
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric 1,3-dipolar cycloaddition reaction of chiral 1-alkyl-1,2-diphospholes with diphenyldiazomethane†
Yulia Ganushevicha,
Almaz Zagidullin *a,
Svetlana Kondrashovaa,
Shamil Latypova,
Vasili Miluykova,
Peter Lönneckeb and
Evamarie Hey-Hawkinsb
*a,
Svetlana Kondrashovaa,
Shamil Latypova,
Vasili Miluykova,
Peter Lönneckeb and
Evamarie Hey-Hawkinsb
aArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, 420088, Kazan, Russia. E-mail: almaz.zagidullin@gmail.com
bInstitute of Inorganic Chemistry, Leipzig University, 04103, Leipzig, Germany
First published on 27th October 2020
Abstract
The 1,3-dipolar cycloaddition of chiral 1-alkyl-1,2-diphosphacyclopenta-2,4-dienes ((1-(−)-menthyl)oxymethyl-1,2-diphosphole and 1-(+)-neomenthyl-1,2-diphosphole) with diphenyldiazomethane leads to novel P-chiral bicyclic phosphiranes having six chiral centers. The degree of diastereoselectivity depends on the substituent at phosphorus, and dramatically increases in the case of (+)-neomenthyl group (de up to 71%). DFT calculations indicate that the cycloaddition is thermodynamically controlled.
Introduction
The addition of a 1,3-dipole to a dipolarophile is a fundamental reaction in organic chemistry, and its high utility spreads from natural product synthesis and chemical biology to materials science, drug discovery and agrochemistry, indicating its diversity.1–3 Different substituents can be included in the 1,3-dipole and the dipolarophile resulting in a wide range of possible cycloadducts, which can serve as useful synthetic building blocks. The dipolarophile can be almost any multiple bond, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO, CN, CS and C
C, CO, CN, CS and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, CN bonds. Organophosphorus compounds with
C, CN bonds. Organophosphorus compounds with ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CP– functionality (heterophospholes,4,5 1,3-diphospholes,6 1,2-diphospholes7) have also been used as dipolarophiles for construction of five-membered P-heterocycles with useful properties.
CP– functionality (heterophospholes,4,5 1,3-diphospholes,6 1,2-diphospholes7) have also been used as dipolarophiles for construction of five-membered P-heterocycles with useful properties.
The asymmetric version of 1,3-dipolar cycloaddition reactions (1,3-DC) is one of the most powerful and useful tools for the construction of enantiomerically pure heterocycles.8–10 Up to 4 stereocenters can be created in a stereoselective manner in one single step. The high regio- and stereoselectivity of 1,3-DC arouse interest to develop new types of dipolarophiles leading to novel heterocycles. Compared to a multitude of literature data on the asymmetric 1,3-DC of C-, N-, O-, or S-containing dipolarophiles, the phospha-1,3-DC reaction employing a CP moiety as dipolarophile is still unknown, despite its potential utility to obtain P-chiral cyclic phosphines for use in asymmetric homogeneous catalysis.11–14 Moreover, the principle of diastereotopic face differentiation by employing a PC double bond of phosphaalkenes,15 2H-monophospholes,16,17 1,2-diphospholes18–21 and heterophospholes22 as prochiral motif in [4 + 2] cycloaddition reactions was successfully used in the synthesis of P-chiral polycyclic phosphines. Herein, we describe the first example of asymmetric 1,3-DC reaction of chiral 1-alkyl-1,2-diphospholes with diphenyldiazomethane.
Results and discussion
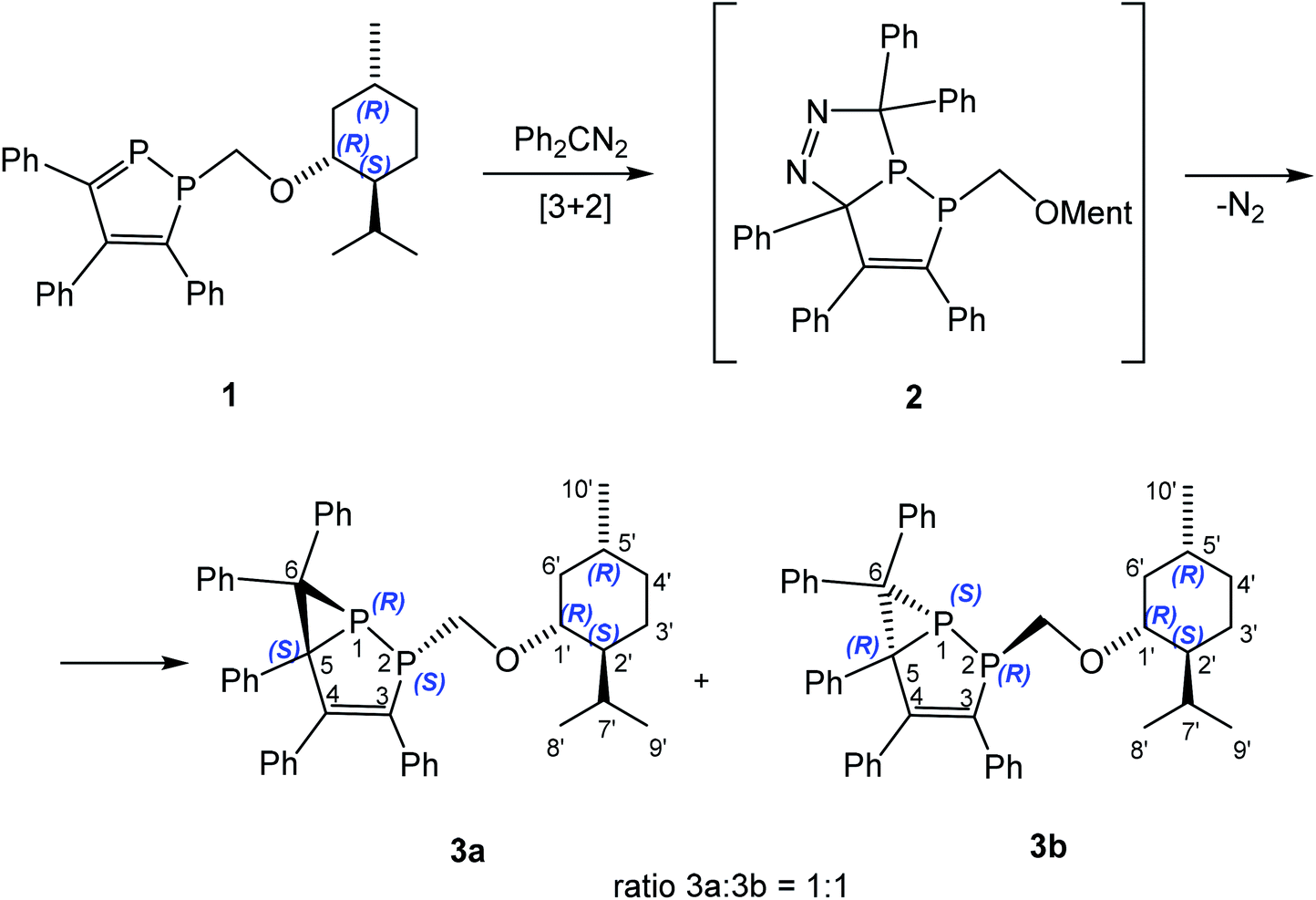
The reactions of heterophospholes as well as 1,2-diphospholes with diazoalkanes proceed with formation of either five-membered bicyclic diazophospholanes23,24 or bicyclic phosphiranes formed by loss of nitrogen.7,25,26 To the best of our knowledge there are no examples of an asymmetric 1,3-DC reaction of diazoalkanes with aCP– group. Chiral 1-alkyl-1,2-diphospholes 127 and 418,28 contain a stable PC bond and a chiral substituent which makes them promising for different cycloaddition reactions. Indeed 1-((1R,2S,5R)-menthyl)oxymethyl-1,2-diphosphole (1) reacts with diphenyldiazomethane in toluene to form exclusively a mixture of two diastereomers, namely 2-((1R,2S,5R-menthyl)oxymethyl)-3,4,5,6,6-pentaphenyl-1,2-diphosphabicyclo[3.1.0]hex-3-enes 3a and 3b with a ratio 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3b = 1:1 (Scheme 1). The small diastereomeric excess (de) is probably caused by the far distance between (1R,2S,5R)-menthyl fragment and the reaction center. Decreasing the reaction temperature to −30 °C does not lead to any change in the isomer ratio.
:3b = 1:1 (Scheme 1). The small diastereomeric excess (de) is probably caused by the far distance between (1R,2S,5R)-menthyl fragment and the reaction center. Decreasing the reaction temperature to −30 °C does not lead to any change in the isomer ratio.
 | ||
| Scheme 1 Reaction of 1-((1R,2S,5R)-menthyl)oxymethyl-1,2-diphosphole 1 with diphenyldiazomethane. | ||
Presumably, first an unstable [3 + 2] cycloaddition product 2 is formed as intermediate, which undergoes further rearrangement and loses of N2 with formation of 3a and 3b. Although we were unable to detect unstable 2 by low temperature NMR spectroscopy, a similar intermediate is proposed for 1,3-DC reactions of heterophospholes5 or chiral alkenes29 with diazomethane derivatives. The 31P{1H} NMR spectrum of the reaction mixture showed only four doublets – two doublets for each diastereomer at +6.8 and −122.3 ppm with 1JPP = 263.8 Hz, and at +6.4 and −119.2 ppm with 1JPP = 264.9 Hz, shifted upfield in comparison to free 1 by ca. 50 and 300 ppm, respectively. The mixture of diastereomeric bicyclic phosphiranes 3a and 3b was separated by chromatography on silica gel with a petroleum ether/toluene mixture to give diastereo- and enantiopure 3a and 3b in 37% and 32% yield, respectively.
Most of the signals in the 1H, 13C and 31P NMR spectra of compounds 3a and 3b were unequivocally assigned employing a variety of NMR correlation experiments30,31 (see ESI† for details). The structure of the skeleton of the bicyclic phosphirane, the menthyl fragment, and their connection through the spacer were unambiguously established. From the point of view for an absolute stereochemistry determination, the ortho-phenyl protons at C3 (o-Ph3), H7′, H5′, H6′, Me-8′, Me-9′ and Me-10′ resonances are the most important. To this end, there is 1H–13C HMBC connectivity from the P2–CH2O protons to C3. The exact assignment of the o-Ph3 protons is based on correlations from these protons to C3 (1H–13C HMBC) and to P2 (1H–31P HMBC). NOE between the o-Ph3 protons and the P2–CH2O protons additionally provide support for this assignment. Good agreement between the calculated (PBE0/6-311G(2d,2p)//PBE0/6-31+G(d)) and experimental 13C and 31P chemical shifts32 further supports this conclusion.
In general, an absolute configuration at one chiral center of a diastereomer can be determined by correlation to another chiral center with known stereochemistry. To this end, one needs to know the geometry of the chain linking two chiral centers, and there should be experimentally observable stereoselective NMR effects (NOE or shielding) between nuclei in these two chiral fragments.33–35
In 3a and 3b, two chiral fragments are linked by the C1′–O–CH2–P2 chain. Conformational preference around O–C1′ and CH2–O bonds is well established: in the former the gauche (G) orientation of C1′–H and CH2–O bonds is preferred,36–38 while the trans (T) conformation should be favored around the later.39 Conformational preference around the P–CH2 bond is not obvious. Therefore, first, the conformational preference around this bond was analyzed. DFT calculations on a simpler model (methyl instead of menthyl group) for three different orientations around the P2–CH2 bond demonstrated that a trans orientation of CH2–O with respect to the lone pair of electrons at P2 should be essentially lower in energy (1.4 kcal mol−1) than other orientations (Table S1†). Thus, the preferential conformation of the C1′–O–CH2–P2 chain is expected to be GTT (Fig. S1a†). Taking into account these results, the structures of both diastereomers were generated and optimized (Fig. 1a and b).
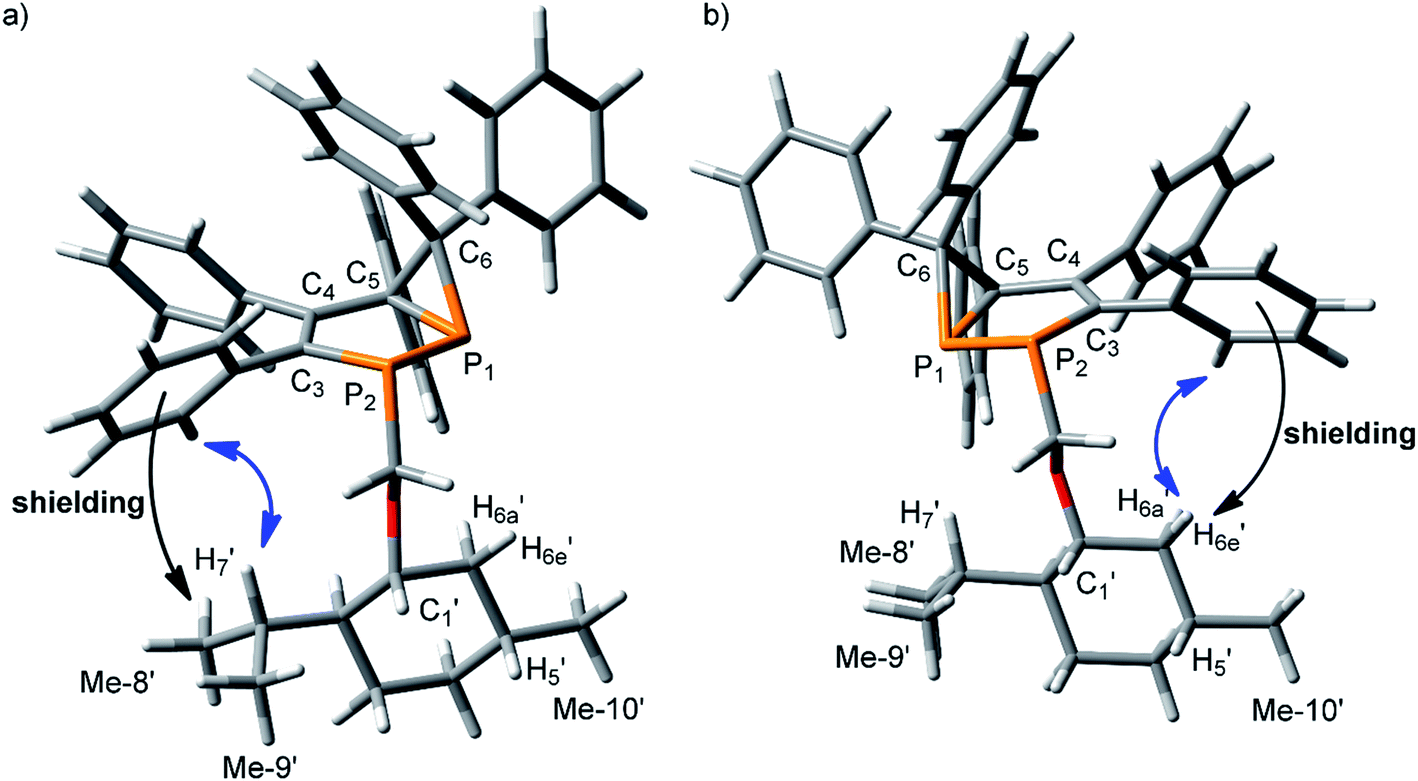 | ||
| Fig. 1 (a) Optimized structures of the isomers 3a (P1:(R), P2:(S), C5:(S)) and (b) 3b (P1:(S), P2:(R), C5:(R)), with indicative NMR effects: aromatic shielding (black arrow) and NOE (blue arrow). | ||
Qualitative analysis of these structures demonstrates that high field shifts may be expected for H7′, Me-8′ and Me-9′ protons due to shielding effects from o-Ph3 protons in 3a, while similar effects should be observed for H5′, Me-10′, H6e′ and H6a′ protons in 3b. Thus, these effects might be used to establish an absolute stereochemistry of the diastereomers. NMR experiments are in line with these conclusions. Indeed, these protons are largely different in the 1H NMR spectra of these two products, 3a and 3b (Fig. S2a and c†). It is important that H7′, Me-8′ and Me-9′ are observed at higher field in, as are 3a H6e′, H6a′, H5′ and Me-10′ in 3b. Moreover, at low temperature these NMR shielding's increase (Fig. S2b and d†) presumably due to the greater contribution of the energetically favored conformer. In addition, there are NOE's between the o-Ph3 and H7′ protons in 3a, while in 3b, such effects are observed between the o-Ph3 and H6e′ protons. Based on these findings, the absolute configuration of 3a can be ascribed as P1:(R), P2:(S), C5:(S) and of 3b as P1:(S), P2:(R), C5:(R).
Similarly, 3,4,5-triphenyl-1-(+)-neomenthyl-1,2-diphosphole (4) reacts with diphenyldiazomethane at 25 °C to form a mixture of two diastereomers, namely 2-(+)-neomenthyl-3,4,5,6,6-pentaphenyl-1,2-diphosphabicyclo[3.1.0]hex-3-enes 5a (major) and 5b (minor) with higher diastereoselectivity de = 71% (ratio 5a:5b = 6:1) than observed for 1 (Scheme 2).
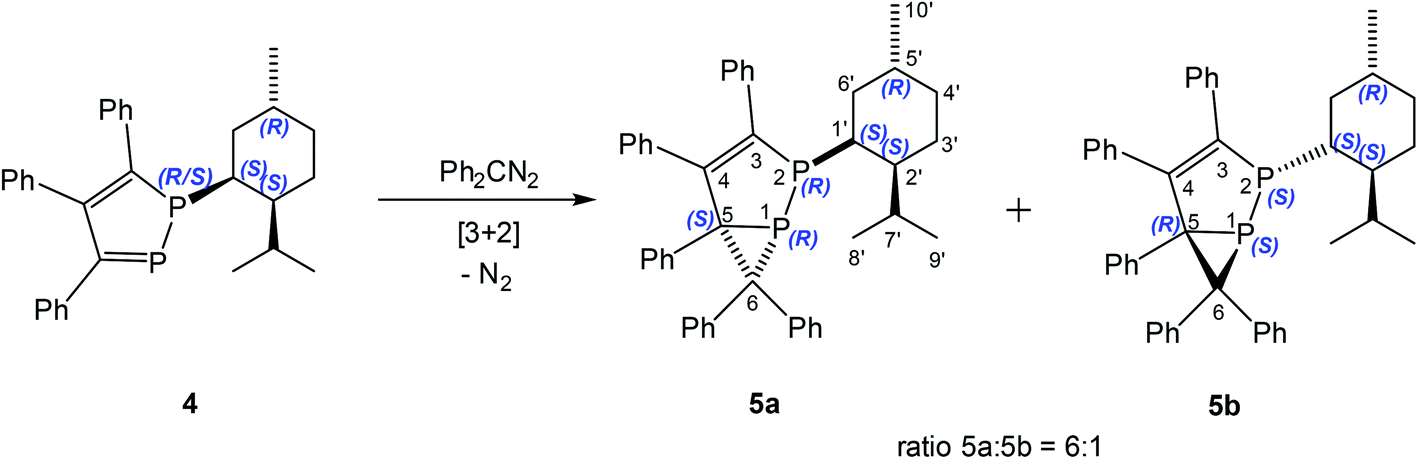 | ||
| Scheme 2 Reaction of 3,4,5-triphenyl-1-(+)-neomenthyl-1,2-diphosphole (4) with diphenyldiazomethane. | ||
An analysis of the structure of 418 shows that two different reactions pathways may be considered for the 1,3-DC reaction (Fig. 2). Steric shielding of one side by the bulky isopropyl group causes a preferential approach of the diphenyldiazomethane from the opposite side resulting in one attractive and one repulsive pathway for the 1,3-dipolar cycloaddition reaction.
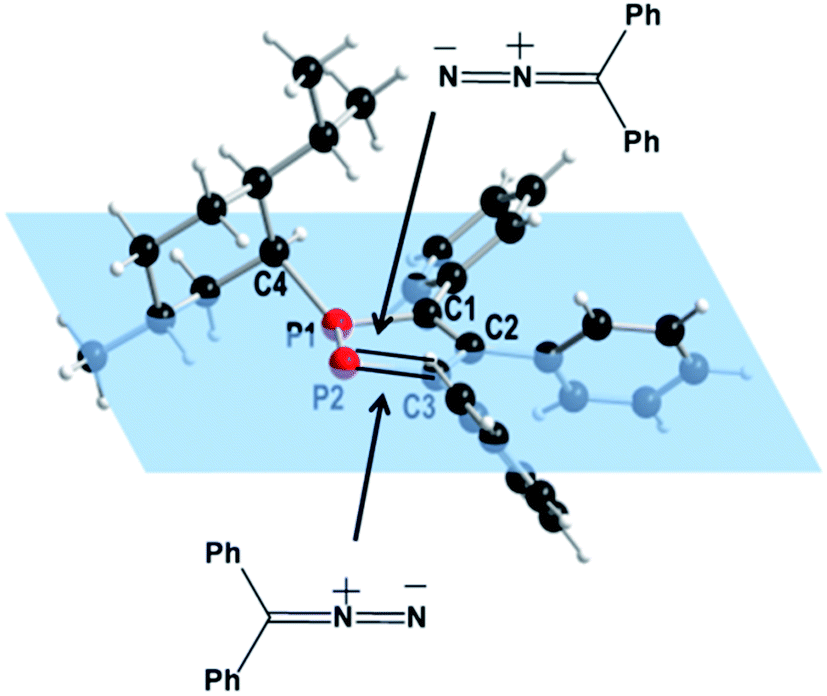 | ||
| Fig. 2 Possible routes for the reaction of 3,4,5-triphenyl-1-(+)-neomenthyl-1,2-diphosphole (4) with diphenyldiazomethane. | ||
Bicyclic chiral phosphirane 5a was isolated by crystallization and fully characterized by 1H, 31P{1H}, 13C{1H} NMR spectroscopy and X-ray structure analysis. In the 31P{1H} NMR spectrum two doublets are observed at +16.0 and −103.3 ppm with 1JPP = 298.1 Hz. Only one set of signals was observed in the 1H and 13C{1H} NMR spectra of the bicyclic phosphine 5a indicating an enantiomerically pure compound.
Most of the signals in the NMR spectra of the major isomer 5a were unequivocally assigned employing NMR correlation experiments. For the minor isomer 5b, the 31P and only some 1H and 13C NMR signals can be assigned with confidence. In the major isomer, the key signals that can be used to establish the absolute configuration are due to the ortho-phenyl protons at C3 (o-Ph3), H5′, and Me-8′ protons. Namely, there is a strong NOE between the o-Ph3 and Me-8′ protons, and a large downfield shift of H5′ (ca. 3.05 ppm) relative to its “normal” position (1.4–1.2 ppm) and an upfield shift of the Me-8′ protons (see ESI† for details).
If low energy structures for these two isomers are considered (Fig. S3†), only the structure 5a can explain these NMR effects. Thus, the close proximity and orientation of o-Ph3 with respect to the Me-8′ group explains the observed NOE and upfield shift of the Me-8′ due to its aromatic shielding effect; the opposite is observed for H5′, which is observed in the deshielding region of the lone pair of electrons at P1. Thus, the major product 5a can be ascribed as the P1:(R), P2:(R), C5:(S) isomer. Given the similarity in changes in the calculated and experimental 31P NMR chemical shifts for these two isomers, the minor product 5b can be assigned to the structure with configuration P1:(S), P2:(S), C5:(R). It is also interesting that in this case the major isomer 5a is lower in energy than 5b (Table S3†). Thus, the observed stereoselectivity of the cycloaddition reaction may be due to the relative thermodynamic stability of the two possible isomers.
A crystal structure analysis of 5a (Fig. 3) confirmed the identity and stereochemistry and showed that only one diastereomer was presented with the neomenthyl group in an anti orientation to the 3-membered P2–C3–C4(Ph2) fragment. Compound 5a crystallizes in the monoclinic space group C2 with a Flack parameter of −0.01(3). There are 6 chiral centers in 5a, each phosphorus-atom has a typical pyramidal environment, the menthyl fragment has the configuration of (+)-neomenthol or (1S,2S,5R)-2-isopropyl-5-methylcyclohexanol.
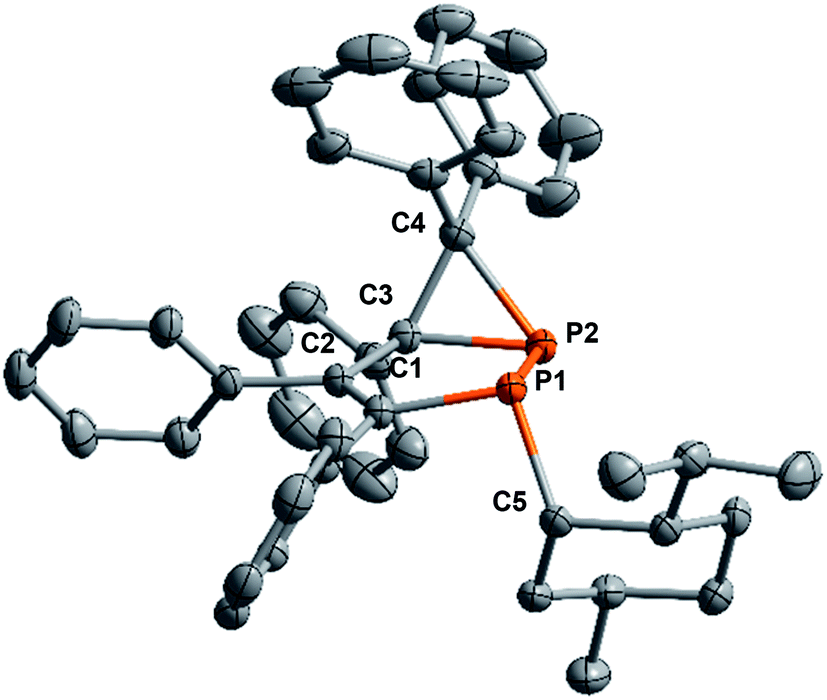 | ||
| Fig. 3 Molecular structure of 2-((+)-neomenthyl)-3,4,5,6,6-pentaphenyl-1,2-(P1RP2RC3S)-diphosphabicyclo[3.1.0]hex-3-ene (5a). Hydrogen atoms are omitted for clarity. Configuration of chiral atoms in the five-membered ring: P1:(R), P2:(R), C3:(S). Selected bond lengths [Å] and angles [°]: P1–C1 1.818(3); P1–C5 1.888(3); P1–P2 2.194(1); P2–C4 1.877(3); P2–C3 1.879(3); C1–C2 1.354(4); C2–C3 1.513(4); C3–C4 1.554(4); C1–P1–C5 99.9(1); C1–P1–P2 93.4(1); C4–P2–C3 48.9(1); C4–P2–P1 100.8(1); C3–P2–P1 94.4(1). | ||
The P–C bond lengths (1.877 and 1.879 Å) of the phosphirane ring of 5a are in the range typical for other phosphiranes (1.78–1.89 Å).40 The sum of bond angles around P2 (Σ(∠P2) = 243.1°) of 5a reflects strong pyramidalization of the environment at the phosphorus atom comparable with those for other bicyclic phosphiranes.26,41,42
Phosphiranes are 3-membered cyclic phosphines with unique stereoelectronic properties, (smaller cone angles and higher inversion barriers compared with their acyclic analogues) and are thought to be poorer σ donors and better π acceptors,40 but their use as ligands in metal-catalyzed reactions is rare.43 Chiral phosphiranes are especially rare;44–47 therefore, the convenient asymmetric cycloaddition reaction reported here is potentially useful for expanding their limited applications in asymmetric catalysis.48–50
In conclusion, we have shown that a diastereoselective 1,3-dipolar cycloaddition reaction of 1-alkyl-1,2-diphospholes 1 and 4 with a chiral substituent at phosphorus with diphenyldiazomethane is a new way for the selective synthesis of P-chiral bicyclic phosphiranes from cheap and readily available starting materials. The formation of two diastereomers 3a and 3b (1:1) was observed in the 1,3-dipolar cycloaddition of 1-((1R,2S,5R)-menthyl)oxymethyl-1,2-diphosphole (1) with diphenyldiazomethane, while the reaction between 1-(+)-neomenthyl-1,2-diphosphole 4 and diphenyldiazomethane proceeded with high de (71%). Enantiopure 2-(+)-neomenthyl-3,4,5,6,6-pentaphenyl-1,2-diphosphabicyclo[3.1.0]hex-3-ene (5a) could be obtained by crystallization. This study proves that combination of a chiral auxiliary with the CP– group of 1,2-diphospholes facilitates stereoselective 1,3-dipolar cycloaddition reactions, which is important for further developments of asymmetric cycloaddition reactions in the synthesis of chiral P-stereogenic phosphines.
Experimental part
NMR spectroscopy
All NMR experiments were performed with a Bruker AVANCE-600 and 500 MHz (600.1 and 500.1 MHz for 1H NMR; 150.9 and 125.7 MHz for 13C NMR; 242.9 and 202.5 MHz for 31P NMR, respectively) spectrometers equipped with a 5 mm diameter gradient inverse broad band probehead and a pulsed gradient unit capable of producing magnetic field pulse gradients in the z direction of 53.5 G cm−1. Most of the NMR experiments were carried out at 303 K. For 1H–31P long range correlations, HMBC experiments were optimized for J = 10 Hz. NOE experiments were performed with 1D DPFGNOE techniques.51 Chemical shifts are reported in the δ (ppm) scale relative to the 1H (7.27 ppm) and 13C (77.0 ppm) signals of CDCl3. 31P chemical shifts were referred to external 85% H3PO4 (0.00 ppm).DNMR experiments were carried out using a Bruker BVT3000 variable-temperature unit (with BTO2000, accuracy ±0.1 K calibrated using a methanol reference). The samples were allowed to equilibrate for 15 min at target temperature.
Elemental analyses were carried out at the microanalysis laboratory of the Arbuzov Institute of Organic and Physical Chemistry, Russian Academy of Sciences.
Synthesis
All reactions and manipulations were carried out under dry pure N2 in standard Schlenk devices. All solvents were distilled from sodium/benzophenone or P2O5 and stored under nitrogen before use. Starting materials: 1-((1R,2S,5R)-menthyl)oxymethyl-1,2-diphosphole 1,27 3,4,5-triphenyl-1-(+)-neomenthyl-1,2-diphosphole 418 and diphenyldiazomethane52 were obtained according to literature procedures.:1, v/v)) to give 0.30 g (37%) of 3a as yellowish powder and 0.26 g (32%) of 3b as yellowish powder. ), 1.76–1.67 (m, 2H, C4′–H, C3′–H), 1.49–1.42 (m, 1H, C2′–H), 1.40–1.31 (m, 1H, C5′–H), 1.04–0.95 (m, 3H, C3′–H, C4′–H,
), 1.76–1.67 (m, 2H, C4′–H, C3′–H), 1.49–1.42 (m, 1H, C2′–H), 1.40–1.31 (m, 1H, C5′–H), 1.04–0.95 (m, 3H, C3′–H, C4′–H,  ), 1.00 (d, 3H, 3JHH = 6.5 Hz, C10′–H), 0.92 (d, 3H, 3JHH = 7.0 Hz, C8′–H), 0.68 (d, 3H, 3JHH = 7.0 Hz, C9′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 151.1 (s, C4), 144.5 (d, 1JCP = 24.2 Hz, C3), 142.7 (br, ipso-C6Ph′), 139.6 (d, 2JCP = 5.4 Hz, ipso-C5Ph), 139.1 (s, ipso-C4Ph), 139.0 (d, 2JCP = 17.7 Hz, ipso-C3Ph), 138.3 (br, ipso-C6Ph), 134.8 (s, o-C6Ph), 134.2 (d, 3JCP = 7.3 Hz, o-C5Ph), 131.2 (s, o-C4Ph), 129.5 (d, 3JCP = 14.2 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.6 Hz, o-C3Ph), 127.3 (br, p-CPh + m-CPh + m-CPh), 127.0 (s, p-CPh), 126.9 (s, m-CPh), 126.8 (s, m-CPh), 126.5 (s, m-CPh), 125.7 (s, p-CPh), 125.6 (s, p-CPh), 125.3 (s, p-CPh), 80.5 (s, C1′), 73.1 (d, 1JCP = 37.8 Hz, C5), 64.8 (dd, 1JCP = 25.4 Hz, 2JCP = 6.2 Hz, P
), 1.00 (d, 3H, 3JHH = 6.5 Hz, C10′–H), 0.92 (d, 3H, 3JHH = 7.0 Hz, C8′–H), 0.68 (d, 3H, 3JHH = 7.0 Hz, C9′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 151.1 (s, C4), 144.5 (d, 1JCP = 24.2 Hz, C3), 142.7 (br, ipso-C6Ph′), 139.6 (d, 2JCP = 5.4 Hz, ipso-C5Ph), 139.1 (s, ipso-C4Ph), 139.0 (d, 2JCP = 17.7 Hz, ipso-C3Ph), 138.3 (br, ipso-C6Ph), 134.8 (s, o-C6Ph), 134.2 (d, 3JCP = 7.3 Hz, o-C5Ph), 131.2 (s, o-C4Ph), 129.5 (d, 3JCP = 14.2 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.6 Hz, o-C3Ph), 127.3 (br, p-CPh + m-CPh + m-CPh), 127.0 (s, p-CPh), 126.9 (s, m-CPh), 126.8 (s, m-CPh), 126.5 (s, m-CPh), 125.7 (s, p-CPh), 125.6 (s, p-CPh), 125.3 (s, p-CPh), 80.5 (s, C1′), 73.1 (d, 1JCP = 37.8 Hz, C5), 64.8 (dd, 1JCP = 25.4 Hz, 2JCP = 6.2 Hz, P![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2O), 54.1 (d, 1JCP = 36.3 Hz, C6), 48.9 (s, C2′), 39.9 (s, C6′), 34.7 (s, C4′), 31.6 (s, C5′), 24.8 (s, C7′), 23.0 (s, C3′), 22.4 (s, C10′), 21.3 (s, C8′), 16.1 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 6.2 (d, 1JCP = 264.6 Hz, P2), −121.4 (d, 1JCP = 264.6 Hz, P1). Anal. calcd for C45H46OP2 (M 665): C, 81.30; H, 6.97; O, 2.41; P 9.32. Found: C, 81.14; H, 7.10; O, 2.18; P 9.58.
H2O), 54.1 (d, 1JCP = 36.3 Hz, C6), 48.9 (s, C2′), 39.9 (s, C6′), 34.7 (s, C4′), 31.6 (s, C5′), 24.8 (s, C7′), 23.0 (s, C3′), 22.4 (s, C10′), 21.3 (s, C8′), 16.1 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 6.2 (d, 1JCP = 264.6 Hz, P2), −121.4 (d, 1JCP = 264.6 Hz, P1). Anal. calcd for C45H46OP2 (M 665): C, 81.30; H, 6.97; O, 2.41; P 9.32. Found: C, 81.14; H, 7.10; O, 2.18; P 9.58.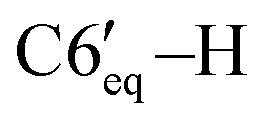 ), 1.70–1.62 (m, 2H, C4′–H, C3′–H), 1.37–1.30 (m, 1H, C2′–H), 1.27–1.19 (m, 1H, C5′–H), 1.02 (d, 3H, 3JHH = 7.2 Hz, C8′–H), 0.88–0.84 (m, 6H, C9′–H, C10′–H), 1.00–0.70 (m, 3H, C3′–H, C4′–H,
), 1.70–1.62 (m, 2H, C4′–H, C3′–H), 1.37–1.30 (m, 1H, C2′–H), 1.27–1.19 (m, 1H, C5′–H), 1.02 (d, 3H, 3JHH = 7.2 Hz, C8′–H), 0.88–0.84 (m, 6H, C9′–H, C10′–H), 1.00–0.70 (m, 3H, C3′–H, C4′–H, 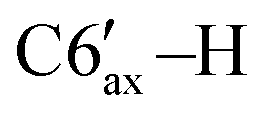 ). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 150.8 (s, C4), 145.35 (d, 1JCP = 24.1, C3), 142.6 (d, 2JCP = 8.8 Hz, ipso-C6Ph′), 139.5 (d, 2JCP = 4.4 Hz, ipso-C5Ph), 139.1 (s, ipso-C4Ph), 139.0 (d, 2JCP = 16.3 Hz, ipso-C3Ph), 138.1 (br, ipso-C6Ph), 134.9 (s, o-C6Ph), 134.1 (d, 3JCP = 6.8 Hz, o-C5Ph), 131.2 (s, o-C4Ph), 129.5 (d, 3JCP = 14.3 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.9 Hz, o-C3Ph), 127.3 (s, p-CPh + m-CPh + m-CPh), 127.0 (s, p-CPh), 126.9 (s, m-CPh), 126.8 (s, m-CPh), 126.6 (s, m-CPh), 125.8 (s, p-CPh), 125.7 (s, p-CPh), 125.3 (s, p-CPh), 80.2 (s, C1′), 73.5 (d, 1JCP = 37.8 Hz, C5), 66.1 (dd, 1JCP = 23.1 Hz, 2JCP = 9.3 Hz, PH2O), 54.8 (d, 1JCP = 35.9 Hz, C6), 48.6 (s, C2′), 40.0 (s, C6′), 34.6 (s, C4′), 31.5 (s, C5′), 25.4 (s, C7′), 23.1 (s, C3′), 22.3 (s, C10′), 21.3 (s, C8′), 16.2 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 5.3 (d, 1JCP = 265.3 Hz, P2), −118.3 (d, 1JCP = 264.6 Hz, P1). Anal. calcd for C45H46OP2 (M 665): C, 81.30; H, 6.97; O, 2.41; P, 9.32. Found: C, 81.34; H, 7.05; O, 2.17; P, 9.44.:2, v/v), this solution was passed through a silica-gel pad (2 cm). Removal of solvents gave 0.34 g (87%) of a mixture of isomers 5a and 5b. The major diastereomer 5a was crystallized from the isomer mixture from methanol as a yellowish crystals.
). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 150.8 (s, C4), 145.35 (d, 1JCP = 24.1, C3), 142.6 (d, 2JCP = 8.8 Hz, ipso-C6Ph′), 139.5 (d, 2JCP = 4.4 Hz, ipso-C5Ph), 139.1 (s, ipso-C4Ph), 139.0 (d, 2JCP = 16.3 Hz, ipso-C3Ph), 138.1 (br, ipso-C6Ph), 134.9 (s, o-C6Ph), 134.1 (d, 3JCP = 6.8 Hz, o-C5Ph), 131.2 (s, o-C4Ph), 129.5 (d, 3JCP = 14.3 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.9 Hz, o-C3Ph), 127.3 (s, p-CPh + m-CPh + m-CPh), 127.0 (s, p-CPh), 126.9 (s, m-CPh), 126.8 (s, m-CPh), 126.6 (s, m-CPh), 125.8 (s, p-CPh), 125.7 (s, p-CPh), 125.3 (s, p-CPh), 80.2 (s, C1′), 73.5 (d, 1JCP = 37.8 Hz, C5), 66.1 (dd, 1JCP = 23.1 Hz, 2JCP = 9.3 Hz, PH2O), 54.8 (d, 1JCP = 35.9 Hz, C6), 48.6 (s, C2′), 40.0 (s, C6′), 34.6 (s, C4′), 31.5 (s, C5′), 25.4 (s, C7′), 23.1 (s, C3′), 22.3 (s, C10′), 21.3 (s, C8′), 16.2 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 5.3 (d, 1JCP = 265.3 Hz, P2), −118.3 (d, 1JCP = 264.6 Hz, P1). Anal. calcd for C45H46OP2 (M 665): C, 81.30; H, 6.97; O, 2.41; P, 9.32. Found: C, 81.34; H, 7.05; O, 2.17; P, 9.44.:2, v/v), this solution was passed through a silica-gel pad (2 cm). Removal of solvents gave 0.34 g (87%) of a mixture of isomers 5a and 5b. The major diastereomer 5a was crystallized from the isomer mixture from methanol as a yellowish crystals.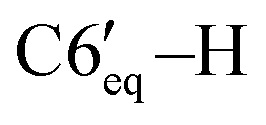 ), 2.11–2.00 (m, 2H, C4′–H, C1′–H), 1.98–1.87 (m, 2H, C3′–H, C7′–H), 1.43 (q, 1H, 3JHH = 10.4 Hz, C3′–H), 1.24–1.13 (m, 1H,
), 2.11–2.00 (m, 2H, C4′–H, C1′–H), 1.98–1.87 (m, 2H, C3′–H, C7′–H), 1.43 (q, 1H, 3JHH = 10.4 Hz, C3′–H), 1.24–1.13 (m, 1H, 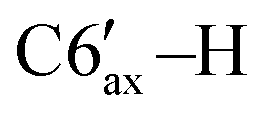 ), 1.17 (d, 3H, 3JHH = 6.3 Hz, C10′–H), 1.14–1.04 (m, 1H, C2′–H), 1.07–0.96 (m, 1H, C4′–H), 0.95 (d, 3H, 3JHH = 6.3 Hz, C9′–H), 0.57 (d, 3H, 3JHH = 6.5 Hz, C8′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 149.5 (d, 2JCP = 5.8 Hz, C4), 148.1 (d, 1JCP = 26.2 Hz, C3), 143.3 (d, 2JCP = 10.8 Hz, ipso-C6Ph′), 140.2 (d, 2JCP = 6.5 Hz, ipso-C5Ph), 139.5 (d, 2JCP = 15.5 Hz, ipso-C3Ph), 139.2 (d, 2JCP = 3.6 Hz, ipso-C4Ph), 136.7 (br, ipso-C6Ph), 135.3 (d, 3JCP = 2.9 Hz, o-C6Ph), 133.9 (d, 3JCP = 6.5 Hz, o-C5Ph), 131.4 (d, 3JCP = 11.5 Hz, o-C4Ph), 129.5 (d, 3JCP = 14.3 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.9 Hz, o-C3Ph), 128.4 (s, m-C3Ph), 127.7–126.3 (p-C6Ph + p-C5Ph + m-C6Ph + m-C6Ph′ + m-C4Ph), 125.6 (s, p-C4Ph), 125.5 (s, p-C3Ph), 125.1 (s, p-C6Ph′), 75.2 (d, 1JCP = 40.7 Hz, C5), 55.3 (dd, 1JCP = 38.6 Hz, 2JCP = 2.3 Hz, C6), 48.8 (d, 2JCP = 11.8 Hz, C2′), 38.5 (d, 2JCP = 4.3 Hz, C6′), 36.0 (s, C4′), 35.8 (d, 1JCP = 34.8 Hz, C1′), 30.5 (d, 3JCP = 17.6 Hz, C5′), 30.3 (d, 3JCP = 17.6 Hz, C7′), 28.3 (s, C3′), 22.8 (s, C10′), 21.3 (s, C9′), 20.7 (d, 4JCP = 3.3 Hz, C8′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 16.0 (d, 1JCP = 298.1 Hz, P2), −103.3 (d, 1JCP = 298.1 Hz, P1). Anal. calcd for C44H44P2 (M 635): C, 83.25; H, 6.99; P, 9.76. Found: C, 83.41; H, 6.85; P 9.69.
), 1.17 (d, 3H, 3JHH = 6.3 Hz, C10′–H), 1.14–1.04 (m, 1H, C2′–H), 1.07–0.96 (m, 1H, C4′–H), 0.95 (d, 3H, 3JHH = 6.3 Hz, C9′–H), 0.57 (d, 3H, 3JHH = 6.5 Hz, C8′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 149.5 (d, 2JCP = 5.8 Hz, C4), 148.1 (d, 1JCP = 26.2 Hz, C3), 143.3 (d, 2JCP = 10.8 Hz, ipso-C6Ph′), 140.2 (d, 2JCP = 6.5 Hz, ipso-C5Ph), 139.5 (d, 2JCP = 15.5 Hz, ipso-C3Ph), 139.2 (d, 2JCP = 3.6 Hz, ipso-C4Ph), 136.7 (br, ipso-C6Ph), 135.3 (d, 3JCP = 2.9 Hz, o-C6Ph), 133.9 (d, 3JCP = 6.5 Hz, o-C5Ph), 131.4 (d, 3JCP = 11.5 Hz, o-C4Ph), 129.5 (d, 3JCP = 14.3 Hz, o-C6Ph′), 129.0 (d, 3JCP = 6.9 Hz, o-C3Ph), 128.4 (s, m-C3Ph), 127.7–126.3 (p-C6Ph + p-C5Ph + m-C6Ph + m-C6Ph′ + m-C4Ph), 125.6 (s, p-C4Ph), 125.5 (s, p-C3Ph), 125.1 (s, p-C6Ph′), 75.2 (d, 1JCP = 40.7 Hz, C5), 55.3 (dd, 1JCP = 38.6 Hz, 2JCP = 2.3 Hz, C6), 48.8 (d, 2JCP = 11.8 Hz, C2′), 38.5 (d, 2JCP = 4.3 Hz, C6′), 36.0 (s, C4′), 35.8 (d, 1JCP = 34.8 Hz, C1′), 30.5 (d, 3JCP = 17.6 Hz, C5′), 30.3 (d, 3JCP = 17.6 Hz, C7′), 28.3 (s, C3′), 22.8 (s, C10′), 21.3 (s, C9′), 20.7 (d, 4JCP = 3.3 Hz, C8′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 16.0 (d, 1JCP = 298.1 Hz, P2), −103.3 (d, 1JCP = 298.1 Hz, P1). Anal. calcd for C44H44P2 (M 635): C, 83.25; H, 6.99; P, 9.76. Found: C, 83.41; H, 6.85; P 9.69.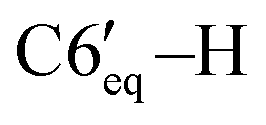 ), 1.61–1.55 (m, 1H, C3′–H), 1.52–1.46 (m, 1H, C4′–H), 1.27–1.2 (m, 1H, C2′–H), 1.21 (d, 3H, 3JHH = 6.3 Hz, C8′–H), 1.19–1.14 (m, 3H, C3′–H, C5′–H,
), 1.61–1.55 (m, 1H, C3′–H), 1.52–1.46 (m, 1H, C4′–H), 1.27–1.2 (m, 1H, C2′–H), 1.21 (d, 3H, 3JHH = 6.3 Hz, C8′–H), 1.19–1.14 (m, 3H, C3′–H, C5′–H, 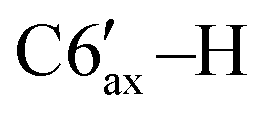 ), 0.99 (d, 3H, 3JHH = 6.7 Hz, C9′–H), 0.75–0.7 (m, 1H, C4′–H), 0.37 (d, 3H, 3JHH = 5.8 Hz, C10′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 75.1 (d, 1JCP = 39.5 Hz, C5), 54.7 (dd, 1JCP = 37.0 Hz, 2JCP = 1.3 Hz, C6), 50.7 (d, 2JCP = 11.5 Hz, C2′), 42.6 (dd, 1JCP = 30.6 Hz, 2JCP = 10.2 Hz, C1′), 40.7 (dd, 2JCP = 5.7 Hz, 3JCP = 5.7 Hz, C6′), 35.8 (s, C4′), 30.3 (d, 3JCP = 17.6 Hz, C7′), 28.3 (C5′), 26.0 (d, 3JCP = 6.4 Hz, C3′), 23.0 (d, 4JCP = 6.1 Hz, C8′), 22.0 (s, C10′), 21.2 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 8.1 (d, 1JCP = 279.9 Hz, P2), −114.2 (d, 1JCP = 279.9 Hz, P1).
), 0.99 (d, 3H, 3JHH = 6.7 Hz, C9′–H), 0.75–0.7 (m, 1H, C4′–H), 0.37 (d, 3H, 3JHH = 5.8 Hz, C10′–H). 13C{1H} NMR (CDCl3, δ, ppm, J, Hz): 75.1 (d, 1JCP = 39.5 Hz, C5), 54.7 (dd, 1JCP = 37.0 Hz, 2JCP = 1.3 Hz, C6), 50.7 (d, 2JCP = 11.5 Hz, C2′), 42.6 (dd, 1JCP = 30.6 Hz, 2JCP = 10.2 Hz, C1′), 40.7 (dd, 2JCP = 5.7 Hz, 3JCP = 5.7 Hz, C6′), 35.8 (s, C4′), 30.3 (d, 3JCP = 17.6 Hz, C7′), 28.3 (C5′), 26.0 (d, 3JCP = 6.4 Hz, C3′), 23.0 (d, 4JCP = 6.1 Hz, C8′), 22.0 (s, C10′), 21.2 (s, C9′). 31P{1H} NMR (CDCl3, δ, ppm, J, Hz): 8.1 (d, 1JCP = 279.9 Hz, P2), −114.2 (d, 1JCP = 279.9 Hz, P1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study of the synthesis of bicyclic phosphiranes was funded by Russian Foundation of Basic Research (RFBR, project number 20-33-70255). The theoretical calculations and NMR experiments were carried out by the government assignment for FRC Kazan Scientific Center of RAS. The authors gratefully acknowledge the Assigned Spectral-Analytical Center of FRC Kazan Scientific Center of RAS.References
- A. J. M. Burrell and I. Coldham, Curr. Org. Synth., 2016, 7, 312–331 CrossRef.
- A. Padwa and W. H. Pearson, Synthetic Applications of 1, 3-dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, Wiley-Interscience, 2002, ISBN 978-0471280613 Search PubMed.
- Y. Joo-Yong, L. Sang-gi and S. Hyunik, Curr. Org. Chem., 2011, 15, 657–674 Search PubMed.
- R. K. Bansal and J. Heinicke, Chem. Rev., 2001, 101, 3549–3578 CrossRef CAS.
- R. K. Bansal, N. Gupta and N. Gupta, Heteroat. Chem., 2004, 15, 271–287 CrossRef CAS.
- J. Steinbach, P. Binger and M. Regitz, Synthesis, 2003, 17, 2720–2724 Search PubMed.
- A. Zagidullin, Y. Ganushevich, V. Miluykov, D. Krivolapov, O. Kataeva, O. Sinyashin and E. Hey-Hawkins, Org. Biomol. Chem., 2012, 10, 5298–5306 RSC.
- K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–910 CrossRef CAS.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2007, 63, 3235–3285 CrossRef CAS.
- L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef.
- M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
- J. Holz, M.-N. Gensow, O. Zayas and A. Börner, Curr. Org. Chem., 2007, 11, 61–106 CrossRef CAS.
- A. Zagidullin, I. Bezkishko, V. Miluykov and O. Sinyashin, Mendeleev Commun., 2013, 23, 117–130 CrossRef CAS.
- S. C. Serin, B. O. Patrick, G. R. Dake and D. P. Gates, Organometallics, 2014, 33, 7215–7222 CrossRef CAS.
- T. Möller, M. Sarosi and E. Hey-Hawkins, Chem. – Eur. J., 2012, 18, 16604–16607 CrossRef.
- T. Möller, P. Wonneberger, M. B. Sárosi, P. Coburger and E. Hey-Hawkins, Dalton Trans., 2016, 45, 1904–1917 RSC.
- A. Zagidullin, V. Miluykov, F. Poyancev, Sh. Latypov, O. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Eur. J. Org. Chem., 2015, 24, 5326–5329 CrossRef.
- E. Oshchepkova, A. Zagidullin, V. Miluykov and O. Sinyashin, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1530–1532 CrossRef CAS.
- A. Zagidullin, V. Miluykov, O. Sinyashin and E. Hey-Hawkins, Catal. Today, 2016, 279, 142–146 CrossRef.
- A. Zagidullin, E. Oshchepkova, I. Chuchelkin, S. Kondrashova, V. Miluykov, Sh. Latypov, K. Gavrilov and E. Hey-Hawkins, Dalton Trans., 2019, 48, 4677–4684 RSC.
- R. Jangid, N. Sogani, N. Gupta, R. Bansal, M. Hopffgarten and G. Frenking, Beilstein J. Org. Chem., 2013, 9, 392–400 CrossRef CAS.
- J. Kerth, T. Jikyo and G. Maas, Eur. J. Org. Chem., 2003, 10, 1894–1903 CrossRef.
- J. Kerth and G. Maas, Eur. J. Org. Chem., 2001, 8, 1581–1587 CrossRef.
- X. Guo, L. Feng, Q. Wang, Z. Li and F. Tao, J. Heterocycl. Chem., 2006, 43, 353–359 CrossRef CAS.
- S. Maurer, T. Jikyo and G. Maas, Eur. J. Org. Chem., 2009, 13, 2195–2207 CrossRef.
- A. Zagidullin, Y. Ganushevich, V. Miluykov, P. Lönnecke and E. Hey-Hawkins, J. Organomet. Chem., 2020, 914, 121218 CrossRef CAS.
- A. Zagidullin, E. Oshchepkova, T. Burganov, V. Miluykov, S. Katsyuba, O. Sinyashin, P. Lönnecke and E. Hey-Hawkins, J. Organomet. Chem., 2018, 867, 125–132 CrossRef CAS.
- F. J. Impastato, L. Barash and H. M. Walborsky, J. Am. Chem. Soc., 1959, 81, 1514–1515 CrossRef CAS.
- E. Derome, Modern NMR Techniques for Chemistry Research, Pergamon, Cambridge, 1988, ISBN 9781483286426 Search PubMed.
- Atta-ur-Rahman, One and Two Dimensional NMR Spectroscopy, Elsevier, Amsterdam, 1989, ISBN-13 978-0444873163 Search PubMed.
- Sh. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976–6987 RSC.
- Sh. K. Latypov, J. M. Seco, E. Quinoa and R. Riguera, J. Org. Chem., 1995, 60, 1538–1545 CrossRef CAS.
- Sh. K. Latypov, J. M. Seco, E. Quinoa and R. Riguera, J. Org. Chem., 1996, 61, 8569–8577 CrossRef CAS.
- M. J. Ferreiro, Sh. K. Latypov, E. Quinoa and R. Riguera, J. Org. Chem., 2000, 65, 2658–2666 CrossRef CAS.
- Sh. K. Latypov, J. M. Seco, E. Quinoa and R. Riguera, J. Org. Chem., 1995, 60, 504–515 CrossRef CAS.
- Sh. K. Latypov, J. M. Seco, E. Quinoa and R. Riguera, J. Am. Chem. Soc., 1998, 120, 877–882 CrossRef CAS.
- Sh. K. Latypov, M. J. Ferreiro, E. Quinoa and R. Riguera, J. Am. Chem. Soc., 1998, 120, 4741–4751 CrossRef CAS.
- E. Eliel, Conformational Analysis, Interscience Publishers, 1965, ISBN 978-0470237427 Search PubMed.
- F. Mathey, Chem. Rev., 1990, 90, 997–1025 CrossRef CAS.
- S. Maurer, C. Burkhart and G. Maas, Eur. J. Org. Chem., 2010, 13, 2504–2511 CrossRef.
- J. Tendyck, H. Klöcker, L. Schürmann, E.-U. Würthwein, A. Hepp, M. Layh and W. Uhl, J. Org. Chem., 2020 DOI:10.1021/acs.joc.9b03056.
- J. C. Slootweg and K. Lammertsma, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, Wiley, Chichester, 2012, ISBN 978-0470666272 Search PubMed.
- M. Deegan, J. Muldoon, R. Hughes, D. Glueck and A. Rheingold, Organometallics, 2018, 37, 1473–1482 CrossRef CAS.
- J. A. Muldoon, B. R. Varga, M. M. Deegan, T. W. Chapp, A. M. Eordogh, R. P. Hughes, D. S. Glueck, C. E. Moore and A. L. Rheingold, Angew. Chem., Int. Ed., 2018, 57, 5047–5051 CrossRef CAS.
- X. Li, K. Robinson and P. Gaspar, J. Org. Chem., 1996, 61, 7702–7710 CrossRef CAS.
- F. Lang and H. Grutzmacher, Chimia, 2003, 57, 187–190 CrossRef CAS.
- A. Ficks, I. Martinez-Botella, B. Stewart, R. W. Harrington, W. Clegg and L. J. Higham, Chem. Commun., 2011, 47, 8274–8276 RSC.
- A. Ficks, W. Clegg, R. W. Harrington and L. J. Higham, Organometallics, 2014, 33, 6319–6329 CrossRef CAS.
- A. F. Abdel-Magied, M. H. Majeed, M. F. Abelairas-Edesa, A. Ficks, R. M. Ashour, A. Rahaman, W. Clegg, M. Haukka, L. J. Higham and E. Nordlander, J. Organomet. Chem., 2017, 849–850, 71–79 CrossRef CAS.
- K. Stott, J. Stonehouse, J. Keeler, T. L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- M. I. Javed and M. Brewer, Org. Synth., 2008, 85, 189–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all compounds, X-ray data for 5a (CIF) and data of quantum chemical calculations. CCDC 1988239. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08080b |
| This journal is © The Royal Society of Chemistry 2020 |