
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing intrinsic dynamics and conformational transition of HIV gp120 by molecular dynamics simulation
Yi Li ab,
Xiao-Ling Zhanga,
Xue Yuana,
Jiang-Chun Houb,
Peng Sangc and
Li-Quan Yang*c
ab,
Xiao-Ling Zhanga,
Xue Yuana,
Jiang-Chun Houb,
Peng Sangc and
Li-Quan Yang*c
aCollege of Mathematics and Computer Science, Dali University, Dali, China
bScience and Education Department, Second People's Hospital of Yunnan Province, Kunming, China
cCollege of Agriculture and Biological Science, Dali University, Dali, China. E-mail: ylqbioinfo@gmail.com
First published on 18th August 2020
Abstract
The HIV envelope glycoprotein gp120 has evolved two distinct conformational states to balance viral infection and immune escape. One is a closed state resistant to most neutralization antibodies, and the other is an open state responsible for the binding of the receptor and coreceptors. Although the structures of gp120 in these two conformational states have been determined, a detailed molecular mechanism involving intrinsic dynamics and conformational transition is still elusive. In this study, μs-scale molecular dynamics simulation is performed to probe molecular dynamics and conformational transition away from the open state and approach the closed state. Our results reveal that open gp120 shows a larger structural deviation, higher conformational flexibility, and more conformational diversity than the form in the closed state, providing a structural explanation for receptor or coreceptor affinity at the open state and the neutralization resistance of closed conformation. Seven regions with greatly decreased coupled motions in the open states have been observed by dynamic cross-correlation analysis, indicating that conformational transition can be mainly attributed to the relaxation of intrinsic dynamics. Three conformations characterized by the structural orientations of the V1/V2 region and the V3 loop, suggesting gp120 is intrinsically dynamic from the open state to the closed state. Taken together, these findings shed light on the understanding of the conformational control mechanism of HIV.
1. Introduction
As an infection machine of the human immunodeficiency virus (HIV), the envelope glycoprotein gp120 is responsible for the binding of the receptor and coreceptors on the surface of the host cell and triggers a series of infection events, including virus attachment to the host cell, virus-cell membrane fusion, and viral genetic material transfer.1 Compared to standard type I membrane fusion proteins, gp120 takes an unusual two-step strategy to enter target cells via sequential binding to two different proteins, the receptor CD4 and the coreceptor CCR5 or CXCR4.2 To balance viral infection and immune escape, gp120 must carefully conceal the conserved receptor and coreceptor binding sites from the attack of neutralizing antibodies by harnessing its significant structural flexibility, resulting in the division of the infection and evasion functions into two distinct conformational states.3 One is a closed state that is resistant to most neutralization antibodies, and the other is an open state that is responsible for interacting with the receptor and coreceptors.4 In the closed state, gp120 adopts a neutralization-resistant conformation, in which parts of the receptor-binding site are dispersed and the coreceptor-binding site is masked by variable loops. Contributed by the unusually rapid sequence variations in these variable loops, more than 90% of the gp120 surface is inaccessible to most immunoglobulins.5 In the open gp120, the previously separated elements of the receptor binding site are ultimately aggregated, and the coreceptor binding site is exposed and matures to form a thermo-active state that facilitates the final fusion between the viral and cellular membrane.6Early biophysical experiments using isothermal titration calorimetry (ITC)7 and hydrogen–deuterium exchange coupled mass spectrometry (HDX-MS)8 observed a significant difference in the molecular dynamics of gp120 in the closed and open state. The binding of the receptor is thought to induce the conformational transition of gp120 between these two states. The ITC experiment detected unexpectedly large changes in enthalpy, entropy, and heat capacity were before and after the addition of the receptor, implying that receptor binding causes considerable conformational flexibility and extensive structural rearrangements of gp120.7 By measuring the rates of deuterium incorporation into the protein backbone in solution, the HDX-MS experiment explored the dynamic changes of gp120 induced by the binding of the receptor. However, these HDX data can indirectly speculate that major structural reorganizations occur in various loops of gp120.8 Both ITC and HDX-MS experiments are limited to the comparison of the thermodynamic and dynamic characteristics before and after adding the receptor, and their results cannot explain whether or not the conformational transition of gp120 is caused by the receptor-induced structural rearrangement from the closed state to the open state, or gp120 has an intrinsically dynamic equilibrium among various conformational states, and the receptor can only capture the open state to make it become a dominant conformation.
Evidence from a single-molecule fluorescence resonance energy transfer (smFRET) experiment9 explained the conformational dynamics and transition of gp120 from another perspective based on the theory of the conformational selection.10 In this experiment, three conformational states of gp120, including closed, open, and at least one intermediate state, were identified and found to coexist in dynamical equilibrium under different conditions. In the absence of any ligands, the closed state had the highest distribution probability, followed by the open state, and the intermediate state had the lowest. After adding the receptor, the probability distribution of the closed and open state significantly decreased, and the probability of the intermediate state greatly increased. When the coreceptor was further added, there was a significant reduction in the intermediate state, and the open state eventually became the dominant conformation. The smFRET experiment revealed that gp120 does indeed experience a dynamic equilibrium among multiple conformational states, and the conformational transition from the closed state to the open state is achieved by the receptor capturing the intermediate state and the coreceptor stabilizing the open state. This finding indicated that gp120 has a high level of intrinsic dynamics to sample different conformations, and the receptor or coreceptor may only bind to favorable conformations. Despite these advances, the atomic-resolution description of the dynamic, thermodynamic, and kinetic behavior of gp120 during the conformational transition between the closed and open states has remained unanswered.
In this study, we comparatively investigated two systems consisting of the closed and open gp120 employing a series of μs-scale multiple-replica molecular dynamics (MD) simulation11 to probe the intrinsic dynamics and conformational transition of HIV gp120. Our results indicate that open gp120 shows a larger structural deviation, higher conformational flexibility, and more conformational diversity than the form in the closed state. Three conformations characterized by structural orientations of the V1/V2 region and V3 loop describe the conformational transition away from the open state and approach the closed state. We claim that gp120 is intrinsically dynamics to sample multiple conformational states. We believe that this detailed structural descriptions of gp120 will shed light on the understanding of the conformational control mechanism of HIV.
2. Materials and methods
2.1. Molecular dynamics simulations
The structural models of gp120 in the closed and open states are extracted from the atomic coordinates of the HIV envelope in the Protein Data Bank (PDB, http://www.rcsb.org) with accession IDs are 5FYK12 and 5FUU13 at 3.11 Å and 4.19 Å resolution, respectively. The full-length amino acid sequence of these two gp120 structures is the HIV JR-FL strain14 which was also used in the smFRET experiment.9 The sequence segment for partial C-termini has been removed to obtain a uniform sequence length, i.e. residues 30–492 according to Q75760 in the UniProt database (https://www.uniprot.org).The selected structural models of the closed and open gp120 were individually used as the starting structure for the MD simulations performed by the software GROMACS V5.1.4.15 Each starting structure is described with the AMBER99SB-ILDN force field16 and the TIP3P water model17 in a dodecahedron box with a protein-wall minimum distance of 1 nm. Na+ and Cl− are introduced to achieve the electroneutral system at a salt concentration of 150 mM. To effectively soak the solute into the solvent, 200 ps position-restrained simulations are carried out, in which the heavy atoms of protein are restrained by decreasing harmonic potential force. The steepest descent algorithm is used to minimize the energy of each system to eliminate steric conflicts. Before the MD production simulation, a pre-simulation process consisted of 100 ps NVT and NPT ensemble is conducted.
In order to improve the efficiency of the conformational sampling, a multi-replica strategy is adopted in the MD production simulation. For each system, 15 independent 100 ns MD runs are performed, where each run is initialized with random atomic velocities assigned from Maxwell distribution at 300 K. The simulation protocol is as follows: the integration time step is set to 2 fs due to the LINCS algorithm18 is used to constrain the bond length involving hydrogen atoms; the partial-mesh Ewald (PME) method19 with a Fourier grid spacing of 0.135 nm and an interpolation order of 4 is employed to handle long-range electrostatic interactions; the van der Waals interaction is treated using a twin-range cut-off scheme with short-range and long-range cut-offs of 1.0 and 1.4 nm, respectively; solute and solvent are separately coupled with a heat bath at 300 K and a pressure bath at 1 atm; the system coordinates are saved every 2 ps.
2.2. Dynamics analysis
The root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) are calculated using trajectory analysis tools implemented in GROMACS. The complex molecular interactions in gp120 are described by the dynamic cross-correlation maps (DCCM)20 (Fig. 3), in which each element (Cij) represents the coupled molecular motion between residue i and j, and can be calculated by:where rn is the position vectors of residue n, which is obtained from the aligned coordinate of the Cα atom.

2.3. Thermodynamics analysis
All trajectories are discretized by two suitable collective values (CVs) characterizing the structural orientations of the V1/V2 region and the V3 loop. One is an angle defined by the center of mass (COM) of the V1/V2 region, the COM of the β20–β21 hairpin, and the COM of the α0 to describe the structural position of the V1/V2 region relative to the core. The other is a distance between the COM of the β20–β21 hairpin and the COM of the V3 loop.Based on these two CVs, a free energy landscape (FEL) is constructed by using probability density function:
F(s) = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Ni/Nmax) ln(Ni/Nmax) |
3. Results
3.1. A significant structural difference intrinsically exists in the closed and open states of gp120
In both the closed (Fig. 1A) and open (Fig. 1B) states, gp120 consists of a relatively conserved structural core, a bridging sheet, and five variable loops V1–5.21 Although they share a similar core, these two states exhibited a significant structural difference in the orientation of the variable loops (see the backbone superimpose in Fig. 1C). In the core of gp120, there are two folded structure domains, a β-sheet sandwich, and a three-layered architecture. The seven-stranded (β![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , β0, β1, β5–7, and β25) β sandwich is a stable subdomain near the N-/C-termini. It is believed that three topological layers, called layer 1 (from β
, β0, β1, β5–7, and β25) β sandwich is a stable subdomain near the N-/C-termini. It is believed that three topological layers, called layer 1 (from β![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) to β0, mainly involving α0), layer 2 (from β1 to β2, mainly involving α1), layer 3 (from β24 to β25, mainly involving α5), play an important role in the molecular interaction among three gp120 subunits in the context of HIV envelope.22
to β0, mainly involving α0), layer 2 (from β1 to β2, mainly involving α1), layer 3 (from β24 to β25, mainly involving α5), play an important role in the molecular interaction among three gp120 subunits in the context of HIV envelope.22
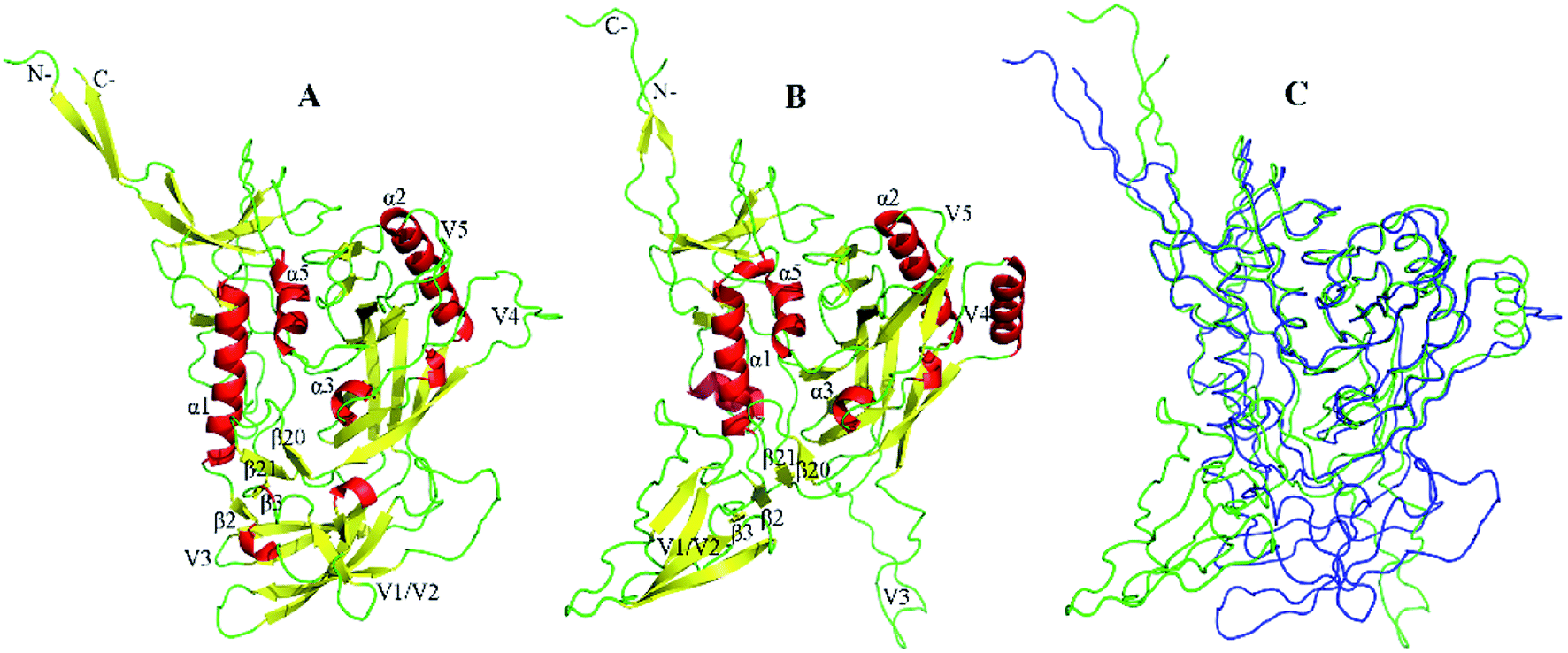 | ||
| Fig. 1 The structural models of gp120. (A and B) Ribbon representation of the closed and open gp120 with PDB IDs of 5FYK and 5FUU, respectively. (C) Backbone superposition of gp120 in the closed (blue) and open (green) states. | ||
The main difference between these two structural models of gp120 is the orientation of the V1/V2 region and the V3 loop. Extending from β2 and β3, a very large variable loops called the V1/V2 region has unique secondary structural characteristics. In the closed state, the V1/V2 region folds into a “Greek key”-like structure composed of four antiparallel β strands (labeled as βA to βD) and three connecting loops: V1 (between βA and βB), L1 (between βB and βC), and V2 (between βC and βD).12 For open gp120, the V1/V2 region undergoes significant conformational rearrangements, resulting in the disintegration of βC–D and the formation of new β stands in the region of V1 and L1 loops. When compared to the closed state, the V1/V2 region and V3 loop move away from gp120 core in the open state, accompanied by disassembly of the V1/V2 region, relaxations of the V3 loop, and rearrangements of the bridging sheet. As the stem of the V1/V2 region, β2–β3 reversal not only directs the V1/V2 region away from the core but also leads to the formation and exposure of the coreceptor binding site.23 It should be noted that the hairpin β20_β21 has been identified as a major regulatory switch for the conformational transition of gp120.24
3.2. The open gp120 experiences larger structural deviation than the conformation of the closed state
To evaluate the structural stability and deviation of gp120 during the simulations, we calculate the time evolution of the gp120 backbone RMSD values (Fig. 2) relative to their respective starting structural models. In the closed state, each replica of gp120 requires about 5 ns to equilibrate the relative stability of the RMSD region between about 0.3 to 0.5 nm. In contrast, more time (about 20 ns) is needed for open gp120 to reach a larger and wider equilibrium region (from about 0.5 to 1.2 nm). There is a more obvious difference in the fluctuation amplitudes of the RMSD curves, such as most of the open replicas exhibit much larger amplitudes than the curves in the closed state. Taken together, the above results reveal that during simulations open gp120 experiences larger structural deviation than the one in the closed state, indicating that the former has higher structural stability. | ||
| Fig. 2 The structural dynamics of gp120. The time evolution of the backbone root-mean-square deviation (RMSD) values of the closed (A) and open (B) gp120 are calculated from 15 replicas (R1–15) of the molecular dynamics simulation. | ||
To ensure that the intrinsic properties of gp120 are reflected, all subsequent analysis was performed based on a 1.2 μs trajectory which is concatenated from the equilibrated portions (20–100 ns) of 15 replicas for each state of gp120.
3.3. Diverse residue subdomains show different conformational flexibility between the closed and open states
As shown in Fig. 3, the RMSF value of Cα atoms with respect to the respective starting structural model in each state was calculated to evaluate and compare the conformational flexibility of gp120. The average RMSF values (dashed line in Fig. 3) of all Cα atoms in the closed and open gp120 are 0.18 and 0.26 nm, respectively, which clearly indicates that the latter has a higher global conformational flexibility than the former. The gp120 structural models in both conformational states show very similar conformational flexibility distributions, where the RMSF values of regular secondary structural regions, especially α0–5, β2–3, and β20–21, are lower than their respective RMSF mean values. Furthermore, N-/C-termini and surface-exposed loops, such as layer 1, V1–5, and L1 loops, exhibit higher values than their corresponding averaged RMSD values. These RMSF curves are consistent with the observations in the HDX-MS experiment,8 indicating that our MD simulations can provide reliable information about the intrinsic dynamics of gp120. | ||
| Fig. 3 The residue dynamics of gp120. The Cα root-mean-square fluctuation (RMSF) value of each residue in the closed (blue line) and open (green line) gp120 during the molecular dynamics simulation. The average RMSF values (dashed line) of all Cα atoms in the closed and open gp120 are 0.18 and 0.26 nm, respectively. The RMSF difference was obtained by subtracting the closed gp120's RMSF value from the open gp120's RMSF value at the same residue position. The differences greater and less than zero are shaded in light red and light blue, respectively. | ||
The regions above the average RMSF value include the N-/C-terminus, the middle area of layer 1, the V1, L1 and V2 loop, the top of the V3 loop, and the V4–V5 loops. Except for the N-/C-terminus, all of the regions above the average are exposed on the surface of the protein. Direct contact with solvents and lack of structural constraints can lead to a high level of conformational flexibility. It should be noted that βA–D divides the V1/V2 region into three loops, i.e. the V1, V2, and L1 loops, with higher conformational flexibility in the closed state. Echoing with the experimental structure, this special RMSD pattern in the V1/V2 region further illustrates that the “Greek key” folded structure inhibits the overall conformational flexibility of the V1/V2 region.25 With the dissociation of the β sheets in the “Greek key”, the RMSF mode in the V1/V2 region has changed in the open state. Despite being covered by the V1/V2 region, the top of the V3 loop still has greater conformational flexibility due to its exposure in the closed state. Compared with the closed gp120, the RMSF values of the V1/V2 region and the V3 loop are significantly increased in the open state. This effect can be attributed to the spatial rearrangement of the V1/V2 region and the changes in the orientation of the V1/V2 region and the V3 loop relative to the structural core of gp120.
To quantitatively compare the conformational flexibility of gp120 between these two states, the RMSF difference at the same residue position was calculated by subtracting the RMSF value of the closed gp120 from the RMSF value of the open gp120. In Fig. 3, the positive and negative differences of the RMSF are shaded in light red and light blue, respectively. In most areas including the V1/V2 region and the V3 loop, the overall conformational flexibility is dramatically improved in the open state. This is because, in the opened gp120, the V1/V2 region and the V3 loop increase their freedom of rotation and translation. It should be noted that the layer 1 and the L1 loop show significantly higher conformational flexibility in the closed state. The layer 1 is partially covered by the displacement of the V1/V2 region, and a part of the α helix is also formed, which greatly reduce its flexibility in the open state.6 The conformational flexibility of the L1 loop is inhibited in the open state because it is located in the middle of the βA–D extended structure.23
3.4. Seven regions with decreased coupled motions in the open states indicate that the conformational transition could be mainly attributed to the relaxation of intrinsic dynamics
The pairwise residue correlated and anticorrelated molecular motions are plotted in the dynamic cross-correlation map (DCCM, Fig. 4), in which the cross-correlation coefficients for closed and open gp120 are shown in the upper and lower triangles, respectively. The coefficient ranging from −1 to 1 indicates the strength of the molecular motions and the positive and negative values indicate the movement of the residue in the same and opposite directions, respectively. It can be seen from DCCM (Fig. 4A) that gp120 exhibits significant collective molecular motions in both states, especially in some subdomains such as the layer 1–3, the V1/V2 region, the V3–4 loops, and their interactive regions. This implies that modular molecular movements with the same trend occurred during the simulation.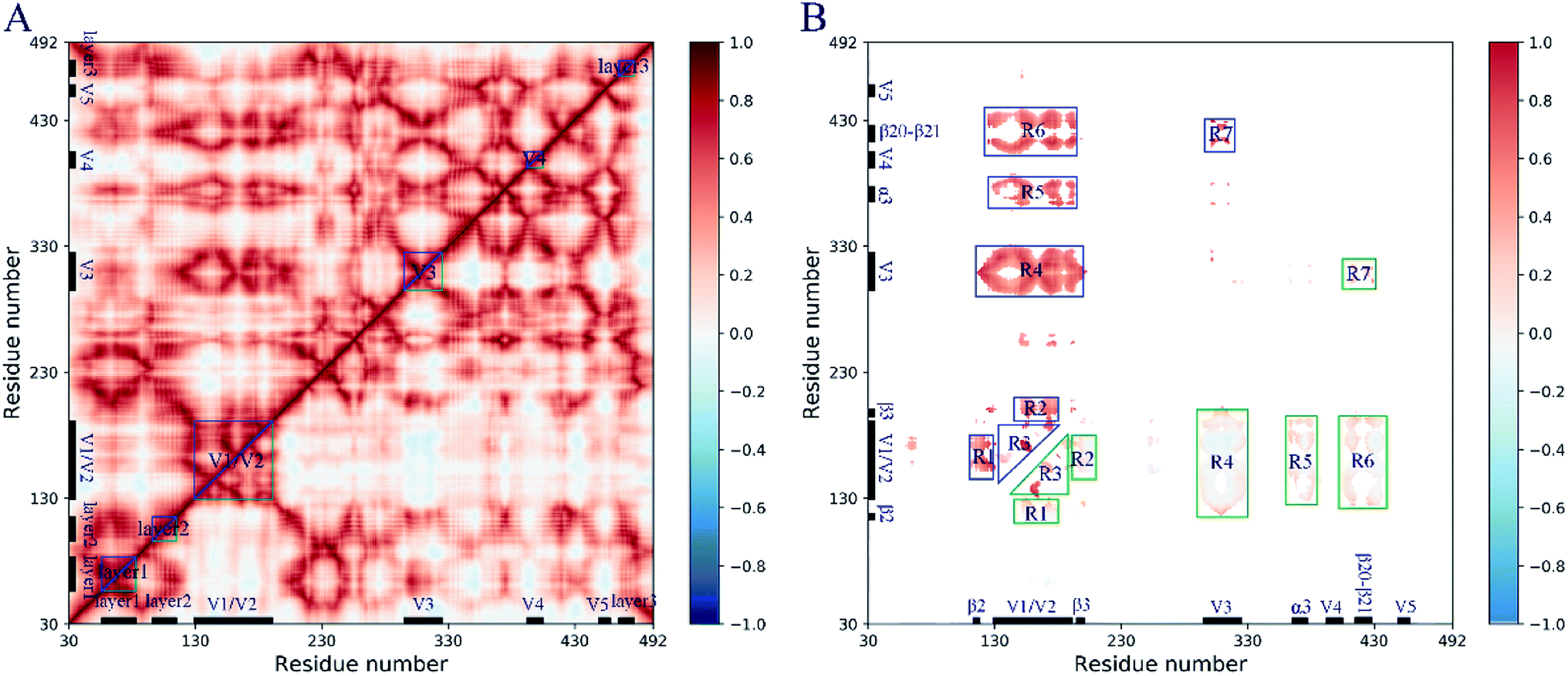 | ||
| Fig. 4 The cross-correlation dynamics of gp120. (A) The dynamic cross-correlation maps (DCCM) of the closed (upper triangle) and open (lower triangle) gp120. The coefficient ranges from −1 to 1. The coefficient value indicates the strength of the motion. The positive and negative values represent the molecular motion in the same and opposite directions, respectively. (B) Seven regions (R1–7) with the coefficient difference between the closed (upper triangle) and open (lower triangle) states is greater than 0.5. | ||
To comparatively investigate the collective molecular motions between these two conformational states, the difference of the cross-correlation coefficient at the same residue position is calculated by subtracting the coefficient of the closed gp120 from the coefficient of the open gp120. Seven regions (R1–7) with differences in coefficient values greater than 0.4 are selected (Fig. 4B). This analysis revealed that the open gp120 has decreased coupled motions in all regions, mainly in the V1/V2 region (R3) or interactive regions with other subdomains, such as the layer 2 (R1), β3 (R2), the V3 loop (R4), α3 (R5), and the β20–21 hairpin (R6). These changes in the collective molecular motions suggest that the relaxation of intrinsic dynamics in the open gp120 could be considered the main contributor of the conformational transitions.
3.5. The conformational transition and thermodynamic distributions of gp120 are characterized by the structural orientation of the V1/V2 region and the V3 loop
Given the significant difference between the closed and open states of gp120 is based primarily on the structural orientation of the V1/V2 region and the V3 loop, all trajectories are discretized by two suitable collective values (CVs). One is an angle defined by the center of mass (COM) of the V1/V2 region, the COM of the β20–β21 hairpin, and the COM of the α0 that describes the structural position of the V1/V2 region relative to the core. The other is a distance between the COM of the β20–β21 hairpin and the COM of the V3 loop. The time evolution of these two CVs for the closed and open gp120 is plotted in Fig. 5 and the different replicas are represented with blue and green lines, respectively. It demonstrates that the closed gp120 is highly stable and the values of angle and distance concentrate at approximately 3 rad and 1.8 nm, respectively. On the contrary, open gp120 shows high dynamics. Its V1/V2 region explores two different areas from the starting position (the angle is about 1.8 rad) relative to the core. One is the area approaching the closed state (to 2.8 rad), and the other is away from the open state with an edge angle of 1.2 rad. This shows that the V1/V2 region of open gp120 could not only transfer into the closed state but also enhance the degree of openness. In the closed state, due to the suppression of the V1/V2 region, the distance from the β20–β21 hairpin to the V3 loop is concentrated around 1.8 nm. This distance in the open gp120 exhibits a decreasing tendency toward the closed state. | ||
| Fig. 5 The conformational orientation of gp120. The time evolution of two suitable collective values (CVs) characterizing the structural orientations of the V1/V2 region and the V3 loop in the closed (blues lines) and open (greens lines) gp120. One is an angle defined by the center of mass (COM) of the V1/V2 region, the COM of the β20–β21 hairpin, and the COM of the α0. The other is a distance between the COM of the β20–β21 hairpin and the COM of the V3 loop. Schematic diagrams of these two CVs are plotted in the structural models of the closed (blue) and open (green) gp120. | ||
As the above two CVs describe the conformational transition of gp120, these trajectory features are used to describe the energy distribution and conformational diversity of gp120 by constructing an FEL (Fig. 6). Overall, the FEL of gp120 at the closed and open states exhibits different conformational distribution. The closed population confines in a narrow area (Fig. 6A). The open gp120 exhibits an irregular and divergent shape (Fig. 6B), presenting a more complicated shape than that in the closed state. Besides, the FEL of open gp120 covers a larger area than that of closed gp120. At the open state, there are at least three free energy minima (≤16 kJ mol−1) which describe the conformational transition away from the open state (locates in the angle of 1.7 rad and the distance of 2.8 nm) and approach the closed state (locates in the angle of 2.5 rad and the distance of 1.5 nm).
 | ||
| Fig. 6 The thermodynamics distribution of gp120 at the closed (A) and open (B) states. A free energy landscape is constructed based on two collective values characterizing the structural orientations of the V1/V2 region and the V3 loop in the open gp120. One is an angle defined by the center of mass (COM) of the V1/V2 region, the COM of the β20–β21 hairpin, and the COM of the α0. The other is a distance between the COM of the β20–β21 hairpin and the COM of the V3 loop. | ||
4. Discussion
Comparative analysis of structural dynamics and conformational flexibility reveal that the closed gp120 exhibits more stable conformational dynamics, while the open form experiences higher structural dynamics, providing a structural explanation for receptor or coreceptor affinity of the open state and the neutralization resistance of the closed conformation. Experimental evidence has proven that gp120 is intrinsically dynamics in the closed and open states.4 In the closed state, gp120 masks its functional centers from attack from by neutralization antibodies, while it can adopt an open state for binding to the receptor and coreceptors. In our study, the RMSF curve is consistent with the observations of the HDX-MS experiment8 and further provides a full-length and quantitative description of the intrinsic dynamics of gp120 in both states. Seven regions with greatly decreased coupled motions in the open states have been observed by dynamic cross-correlation analysis, indicating that conformational transition can be mainly attributed to the relaxation of intrinsic dynamics.Based on the two collective variables that characterize the orientation of the V1/V2 region and the V3 loop, we extract the population distributions of three to five macrostates and the conformational transition from the closed state to the open state. These data, together with the smFRET experiment,9 indicate that capturing the open state is one of the major molecular mechanisms for the conformational transition of gp120. This evidence from the MD simulation and the Markov modeling suggests that gp120 is intrinsically dynamic from the open state to the closed state, and eventually results in a conformational transition between these two states. Compared with our previous research on the CD4-binding effects on the conformational dynamics, molecular motions, and thermodynamics of gp120,26 the comparative MD simulation of these two terminal states of gp120 demonstrates that the molecular dynamics of gp120 are rooted in its structural architecture. The binding of CD4 hinders the conformation transitions of gp120 from the open to the closed state, resulting in gp120 has been restricted in the open state.27
Although gp120 is covered by a dense glycan layer,5 the effect of carbohydrates on the conformational flexibility and transitions of gp120 is limited. Previous MD simulation studies on gp120 with a glycosylated or non-glycosylated V3 loop showed that there were no significant differences in the molecular fluctuations between these two forms.28 In our study, two MD simulation systems of gp120 in the closed and open states are performed to comparatively investigate the intrinsic dynamics and conformational transitions. Therefore, comparative MD simulations without glycans could still reflect the true differences in the dynamics and thermodynamics of gp120 in these two states.
For gp120, another factor that can potentially affect its molecular dynamics is its highly variable residues. Although the role of mutations in the conformational changes of gp120 cannot be ignored, the selection of standard sequences for comparative studies can still reveal the intrinsic dynamics and conformational transition of gp120. Our previous simulation studies on gp120 from two extreme neutralization phenotype HIV strains also revealed the effect of mutations on structural dynamics.29 Our team is preparing a study on more in-depth molecular dynamics of gp120 from different HIV-1 isolates. Like the case study about NPAC,30 a cytokine-like nuclear factor involved in chromatin modification and regulation of gene expression, we expect to identify key mutations that affect the conformational flexibility of gp120 in future studies.
5. Conclusion
In this paper, we perform cumulative 3 μs multiple-replica MD simulations for two full-length structural models of gp120 in the closed and open states to probe the intrinsic dynamics and conformational transitions of gp120. Comparative investigations of the structural dynamics, conformational flexibility, and molecular cross-correlations of gp120 between these two states reveal that gp120 exhibits distinct intrinsic dynamics between the closed and open states. Three conformations characterized by structural orientations of the V1/V2 region and V3 loop describes an obvious conformational transition from the closed state to the open state, which can be considered as direct evidence for conformational selection. In summary, our results have provided the detailed intrinsic dynamics and conformational transitions of gp120, which will facilitate our current understanding of the HIV conformational control mechanism.Author contributions
Y. L. designed the study, performed the simulations, analyzed the data, and drafted the manuscript. X. L. Z., X. Y. and J. C. H. participated in analyzing the data. P. S. and L. Q. Y. revised the manuscript. All authors contributed to and approved the final version of the manuscript.Funding
This study is funded by the Startup Foundation for Advanced Talents of Dali University (No. KYBS2018031), the Yunnan Fundamental Research Projects (No. 2019FD014, 2019FH001-103, 2019FB021, and 2017FH001-032), and the National Natural Sciences Foundation of China (31960198).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the High-Performance Computer Center of Dali University for providing the computing resources.References
- B. Chen, Molecular Mechanism of HIV-1 Entry, Trends Microbiol., 2019, 27, 878–891, DOI:10.1016/j.tim.2019.06.002.
- R. Wyatt and J. Sodroski, The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens, Science, 1998, 280, 1884–1888, DOI:10.1126/science.280.5371.1884.
- X. Wei, J. M. Decker, S. Wang, H. Hui, J. C. Kappes and X. Wu, et al., Antibody neutralization and escape by HIV-1, Nature, 2003, 422, 307–312, DOI:10.1038/nature01470.
- G. Ozorowski, J. Pallesen, N. De Val, D. Lyumkis, C. A. Cottrell and J. L. Torres, et al., Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike, Nature, 2017, 547, 360–361, DOI:10.1038/nature23010.
- M. Pancera, T. Zhou, A. Druz, I. S. Georgiev, C. Soto and J. Gorman, et al., Structure and immune recognition of trimeric pre-fusion HIV-1 Env, Nature, 2014, 514, 455–461, DOI:10.1038/nature13808.
- M. Rasheed, R. Bettadapura and C. Bajaj, Computational Refinement and Validation Protocol for Proteins with Large Variable Regions Applied to Model HIV Env Spike in CD4 and 17b Bound State, Structure, 2015, 23, 1138–1149, DOI:10.1016/j.str.2015.03.026.
- D. G. Myszka, R. W. Sweet, P. Hensley, M. Brigham-Burke, P. D. Kwong and W. A. Hendrickson, et al., Energetics of the HIV gp120-CD4 binding reaction, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9026–9031, DOI:10.1073/pnas.97.16.9026.
- M. Guttman, N. K. Garcia, A. Cupo, T. Matsui, J. P. Julien and R. W. Sanders, et al., CD4-induced activation in a soluble HIV-1 Env trimer, Structure, 2014, 22, 974–984, DOI:10.1016/j.str.2014.05.001.
- J. B. Munro, J. Gorman, X. Ma, Z. Zhou, J. Arthos and D. R. Burton, et al., Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions, Science, 2014, 346, 759–763, DOI:10.1126/science.1254426.
- X. Du, Y. Li, Y.-L. Xia, S.-M. Ai, J. Liang and P. Sang, et al., Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods, Int. J. Mol. Sci., 2016, 17, 144, DOI:10.3390/ijms17020144.
- M. Karplus and J. Kuriyan, Molecular dynamics and protein function, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6679–6685, DOI:10.1073/pnas.0408930102.
- G. B. E. Stewart-Jones, C. Soto, T. Lemmin, G. Y. Chuang, A. Druz and R. Kong, et al., Trimeric HIV-1-Env Structures Define Glycan Shields from Clades A, B, and G, Cell, 2016, 165, 813–826, DOI:10.1016/j.cell.2016.04.010.
- J. H. Lee, G. Ozorowski and A. B. Ward, Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer, Science, 2016, 351, 1043–1048, DOI:10.1126/science.aad2450.
- Y. Koyanagi, S. Miles, R. T. Mitsuyasu, J. E. Merrill, H. V. Vinters and I. S. Y. Chen, Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms, Science, 1987, 236, 819–822, DOI:10.1126/science.3646751.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith and B. Hess, et al., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25, DOI:10.1016/j.softx.2015.06.001.
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics, Proteins: Struct., Funct., Bioinf., 2014, 82, 195–215, DOI:10.1002/prot.24350.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935, DOI:10.1063/1.445869.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A Linear Constraint Solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472, DOI:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092, DOI:10.1063/1.464397.
- A. Ghosh and S. Vishveshwara, A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15711–15716, DOI:10.1073/pnas.0704459104.
- J. Liu, A. Bartesaghi, M. J. Borgnia, G. Sapiro and S. Subramaniam, Molecular architecture of native HIV-1 gp120 trimers, Nature, 2008, 455, 109–113, DOI:10.1038/nature07159.
- M. Pancera, S. Majeed, Y.-E. A. Ban, L. Chen, C.-c. Huang and L. Kong, et al., Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1166–1171, DOI:10.1073/pnas.0911004107.
- Y. Li, L. Deng, J. Liang, G.-H. Dong, Y.-L. Xia and Y.-X. Fu, et al., Molecular dynamics simulations reveal distinct differences in conformational dynamics and thermodynamics between the unliganded and CD4-bound states of HIV-1 gp120, Phys. Chem. Chem. Phys., 2020, 22, 5548–5560, 10.1039/C9CP06706J.
- A. Herschhorn, C. Gu, F. Moraca, X. Ma, M. Farrell and A. B. Smith, et al., The β20–β21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions, Nat. Commun., 2017, 8, 1049, DOI:10.1038/s41467-017-01119-w.
- J. S. McLellan, M. Pancera, C. Carrico, J. Gorman, J. P. Julien and R. Khayat, et al., Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9, Nature, 2011, 480, 336–343, DOI:10.1038/nature10696.
- Y. Li, L. Deng, L.-Q. Yang, P. Sang and S.-Q. Liu, Effects of CD4 Binding on Conformational Dynamics, Molecular Motions, and Thermodynamics of HIV-1 gp120, Int. J. Mol. Sci., 2019, 20, 260, DOI:10.3390/ijms20020260.
- Y. Li, Y.-C. Guo, X.-L. Zhang, L. Deng, P. Sang and L.-Q. Yang, et al., CD4-binding obstacles in conformational transitions and allosteric communications of HIV gp120, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183217, DOI:10.1016/j.bbamem.2020.183217.
- M. Yokoyama, S. Naganawa, K. Yoshimura, S. Matsushita and H. Sato, Structural dynamics of HIV-1 envelope GP120 outer domain with V3 loop, PLoS One, 2012, 7(5), e37530, DOI:10.1371/journal.pone.0037530.
- Y. Li, L. Deng, S.-M. Ai, P. Sang, J. Yang and Y.-L. Xia, et al., Insights into the molecular mechanism underlying CD4-dependency and neutralization sensitivity of HIV-1: a comparative molecular dynamics study on gp120s from isolates with different phenotypes, RSC Adv., 2018, 8, 14355–14368, 10.1039/C8RA00425K.
- M. Montefiori, S. Pilotto, C. Marabelli, E. Moroni, M. Ferraro and S. A. Serapian, et al., Impact of Mutations on NPAC Structural Dynamics: Mechanistic Insights from MD Simulations, J. Chem. Inf. Model., 2019, 59, 3927–3937, DOI:10.1021/acs.jcim.9b00588.
| This journal is © The Royal Society of Chemistry 2020 |