
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile metal-free one-pot synthesis of 3-aminoisoquinolines by intramolecular transannulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles†
Hai Shang ab,
Ling-Yu Lia,
Yu Tiana,
Hong-Mei Jiaa and
Zhong-Mei Zou*a
ab,
Ling-Yu Lia,
Yu Tiana,
Hong-Mei Jiaa and
Zhong-Mei Zou*a
aInstitute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100193, P. R. China. E-mail: zmzou@implad.ac.cn; Tel: +86-10-5783-3290
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, P. R. China
First published on 28th October 2020
Abstract
A metal-free one-pot intramolecular transannulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles has been developed, which enables the facile synthesis of the various 3-aminoisoquinolines as well as relevant scaffolds from readily available starting materials.
Introduction
Isoquinoline represents an important class of heterocyclic skeleton, which is widely distributed in diverse natural products, and some are found to have promising biological performance and medicinal importance.1–3 The unique structure of 3-aminoisoquinolines is well-explored to be commonly used as the unit for synthesizing biologically active representatives, such as compounds 1–3 (Fig. 1a).4–6 In view of the wide application of 3-aminoisoquinolines in medicinal chemistry, a large array of synthetic methods had been developed for the construction of 3-aminoisoquinolines and their structurally similar analogues.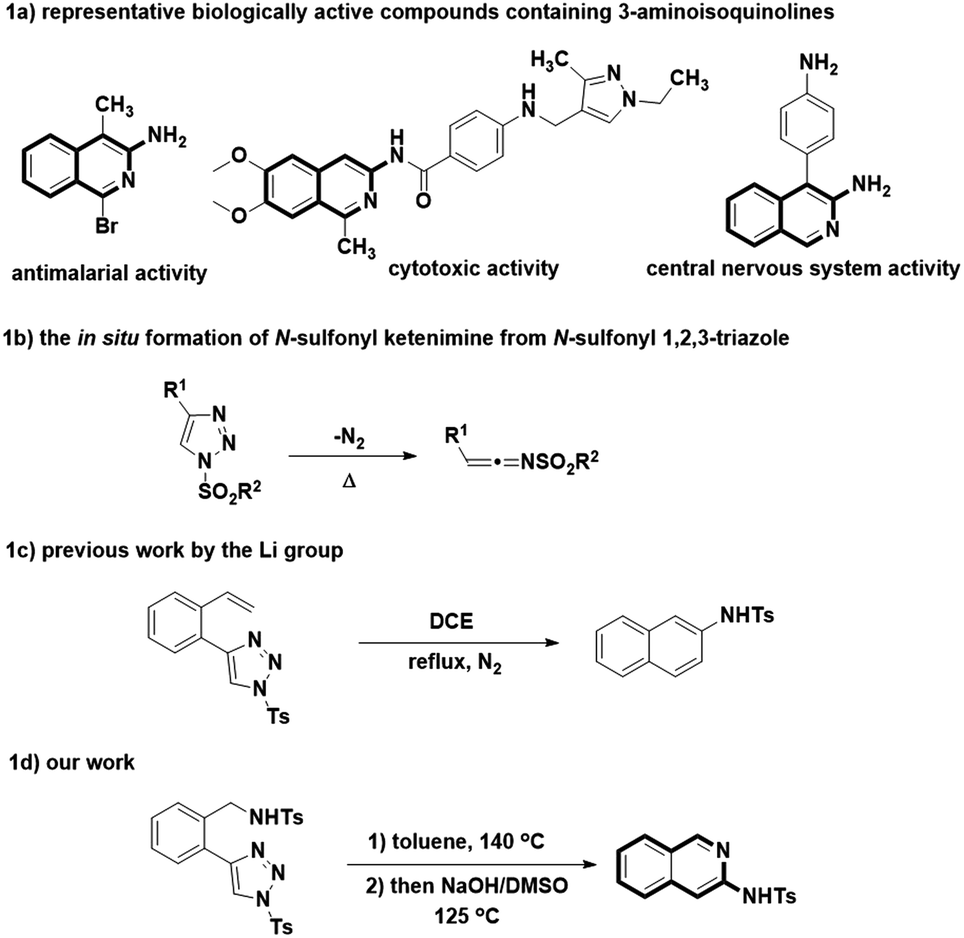 | ||
| Fig. 1 Background and our work. | ||
In general, synthetic approaches towards 3-aminoisoquinolines were historically reported, including the direct amination of 3-haloisoquinoline with sodium amide, aqueous ammonia or cyclic amine.7–9 In order to prove the acceptable reaction efficiency of the above mentioned transformations, some harsh reaction conditions are commonly required, such as high reaction temperature or pressure. Otherwise, the cyclocondensation of 2-cyanomethyl benzoaldehydes or 2-acylphenylacetonitriles with amines has been served as an accessible option for obtaining the 3-aminoisoquinolines, which highly depends on the intrinsic nature of nitrogen substrates used.10,11 For instance, ammonia or primary amines could smoothly afford 3-aminoisoquinolines with good yields whereas the secondary amines were converted slowly, and failed to proceed to the formation of desired product with satisfying results in the most cases. In addition, some alternative procedures have also been reported. These methods include the cyclization of arylmethylazapropenylium perchlorates,12 Cu(I)-catalyzed cyclocondensation of o-iodobenzylamine and acetonitriles13 and the palladium catalyzed α-arylation reaction of tert-butyl cyanoacetate, followed by in situ trapping with an electrophile, and aromatization with ammonium chloride.14 However, these methods are revealed to suffer from difficult access to starting material, the necessity of transition metal catalysts, the limited scope of applicability or low yield. The synthetically practical and operationally convenient method for preparing the 3-aminoisoquinoline is still necessary.
N-Sulfonyl ketenimine, which is readily in situ generated from ring-opening of N-sulfonyl 1,2,3-triazole, has been evolved into a useful synthetic intermediate in recent years for the synthesis of numerous complex organic compounds, particularly the biologically interesting heterocycles (Fig. 1b).15 As a highly active intermediate, N-sulfonyl ketenimine can readily undergo nucleophilic additions to give their corresponding products.16 For example, Li's group developed a method for synthesis of naphthalenes by intramolecular transannulation of 1-sulfonyl-4-(2-alkenylphenyl)-1,2,3-triazoles (Fig. 1c).17 Inspired by this work, we envisaged the N-sulfonyl ketenimine could pave the way of advancing to the aforementioned heterocycles. As a result, we report herein a metal-free intramolecular transannulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles, a reaction enabling the efficient synthesis of 3-aminoisoquinolines (Fig. 1d).
Results and discussion
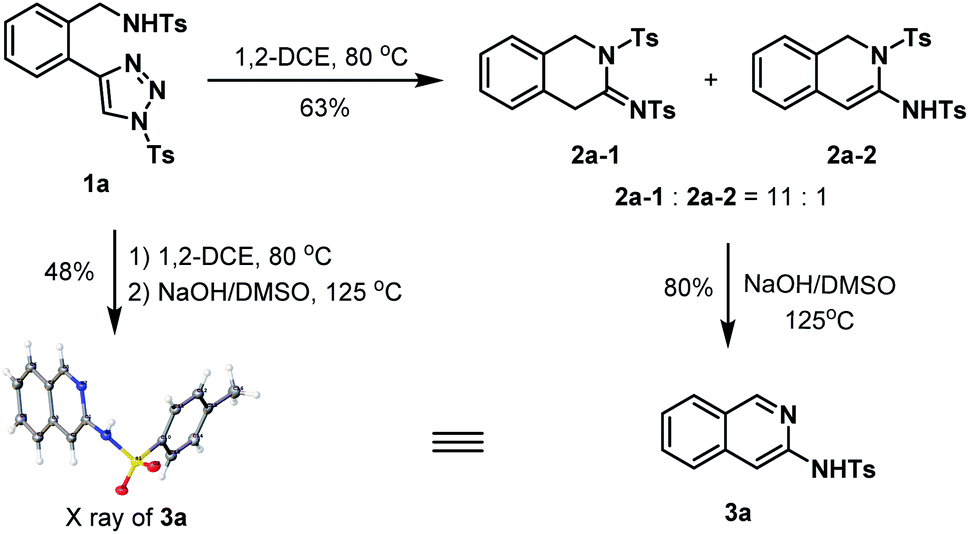
Using 1a as a model substrate, we commenced our investigation by treating it in 1,2-DCE at 80 °C in a sealed tube (Scheme 1). After 36 h, two main products were obtained, which were further identified as the 1,2-dihydroisoquinolin-3(4H)-imine 2a-1, and its regioisomer 1,2-dihydroisoquinoline 2a-2 generated from the enamine-iminium isomerization. The overall isolated yield of 2a-1 and 2a-2 was 63% and the molar ratio 2a-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a-2 = 11:1. Of note, the two isomers were observed as an inseparable mixture that each of them was unable to be respectively obtained by simply using the silica gel column chromatography. In order to unambiguously characterize the presumed structures of the two isomers, the mixture was subject to the treatment of NaOH/DMSO solution that prompting the formation of 3-aminoisoquinoline 3a (80%) as an aromatized product.18 The structure of 3a was unambiguously confirmed by the X-ray crystallographic study.19 Furthermore, we found that the tandem annulation and aromatization could be smoothly achieved in an operationally simple one-pot manner, which allowed for the access of 3a directly from 1a with comparable efficiency.
:2a-2 = 11:1. Of note, the two isomers were observed as an inseparable mixture that each of them was unable to be respectively obtained by simply using the silica gel column chromatography. In order to unambiguously characterize the presumed structures of the two isomers, the mixture was subject to the treatment of NaOH/DMSO solution that prompting the formation of 3-aminoisoquinoline 3a (80%) as an aromatized product.18 The structure of 3a was unambiguously confirmed by the X-ray crystallographic study.19 Furthermore, we found that the tandem annulation and aromatization could be smoothly achieved in an operationally simple one-pot manner, which allowed for the access of 3a directly from 1a with comparable efficiency.
 | ||
| Scheme 1 Initial result of intramolecular annulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles. | ||
To improve the efficiency of the above mentioned transformation, we performed an extensive condition screening (Table 1). First of all, we discovered that the better reactivity could be secured by gradually increasing the reaction temperature from 80 to 140 °C (entries 2–4). A good yield (73%) of 3a was obtained with raising the reaction temperature to 140 °C. Gratifyingly, simply replacing the reaction solvent with non-polar toluene can provide the best result (81%) instead of the rest (THF, CHCl3, CH3CN and EtOAc). In sharp contrast, the lower reaction temperature actually led to inferior results that advance to the desired product 3a with only moderate yield (entries 5 and 6). In addition, the Rh catalyst were also examined, but negative impact on the reaction outcome was observed.
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | T [°C] | Yieldb |
| a Reaction conditions: 1a (0.1 mmol), catalyst (2.0 μmol), and solvent (1.0 mL) in a sealed tube, then NaOH (1.0 mmol) and DMSO (1.0 mL).b Refers to isolated yield. 1,2-DCE = dichloroethane, Oct = octanoate, esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid. | ||||
| 1 | 1,2-DCE | 80 | 48% | |
| 2 | 1,2-DCE | 100 | 56% | |
| 3 | 1,2-DCE | 120 | 65% | |
| 4 | 1,2-DCE | 140 | 73% | |
| 5 | PhMe | 100 | 59% | |
| 6 | PhMe | 120 | 71% | |
| 7 | PhMe | 140 | 81% | |
| 8 | THF | 140 | 77% | |
| 9 | CHCl3 | 140 | 62% | |
| 10 | CH3CN | 140 | 73% | |
| 11 | EtOAc | 140 | 77% | |
| 12 | Rh2(Oct)4 | PhMe | 140 | 45% |
| 13 | Rh2(esp)2 | PhMe | 140 | 39% |
Having the optimal conditions in hands, we then turned to evaluate the substrate scope generality of the above reaction (Table 2). First of all, the substituent variants on the phenyl ring were examined. It was shown that all of them could undergo the desired reactions to provide the corresponding products 3b–k in moderate to excellent yields. 5-Br, 5-Cl and 4-Cl substituted phenyl triazoles delivered 1b–d with satisfactory yield (78–85%), as well as 3e and 3f gave the acceptable results (65% and 63%) when 4-F and 5-F substituted phenyl triazoles were used.
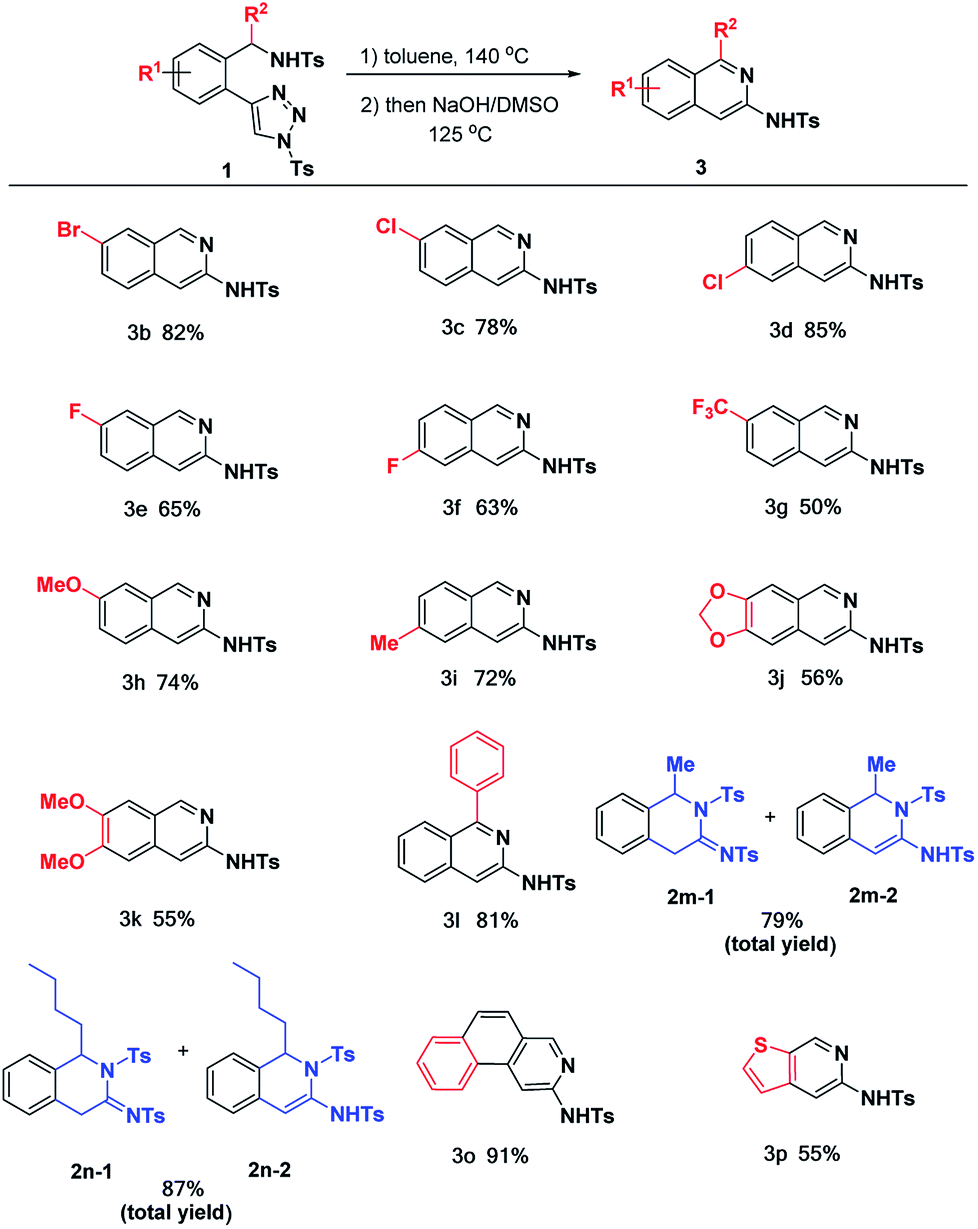
The arene substitution pattern indicates that the bromo and chloro substituents are superior to fluoro substituent. The 5- and 4-functionalized substrates result in comparable reactivity, afford their corresponding products with moderate to good yield. For substrate 1g with trifluoromethyl group, the reaction gave the desired product 3g in moderate yield. The reaction could also be applied to substrates with electron-donating groups (1h–k). As shown, mono-substituted phenyl triazoles with electron-donating groups (1h, 1i) could proceed smoothly to furnish the desired products 3h and 3i in 72% and 74% yields. Comparably, substrates with double electron-donating groups led to relatively lower yields (3j, 3k), most likely due to the stronger electronic effect causing the decomposition of partial substrate under the current reaction conditions.
In addition to the substituent on the phenyl ring, substitution effect on the benzylic position has also been evaluated. 2l bearing Ph group (R2) at the benzylic position underwent the reactions smoothly to afford the desired product 3l in 81% yield. For the alkyl substituted substrates 1m and 1n, the corresponding annulation products were obtained in 79 and 87% total yields, respectively. However, it failed to undergo aromatization to give the corresponding 3-aminoisoquinolines under NaOH condition, presumably due to electron-donating effect of alkyl group. Finally, it was found that the benzene ring unit of the substrates could be replaced by some other aromatic rings, such as naphthalene and thiophene, as shown by the cases of 3o and 3p.
Based on the above results and the previously reported literature, the presumable mechanistic hypothesis of the transannulation using substrate 1a as a model is rationally illustrated in Scheme 2. Substrate 1a undergoes a ring-opening denitrogenative process upon heating to afford carbene intermediate A, which was followed by a 1,2-H migration to give ketenimine intermediate B. Through the intramolecular nucleophilic addition, zwitterionic intermediate C and its resonanced intermediates C′ were generated, which could convert to 2a-1 and 2a-2 via intramolecular proton transfer. Finally, base-mediated elimination/aromatization of sulfinate results in 3-aminoisoquinoline 3a.
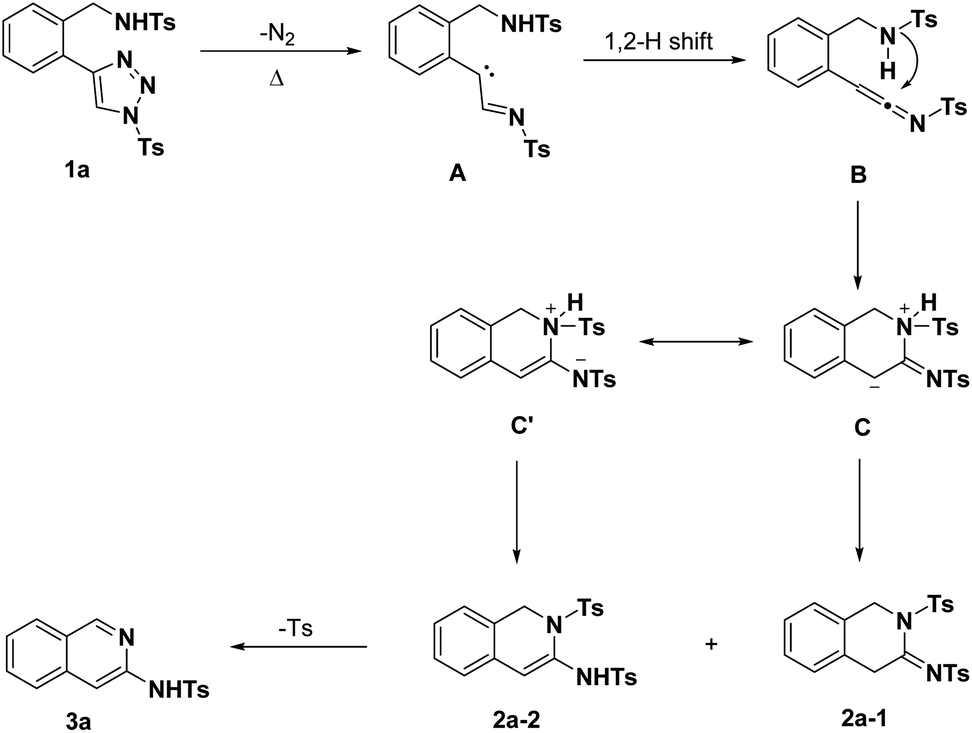 | ||
| Scheme 2 Proposed mechanism of intramolecular annulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles. | ||
Conclusions
In summary, we have developed an efficient method for the construction of 3-aminoisoquinolines through a metal-free one-pot intramolecular transannulation of 1-sulfonyl-4-(2-aminomethylphenyl)-1,2,3-triazoles. This method would provide an alternative to the current toolbox for the synthesis of 3-aminoisoquinolines, and further applications of the method to the syntheses of bioactive molecules are in progress.Experimental
General information
NMR spectra were recorded on Bruker AV III 600 NMR spectrometer and Bruker AV 400 instrument. Solvent signal was used as reference for 1H NMR (CDCl3, 7.26 ppm; DMSO-d6, 2.50 ppm) and 13C NMR (CDCl3, 77.16 ppm; DMSO-d6, 39.52 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, td = triple doublet, m = multiplet. Infrared (IR) spectra were recorded on a FTIR-8400S spectrometer. High-resolution mass spectra (HRMS) were recorded on a Waters SYNAPT G2 HDMS.Reactions were monitored by Thin Layer Chromatography on plates (GF254) supplied by Yantai Chemicals (China). If not specially mentioned, flash column chromatography uses silica gel (200–300 mesh) supplied by Tsingtao Haiyang Chemicals (China). Solvent purification was conducted according to Purification of Laboratory Chemicals (D. D. Peerrin, W. L. Armarego and D. R. Perrins, Pergamon Press, Oxford, 1980).
General procedure for the preparation of 3-aminoisoquinolines
A 10 mL pressure tube, fitted with a rubber septum, was charged with triazole (0.1 mmol, 1.0 eq.). The reaction vessel was added freshly distilled toluene (1.0 mL) and then was sealed with a Teflon screwcap and placed in an oil bath preheated to 140 °C. Upon complete consumption of the substrate (monitored by TLC), the solvent was removed and DMSO (1.0 mL) with an aqueous solution of sodium hydroxide (30% w/w, 133 mg, 1.0 mmol) was added. The mixture was heated to around 125 °C, and then stirred at this temperature for 2 h. The reaction was monitored by TLC. After the reaction was complete, the mixture was allowed to cool to room temperature and diluted with water (10 mL). The aqueous solution was then extracted with ethyl acetate (3 × 15 mL) and the combined extracts were dried with anhydrous Na2SO4. The solvent was evaporated in vacuo and the residue was purified by a silica gel flash column chromatography (eluent: petroleum ether/EtOAc) to give the corresponding products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Drug Innovation Major Project (2018ZX09711-001-005-008); National Natural Science Foundation of China (81502929) and CAMS Innovation Fund for Medical Sciences (2016-I2M-3-015).Notes and references
- M. Iranshahy, R. J. Quinn and M. Iranshahi, RSC Adv., 2014, 4, 15900–15913 CAS.
- K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC.
- P. Joshi, R. A. Vishwakarma and S. B. Bharate, Eur. J. Med. Chem., 2017, 138, 273–292 CrossRef CAS.
- J. L. Neumeuer and K. K. Weinhardt, J. Med. Chem., 1970, 13, 999–1002 CrossRef CAS.
- G. B. Lapa, T. Tsunoda, S. Shirasawa, M. A. Baryshnikova, G. G. Evseev, D. A. Afanasyeva and E. A. Chigorina, Chem. Biol. Drug Des., 2016, 87, 575–582 CrossRef CAS.
- J. L. Neumeyer, K. K. Weinhardt, R. A. Carrano and D. H. McCurdy, J. Med. Chem., 1973, 16, 808–813 CrossRef CAS.
- T. Kametani, K. Kigasawa and M. Hiiragi, Chem. Pharm. Bull., 1974, 93, 704–707 Search PubMed.
- W. Bartmann, E. Konz and W. Rüger, J. Heterocycl. Chem., 1987, 24, 677–682 CrossRef CAS.
- G. M. Sanders, M. van Dijk and H. J. den Hertog, Recl. Trav. Chim. Pays-Bas, 1974, 93, 198–200 CrossRef CAS.
- T. Zdrojewski and A. Jończyk, Tetrahedron, 1995, 51, 12439–12444 CrossRef CAS.
- A. S. Cánepa and R. D. Bravo, J. Heterocycl. Chem., 2006, 43, 235–238 CrossRef.
- G. V. Boyd, P. F. Lindley and G. A. Nicolaou, J. Chem. Soc., Chem. Commun., 1984, 1105–1107 RSC.
- H. Suzuki and H. Abe, Synthesis, 1995, 1995, 763–765 CrossRef.
- B. S. Pilgrim, A. E. Gatland, C. T. McTernan, P. A. Procopiou and T. J. Donohoe, Org. Lett., 2013, 15, 6190–6193 CrossRef CAS.
- (a) P. Lu and Y. Wang, Chem. Soc. Rev., 2012, 41, 5687–5705 RSC; (b) M. Alajarin, M. Marin-Luna and A. Vidal, Eur. J. Org. Chem., 2012, 5637–5653 CrossRef CAS; (c) S. H. Kim, S. H. Park, J. H. Choi and S. Chang, Chem.–Asian J., 2011, 6, 2618–2634 CrossRef CAS; (d) P. Lu and Y. Wang, Synlett, 2010, 2, 165–173 Search PubMed.
- (a) S. H. Cho, E. J. Yoo, I. Bae and S. Chang, J. Am. Chem. Soc., 2005, 127, 16046–16047 CrossRef CAS; (b) E. J. Yoo, I. Bae, S. H. Cho, H. Han and S. Chang, Org. Lett., 2006, 8, 1347–1350 CrossRef CAS; (c) I. Bae, H. Han and S. Chang, J. Am. Chem. Soc., 2005, 127, 2038–2039 CrossRef CAS; (d) S. L. Cui, J. Wang and Y. Wang, Org. Lett., 2007, 9, 5023–5025 CrossRef CAS; (e) S. Chang, M. J. Lee, D. Y. Jung, E. J. Yoo, S. H. Cho and S. K. Han, J. Am. Chem. Soc., 2006, 128, 12366–12367 CrossRef CAS; (f) E. J. Yoo and S. Chang, Org. Lett., 2008, 10, 1163–1166 CrossRef CAS; (g) Y. Jiang, R. Sun, Q. Wang, X. Y. Tang and M. Shi, Chem. Commun., 2015, 51, 16968–16971 RSC; (h) J. Kim, Y. Lee, J. Lee, Y. Do and S. Chang, J. Org. Chem., 2008, 73, 9454–9457 CrossRef CAS; (i) R. Sun, Y. Jiang, X. Y. Tang and M. Shi, Chem.–Eur. J., 2016, 22, 5727–5733 CrossRef CAS; (j) H. D. Xu, Z. J. Jia, K. Xu, M. Han, S. N. Jiang, J. Cao, J. C. Wang and M. H. Shen, Angew. Chem., Int. Ed., 2014, 53, 9284–9288 CrossRef CAS.
- Z. F. Xu, X. Yu, D. Yang and C. Y. Li, Org. Biomol. Chem., 2017, 15, 3161–3164 RSC.
- J. Dong, X. X. Shi, J. J. Yan, J. Xing, Q. Zhang and S. Xiao, Eur. J. Org. Chem., 2010, 6987–6992 CrossRef CAS.
- CCDC 2026878 (3a) contains the ESI† crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of isolated compounds. CCDC 2026878. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06563c |
| This journal is © The Royal Society of Chemistry 2020 |