
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an antimicrobial and antioxidant hydrogel/nano-electrospun wound dressing†
Alejandra Romero-Monteroa,
Pablo Labra-Vázquez b,
Luis J. del Vallec,
Jordi Puiggalícd,
Roeb García-Arrazolaa,
Carmina Montiela and
Miquel Gimeno*a
b,
Luis J. del Vallec,
Jordi Puiggalícd,
Roeb García-Arrazolaa,
Carmina Montiela and
Miquel Gimeno*a
aDepartamento de Alimentos y Biotecnología, Facultad de Química, Universidad Nacional Autónoma de México, 04510 CDMX, Mexico. E-mail: mgimeno@unam.mx
bDepartamento de Química Orgánica, Facultad de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Ciudad de México, 04510, Mexico
cChemical Engineering Department, Escola d'Enginyeria de Barcelona Est-EEBE, Universitat Politècnica de Catalunya, c/Eduard Maristany 10-14, 08019 Barcelona, Spain
dInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, 08028 Barcelona, Spain
First published on 18th August 2020
Abstract
A nanocomposite based on an antibiotic-loaded hydrogel into a nano-electrospun fibre with antimicrobial and antioxidant capacities is investigated. The material is composed of nanofibres of enzymatic PCL grafted with poly(gallic acid) (PGAL), a recently developed enzyme-mediated hydrophilic polymer that features a multiradical and polyanionic nature in a helicoidal secondary structure. An extensive experimental–theoretical study on the molecular structure and morphological characterizations for this nanocomposite are discussed. The hydrogel network is formed by sodium carboxymethylcellulose (CMC) loaded with the broad-spectrum antibiotic clindamycin. This nano electrospun biomaterial inhibits a strain of Staphylococcus aureus, which is the main cause of nosocomial infections. The SPTT assay demonstrates that PGAL side chains also improve the release rates for this bactericide owing to the crosslinking to the CMC hydrogel matrix. The absence of hemolytic activity and the viability of epithelial cells demonstrates that this nanocomposite has no cytotoxicity.
Introduction
Dressings have recently drawn attention in the field of wound healing because they provide a physiological environment compatible with cell development. Other wanted characteristics are the absence of cytotoxicity, and the ability to maintain a moist environment and oxygen permeability. A bioactive dressing also prevents bacterial infections, which are among the most common and severe complications in wounds.1,2 In a typical medical procedure, the initial wound cleaning is followed by antibiotic treatment;3,4 unfortunately, these actions might be inefficient for some patients, and the rapid development of the infections leads to complications in the healing process, associated with pain, erythema, edema, and long-term disabilities.5,6 Furthermore, the administration of antioxidants is highly beneficial as the excessive generation of reactive oxygen species (ROS) at the healing site delays the recovery due to severe damage to the surrounding tissues caused by inflammation and other ailing due to oxidative stress.7–10Currently, polymer films,11–13 hydrogels,14–16 and nanofibrous17–19 have been used effectively as wound dressings. Among all options, polymeric electrospun and hydrogels are appealing owing to their ability to mimic the biological conditions of many tissues.20 Hydrogels are widely used in regenerative medicine as drug and cell carriers, as well as in tissue engineering matrices because they possess similar physicochemical and biological properties of the tissue microenvironment due to their porous and hydrated molecular structure.21,22 The characteristics of the hydrogels, however, may improve when loaded into electrospun polymers to provide mechanical support in a porous 3D structure for cell attachment. This approach offers a high surface-area-to-volume ratio that allows high gas permeation and good conformability.23,24 The electrospinning is in this regard a versatile process for the production of nonwoven nanofibres, which are advantageous in the biomedical field due to their physical properties such as high flexibility, smooth, lack of sharp edges, nonabrasive, high drug-loading capacity, and are easily fabricated into different geometries toward beneficial designs.25 Therefore, electrospinning has been studied in many adequate polymers, including polystyrene (PS), polysaccharide derivatives, and their blends, polylactides (PLA), polylactide-co-glycolides (PLGA), and poly-ε-caprolactone (PCL), among others. The PCL is commonly employed to obtain a continuous morphology in a uniform and controlled distribution of biocompatible micro or nanofibres.25,26 We recently investigated films of an active composite of enzymatically produced PCL grafted with poly(gallic acid) (PGAL).27 The latter is an enzyme-mediated polymer that features a multiradical and polyanionic nature in a helicoidal secondary structure.28 The radical scavenging capacity in PGAL, along with its high solubility in physiological media, null cytotoxicity, electric semi-conductivity and high thermal and photostabilities are promising characteristics for application in wound dressing.28–31 We produce scaffolds of nanofibres of the enzymatic PCL grafted with PGAL, as a nanocomposite dressing with antioxidant capacity composed of an antimicrobial hydrogel loaded into a nano-electrospun fibre. The hydrogel network is formed by sodium carboxymethylcellulose (CMC) loaded with clindamycin, which is a broad-spectrum antibiotic for the treatment of infections of the bones, skin, soft tissues and wounds and can be used to treat penicillin-allergic patients.32 This biomaterial inhibits a strain of Staphylococcus aureus, which is the main cause of nosocomial infections, affecting both mucous membranes and skin, which allows the bacteria to penetrate the patient's bloodstream through wounds. The hydrogels are three-dimensional cross-linked highly hydrated polymeric networks, and they can absorb wound exudates or maintain moisture, and able to swell several times their dry volume without being soluble under physiological conditions.33 This work considers using a CMC hydrogel as a composite complement owing to its high oxygen permeability and its ability to maintain the structural integrity of the drugs. The presence of the hydrogel is essential to achieve a controlled drug release, in addition to the absorbent capacity-achieving a biomimetic environment. In this regard, the CMC is widely compatible with the mucous membranes and with the skin and it maintains an optimal moist environment in the wound region for extracellular matrix formation and re-epithelialization.34 In this work, an extensive experimental–theoretical study on the molecular structure, morphological characterization, and biological activity for the novel nanocomposite are discussed.
Materials and methods
Materials
Laccase from Trametes versicolor (LTV, specific enzyme activity 2.35 U g−1, lyophilized protein stored at −20 °C) was purchased from Fluka (USA). Immobilized lipase B from Candida antarctica (CALB; Lewatit VPOC1600, Lanxess, USA; supported on macroporous poly(methacrylic divinylbenzene) spherical beads (1 mm), specific enzyme activity 5000 U g−1, stored at 5 °C) was purchased from C-Lecta (Germany). Gallic acid (GA), ε-caprolactone (CL), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), clindamycin hydrochloride, commercial PCL (PCLc) (Mn = 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), and CMC (Mn ≈ 700000 g mol−1) were purchased from Sigma-Aldrich (Mexico). Ethyl alcohol (technical grade, >99%) was supplied by Química Barsa (Mexico). Phosphate buffered saline (PBS) was prepared by dissolving reagent grade chemicals, NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·H2O, CaCl2, and Na2SO4, in deionized water, buffered at pH = 7.25. 1,1,1,2-Tetrafluoroethane (R-134a/norflurane Dupont, SUVA, USA) cylinder (60 kg) was supplied by Refracciones Star (Mexico) with 98% purity. Enzyme-mediated PGAL was produced as reported elsewhere30 in 96% yield (Mn = 5800 Da, PDI = 1.1). Enzymatic PCL (PCLe) was produced following a previous report,35 with Mn = 38519 Da (PDI = 2.1), Tm = 55.6 °C, DGTA peak = 475 °C and percentage of crystallinity = 54.1%.
000 g mol−1), and CMC (Mn ≈ 700000 g mol−1) were purchased from Sigma-Aldrich (Mexico). Ethyl alcohol (technical grade, >99%) was supplied by Química Barsa (Mexico). Phosphate buffered saline (PBS) was prepared by dissolving reagent grade chemicals, NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·H2O, CaCl2, and Na2SO4, in deionized water, buffered at pH = 7.25. 1,1,1,2-Tetrafluoroethane (R-134a/norflurane Dupont, SUVA, USA) cylinder (60 kg) was supplied by Refracciones Star (Mexico) with 98% purity. Enzyme-mediated PGAL was produced as reported elsewhere30 in 96% yield (Mn = 5800 Da, PDI = 1.1). Enzymatic PCL (PCLe) was produced following a previous report,35 with Mn = 38519 Da (PDI = 2.1), Tm = 55.6 °C, DGTA peak = 475 °C and percentage of crystallinity = 54.1%.
Synthesis of the grafted copolymer PCLe-g-PGAL
PGAL was grafted to the surface of PCLe strands throughout acidic esterification by reacting PGAL with the hydroxyl groups available on the hydrolysed PCL surface.36 To this end 0.6 g of PCLe were introduced in an aqueous solution of PGAL (0.28 mM) at pH = 3 (adjusted with citric acid), followed by exposure to UV-irradiation (I = 1.35 mW cm−2) during 24 h. After the grafting, the strands were washed with water and ethanol to remove any residual PGAL and other non-reacted compounds. The PCLe-g-PGAL was obtained in 46% yield.Determination of the grafting yield followed the eqn (1).
| GY = [(Wa − Wb)/Wb] × 100% | (1) |
Preparation of PCLe-g-PGAL nanofibers by electrospinning
Electrospun fibres were collected on a target placed at different distances (10–25 cm) from the needle tip (inside diameter 0.84 mm). Applied voltage varied between 10 and 30 kV using a high-voltage supply (Gamma High Voltage Research, ES 30-5W). Polymer solutions were delivered by a single KDS100 infusion syringe pump (KD Scientific, USA) to control the flow rate (from 1 to 5 mL h−1). All electrospinning experiments were carried out at room temperature. PCL-g-PGAL electrospun fibres were prepared using optimized parameters (collector distance of 20 cm, 25 V, flow rate = 2.5 mL h−1) and solvent conditions (proportion of 60 w/v% in chloroform/acetone mixture 2:1 v/v).
Preparation of hydrogel loaded nanocomposites
Aqueous solutions of citric acid at 0.41, 1.05, and 1.62 mg mL−1 were prepared; then clindamycin hydrochloride (6 mg) powder was added to 10 mL each solution and sonicated for 10 min. Then, 200 mg of CMC was added with continuous mechanical stirring to reach a homogeneous viscous mixture hydrogel. To ensure the homogeneity and viscosity of the solution, the mixture was kept at 37 °C under stirring (200 rpm) for 18 h. Then, 1 cm2 electrospun samples were cut and treated with water during 48 h before adding the hydrogel on their surface, then was retired from water and sterilized with UV light for 15 min to which 0.5 mL of the hydrogel precursor was dropped. Subsequently, the crosslinking was carried out at 50 °C for 6 h and then dried at room temperature under high vacuum until complete evaporation of the solvent. Finally, the composites were cured under UV-irradiation for 30 min.Chemical characterizations
CP/MAS 13C-NMR spectra were recorded in a Varian MR-400 spectrometer. FT-IR spectra were acquired in a PerkinElmer (USA) ATR-FTIR Spectrum 400. Size exclusion chromatography (SEC) measurements were conducted in an Agilent 1210 series (USA) liquid chromatograph equipped with a RID detector. Narrow molecular weight polyethylene glycol in deionized water (Millipore, Milli-Q) and polystyrene in THF (HPLC grade Merck) standards were used for calibration of PGAL and PCLe, respectively. The chromatographer was equipped with an Ultrahydrogel-500 (7.8 × 300 mm, Waters, USA) column for PGAL and two columns PLGEL 10 cm MIXED-B (300 × 7.5 mm, Varian, USA) in series for PCLe. Samples were dissolved in the mobile phase (1 mg mL−1) and filtered (0.45 μm) before injection in the chromatographer. Electron Spin Resonance (ESR) analyses were conducted in a Bruker ESR Elexsys E500 in quartz tubes at room temperature in the 80–4080 G interval and 9.8 × 109 Hz microwave frequency.Computational methods
Spin-unrestricted Hartree–Fock (UHF) quantum chemical computations were performed within the framework of the Density Functional Theory (DFT) through the Gaussian-09 software package,37 using the M06-2X functional combined with the triple-ζ 6-311++G(d,p) polarized basis set,28 to take into account non-covalent interactions, for which meta-GGA functionals usually outperform many other hybrid functionals.38,39 Water solvation effects were incorporated through the Polarizable Continuum Model (PCM).40 We used a large basis set comprising diffuse functions, as their inclusion usually improves the reliability of the quantum chemical description for open-shell and anionic systems.41,42 Equilibrium geometries were optimized without any constraint, and their vibrational frequencies were inspected to confirm true local minimum, which was found by the absence of imaginary frequencies. Transition states were searched and fully optimized at the same level of theory using the eigenvector following (EF) method through the Berny optimization algorithm; the resulting optimized geometries were confirmed as transition states by the presence of a single imaginary vibrational frequency at ν = −425.1 cm−1 and ν = −1312.2 cm−1 for S1-TS and S2-TS, respectively.DPPH radical scavenging of PCL-g-PGAL
Inhibition of the DPPH radical by PCL-g-PGAL samples was conducted in a Genesys 10S UV-vis spectrophotometer (Thermo Scientific) at 517 nm. Stock solutions of PCL-g-PGAL (25–500 ppm) were diluted in chloroform and then 100 μL aliquots of each dilution were added into the assay tubes containing 2.9 mL of DPPH solution (6.34 × 10−5 M). Samples were incubated for 30 min at room temperature before UV analyses. The relative percentage of DPPH radical scavenging activity (RSA) was measured according to eqn (2):
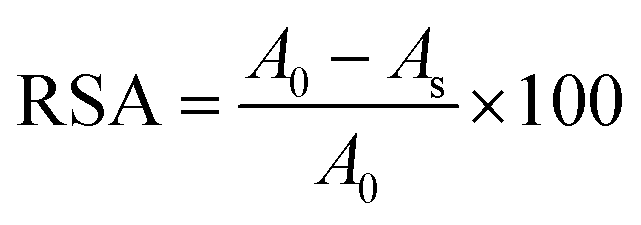 | (2) |
Morphology analysis of samples
Morphology of electrospun samples was determined in a Focused Ion Beam Zeiss Neon 40 microscope (Carl Zeiss MicroImaging GmbH, Germany) operating at 5 kV. Samples of electrospun were deposited on pin stubs of aluminium, and sputter-coated with a thin layer of carbon. The diameter of electrospun threads was measured with SmartTiff software from Carl Zeiss SMT Ltd. Energy dispersive X-ray (EDX) SEM microanalysis was recorded in a JEOL JSM-5900-LV scanning electron microscope (Japan).Contact angle measurements
Static contact angle measurements with the sessile drop method were recorded and analysed at room temperature on OCA-15EC equipment from DataPhysics Instruments GmbH (Germany) with SCA20 software (DataPhysics Instruments GmbH, Germany). Contact angle values of the right and left sides of the solvent drop were measurement and averaged. Measurements were performed 5 s after the deposition of the drop (1 μL) on the surface. All data were an average of ten measurements on different surface locations.Antimicrobial studies
Serial Plate Transfer Test (SPTT) was used to study the electrospun nanocomposites behaviour against Staphylococcus aureus (ATCC, USA), thus monitoring antimicrobial activity and the duration of antibiotic release. Plates of Müller–Hinton agar were inoculated using a sterile swab with a suspension of bacteria with turbidity equivalent to 0.5 McFarland standard (1 × 108 colony-forming units per mL). Composites with and without antibiotics were placed in the centre of an agar plate (n = 3) and incubated at 37 °C for 18 h. Inhibition, determined as the zone diameter minus composite diameter in mm, was measured using a Vernier. Composites were transferred daily to a fresh seeded plate to ensure that the same material surface was in contact with the agar until a zone of inhibition was absent. Composites that ceased to produce a zone of inhibition were split lengthways and placed onto seeded agar plates with the surface in contact with the agar and incubated overnight at 37 °C to ensure that no further antibiotics were being released. Composites without loaded clindamycin did not show any antibacterial effect.Drug delivery kinetic
To evaluate the drug delivery kinetics Cly-loaded and unloaded (controls) hydrogel–electrospun fiber mat composites (1 cm2) and CMC hydrogels (equivalent to 1 mL in wet) were placed in Falcon tubes. Phosphate buffered saline (PBS) solution of pH 7.4 was considered as the release medium. Assays, which were performed in triplicate, were carried out by immersing sample materials in 20 mL of the release medium using a rotating agitator. The medium (0.5 mL) was removed at predetermined time intervals and replaced by an equal volume of fresh medium. The removed medium was used to quantify the release of Cly by measuring its absorbance in a Genesys 10S UV-vis spectrophotometer (Thermo Scientific) at λ = 210 nm. The calibration curve for Cly (y = 3.997x − 0.0314, R2 = 0.9996) was obtained by plotting the absorbance measured at 210 nm against Cly concentration. All assays were performed in triplicate and the results averaged. The original amount of the entrapped drug in each composite and hydrogel was obtained by soaking the sample in water (20 mL) at 37 °C for 5 days and measured as mentioned above. The power-law model (eqn (3)) developed by Ritger and Peppas43 was applied to analyze the first 60% of drug fractional release
 | (3) |
Hemolytic activity
Human blood agar plates were prepared by adding 10 v/v% of healthy human blood from consent donors in sterile blood agar base medium on sterile Petri plates. After solidification, wells were made on the agar plate using a sterile cork borer (6 mm diameter) for water positive control (100 μL). Composites with and without antibiotics were placed in the centre of the agar plate (n = 3) and incubated at 37 °C for 24 h. After the incubation period, the hemolysis was observed by a clear zone of inhibition (zone diameter minus composite diameter in mm) which was measured using a Vernier.Cell viability
Vero cell line (African green monkey, Cercopithecus aethiops, kidney) (ATCC, USA) was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mM of L-glutamine at 37 °C in a humidified atmosphere with 5% CO2 and 95% air. Culture medium was changed every two days and, for subculture, cell monolayers were rinsed with phosphate-buffered saline (PBS) and detached by incubation with trypsin EDTA (0.25%) at 37 °C for 2–5 min. Cell concentration was determined by counting the number of cells with a Neubauer chamber employing 4% trypan blue as a dye vital. Detached cells with viability ≥95% were used for cell culture assays. For cytotoxicity assays, samples were washed with PBS and ethanol (70%) and UV-sterilized for 15 min. Samples were adhered to the wells (24-well culture plate) using a small drop of silicone (Silbione® MED ADH 4300 TRV, Bluestar Silicones France SAS, Lyon, France). Cell viability was determined at the endpoint for cell growth (96 h). The final count of cells was obtained by the MTT assay. Specifically, 50 μL of MTT solution (5 mg mL−1 in PBS) was added to each well. After 3 h of incubation, samples were washed twice with PBS and stored in clean wells. To dissolve formazan crystals, 1 mL of DMSO/methanol/water (70/20/10% v/v) was added. Finally, the absorbance at 570 mm was measured using a UV-vis spectrophotometer. The resulting viability results were normalized to control as relative percentages. Each sample was evaluated in four replicates. Cell proliferation (or growth) was determined by its relative viability; this value was obtained by normalizing the number of cells in the samples to the number of cells in the control (i.e., polystyrene plate with the treated surface for tissue culture, named TCPS).Results and discussion
The successful grafting of PGAL onto PCLe by Fischer esterification reaction in acidic conditions using UV-photo-induction is corroborated by CP/MAS 13C-NMR (Fig. 1) and FT-IR spectroscopies (ESI†). | ||
| Fig. 1 Representative CP/MAS 13C-NMR spectrum of the PCLe-g-PGAL, taken at 400 MHz with a spinning frequency of 10 kHz. | ||
Comparing this spectrum with that for PGAL reported elsewhere,29 where the carboxyl carbon signal appeared at δ = 177 ppm, this signal is now shifted to a higher field (165.74 ppm) due to grafting. The absence of a signal at δ = 177 ppm for PCLe-g-PGAL confirms all the PGAL molecules are grafted onto PCL. The δ = 120–140 ppm region displays resonances assigned to the aromatic carbons of the phenyl ring; 4 different signals are expected due to the C2 symmetry axis across the phenyl ring. The complexity and paramagnetic nature of the molecule do not allow further resolution, however, three different resonances at δ = 126.41, 129.53, and 133.79 ppm are observed, as well as an additional signal with a lower intensity at δ = 132.15 ppm, assigned to the quaternary carbon directly attached to the carboxyl group, for which long relaxation times are expected. The theoretical investigation on the connectivity between PGAL and the PCLe surface also confirms the target molecule. These polymers feature COOH and OH functional groups, therefore, two different reaction pathways are considered, as shown in Fig. 2, employing GA and 6-hydroxyhexanoic acid units as model compounds. A nucleophilic attack of phenol from PGAL to a carboxyl pending group on the surface activated PCLe yields S1 through the S1-TS transition state; alternatively, the hydroxyl group from PCLe could attack the carboxyl group of PGAL to yield the S2 connectivity through the S2-TS transition state. Fig. 3 shows the results from scanning the energy reaction paths towards S1 and S2, depicting ΔGR values of 9.28 and −4.06 kcal mol−1 for S1 and S2, respectively.
 | ||
| Fig. 2 Potential reaction pathways for the PCLe-g-PGAL employing GA and 6-hydroxyhexanoic acid units as model compounds. | ||
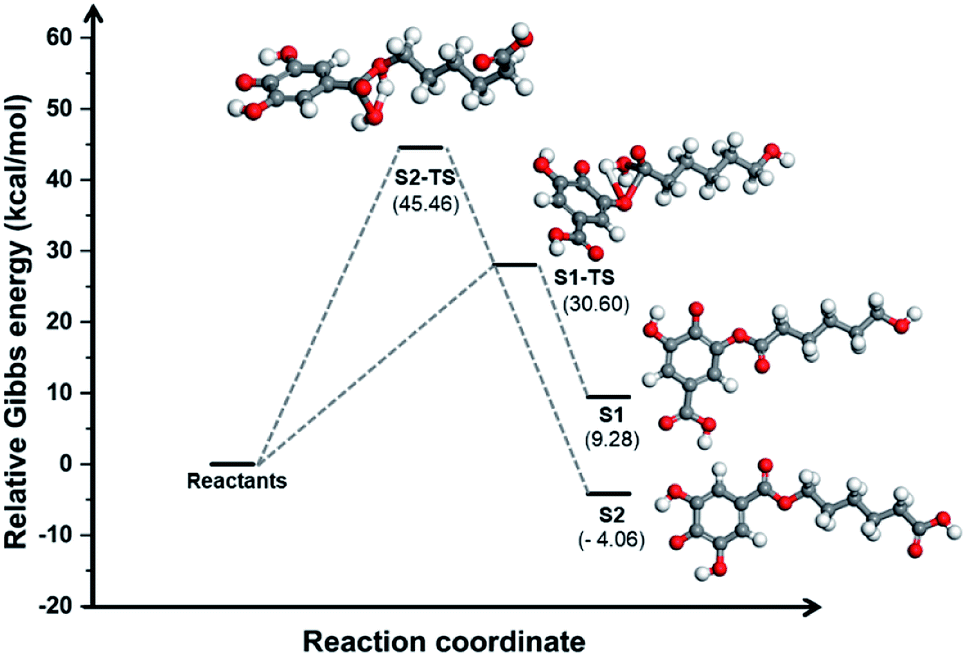 | ||
| Fig. 3 Energy profile along two possible reaction paths for the PCLe-g-PGAL at the M06-2X/6-311++G(d,p) (PCM:water) level of theory employing GA and 6-hydroxyhexanoic acid units as model compounds. | ||
The energetic difference of ca. 13 kcal mol−1 between S1 and S2 might reasonably be accounted to improved stabilization of the radical in S2, as the radicalarian oxygen atom has two ortho-phenol groups which we recently found to participate extensively in hydrogen bonding within the PGAL backbone with stabilizing interaction energies in the 3–11 kcal mol−1 regime.28
It should be noted that although the energy of S1-TS is lowered with respect of S2-TS due to intramolecular hydrogen bonding, the reaction path towards S1 is endergonic and consequently shifted to the reactants while the formation of S2 is exergonic; thus for this process, S1 and S2 are kinetically and thermodynamically favoured, respectively.
Nonetheless, this reaction is expected to yield S2 selectively at room temperature as the low activation energy for the backward reaction from S1 to reactants is easily overcome at this temperature.
Moreover, the activation energy predicted for S2 should be lowered due to the acidic conditions of the reaction (pH = 3),44,45 further favouring its selective formation over S1.
In summary, our DFT computations showed that under the conditions employed, the connectivity for the grafting of PGAL on the PCLe surface is largely that of hydroxyls from activated PCLe surface linked to the carboxyl in PGAL units, described as S2 in this model.
Electrospinning of PCLc, PCLe, and PCLe-g-PGAL
In the present work, the electrospinning parameters are optimized to attain fibres with an average diameter of ca. 200 nm, for PCLc these parameters are relatively well known but the stabilization of the spinning jet in the electrospinning of PCLe and PCLe-g-PGAL require increasing the polymer concentration in the solution. The best conditions for the electrospinning of these materials are gathered in Table 1.| Sample | Concentration (w/v%) | Flow rate (mL h−1) | Voltage (kV) | Distance (cm) |
|---|---|---|---|---|
| PCLc | 25 | 5 | 15 | 15 |
| PCLe | 30 | 4 | 15 | 15 |
| PCLe-g-PGAL | 60 | 2.5 | 25 | 20 |
All samples are dissolved in a chloroform/acetone mixture (2:1 v/v). A high concentration of 60 w/v% for PCLe-g-PGAL renders adequate viscosity in a relatively lower flow rate (2.5 mL h−1) compared to that required for PCLe (4 mL h−1), and in high voltage. These required parameters might be attributed to the relatively low molar mass in PCLe-g-PGAL.
Characterization of the electrospun fibers
SEM micrographs in Fig. 4 show the morphology of the electrospun scaffolds, revealing mostly homogeneous, continuous, and straight fibres with smooth surfaces for all samples. The electrospinning renders nanofibers with an average diameter near 200 nm, which is an adequate size for exhibiting a high level of porosity in the mat, with adequate gas permeation, and high surface-to-volume ratio. Of note is that these properties promote cell respiration, skin regeneration, moisture retention, removal of exudates, and homeostasis.46 | ||
| Fig. 4 SEM images of PCLc (a), PCLe (b), and PCLe-g-PGAL (c–f) electrospun fibres prepared under the optimized conditions. | ||
Fig. 5 describes representative histograms and Gaussian curves of diameter distributions for the different electrospun fibres to provide a quantification of the variability in samples.
 | ||
| Fig. 5 Frequency distribution of the fibre diameter and Gaussian functions for PCLc (a), PCLe (b), and PCLe-g-PGAL (c) fibres. | ||
The diameters for the PCLe and their derivative PCLe-g-PGAL fibres are similar, i.e. 218.45 ± 3 and 232.30 ± 8 nm, respectively, despite being obtained under different conditions. Moreover, the distribution of the enzyme-mediated polyester is the narrowest, while PCLe-g-PGAL has a wide distribution in the range of 100–400 nm. It is notorious that the population of fibres with a diameter >300 nm has a frequency close to 20%.
This trend toward large thread diameters is due to the high concentration of the polymer solution and the relatively low flow applied during the electrospinning. SEM micrographs and the histogram of the PCLc electrospun sample, on the other hand, show that the population has a smaller diameter size on average (i.e. 183.83 ± 8 nm) than the other products with only 10% of the distribution displaying diameters higher than 250 nm.
Antioxidant capacity
The solid-state ESR analysis for PCLe-g-PGAL fibres confirms the presence of the unpaired electron in the PGAL backbone. The characteristic ESR signal for PGAL shows a featureless hyperfine structure, as it depends on the magnetic moment of the unpaired electron and the neighbouring nuclei with a spin number other than zero in agreement to previous work.28 The ESR results for PCLe-g-PGAL are compared to that for PGAL measured under the same conditions. The analyses display an intense and narrow signal for the latter (i.e., ΔH = 2.59 G), which broadens for PCLe-g-PGAL (i.e., ΔH = 13.25 G) due to the decrease of the polyphenol concentration in the sample, which agrees to the Bleaney and Rubins relation (Fig. 6).47 The Landé g-factor for PGAL is near 2.00, which is close to that for a free electron, and this value is preserved for the graft polymer. This leads to the conclusion that the multiradical character of the PGAL is preserved in the nanocomposite fibres. To make an approximation of antioxidant capacity, the IC50 of the PCL–PGAL nanofibers is determined by DPPH. The RSA measured by UV-spectroscopy, shown in Fig. S3,† demonstrates the proportional inhibition of DPPH radical with the increase of PCL-g-PGAL concentration. The PCL-g-PGAL retained the antioxidant capacity of PGAL (i.e., IC50 = 23.48 μg mL−1) with IC50 ca. 104 μg mL−1. The addition of CMC hydrogel could affect this property; notwithstanding previous works confirmed the antioxidant capacity of PGAL when crosslinked with CMC.29 | ||
| Fig. 6 Solid-state ESR spectra for PCLe (blue), PGAL (purple), and PCLe-g-PGAL (green) at 298 K. All measurements are at 9.78 GHz with a microwave power of 5 mW. | ||
Hybrid nanocomposite characterization
The surface morphology of the hybrid composite by SEM, shown in Fig. 7 (left), exemplifies that the hydrogel permeates differently for each sample. This observation might be ascribed to the porosities; the PCLc sample has a hydrogel coating on the surface, whereas PCLe and PCLe-g-PGAL display the best flow within the scaffold (Fig. 7-right). | ||
| Fig. 7 SEM images of the hybrid composites. PCLc/CMC (a), PCLe/CMC (b) (scale bars = 2 μm) and PCLe-g-PGAL/CMC (c) composites surface (left) and qualitative EDX mapping for chloride element in cyan blue (center), transversal cut (right). | ||
This suggests that the hydrogel network is formed at greater depth in PCLe and PCLe-g-PGAL, while for PCLc, there is a deposition of hydrogel lumps on the surface of the fibres, thus causing insufficient permeation to form a network. The EDX mapping allows observing the distribution of the clindamycin in these composites, and Fig. 7 (centre) marks the chlorine with cyan blue, with a homogeneous scattering of the drug in the material. This information is relevant because the release of the drug can be controlled, and it does not accumulate in a single region of the nanocomposite.
The water drop measurements indicate that the PCLc, PCLe, and PCLe-g-PGAL electrospun materials are hydrophobic (i.e., θ > 115°) with a slightly lowest value for PCLe-g-PGAL owing to the hygroscopic hydroxyl groups in PGAL capable of forming hydrogen bonds and dipole–dipole interactions (Fig. 8). This wettability in the dressings is an important characteristic that influences the initial cell adhesion, and consequently, cell migration.48 Once the hydrogel is embedded within the nanofibre, there is a reduction in the contact angle (Δθ ≤ 82°), which is expected for matrices of hygroscopic materials. The wettability of the composites increases largely for those obtained from PClc at the observation of fibres superficially covered with the hydrogel, which agrees to the SEM analyses. However, the diameter size of the fibres also affects the materials, as PCLe presents higher hydrophobicity than PCLc, which might be explained by the slightly higher average nanofiber diameters. Specifically, the increase of the fibre diameter size leads to increasingly hydrophobic surfaces according to the Wenzel equation.49
 | ||
| Fig. 8 Contact angles for PCLc, PCLe, and PCLe-g-PGAL scaffolds and PCLc/CMC PCLe/CMC and PCLe-g-PGAL/CMC samples. | ||
Therefore, the contact angles are influenced not only by the chemical structure on the surface but also by the fibre morphology. Nonetheless, the surface wettability increases by the grafting of PGAL, thus evidencing the impact of this polyphenol on the surface energy.
In summary, these results show that the physical properties (e.g. hydrophobic/hydrophilic surface characteristics) of this dressing can be tuned by changing the chemical composition and the fibre morphology, with the hybrid composites showing advantageous characteristics for potential biomedical applications.
Antimicrobial activity for PCLe-g-PGAL
The SPTT assay allows the determination of the antimicrobial activity of the composite by measuring the release rates of clindamycin. This assay determines the duration of activity under static conditions as a means of evaluating the potential of materials for clinical trials, and noteworthy, its ability to indicate duration because its serial procedure is superior to the one-day zone plate test.50 This study, which is conducted until no inhibition zone is detected around the materials, shows no resistance to the antibiotic during the assay, and there is no inhibition around the non-antibiotic materials. The hydrogel/electrospun composites have three different degrees of citric acid crosslinking (2, 5, and 7.5 w/w%), which varies their ability to release clindamycin, as shown in Fig. 9.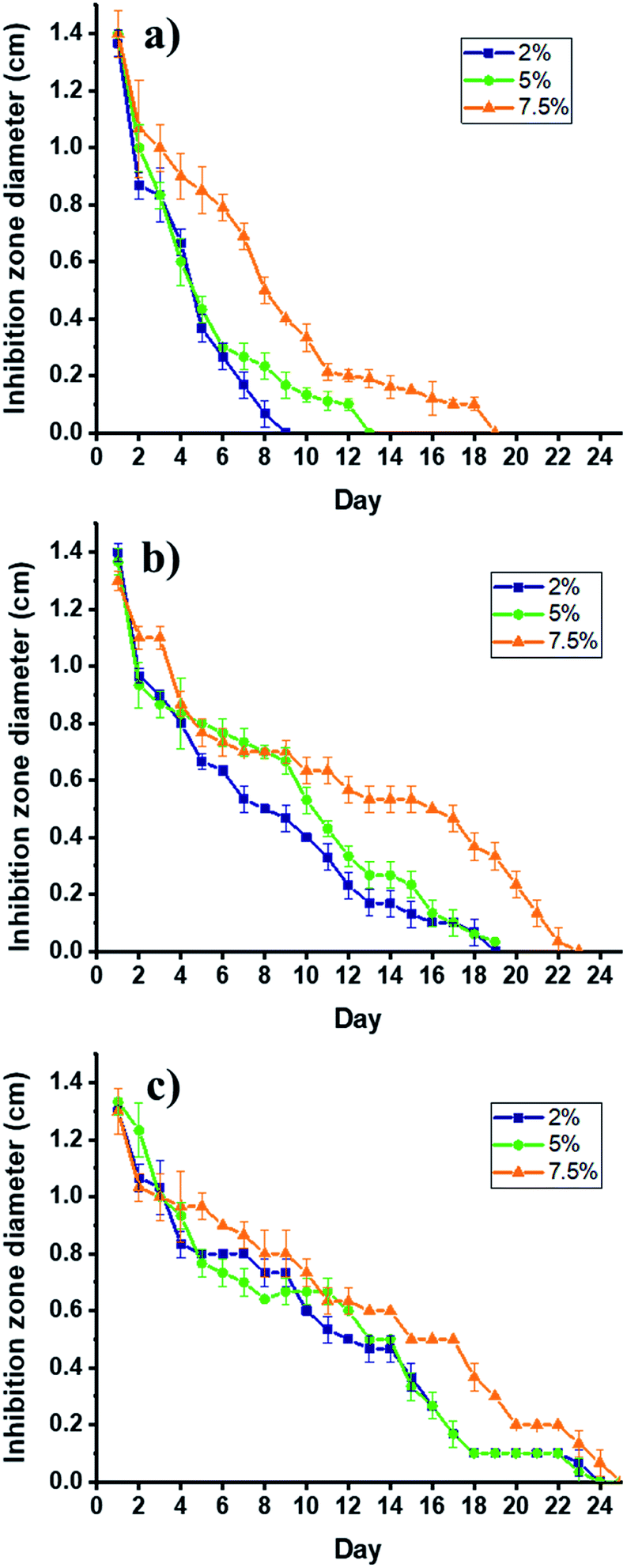 | ||
| Fig. 9 Bactericidal activity PCLc/CMC (a); PCLe/CMC (b); and PCLe-g-PGAL/CMC (c) composites loaded with 2% (■), 5% (●) and 7.5% (▲) of clindamycin against S. aureus by SPTT assay. | ||
Several factors are involved in the release of drugs from matrices, these include the drug molecular weight and its solubility in the matrix. Remarkably, the diffusivity of the drug decreases with high molecular weights but the time of release increase with that of the molar mass distribution in the polymeric matrix.51 Of note is that the determination of the solubility of clindamycin, and any other drug, in a crosslinked matrix is experimentally challenging, therefore, this is generally inferred indirectly.51
In this regard, the clindamycin is highly soluble in the non-crosslinked matrix in water, so the amounts of the drug in each material (i.e. 0.6 mg) can be inferred considering that the hydrogel precursor displays a homogeneous distribution of the drug, which is corroborated by SEM analyses. The initial bactericidal activity may be due to the dissolution of the particles directly accessible from the surface of the dressings. The high degree of persistent activity over time suggests the ability of the carrier hydrogel to diffuse through the matrix, as expected. However, there is a rapid release for PCLc-based dressings (i.e. 9, 13, and 19 days maximum), which correlates with the permeation of the hydrogel into the matrix, thereby producing the agglomeration of the drug on the surface. This behaviour is attributed to the low porosity of the scaffold, as the size of the fibres tends toward smaller sizes than those for PCLe and PCLe-g-PGAL. On the other hand, PCLe-g-PGAL shows longer retention time of the drug than that for PCLe.
This suggests the presence of available carbonyl groups of the PGAL for crosslinking with CMC, in agreement with previous work.28 Consequently, the hydrogel is crosslinked to some extent to PGAL and the drug particles are likely imprisoned within the crosslinked network. This indicates that PGAL side chains improve the release time of the drug in addition to the antioxidant property. On the effect of the crosslinker, there are no significant differences between 2 and 5%, whereas the incorporation of 7.5% and beyond (data not shown) lengthen the release times due to crosslinking.21,52 Noteworthy, composites without loaded clindamycin show no antibacterial effect.
Drug delivery kinetics
Results obtained from the Cly release from the PCL/CMC composites and CMC hydrogels in PBS medium are shown in Fig. 10, and the corresponding kinetic parameters gathered in Table 2. The release mechanism of the hydrogel/electrospun composites is rather complex and influenced by several parameters as polymer degradation, hydrophobicity/philicity, hydrogel swelling, and solute diffusion. An approach, the power-law model (eqn (3)) developed by Ritger and Peppas43 was applied to analyze the first 60% of drug fractional release.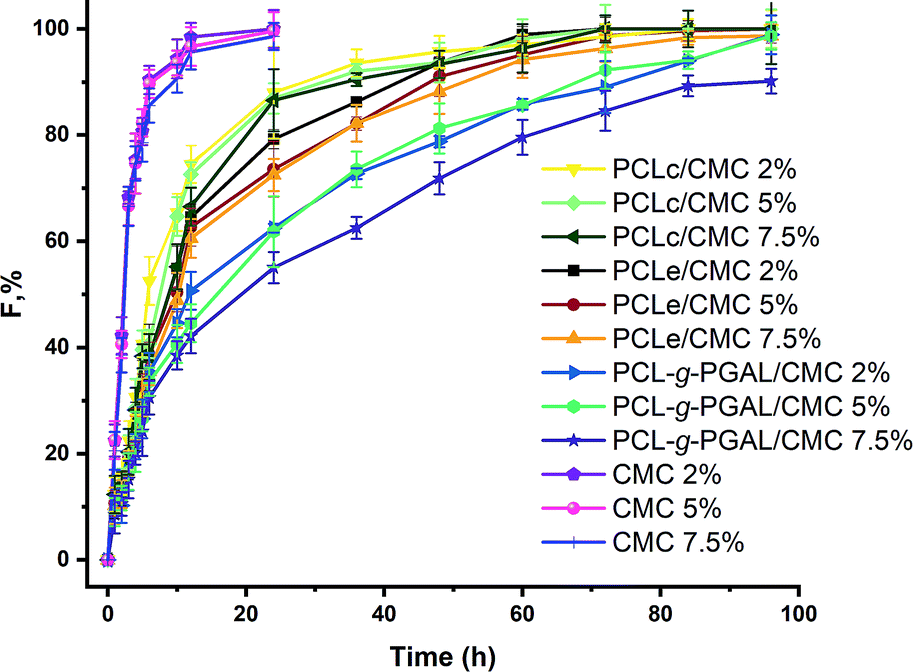 | ||
| Fig. 10 Cly release profiles from composites and hydrogels in PBS, pH = 7.4. Values correspond to the average while the error bars indicate the standard deviations (i.e. assays were carried out for triplicate). | ||
| Sample | Citric acid (%) | n | k | r2 | T1/2 |
|---|---|---|---|---|---|
| PCLc/CMC | 2 | 0.483 ± 0.020 | 0.292 | 0.9946 | 12.87 |
| PCLc/CMC | 5 | 0.489 ± 0.020 | 0.292 | 0.9861 | 14.08 |
| PCLc/CMC | 7.5 | 0.496 ± 0.016 | 0.249 | 0.9867 | 17.75 |
| PCLe/CMC | 2 | 0.486 ± 0.015 | 0.262 | 0.9981 | 19.21 |
| PCLe/CMC | 5 | 0.482 ± 0.013 | 0.246 | 0.9972 | 23.13 |
| PCLe/CMC | 7.5 | 0.463 ± 0.013 | 0.228 | 0.9927 | 24.01 |
| PCL-g-PGAL/CMC | 2 | 0.455 ± 0.013 | 0.198 | 0.9851 | 26.035 |
| PCL-g-PGAL/CMC | 5 | 0.431 ± 0.007 | 0.157 | 0.9888 | 50.83 |
| PCL-g-PGAL/CMC | 7.5 | 0.440 ± 0.007 | 0.144 | 0.9848 | 45.85 |
| CMC | 2 | 0.226 ± 0.019 | 0.618 | 0.9764 | 6.6 |
| CMC | 5 | 0.231 ± 0.020 | 0.619 | 0.9875 | 6.31 |
| CMC | 7.5 | 0.234 ± 0.022 | 0.564 | 0.9802 | 6.17 |
The power n in eqn (3) was determined from the slope of the release fraction (F) vs. time log–log plot. The n values are used together with eqn (3) to calculate the release kinetic constant (k), which are also reported in Table 2. These values indicate that the Cly release from the nanocomposites follows a Fickian diffusion for all the crosslinking agent concentrations. The CMC hydrogels without electrospun fiber mat have the lowest release index n value (i.e., 0.23), the largest release constant k (i.e. 0.6), and the fastest release time T1/2 (6 h).
According to Gaharwar et al.,53 the release of drugs through the water-filled pores of the hydrogel should have a release index n smaller than 0.5. In our results, a value of n = 0.23 for the CMC hydrogels suggests a rapid release because the pores in the hydrogel structure are rapidly filled with water, thereby causing swelling of the matrix.
This agrees with previous reports of drug delivery from a hydrogel.54 On the other hand, the fitting results to Peppas power-law model indicate that the composite has a similar diffusional index n value, which means that the release Fickian mechanism is maintained. Instead, the constant k values are significantly lowered in comparison with pure CMC hydrogels, and they decrease with the increase of crosslinking agent (CA). The dependence of the constant k on diffusion path length and drug diffusivity could indicate that the presence of PCL nanofiber significantly increased the diffusion path length and tortuosity for drug release.
Additionally, the hydrophobic PCL electrospun fiber mats serve as an additional diffusion barrier to decrease the effective diffusion coefficients, thus reducing hydrogel swelling and water penetration rates as previously demonstrated by Han et al.55 Of note is that the PGAL also reduces the constant k, which might be ascribed to the drastic reduction of the swelling behavior for CMC when crosslinked to this polyphenol, in agreement to the previous study,29 thereby the swelling is abated up to 90%. Noteworthy, the influence of PGAL is also evidenced by the mean release time (T1/2), which increases significantly.
Hemolytic activity and cell viability
The nanocomposites behaviour against red blood human cells in the agar well diffusion method shows no hemolysis.56 The clear formation of an inhibition halo (ca. 5 mm) around the water-filled well was observed, which was used as a positive control. Conversely, none of the composites induced the formation of a zone of hemolysis. These data of human erythrocytes confirm that the materials are not cytotoxic, and therefore, suitable for application in wound treatment because they are not affecting the integrity of the membrane and consequently cell lysis does not occur. On the other hand, viability experiments with seeded cells on PCLc, PCLe, and PCLe-g-PGAL electrospun fibres (see ESI data† for Vero cell viability on PCLc, PCLe and PCLe-g-PGAL electrospun, cells at 96 h of culture) demonstrate no cytotoxic effects on cell surfaces, and it displays viability entirely comparable with that for the surface of the culture plate (TCPS).In summary, these materials are not cytotoxic and do not affect cell growth, thus demonstrating that PCLe-g-PGAL-based material is safe for use in biomedical applications.
Conclusions
The clindamycin loaded in a CMC hydrogel crosslinked to electrospun PCL-g-PGAL shows improved characteristics for a wound dressing regarding its antioxidant, antibacterial, and morphological characteristics. Analytical and theoretical studies sustain the consecution of the grafting of PGAL onto PCLe, were the esterification from a PGAL carboxyl group to a hydroxyl of the activated polyester surface is kinetically favoured. The formed nanofibres show average diameters slightly above 200 nm for this polymer in a narrow distribution. Besides the antioxidant effect, PGAL also increases the hydrophilicity of the biomaterial and the results evidence the crosslinking of this polyphenol to CMC which improves drug diffusion rates. Finally, the PCL-g-PGAL shows no hemolytic activity in cells and it does not affect the viability of epithelial cells. Further progress will require continued assessment in vivo to identify ways to use this hybrid nanocomposite for wound healing.Live subject statement
We declare that human consent was obtained for all blood donations following the Code of Ethics stated in the International Society of Blood Transfusion, and approved by the committee of human blood transfusions under the supervision of the Head of the Food and Biotechnology Department of the Faculty of Chemistry at the National Autonomous University of Mexico.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DGAPA-UNAM for funding (PAPIIT IN200520) and CONACyT for doctoral scholarships for A. R.-M. (336576) and P. L.-V. (337958). The authors are in debt to support from MINECO and FEDER (RTI2018-101827-B-I00) and the Generalitat de Catalunya (2017SGR373).References
- J. Tavakoli and Y. Tang, Mater. Sci. Eng., C, 2017, 77, 318–325 CrossRef CAS PubMed.
- M. Contardi, J. A. Heredia-Guerrero, G. Perotto, P. Valentini, P. P. Pompa, R. Spanò, L. Goldoni, R. Bertorelli, A. Athanassiou and I. S. Bayer, Eur. J. Pharm. Sci., 2017, 104, 133–144 CrossRef CAS PubMed.
- K. Totoraitis, J. L. Cohen and A. Friedman, J. Drugs Dermatol., 2017, 16, 209–212 CAS.
- H. A. Tran, K. L. Ly, K. E. Fox, P. A. Tran and T.-H. Nguyen, Int. J. Nanomed., 2019, 14, 9929 CrossRef PubMed.
- I. Ludolph, F. W. Fried, K. Kneppe, A. Arkudas, M. Schmitz and R. E. Horch, Int. Wound J., 2018, 15, 978–984 CrossRef PubMed.
- M. Zhai, Y. Xu, B. Zhou and W. Jing, J. Photochem. Photobiol., B, 2018, 180, 253–258 CrossRef CAS PubMed.
- L. Mei, R. Fan, X. Li, Y. Wang, B. Han, Y. Gu, L. Zhou, Y. Zheng, A. Tong and G. Guo, Polym. Chem., 2017, 8, 1664–1671 RSC.
- P. Zahedi, I. Rezaeian, S. O. Ranaei-Siadat, S. H. Jafari and P. Supaphol, Polym. Adv. Technol., 2010, 21, 77–95 CrossRef CAS.
- M. di Luca, M. Curcio, E. Valli, G. Cirillo, M. E. Butini, A. Farfalla, E. Pantuso, A. Leggio, F. P. Nicoletta and A. Tavanti, J. Mater. Chem. B, 2019, 7, 4361–4370 RSC.
- T. Amna, F. N. Gharsan, K. Shang, M. S. Hassan, M.-S. Khil and I. Hwang, Macromol. Res., 2019, 27, 663–669 CrossRef CAS.
- S. M. Lee, I. K. Park, Y. S. Kim, H. J. Kim, H. Moon, S. Mueller and Y.-I. Jeong, Biomater. Res., 2016, 20, 15 CrossRef PubMed.
- M. Yoshimura, N. Ohura, J. Tanaka, S. Ichimura, Y. Kasuya, O. Hotta, Y. Kagaya, T. Sekiyama, M. Tannba and N. Suzuki, Int. Wound J., 2018, 15, 188–197 CrossRef PubMed.
- C.-Y. Chin, J. Jalil, P. Y. Ng and S.-F. Ng, J. Ethnopharmacol., 2018, 212, 188–199 CrossRef CAS PubMed.
- S. Tyeb, N. Kumar, A. Kumar and V. Verma, Carbohydr. Polym., 2018, 200, 572–582 CrossRef CAS PubMed.
- D. S. Yoon, Y. Lee, H. A. Ryu, Y. Jang, K.-M. Lee, Y. Choi, W. J. Choi, M. Lee, K. M. Park and K. D. Park, Acta Biomater., 2016, 38, 59–68 CrossRef CAS PubMed.
- M. Comotto, S. Saghazadeh, S. Bagherifard, B. Aliakbarian, M. Kazemzadeh-Narbat, F. Sharifi, S. A. Mousavi Shaegh, E. Arab-Tehrany, N. Annabi and P. Perego, J. Biomater. Appl., 2019, 33, 1265–1276 CrossRef CAS PubMed.
- I. Garcia-Orue, G. Gainza, F. B. Gutierrez, J. J. Aguirre, C. Evora, J. L. Pedraz, R. M. Hernandez, A. Delgado and M. Igartua, Int. J. Pharm., 2017, 523, 556–566 CrossRef CAS PubMed.
- S. Gilotra, D. Chouhan, N. Bhardwaj, S. K. Nandi and B. B. Mandal, Mater. Sci. Eng., C, 2018, 90, 420–432 CrossRef CAS PubMed.
- J. Haik, R. Kornhaber, B. Blal and M. Harats, Adv. Wound Care, 2017, 6, 166–174 CrossRef PubMed.
- L. A. Bosworth, L.-A. Turner and S. H. Cartmell, Nanomedicine, 2013, 9, 322–335 CrossRef CAS PubMed.
- H. Namazi, R. Rakhshaei, H. Hamishehkar and H. S. Kafil, Int. J. Biol. Macromol., 2016, 85, 327–334 CrossRef CAS PubMed.
- A. Francesko, P. Petkova and T. Tzanov, Curr. Med. Chem., 2018, 25, 5782–5797 CrossRef CAS PubMed.
- R. Lalani and L. Liu, Biomacromolecules, 2012, 13, 1853–1863 CrossRef CAS PubMed.
- X. Yang, L. Fan, L. Ma, Y. Wang, S. Lin, F. Yu, X. Pan, G. Luo, D. Zhang and H. Wang, Mater. Des., 2017, 119, 76–84 CrossRef CAS.
- Z. Tang, C. He, H. Tian, J. Ding, B. S. Hsiao, B. Chu and X. Chen, Prog. Polym. Sci., 2016, 60, 86–128 CrossRef CAS.
- G. Amariei, V. Kokol, K. Boltes, P. Letón and R. Rosal, RSC Adv., 2018, 8, 28013–28023 RSC.
- A. Romero-Montero, L. J. del Valle, J. Puiggalí, C. Montiel, R. García-Arrazola and M. Gimeno, Mater. Sci. Eng., C, 2020, 115, 111154 CrossRef CAS PubMed.
- A. Romero-Montero, M. Gimeno, N. Farfán and P. Labra-Vázquez, J. Mol. Struct., 2019, 1197, 326–335 CrossRef CAS.
- A. Romero-Montero, A. Tecante, R. García-Arrazola, C. Montiel, L. J. del Valle, J. Puiggalí and M. Gimeno, RSC Adv., 2017, 7, 17660–17669 RSC.
- J. López, J. M. Hernández-Alcántara, P. Roquero, C. Montiel, K. Shirai, M. Gimeno and E. Bárzana, J. Mol. Catal. B: Enzym., 2013, 97, 100–105 CrossRef.
- R. Sánchez-Sánchez, A. Romero-Montero, C. Montiel, Y. Melgarejo-Ramírez, C. Sánchez-Ortega, H. Lugo-Martínez, B. Cabello-Arista, R. García-Arrazola, C. Velasquillo and M. Gimeno, Mater. Sci. Eng., C, 2017, 76, 417–424 CrossRef PubMed.
- A. Karczewski, S. A. Feitosa, E. I. Hamer, D. Pankajakshan, R. L. Gregory, K. J. Spolnik and M. C. Bottino, J. Endod., 2018, 44, 155–162 CrossRef PubMed.
- A. Hebeish, M. Hashem, M. M. Abd El-Hady and S. Sharaf, Carbohydr. Polym., 2013, 92, 407–413 CrossRef CAS PubMed.
- T. W. Wong and N. A. Ramli, Carbohydr. Polym., 2014, 112, 367–375 CrossRef CAS PubMed.
- R. García-Arrazola, M. Gimeno and E. Bárzana, Macromolecules, 2007, 40, 4119–4120 CrossRef.
- F. Xu, Z. Wang and W. Yang, Biomaterials, 2010, 31, 3139–3147 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. E. Ochoa, P. Labra-Vázquez, N. Farfán and R. Santillan, Cryst. Growth Des., 2018, 18, 2795–2803 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- N. Treitel, R. Shenhar, I. Aprahamian, T. Sheradsky and M. Rabinovitz, Phys. Chem. Chem. Phys., 2004, 6, 1113–1121 RSC.
- Y. Inada and H. Orita, J. Comput. Chem., 2008, 29, 225–232 CrossRef CAS.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 37–42 CrossRef CAS.
- T. Yu, H.-B. Chang, W.-P. Lai and X.-F. Chen, Polym. Chem., 2011, 2, 892–896 RSC.
- M. Vafaeezadeh and A. Fattahi, Comput. Theor. Chem., 2015, 1071, 27–32 CrossRef CAS.
- K. A. Rieger, N. P. Birch and J. D. Schiffman, J. Mater. Chem. B, 2013, 1, 4531–4541 RSC.
- B. Bleaney and R. Rubins, Proc. Phys. Soc., 1961, 77, 103 CrossRef CAS.
- M. Nakamura, N. Hori, H. Ando, S. Namba, T. Toyama, N. Nishimiya and K. Yamashita, Mater. Sci. Eng., C, 2016, 62, 283–292 CrossRef CAS PubMed.
- M. Lampin, R. Warocquier-Clérout, C. Legris, M. Degrange and M. Sigot-Luizard, J. Biomed. Mater. Res., 1997, 36, 99–108 CrossRef CAS PubMed.
- R. Bayston, L. E. Fisher and K. Weber, Biomaterials, 2009, 30, 3167–3173 CrossRef CAS PubMed.
- R. Bayston and R. Milner, J. Clin. Pathol., 1981, 34, 1057–1062 CrossRef CAS PubMed.
- N. S. Capanema, A. A. Mansur, A. C. de Jesus, S. M. Carvalho, L. C. de Oliveira and H. S. Mansur, Int. J. Biol. Macromol., 2018, 106, 1218–1234 CrossRef CAS PubMed.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441–453 CrossRef CAS PubMed.
- T. L. Tsou, S. T. Tang, Y. C. Huang, J. R. Wu, J. J. Young and H. J. Wang, J. Mater. Sci.: Mater. Med., 2005, 16, 95–100 CrossRef CAS PubMed.
- N. Han, J. Johnson, J. J. Lannutti and J. O. Winter, J. Controlled Release, 2012, 158, 165–170 CrossRef CAS PubMed.
- S. Prakash, R. Ramasubburayan, V. S. Ramkumar, E. Kannapiran, A. Palavesam and G. Immanuel, Biomed. Pharmacother., 2016, 83, 648–657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05935h |
| This journal is © The Royal Society of Chemistry 2020 |