
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA [3 + 2] cycloaddition/C-arylation of isatin N,N′-cyclic azomethine imine 1,3-dipole with arynes†
Qiaomei Jin‡
 *ab,
Dongjian Zhang‡ab and
Jian Zhang*ab
*ab,
Dongjian Zhang‡ab and
Jian Zhang*ab
aAffiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing 210028, Jiangsu, China. E-mail: jqmxy@163.com; zjwonderful@hotmail.com
bLaboratories of Translational Medicine, Jiangsu Province Academy of Traditional Chinese Medicine, Nanjing 210028, Jiangsu, China
First published on 19th August 2020
Abstract
A [3 + 2] annulation/C-arylation of isatin N,N′-cyclic azomethine imine 1,3-dipole 1 with in situ generated arynes has been established for the synthesis of 3,3-disubstituted oxindole scaffolds. These highly functionalized scaffolds were assembled in moderate yields (up to 85% yield). The novel spirooxindole scaffolds displayed moderate antitumor activities, which represented promising lead compounds for antitumor drug discovery.
The construction of new heterocyclic architectures is of great importance because such privileged scaffolds that widely occur in natural products and drugs increase the returns of drug-discovery studies.1 Among these scaffolds, the 3,3-disubstituted oxindoles are associated with interesting biological properties (Fig. 1). For instance, nelivaptan could be used as an orally active non-peptide vasopressin receptor antagonist.2 NITD609 has been identified for potential treatment of malaria based on in vivo activity, having single dose efficacy in a rodent malaria model.3 Moreover, other bioactive 3,3-disubstituted oxindoles have also attracted considerable attention due to their high activities, such as poliovirus inhibitor,4 MDM2 inhibitor SAR405838 (ref. 5) and anti-bacterial agents.6 Given the biological significance of functionalized 3,3-disubstituted oxindoles, synthesis of this class of compounds with high structural diversity from readily available starting materials by using simple manipulations is highly desirable.
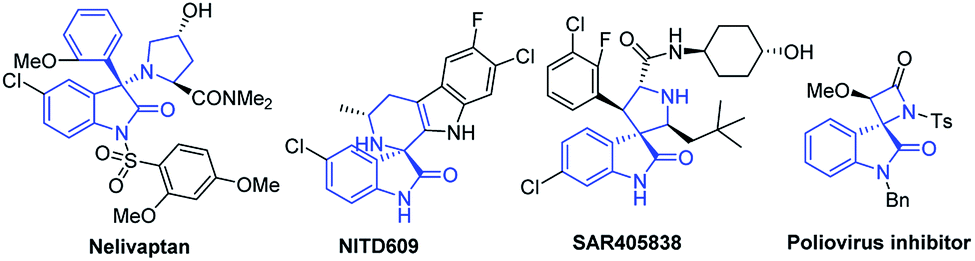 | ||
| Fig. 1 Representative biologically active compounds containing the 3,3-disubstituted oxindole skeleton. | ||
Recently, 1,3-dipolar cycloadditions (1,3-DCs) are among the most powerful approaches for the construction of carbon–carbon bonds and heterocycles.7 And the Wang's group reported an abnormal [3 + 2] cycloaddition of a new isatin N,N′-cyclic azomethine imine 1,3-dipoles with maleimides, an unusual Michael reaction between these 1,3-dipoles with β-nitrostyrenes and an abnormal [3 + 2] cycloaddition between these 1,3-dipoles and 3-methyleneoxindole, which are very scarce examples of 1,3-dipolar cycloaddition reaction (Scheme 1a).8 After that, we disclosed an DMAP-catalyzed direct alkylation at the a-position of the cyclic amine of these isatin N,N′-cyclic azomethine imine 1,3-dipoles with Morita–Baylis–Hillman carbonates and developed an efficient way to synthesize seven-membered heterocyclic spirooxindoles via a [3 + 4] cycloaddition reaction of these 1,3-dipoles with N-(ortho-chloromethyl)aryl amides (Scheme 1a).9 Furthermore, the Moghaddam's group reported an unexpected abnormal [3 + 3] tandem Michael addition/N-cyclization of these 1,3-dipoles and 2-arylidenemalononitrile under DABCO catalysis (Scheme 1a).10 During the course of the studies on these isatin N,N′-cyclic azomethine imine 1,3-dipoles, the related studies envisioned that these 1,3–dipoles have been utilized as valuable building blocks in cycloaddition reactions with alkenes. Given our ongoing interest in 1,3-dipolar cycloaddition and spirooxindole alkaloids, we envisioned the reaction of the isatin N,N′-cyclic azomethine imine 1,3-dipoles and arynes would be one approach to obtaining new pyrazole-spirooxindole derivatives via [3 + 2] cycloadditions (Scheme 1b).
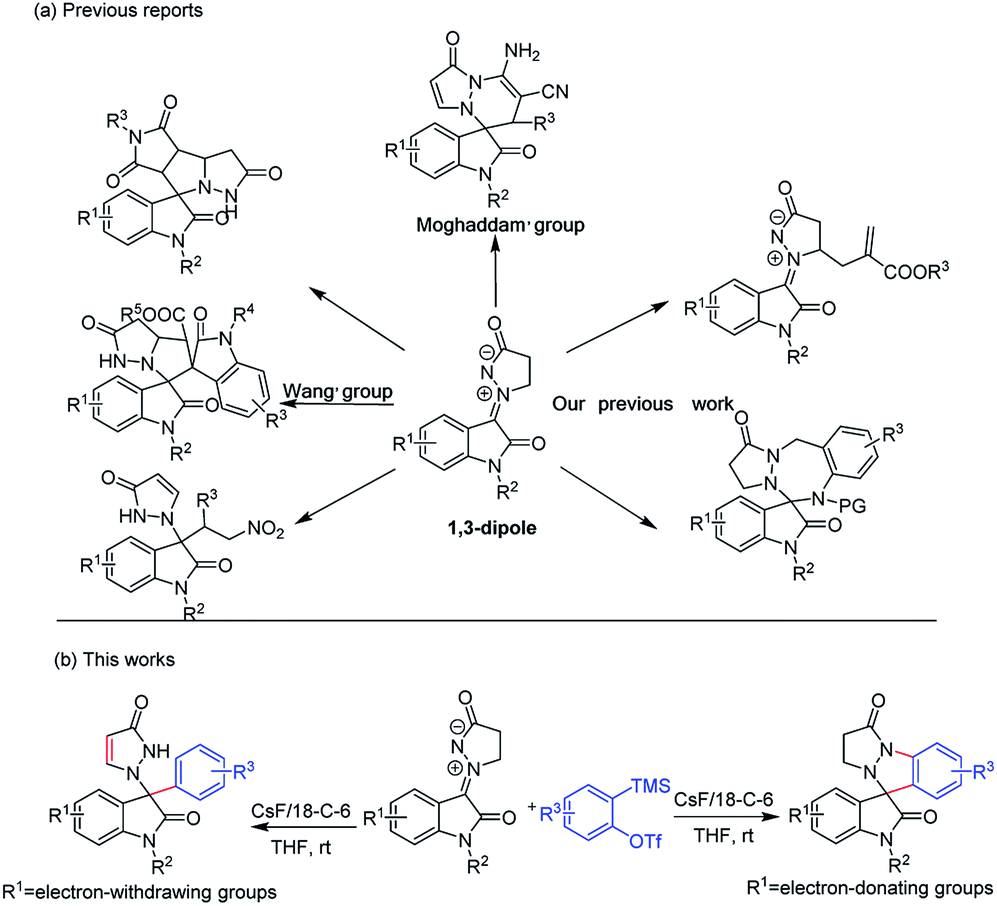 | ||
| Scheme 1 (a) Previous reports of isatin N,N′-cyclic azomethine imine 1,3-dipoles. (b) Design of the new [3 + 2] cycloaddition. | ||
Our investigations started with the screening of the reaction between the isatin N,N′-cyclic azomethine imine 1,3-dipole 1a and the unsubstituted aryne precursor 2a in the presence of CsF in CH3CN at room temperature for 2 h. To our delight, the desired product 3a was obtained in 20% yield (entry 1, Table 1). In order to improve the yield of the reaction product we used TBAF and KF instead of CsF as the fluoride source. Unfortunately, these two fluoride sources did not perform well (entries 2 and 3, Table 1). Performing the reaction in the presence of 18-crown-6 and CsF increased the yield to 55% (entry 4, Table 1). This result indicates that the 18-crown-6 has a remarkable influence on the yield. When the reaction was carried out at 50 °C, the reaction yields were not improved (entry 5, Table 1). Subsequently, a few more experiments were performed by changing the amount of CsF and 18-crown-6 (entries 6–9, Table 1). Interestingly, an improved yield of 67% was observed when the reaction was performed using 2.5 equiv. of CsF as a fluoride source and 3.0 equiv. of 18-crown-6 as an additive (entry 9, Table 1). And no improvements were observed with increasing the loadings of CsF (entries 7 and 8, Table 1). Finally, we investigated the reaction media by screening some common solvents, such as DMF, CH2Cl2, THF, 1,4-dioxane, EA, DCE and MeOH (entries 10–17, Table 1). The brief screening of the reaction solvent proved that THF was the best choice with respect to yields (entry 12, Table 1). Thus, the best reaction conditions utilized the isatin N,N′-cyclic azomethine imine 1,3-dipole 1a (1.0 equiv.), aryne precursor 2a (1.25 equiv.), CsF (2.5 equiv.) as the fluoride source and 18-crown-6 (3.0 equiv.) as the additive at room temperature.
|
||||||
|---|---|---|---|---|---|---|
| Entry | F-source (equiv.) | Additive (equiv.) | Solvent | Temp (°C) | Time (h) | Yield of 3ab (%) |
| a Unless noted otherwise, reaction of 1a (0.2 mmol), 2a (0.25 mmol), fluoride source (0.5 mmol) and 18-C-6 (0.6 mmol) was performed in 3.0 mL of solvent under Ar.b Isolated yield based on 1a. | ||||||
| 1 | CsF (2.0) | — | MeCN | rt | 2 | 20 |
| 2 | TBAF (2.0) | — | MeCN | rt | 2 | Trace |
| 3 | KF (2.0) | — | MeCN | rt | 2 | 15 |
| 4 | CsF (2.0) | 18-C-6 (2.0) | MeCN | rt | 2 | 55 |
| 5 | CsF (2.0) | 18-C-6 (2.0) | MeCN | 50 | 1 | 53 |
| 6 | CsF (2.5) | 18-C-6 (2.0) | MeCN | rt | 2 | 64 |
| 7 | CsF (3.0) | 18-C-6 (2.0) | MeCN | rt | 2 | 51 |
| 8 | CsF (4.0) | 18-C-6 (2.5) | MeCN | rt | 2 | 53 |
| 9 | CsF (2.5) | 18-C-6 (3.0) | MeCN | rt | 2 | 67 |
| 10 | CsF (2.5) | 18-C-6 (3.0) | DMF | rt | 2 | 45 |
| 11 | CsF (2.5) | 18-C-6 (3.0) | CH2Cl2 | rt | 2 | 33 |
| 12 | CsF (2.5) | 18-C-6 (3.0) | THF | rt | 2 | 72 |
| 13 | CsF (2.5) | 18-C-6 (3.5) | THF | rt | 2 | 69 |
| 14 | CsF (2.5) | 18-C-6 (3.0) | 1,4-Dioxane | rt | 2 | Trace |
| 15 | CsF (2.5) | 18-C-6 (3.0) | EA | rt | 4 | 55 |
| 16 | CsF (2.5) | 18-C-6 (3.0) | DCE | rt | 4 | 27 |
| 17 | CsF (2.5) | 18-C-6 (3.0) | MeOH | rt | 4 | 0 |
With the favorable reaction conditions established, the substrate scope and limitations of this catalyst-free self [3 + 2] cycloaddition of isatin N,N′-cyclic azomethine imine 1,3-dipole 1 with aryne precursors 2 were explored (Scheme 2). To our delight, most isatin N,N′-cyclic azomethine imines 1 and aryne precursors 2 were well tolerated. First, we evaluated the different 1,3-dipoles 1 with diverse substituents. It seems that the electronic nature of the substituents had intriguingly impact on the reaction. In general, substrates with electron-donating groups at the 5-position of 1 resulting in the formation of the cycloaddition products in good yields (3f: 78%; 3g: 85%). In addition, a few 1,3-dipoles 1 with diverse N-substituted groups, including that with a free NH group, showed inert reactivity and failed to deliver the expected product. Only N-methyl could smoothly afford the desired products with good results (3b: 57%). Interestingly, reactions carried out using the 1,3-dipoles 1 with electron-withdrawing groups resulted in the formation of the 3,3-disubstituted oxindole products in moderate yields (products 3h–3k), probably because the latter substituent would lower the nucleophilicity of the 1,3-dipoles 1. Next, the tolerance of substituents on the aryne moiety was also studied.
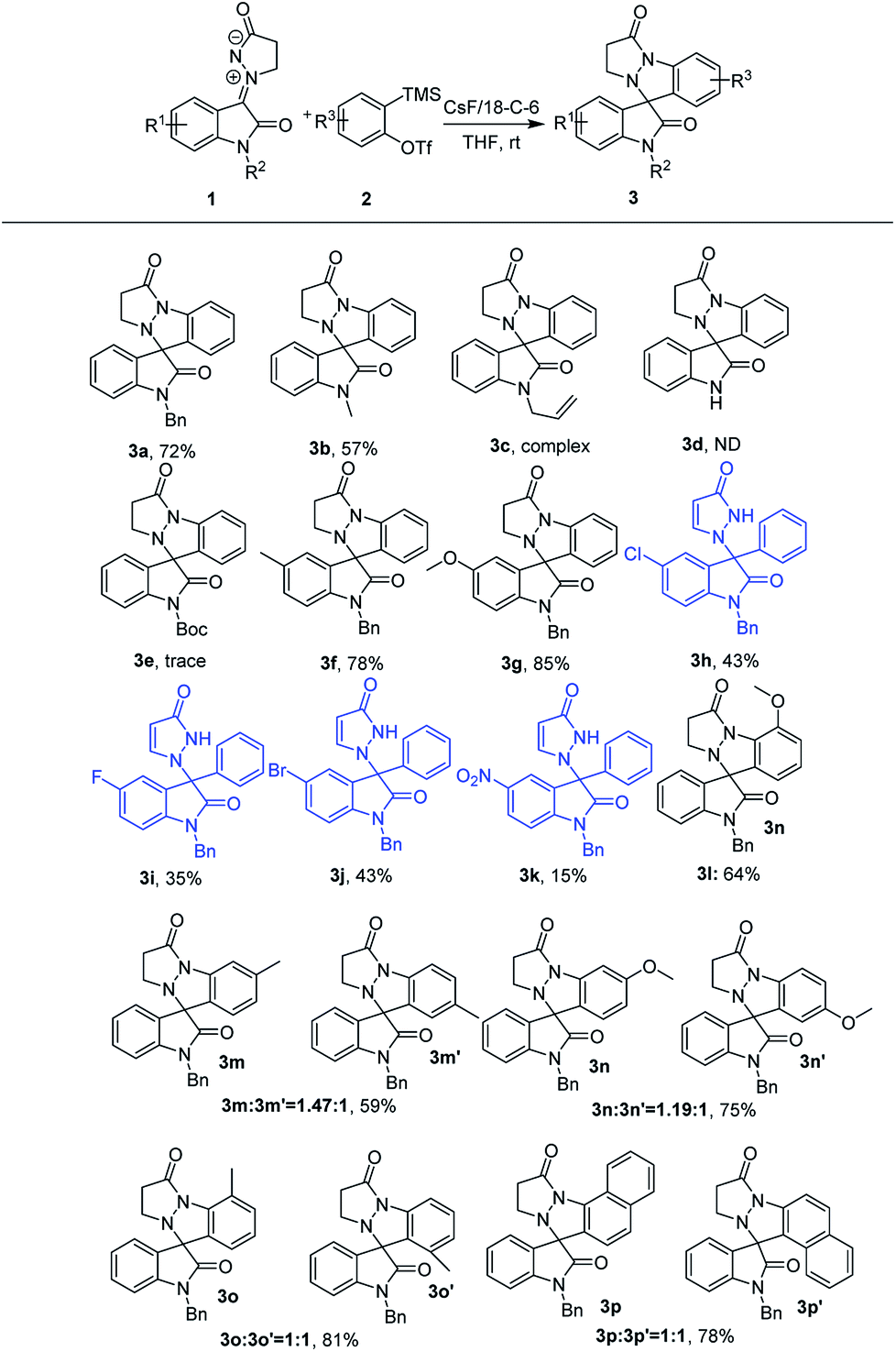 | ||
| Scheme 2 The reaction scope.a,b aTypical conditions: reaction of 1 (0.2 mmol), 2 (0.25 mmol), CsF (0.5 mmol) and 18-C-6 (0.6 mmol) was performed in 3.0 mL of THF at room temperature under Ar. bIsolated yield based on 1. | ||
The cycloaddition products 3l–3o are formed in moderate to good yields when this reaction was performed using unsymmetrical 3-substituted or 4-substituted arynes generated from the precursors. The unsymmetrical 4-methyl aryne afforded an inseparable mixture of regioisomers 3m and 3m′ in 59% yield and a 1.47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioisomer ratio. Similarly, inseparable regioisomeric products were formed when the reaction was carried out using unsymmetrical 4-methoxy, 3-methyl and 3-methoxy arynes. Moreover, the unsymmetrical naphthalyne was well tolerated to furnish the separable regioisomeric products 3p and 3p′ in 78%.
:1 regioisomer ratio. Similarly, inseparable regioisomeric products were formed when the reaction was carried out using unsymmetrical 4-methoxy, 3-methyl and 3-methoxy arynes. Moreover, the unsymmetrical naphthalyne was well tolerated to furnish the separable regioisomeric products 3p and 3p′ in 78%.
In order to address the viability and potential synthetic application of this reaction, a gram-scale scale-up and several applications were carried out (Scheme 3). Under identified conditions, product 3a was obtained without a significant loss of efficiency (69%) with a 4 mmol scale (Scheme 3a). To further illustrate synthetic applications of this method, we conducted a Suzuki coupling of product 3j with N-Boc-1,2,5,6- tetrahydropyridine-4-boronic acid pinacol ester, and then deprotection of the Boc group, which afforded product 4 in 52% yield (Scheme 3b).
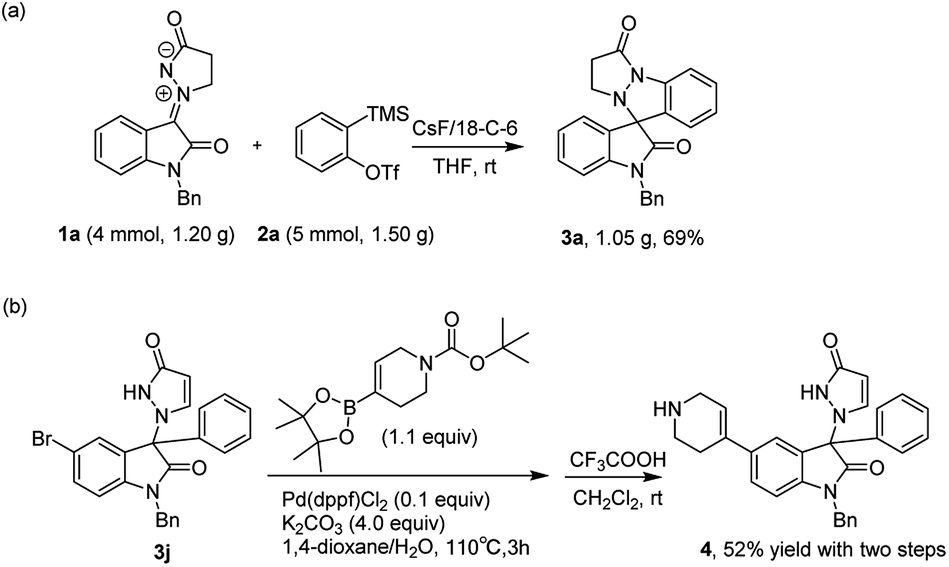 | ||
| Scheme 3 Follow-up Chemistry. | ||
On the basis of our results and the previous studies,7d,8–10 two plausible mechanisms was proposed as illustrated in Scheme 4. One approach, the in situ generated aryne (formed by the fluoride induced 1,2-elimination from 2) reacts with the isatin N,N′-cyclic azomethine imines 1 to generate the final product 3 through a thermal [3 + 2] annulation. The other, the more stable intermediate I was formed by the tautomerism of 1 in the presence of a base. Then intermediate I underwent C-arylation reaction with the in situ generated aryne with a double bond shift to generate the final product 3h, 3i and 3j (Scheme 4).
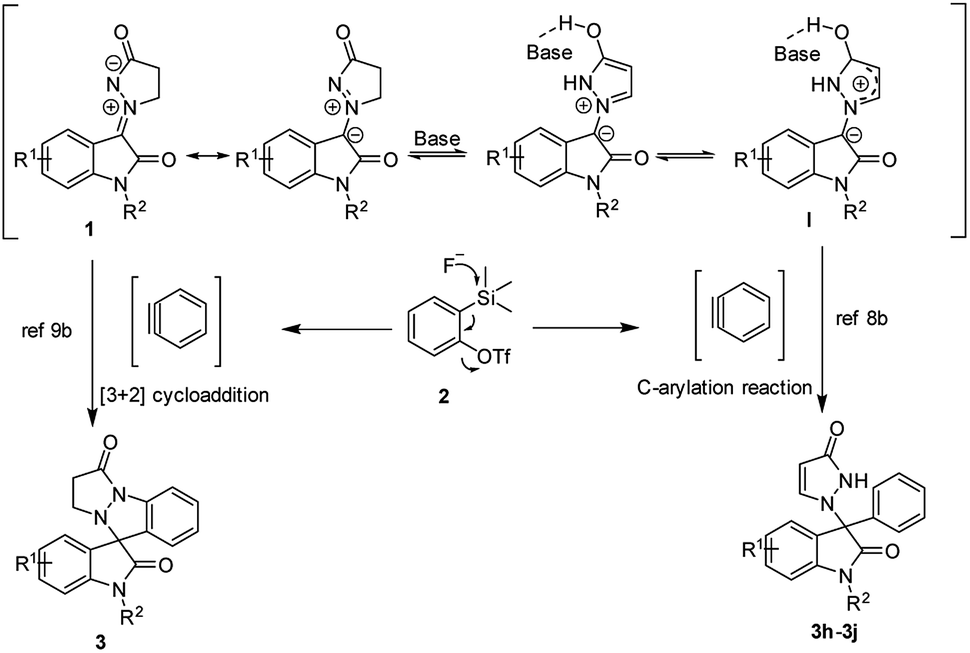 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Drawing inspiration from these 3,3-disubstituted oxindoles, a number of new drugs and lead compounds have been developed, especially in the field of anti-tumor.11 Therefore, the target compounds were assayed for in vitro antitumor activity against hepatocellular carcinoma (Hep3B) using the standard MTT method (Table 2). Unfortunately, most compounds showed little bioactivity against Hep3B cells. Among all the compounds, only 3j (4986.0 nM) and 4 (601.7 nM) showed good antitumor activities against Hep3B cells. And the results maybe suggest that these new 3,3-disubstituted oxindoles framework could be a new template for the design of novel anticancer molecules.
In summary, we have established an efficient method for the [3 + 2] annulation/C-arylation of isatin N,N′-cyclic azomethine imine 1,3-dipole 1 with in situ generated arynes, which constructed biologically important 3,3-disubstituted oxindoles in average good yields (up to 85% yield). The present methodology is both concise and mild, practical and one-pot method. The in vitro antitumor activity assay indicated that these scaffolds displayed moderate antitumor activities. Further chemical modification and biological exploration of these compounds are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21907053) and we also sincerely thank the China Pharmaceutical University for providing spectra.Notes and references
- (a) V. Polshettiwar and R. S. Varma, Curr. Opin. Drug Discovery Dev., 2007, 10, 723 CAS; (b) I. Sharma and D. S. Tan, Nat. Chem., 2013, 5, 157 CrossRef CAS PubMed; (c) A. Padwa and S. K. Bur, Tetrahedron, 2007, 63, 5341 CrossRef CAS PubMed; (d) D. M. D'Souza and T. J. Muller, Chem. Soc. Rev., 2007, 36, 1095 RSC; (e) S. Dadiboyena, Eur. J. Med. Chem., 2013, 63, 347 CrossRef CAS PubMed; (f) R. W. DeSimone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Comb. Chem. High Throughput Screening, 2004, 7, 473 CrossRef CAS PubMed; (g) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS PubMed.
- (a) K. Bernard, S. Bogliolo and J. Ehrenfeld, Br. J. Pharmacol., 2005, 144, 1037 CrossRef CAS PubMed; (b) C. S. L. Gal, D. Sylvain, G. Brossard, M. Manning, J. Simiand, R. Gaillard, G. Griebel and G. Guillon, Stress, 2003, 6, 199 CrossRef PubMed.
- M. Rottmann, C. McNamara, B. S. K. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. Gonzalez-Paez, L. Lakshiminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H. P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed.
- J. W. Skiles and D. McNeil, Tetrahedron Lett., 1990, 31, 7277 CrossRef CAS.
- (a) Y. Zhao, S. Yu, W. Sun, L. Liu, J. Lu, D. McEachern, S. Shargary, D. Bernard, X. Li, T. Zhao, P. Zou, D. Sun and S. Wang, J. Med. Chem., 2013, 56, 5553 CrossRef CAS PubMed; (b) A. Aguilar, J. Lu, L. Liu, D. Du, D. Bernard, D. McEachern, S. Przybranowski, X. Li, R. Luo, B. Wen, D. Sun, H. Wang, J. Wen, G. Wang, Y. Zhai, M. Guo, D. Yang and S. Wang, J. Med. Chem., 2017, 60, 2819 CrossRef CAS PubMed; (c) C. J. A. Ribeiro, J. D. Amaral, C. M. P. Rodrigues, R. Moreira and M. M. M. Santos, MedChemComm, 2016, 7, 420 RSC; (d) A. Bertamino, M. Soprano, S. Musella, M. R. Rusciano, M. Sala, E. Vernieri, V. Di Sarno, A. Limatola, A. Carotenuto, S. Cosconati, P. Grieco, E. Novellino, M. Illario, P. Campiglia and I. Gomez-Monterrey, J. Med. Chem., 2013, 56, 5407 CrossRef CAS PubMed.
- G. Periyasami, R. Raghunathan, G. Surendiran and N. Mathivanan, Bioorg. Med. Chem. Lett., 2008, 18, 2342 CrossRef CAS PubMed.
- For selected 1,3-dipolar cycloaddition reviews, see: (a) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887 CrossRef CAS PubMed; (b) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed; (c) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (d) Y. Shi, G. G. Wang, Z. Y. Chen, M. H. Wu, J. Wang, L. Trigoura, H. B. Guo, Y. L. Xing and S. F Sun, J. Heterocycl. Chem., 2020, 57, 2044 CrossRef CAS; (e) T. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211 CrossRef.
- (a) X. Wang, P. Yang, Y. Zhang, C. Z. Tang, F. Tian, L. Peng and L. X. Wang, Org. Lett., 2017, 19, 646 CrossRef CAS PubMed; (b) X. Wang, L. Wu, P. Yang, X. J. Song, H. X. Ren, L. Peng and L. X. Wang, Org. Lett., 2017, 19, 3051 CrossRef CAS PubMed; (c) X. J. Song, H. X. Ren, M. Xiang, C. Y. Li, F. Tian and L. X. Wang, J. Org. Chem., 2020, 85, 3921 CrossRef CAS PubMed.
- (a) S. Hu, J. Zhang and Q. Jin, New J. Chem., 2018, 42, 7025 RSC; (b) Q. Jin, J. Zhang, C. Jiang, D. Zhang, M. Gao and S. Hu, J. Org. Chem., 2018, 83, 8410 CrossRef CAS PubMed.
- F. M. Moghaddam, M. Eslami, A. Siahpoosh and G. Hoda, New J. Chem., 2019, 43, 10318 RSC.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (b) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/d0ra06404a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |