) is smaller than 12.0 kcal mol−1, XRH is an excellent alkylation reagent.
It is known that the reaction conditions of XRH using as alkylation reagents are quite complex, and this paper focuses on the intrinsic properties of key intermediate XRH˙+ releasing R˙. Since ΔG≠RD(XRH˙+) has no simple correlation with the XRH's structure, the electronegativity and steric resistance, it is difficult to judge whether XRH can be used as an alkylation reagent in chemical reactions solely by experience. The calculation method and results provided in this work are helpful to guide the application of XRH as a free radical donor in organic synthesis.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This study was supported by NSFC cultivation project of Jining Medical University (JYP2018KJ18 and JYP2019KJ25) and College Students' Innovative Training Plan Program of Jining Medical University (cx2020106).
References
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637 CrossRef.
- Reviews:
(a) C. Zheng and S.-L. You, Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC;
(b) S.-L. You, Recent Developments in Asymmetric Transfer Hydrogenation with Hantzsch Esters: A Biomimetic Approach, Chem.–Asian J., 2007, 2, 820–827 CrossRef CAS PubMed;
(c) S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Enantioselective Organocatalytic Transfer Hydrogenation Reactions using Hantzsch Esters, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed;
(d) M. Rueping, J. Dufour and F. R. Schoepke, Advances in Catalytic Metal-free Reductions: from Bio-inspired Concepts to Applications in the Organocatalytic Synthesis of Pharmaceuticals and Natural Products, Green Chem., 2011, 13, 1084–1105 RSC.
-
(a) M. O. Konev, L. Cardinale and A. J. Wangelin, Catalyst-Free N-Deoxygenation by Photoexcitation of Hantzsch Ester, Org. Lett., 2020, 22, 1316–1320 CrossRef CAS PubMed;
(b) S. Zhang, G. Li, L. Li, X. Deng, G. Zhao, X. Cui and Z. Tang, Alloxan-Catalyzed Biomimetic Oxidations with Hydrogen Peroxide or Molecular Oxygen, ACS Catal., 2020, 10, 245–252 CrossRef CAS;
(c) C. T. Ser, H. Yang and M. W. Wong, Iodoimidazolinium-Catalyzed Reduction of Quinoline by Hantzsch Ester: Halogen Bond or Brønsted Acid Catalysis, J. Org. Chem., 2019, 84, 10338–10348 CrossRef CAS PubMed;
(d) Q.-A. Chen, M.-W. Chen, C.-B. Yu, L. Shi, D.-S. Wang, Y. Yang and Y.-G. Zhou, Biomimetic Asymmetric Hydrogenation: In Situ Regenerable Hantzsch Esters for Asymmetric Hydrogenation of Benzoxazinones, J. Am. Chem. Soc., 2011, 133, 16432–16435 CrossRef CAS PubMed.
-
(a) S. B. Nagode, R. Kant and N. Rastogi, Hantzsch Ester-Mediated Benzannulation of Diazo Compounds under Visible Light Irradiation, Org. Lett., 2019, 21, 6249–6254 CrossRef CAS PubMed;
(b) G. Hamasaka, H. Tsuji, M. Ehara and Y. Uozumi, Mechanistic Insight into the Catalytic Hydrogenation of Nonactivated Aldehydes with a Hantzsch Ester in the Presence of a Series of Organoboranes: NMR and DFT Studies, RSC Adv., 2019, 9, 10201–10210 RSC;
(c) R. Tang, Z. Shao, J. Wang, Z. Liu, Y.-M. Li and Y. Shen, Iron(II)-Catalyzed Radical Addition to Aldimines with Hantzsch Ester as a Two-Hydrogen Atom Donor, J. Org. Chem., 2019, 84, 8177–8184 CrossRef CAS PubMed;
(d) J. A. Leitch, T. Rogova, F. Duarte and D. J. Dixon, Dearomative Photocatalytic Construction of Bridged 1,3-Diazepanes, Angew. Chem., Int. Ed., 2020, 59, 4121–4130 CrossRef CAS PubMed.
- G. Li, R. Chen, L. Wu, Q. Fu, X. Zhang and Z. Tang, Alkyl Transfer from C-C Cleavage, Angew. Chem., Int. Ed., 2013, 52, 8432–8436 CrossRef CAS PubMed.
- Reviews:
(a) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Hantzsch esters: an emerging versatile class of reagents in photoredox catalyzed organic synthesis, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC;
(b) S. Ye and J. Wu, 4-Substituted Hantzsch Esters as Alkylation Reagents in Organic Synthesis, Acta Chim. Sin., 2019, 77, 814–831 CrossRef.
-
(a) K. Nakajima, S. Nojima and Y. Nishibayashi, Nickel- and Photoredox-Catalyzed Cross-Coupling Reactions of Aryl Halides with 4-Alkyl-1,4-dihydropyridines as Formal Nucleophilic Alkylation Reagents, Angew. Chem., Int. Ed., 2016, 55, 14106–14110 CrossRef CAS PubMed;
(b) D.-L. Zhu, Q. Wu, H.-Y. Li, H.-X. Li and J.-P. Lang, Hantzsch Ester as a Visible-Light Photoredox Catalyst for Transition-Metal-Free Coupling of Arylhalides and Arylsulfinates, Chem.–Eur. J., 2020, 26(16), 3484–3488 CrossRef CAS PubMed;
(c) J. Li, X.-E. Yang, S.-L. Wang, L.-L. Zhang, X.-Z. Zhou, S.-Y. Wang and S.-J. Ji, Visible-Light-Promoted Cross-Coupling Reactions of 4-Alkyl-1,4-dihydropyridines with Thiosulfonate or Selenium Sulfonate: A Unified Approach to Sulfides, Selenides, and Sulfoxides, Org. Lett., 2020, 22(12), 4908–4913 CrossRef CAS PubMed.
-
(a) Z.-Y. Song, C.-L. Zhang and S. Ye, Visible light promoted coupling of alkynyl bromides and Hantzsch esters for the synthesis of internal alkynes, Org. Biomol. Chem., 2019, 17, 181–185 RSC;
(b) H. Chen, D. Anand and L. Zhou, Photoredox Defluorinative Alkylation of 1-Trifluoromethyl Alkenes and 1,3-Butadienes with 1,4-Dihydropyridines as Alkylation Reagents, Asian J. Org. Chem., 2019, 8(5), 661–664 CrossRef CAS;
(c) H.-W. Du, J. Sun, Q.-S. Gao, J.-Y. Wang, H. Wang, Z. Xu and M.-D. Zhou, Synthesis of Monofluoroalkenes through Visible-Light-Promoted Defluorinative Alkylation of gem-Difluoroalkenes with 4-Alkyl-1,4-dihydropyridines, Org. Lett., 2020, 22(4), 1542–1546 CrossRef CAS PubMed.
-
(a) Z.-J. Wang, S. Zheng, J. K. Matsui, Z. Lu and G. A. Molander, Desulfonative photoredox alkylation of N-heteroaryl sulfones- an acid-free approach for substituted heteroarene synthesis, Chem. Sci., 2019, 10, 4389–4393 RSC;
(b) Á. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, Late-Stage C–H Alkylation of Heterocycles and 1,4-Quinones via Oxidative Homolysis of 1,4-Dihydropyridines, J. Am. Chem. Soc., 2017, 139(35), 12251–12258 CrossRef PubMed;
(c) L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Radical-Based C-C Bond-Forming Processes Enabled by the Photoexcitation of 4-Alkyl-1,4-dihydropyridines, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed.
-
(a) A. Gutierrez-Bonet, J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis, ACS Catal., 2016, 6, 8004–8008 CrossRef CAS PubMed;
(b) A. Dumoulin, J. K. Matsui, A. Gutierrez-Bonet and G. A. Molander, Synthesis of Non-Classical Arylated C-Saccharides through Nickel/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef CAS PubMed;
(c) S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Synthesis of Reversed C-Acyl Glycosides through Ni/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS PubMed.
-
(a) H.-H. Zhang, J.-J. Zhao and S. Yu, Enantioselective Allylic Alkylation with 4-Alkyl-1,4-dihydro-pyridines Enabled by Photoredox/Palladium Cocatalysis, J. Am. Chem. Soc., 2018, 140(49), 16914–16919 CrossRef CAS PubMed;
(b) S. Liang, T. Kumon, R. A. Angnes, M. Sanchez, B. Xu and G. B. Hammond, Synthesis of Alkyl Halides from Aldehydes via Deformylative Halogenation, Org. Lett., 2019, 21(10), 3848–3854 CrossRef CAS PubMed.
-
(a) R. A. Angnes, C. Potnis, S. Liang, C. R. D. Correia and G. B. Hammond, Photoredox-Catalyzed Synthesis of Alkylaryldiazenes: Formal Deformylative C-N Bond Formation with Alkyl Radicals, J. Org. Chem., 2020, 85(6), 4153–4164 CrossRef CAS PubMed;
(b) S. Zhang, Y. Li, J. Wang, X. Hao, K. Jin, R. Zhang and C. Duan, A photocatalyst-free photo-induced denitroalkylation of β-nitrostyrenes with 4-alkyl substituted Hantzsch esters at room temperature, Tetrahedron Lett., 2020, 61(14), 151721 CrossRef CAS;
(c) J. Wang, Y.-B. Pang, N. Tao, R.-S. Zeng and Y. Zhao, Mn-Enabled Radical-Based Alkyl-Alkyl Cross-Coupling Reaction from 4-Alkyl-1,4-dihydropyridines, J. Org. Chem., 2019, 84(23), 15315–15322 CrossRef CAS PubMed.
-
(a) S. Liang, R. A. Angnesa, C. S. Potnisb and G. B. Hammond, Photoredox catalyzed C(sp3)-C(sp) coupling of dihydropyridines and alkynylbenziodoxolones, Tetrahedron Lett., 2019, 60(45), 151230 CrossRef;
(b) X. Shen, L. Qian and S. Yu, Photoredox/palladium-cocatalyzed enantioselective alkylation of secondary
benzyl carbonates with 4-alkyl-1,4-dihydropyridines, Sci. China: Chem., 2020, 63(5), 687–691 CrossRef CAS;
(c) K. Zhang, L.-Q. Lu, Y. Jia, Y. Wang, F.-D. Lu, F. Pan and W.-J. Xiao, Exploration of a Chiral Cobalt Catalyst for Visible-Light-Induced Enantioselective Radical Conjugate Addition, Angew. Chem., Int. Ed., 2019, 131(38), 13509–13513 CrossRef.
-
(a) Q.-Y. Wu, Q.-Q. Min, G.-Z. Ao and F. Liu, Radical alkylation of para-quinone methides with 4-substituted Hantzsch esters/nitriles via organic photoredox catalysis, Org. Biomol. Chem., 2018, 16, 6391–6394 RSC;
(b) H.-H. Zhang and S. Yu, Radical Alkylation of Imines with 4-Alkyl-1,4-dihydropyridines Enabled by Photoredox/Brønsted Acid Cocatalysis, J. Org. Chem., 2017, 82, 9995–10006 CrossRef CAS PubMed;
(c) F. F. Assis, X. Huang, M. Akiyama, R. A. Pilli and E. Meggers, Visible-Light-Activated Catalytic Enantioselective β-Alkylation of α,β-Unsaturated 2-Acyl Imidazoles Using Hantzsch Esters as Radical Reservoirs, J. Org. Chem., 2018, 83, 10922–10932 CrossRef PubMed.
-
(a) X. Liu, R. Liu, J. Dai, X. Cheng and G. Li, Application of Hantzsch Ester and Meyer Nitrile in Radical Alkynylation Reactions, Org. Lett., 2018, 20, 6906–6909 CrossRef CAS PubMed;
(b) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed;
(c) F. Gu, W. Huang, X. Liu, W. Chen and X. Cheng, Substituted Hantzsch Esters as Versatile Radical Reservoirs in Photoredox Reactions, Adv. Synth. Catal., 2018, 360(5), 925–931 CrossRef CAS.
-
(a) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, Building Congested Ketone: Substituted Hantzsch Ester and Nitrile as Alkylation Reagents in Photoredox Catalysis, J. Am. Chem. Soc., 2016, 138, 12312–12315 CrossRef CAS PubMed;
(b) X. Wang, M. Yang, W. Xie, X. Fan and J. Wu, Photoredox-catalyzed hydrosulfonylation reaction of electron deficient alkenes with substituted Hantzsch esters and sulfur dioxide, Chem. Commun., 2019, 55, 6010–6013 RSC;
(c) B. R. McDonald and K. A. Scheidt, Intermolecular Reductive Radical-Radical Couplings of Arylidene Malonates via Lewis Acid/Photoredox Cooperative Catalysis, Org. Lett., 2018, 20(21), 6877–6881 CrossRef CAS PubMed.
-
(a) X. Wang, H. Li, G. Qiu and J. Wu, Substituted Hantzsch esters as radical reservoirs with the insertion of sulfur dioxide under photoredox catalysis, Chem. Commun., 2019, 55, 2062–2065 RSC;
(b) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed;
(c) T. Leeuwen, L. Buzzetti, L. A. Perego and P. Melchiorre, A Redox-Active Nickel Complex that Acts as an Electron Mediator in Photochemical Giese Reactions, Angew. Chem., Int. Ed., 2019, 58, 4953–4957 CrossRef PubMed.
-
(a) C. Verrier, N. Alandini, C. Pezzetta, M. Moliterno, L. Buzzetti, H. B. Hepburn, A. Vega-Peñaloza, M. Silvi and P. Melchiorre, Direct Stereoselective Installation of Alkyl Fragments at the β-Carbon of Enals via Excited Iminium Ion Catalysis, ACS Catal., 2018, 8, 1062–1066 CrossRef CAS;
(b) G. Goti, B. Bieszczad, A. Vega-Penaloza and P. Melchiorre, Stereocontrolled Synthesis of 1,4-Dicarbonyl Compounds by Photochemical Organocatalytic Acyl Radical Addition to Enals, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed;
(c) K. Nakajima, Y. Zhang and Y. Nishibayashi, Alkylation Reactions of Azodicarboxylate Esters with 4-Alkyl-1,4-Dihydropyridines under Catalyst-Free Conditions, Org. Lett., 2019, 21(12), 4642–4645 CrossRef CAS PubMed.
-
(a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, 2013 Search PubMed;
(b) T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
-
(a) A. D. Becke, Density-Functional Thermochemistry. 3. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS;
(b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab-initio Calculation of Vibrational Absorption and Circular-dichroism Spectra using Density-functional Force-fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
-
(a) R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type Basis for Molecular-orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724 CrossRef CAS;
(b) W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. 12. Further Extensions of Gaussian-type Basis Sets for Use in Molecular-orbital Studies of Organic-molecules, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS;
(c) P. C. Hariharan and J. A. Pople, Influence of Polarization Functions on Molecular-orbital Hydrogenation Energies, Theor. Chem. Acc., 1973, 28, 213–222 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
-
(a) X.-Q. Zhu, H.-R. Li, Q. Li, T. Ai, J.-Y. Lu, Y. Yang and J.-P. Cheng, Determination of the C4-H Bond Dissociation Energies of NADH Models and Their Radical Cations in Acetonitrile, Chem.–Eur. J., 2003, 9(4), 871–880 CrossRef CAS PubMed;
(b) X.-Q. Zhu, Q.-Y. Liu, Q. Chen and L.-R. Mei, Hydride, Hydrogen, Proton, and Electron Affinities of Imines and Their Reaction Intermediates in Acetonitrile and Construction of Thermodynamic Characteristic Graphs (TCGs) of Imines as a “Molecule ID Card”, J. Org. Chem., 2010, 75, 789–808 CrossRef CAS PubMed.
-
(a) Y.-H. Fu, G.-B. Shen, Y. Li, L. Yuan, J.-L. Li, L. Li, A.-K. Fu, J. Chen, B.-L. Chen, L. Zhu and X.-Q. Zhu, Realization of Quantitative Estimation for Reaction Rate Constants Using only One Physical Parameter for Each Reactant, ChemistrySelect, 2017, 2, 904–925 CrossRef CAS;
(b) X.-Q. Zhu, F.-H. Deng, J.-D. Yang, X.-T. Li, Q. Chen, N.-P. Lei, F.-K. Meng, X.-P. Zhao, S.-H. Han, E.-J. Hao and Y.-Y. Mu, A classical but new kinetic equation for hydride transfer reactions, Org. Biomol. Chem., 2013, 11, 6071–6089 RSC.
-
(a) S.-M. Si, Y.-H. Fu and X.-Q. Zhu, Determination and Comparison of Thermodynamic Driving Forces of Elementary Steps for the Reductions of Alkynes and the Corresponding Alkenes in Acetonitrile, J. Phys. Chem. C, 2015, 119, 62–74 CrossRef CAS;
(b) X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, Hydride, Hydrogen Atom, Proton, and Electron Transfer Driving Forces of Various Five-Membered Heterocyclic Organic Hydrides and Their Reaction Intermediates in Acetonitrile, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed;
(c) Y. Cao, S.-C. Zhang, M. Zhang, G.-B. Shen and X.-Q. Zhu, Determination of Thermodynamic Affinities of Various Polar Olefins as Hydride, Hydrogen Atom, and Electron Acceptors in Acetonitrile, J. Org. Chem., 2013, 78, 7154–7168 CrossRef CAS PubMed.
- B. C. Garrett and D. G. Truhlar, Transition State Theory, in Encyclopedia of Computational Chemistry, John Wiley & Sons, Hoboken, NJ, USA, 1998, pp. 3094–3104 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Li Xiea,
Hao-Yun Yua,
Jie Liua,
Yan-Hua Fub and
Maocai Yan
a,
Li Xiea,
Hao-Yun Yua,
Jie Liua,
Yan-Hua Fub and
Maocai Yan