
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MIDA boronate allylation – synthesis of ibuprofen†
David Phillips,
Glen Brodie,
Sarah Memarzadeh,
Gi Lum Tang and
David J. France *
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: david.france@glasgow.ac.uk
First published on 18th August 2020
Abstract
MIDA boronates are among the most useful reagents for the Suzuki–Miyaura reaction. This chemistry typically generates new bonds between two aromatic rings, thereby restricting access to important areas of chemical space. Here we demonstrate the coupling of MIDA boronates to allylic electrophiles, including a new synthesis of the well-known COX inhibitor ibuprofen.
Introduction
N-Methyliminodiacetic acid (MIDA) boronates are one of the most useful organoboranes for the Suzuki–Miyaura cross-coupling strategy, as pioneered by Burke et al. (Fig. 1).1 In addition to helping to suppress undesired side reactions such as protodeboronation, these BMIDA compounds have shown promise in enabling carbon–carbon bond formation through an iterative “building block” approach. This chemistry takes advantage to two key properties of MIDA boronates to enable iterative couplings: the orthogonal reactivity of these boronates under aqueous vs. anhydrous conditions, and crucially a “catch-and-release” purification based on the chromatographic properties of the BMIDA functionality. By deprotecting MIDA boronates under aqueous conditions, followed by Pd-catalyzed coupling, a wide array of targets have been prepared using this methodology including natural products, pharmaceuticals, biological probes, and materials components.2 Related work by Watson et al. has used a controlled speciation approach for iterative biaryl coupling of boronic acid pinacol esters with MIDA boronates.3 While iterative strategies are widely used for generating biopolymers like polypeptides and polynucleic acids, the diversity of chemical bonds found in small molecules has hindered the development of an approach to preparing these compounds that is both modular and general. Iterative synthesis strategies that can address the challenging structural complexity posed by small molecules thus have the potential to transform the way synthetic chemistry is conducted.4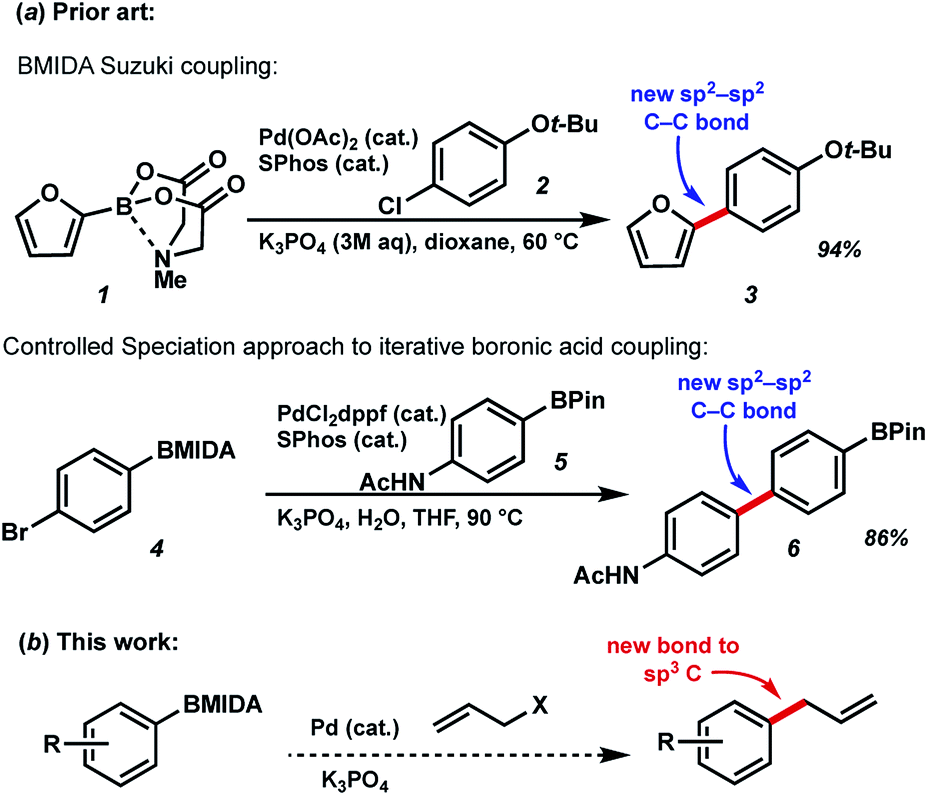 | ||
| Fig. 1 (a) Previous BMIDA coupling strategies (b) proposed BMIDA allylation to expand scope to generating new bonds to sp3-hybridized carbons. | ||
Use of MIDA boronates in Pd-catalyzed coupling has enabled the synthesis of a wide array of targets.2,4 A hallmark of these Pd-catalyzed C–C bond formations is that they are used almost universally to couple two carbons that are sp2 hybridised, most commonly 2 aromatic rings. The relative ease with which flat aromatic rings can be coupled has contributed to the proliferation of these structures in MedChem programmes, however it is now deemed partially responsible for the current crisis in compound attrition rate.5 This drawback is also important because increased sp3-C content correlates to clinical success.6 Very few examples of coupling MIDA boronates to sp3 hybridised carbons are to be found in the literature,7 and we chose to further explore this chemistry (Fig. 1b), including in the context of drug synthesis. Our approach builds upon the well precedented allylation of boronic acids,8 while enabling the synthesis of new targets that could not be accessed using standard Csp2–Csp2 MIDA boronate coupling chemistry (Fig. 2).
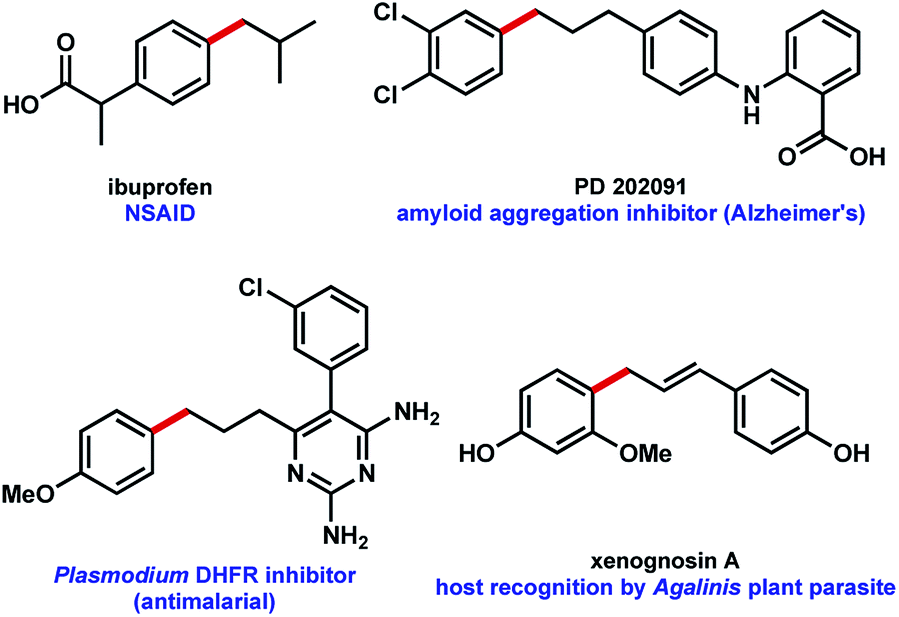 | ||
| Fig. 2 Potential bioactive targets for a MIDA boronate allylation approach to C–C coupling.9 | ||
Results and discussion
Our initial efforts to allylate aryl BMIDA compounds were quickly rewarded, as 2-naphthyl MIDA boronate could be efficiently coupled with allyl bromide using standard hydrolysis conditions (Fig. 3). Pleasingly, the reaction could be extended to aryl halide containing substrates functionalized at each position of a phenyl ring. Clearly these functional handles allow for further couplings. Furthermore, electron withdrawing nitro and cyano substituents were well tolerated, and a vinyl MIDA boronate could also be coupled. Use of an unprotected phenol resulted in a decreased yield of the desired product of C–C bond formation, due in large part to competing O-allylation, even when K3PO4 was replaced with the milder base NaHCO3. 2-MIDA boronate substituted furan and N-Boc-pyrrole could also be allylated in moderate yield. Attempted allylation of other heterocycles including 3-pyridyl MIDA boronate was less successful, due in part to competing side reactions such as N-allylation. Interestingly, we were able to couple neo-pentyl BMIDA to cinnamyl chloride resulting in a new sp3–sp3 C–C bond (9l). This process did not prove general, with other sp3–sp3 candidates such as methyl BMIDA and cyclobutyl BMIDA having limited success (data not shown). Finally, we were able to couple a BMIDA-modified phenylalanine to cinnamyl chloride in excellent yield, demonstrating this methodology's potential use in peptide modification.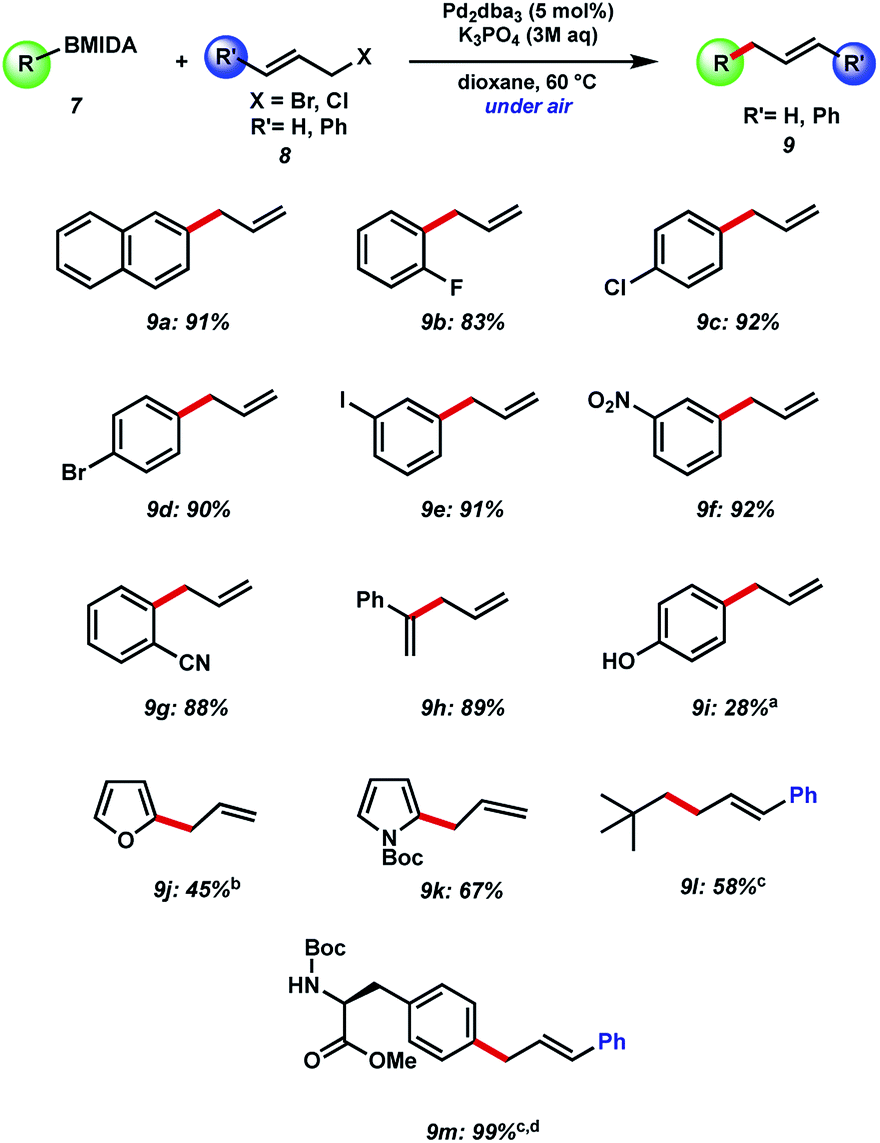 | ||
| Fig. 3 Substrate scope of BMIDA precursor with allyl bromide and cinnamyl chloride. Yields reported are by NMR unless stated. aK3PO4 replaced with NaHCO3. bReaction carried out at room temperature. cDioxane replaced with benzene. dIsolated yield. | ||
Next, we chose to examine the substrate tolerance on the allyl halide component by coupling 2-naphthyl MIDA boronate 7a with a range of commercial allyl halides (Fig. 4). 2-Methyl allyl bromide reacted successfully in comparable yield to the unfunctionalized system above. Coupling of cinnamyl chloride also proceeded well, and with excellent regiocontrol. BMIDA allylation with 1-methyallyl chloride and prenyl chloride resulted in decreased regioselectivity, though still good isolated yields.
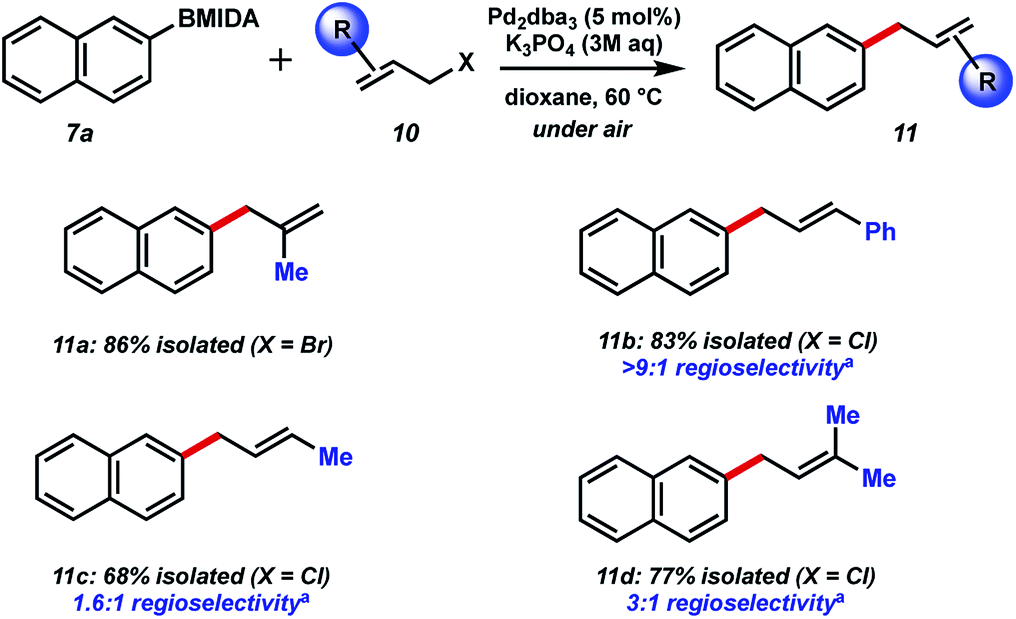 | ||
| Fig. 4 Substrate scope of allyl halide partner. aMajor regioisomer shown. | ||
In order to study the mechanism of the BMIDA allylation, we undertook a deuterium labelling study using dideuterioallyl bromide (Fig. 5). Coupling with 2-naphthyl MIDA boronate proceeded well, providing deuterated products 13 and 14 in 83% combined yield, but more importantly in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This outcome is consistent with the intermediacy of π-allyl species 12, formed by oxidative addition to the allyl halide, and transmetallation with the boronic acid generated in situ.
:1 ratio. This outcome is consistent with the intermediacy of π-allyl species 12, formed by oxidative addition to the allyl halide, and transmetallation with the boronic acid generated in situ.
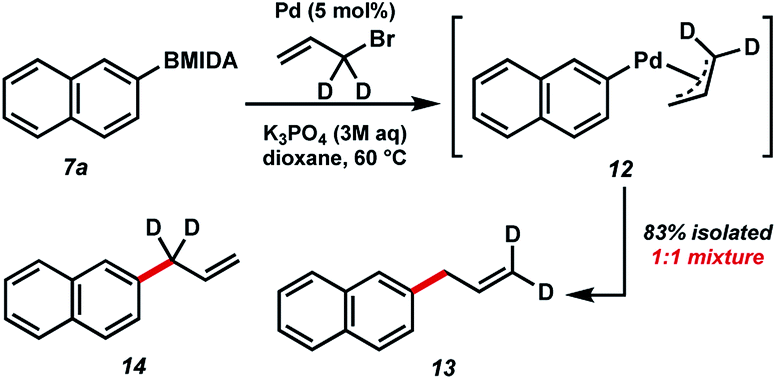 | ||
| Fig. 5 Isotopically labelled mechanistic probe. | ||
Finally, we chose to exemplify the utility of the BMIDA allylation methodology by featuring it in the synthesis of the well-known NSAID ibuprofen (Fig. 6). Our initial approach called for an enolate arylation reaction of a propionate ester with p-bromo aryl BMIDA 4.10 Several conditions proved unsuccessful, likely due to complications arising from the MIDA functional group. Recourse to a Negishi arylation11 of α-bromo-t-butyl propionate however proved successful using QPhos as a ligand for Pd.12 Pleasingly, BMIDA allylation of 16 proceeded well, providing the coupled product in 73% isolated yield. Completion of the synthesis of ibuprofen was realized by alkene hydrogenation and deprotection of the carboxylic acid. Attempts to use the same Pd source to effect multiple transformations in this synthesis met with limited success.
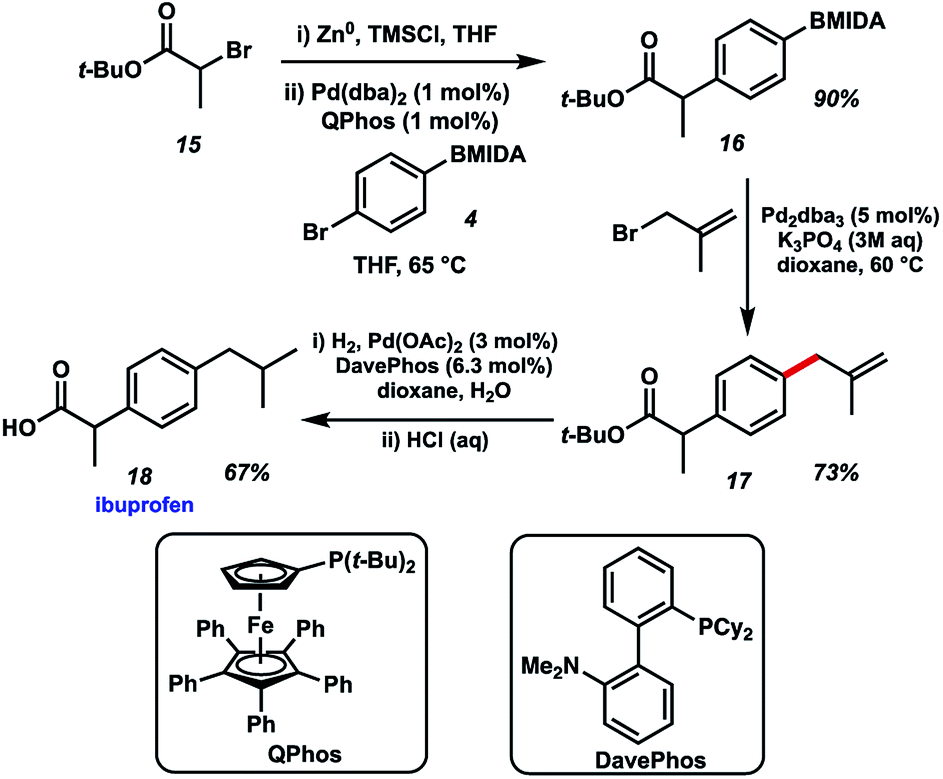 | ||
| Fig. 6 Synthesis of ibuprofen using BMIDA allylation. | ||
Conclusions
We have expanded the scope of MIDA boronate coupling to generate new bonds to sp3-hybridized carbons directly. The coupling is tolerant of a range of functional groups, including aryl halides, can be used with different allyl halide electrophiles, and is competent for sp3–sp3 C–C bond formation. This transformation can serve as a useful addition to the BMIDA-based approach to generating small molecules, as shown by a new synthesis of the widely used drug ibuprofen.Experimental section
Reactions involving air-sensitive agents and dry solvents were performed in glassware that had been dried in an oven (150 °C) or flame-dried prior to use. These reactions were carried out with the exclusion of air using an argon atmosphere. NMR spectra were recorded on a Bruker DPX-400 spectrometer (1H NMR at 400 MHz and 13C NMR at 100 MHz) or a Bruker DPX-500 spectrometer (1H NMR at 500 MHz and 13C NMR at 125 MHz). Chemical shifts are reported in ppm. 1H NMR spectra were recorded with CDCl3 or d6-DMSO as the solvent using residual proton-containing solvent as internal standard, and for 13C NMR spectra the chemical shifts are reported relative to the central resonance of CDCl3 or d6-DMSO. Signals in NMR spectra are described as singlet (s), doublet (d), triplet (t), multiplet (m), etc. or combination of these, which refers to the spin–spin coupling pattern observed. Spin–spin coupling constants reported are uncorrected. Two-dimensional (COSY, HSQC, HMBC, NOESY) NMR spectroscopy was used where appropriate to assist the assignment of signals in the 1H and 13C NMR spectra. IR spectra were obtained employing a Shimadzu FTIR-8400 instrument with a Golden Gate™ attachment that uses a type IIa diamond as a single reflection element so that the IR spectrum of the compound (solid or liquid) could be detected directly (thin layer). High resolution mass spectra were recorded under FAB, ESI, EI and CI conditions by the analytical services at the University of Glasgow. Flash column chromatography was performed using forced flow of the indicated solvent system on EMD Geduran Silica Gel 60 as solid support and HPLC graded solvents as eluant. Reactions were monitored by thin layer chromatography (TLC) on Merck silica gel 60 covered aluminium sheets. TLC plates were developed under UV-light and/or with an acidic ethanolic anisaldehyde solution or a KMnO4-solution. All reagents were purchased from commercial suppliers and used without further purification unless otherwise stated.2-Naphthyl boronic acid MIDA ester (7a)
Following a literature procedure,13 naphthyl boronic acid (0.86 g, 5.0 mmol) and MIDA (0.74 g, 5.0 mmol) were stirred in DMF (15 mL) at 85 °C for 16 h then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (100 mL) and H2O (100 mL) then separated. The organic phase was washed with H2O (50 mL) and brine (3 × 50 mL) then dried (Na2SO4), filtered and concentrated in vacuo. Purification by flash chromatography (diethyl ether:methanol, 98.5:1.5 then THF) gave a yellow oil. Addition of Et2O formed a white precipitate which was collected by vacuum filtration affording the title compound as a white solid (1.3 g, 93%). Analytical data were in accordance with literature values.13 1H NMR (400 MHz, d6-DMSO) δ (ppm): 7.99 (1H, s, Ar-H), 7.94–7.88 (3H, m, Ar-H), 7.57–7.50 (3H, m, Ar-H), 4.38 (2H, d, J = 17.2 Hz, 2× NCHH), 4.17 (2H, d, J = 17.1 Hz, 2× NCHH), 2.52 (3H, s, NCH3).
Neopentyl boronic acid MIDA ester (7l)
Neopentyl boronic acid (200 mg, 1.72 mmol) and MIDA (253 mg, 1.72 mmol) were stirred in DMF (5 mL) at 85 °C for 16 h then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (30 mL) and H2O (30 mL) then separated. The organic phase was washed 3 times with 5% LiCl solution then dried over Na2SO4, filtered and concentrated in vacuo. The residue was dissolved in a minimal amount of MeCN and dropped into ice-cold Et2O forming a white precipitate. The solvent was removed by filtration affording the title compound as a white solid (226 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 3.76 (d, J = 16.2 Hz, 2H, NCHH), 3.60 (d, J = 16.1 Hz, 2H, NCHH), 2.84 (s, 3H, NCH3), 1.04 (s, 9H, 3× CH3), 0.58 (s, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 166.9 (2× C), 110.1 (CH2), 61.4 (2× CH2), 45.5 (CH3), 32.4 (3× CH3), 30.3 (C); IR (solid) 2949, 2862, 1739, 1464, 1109, 1032 cm−1; HRMS (ESI) exact mass calculated for C10H18NO410BNa [M + Na]+ m/z 249.1257, found m/z 249.1248.(S)-(4-(2-((tert-Butoxycarbonyl)amino)-3-methoxy-3-oxopropyl)phenyl)boronic acid MIDA ester (7m)
The boronic acid (200 mg, 0.620 mmol) and MIDA (91 mg, 0.62 mmol) were stirred in DMF (5 mL) at 85 °C for 16 h then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (30 mL) and H2O (30 mL) then separated. The organic phase was washed 3 times with 5% LiCl solution then dried over Na2SO4, filtered and concentrated in vacuo. The residue was dissolved in a minimal amount of MeCN and dropped into ice-cold Et2O forming a white precipitate. The solvent was removed by filtration affording the title compound as a white solid (223 mg, 82%).1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 7.8 Hz, 2H, Ar-H), 7.16 (d, J = 7.9 Hz, 2H, Ar-H), 5.00 (d, J = 8.1 Hz, 1H, NH), 4.62–4.52 (m, 1H, CH), 3.95 (d, J = 16.4 Hz, 2H, NCHH), 3.76 (dd, J = 16.4, 4.7 Hz, 2H, NCHH), 3.71 (s, 3H, OCH3), 3.13 (dd, J = 13.4, 5.4 Hz, 1H, CHH), 3.03 (dd, J = 13.4, 6.3 Hz, 1H, CHH), 2.54 (s, 3H, NCH3), 1.41 (s, 9H, 3× CH3); 13C NMR (100 MHz, CDCl3) δ 172.3 (C), 167.8 (2× C), 155.2 (C), 138.2 (C), 132.6 (2× CH), 129.5 (2× CH), 80.1 (C), 61.9 (2× CH2), 54.6 (CH), 52.4 (CH3), 47.7 (CH3), 38.5 (CH2), 28.4 (3× CH3). The 13C peak corresponding to the carbon atom adjacent to the boron atom was not observed. IR (thin film) 2980, 1743, 1705, 1612, 1500, 908 cm−1; HRMS (ESI) exact mass calculated for C20H27N2O810BNa [M + Na]+ m/z 457.1753, found m/z 457.1751.General procedure for allylation of MIDA boronates
A screw-top glass vial was charged with boronic acid MIDA ester (1 equiv.), dioxane (0.08 M), allyl bromide (2 equiv.) and Pd2dba3 (5 mol%). A 3 M aq. solution of K3PO4 (1:5 v/v ratio to dioxane) was added and the vial sealed under ambient atmosphere. The resulting mixture was heated to 60 °C by immersion of the entire vial into a preheated aluminium block until the substrate had been consumed, as judged by TLC analysis. The reaction mixture was cooled to room temperature, quenched with 1 M aq. NaOH then extracted with Et2O (3×). The combined organic extracts were dried (Na2SO4), filtered and concentrated in vacuo. Addition of 1,3,5-trimethoxybenzene (17 mg, 0.10 mmol) allowed determination of the yield by 1H NMR.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.19 (1H, dq, J = 10.5, 1.7 Hz, CCHa), 5.17–5.15 (1H, m, CCHb), 3.59 (2H, d, J = 6.6 Hz, CH2).CH2), 5.10–5.05 (2H, m, CHCH2), 3.41 (2H, dd, J = 6.6, 1.5 Hz, CH2).CH2), 5.11–5.10 (1H, m, CHCHa), 5.09–5.05 (1H, m, CHCHb), 3.36 (2H, d, J = 6.7 Hz, CH2).CH2), 5.11–5.03 (2H, m, CHCH2), 3.34 (2H, d, J = 6.6 Hz, CH2).CH2), 5.13–5.06 (2H, m, CHCH2), 3.33 (2H, d, J = 6.6 Hz, CH2).CH2), 5.17 (1H, dq, J = 10.3, 1.4 Hz, CHCHa), 5.13 (1H, dq, J = 17.3, 1.6 Hz, CHCHb), 3.50 (2H, d, J = 6.6 Hz, CH2).CH2), 5.17–5.08 (2H, m, CHCH2), 3.61 (2H, d, J = 6.6 Hz, CH2).CH2), 5.42–5.41 (1H, m, CCHa), 5.16–5.04 (3H, m, CCHb, CHCH2), 3.27–3.25 (2H, m, CH2).CH2), 5.08–5.03 (2H, m, CHCH2), 4.86 (1H, s, OH), 3.32 (2H, d, J = 6.7 Hz, CH2).CH2), 5.16–5.10 (2H, m, CHCH2), 3.39 (2H, d, J = 6.5 Hz, CH2).CH2), 5.98–5.96 (1H, m, Ar-H), 5.10–5.02 (2H, m, CHCH2), 3.61 (2H, dd, J = 6.3, 1.2 Hz, CH2), 1.58 (9H, s, C(CH3)3).CH), 6.33 (dt, J = 15.8, 6.7 Hz, 1H, CH2CHCH), 4.96 (d, J = 8.4 Hz, 1H, NH), 4.58 (q, J = 6.8 Hz, 1H, CH), 3.72 (s, 3H, OCH3), 3.52 (d, J = 6.8 Hz, 2H, CH2), 3.06 (m, 2H, CH2), 1.41 (s, 9H, 3× CCH3). 13C NMR (101 MHz, CDCl3) δ 172.5 (C), 155.2 (C), 139.1 (C), 137.6 (C), 133.9 (C), 131.3 (CH), 129.6 (2× CH), 129.2 (CH), 129.0 (2× CH), 128.6 (2× CH), 127.3 (CH), 126.3 (2× CH), 80.1 (C), 54.6 (CH), 52.3 (CH3), 39.1 (CH2), 38.1 (CH2), 28.5 (3× CH3). IR (thin film): 1719, 1366, 1165, 692 cm−1. HRMS (ESI) exact mass calculated for C24H29NO4Na [M + Na]+ m/z 418.1989, found m/z 418.1979.CHa), 4.81–4.80 (1H, m, CCHb), 3.50 (2H, s, CH2), 1.72 (3H, s, CH3).CH), 6.45 (1H, dt, J = 15.8, 6.3 Hz, CH2CHCH), 3.73 (2H, d, J = 6.2 Hz, CH2CHCH).:1). Analytical data were in accordance with literature values.24 1H NMR (400 MHz, CDCl3) δ 7.82–7.77 (3H, m, Ar-H), 7.66 (1H, s, Ar-H), 7.62 (1H, s, Ar-H), 7.48–7.40 (2H, m, Ar-H), 7.39–7.32 (1H, m, Ar-H), 6.10 (1H, ddd, J = 16.9, 10.3, 6.4 Hz, CHCH2), 5.72–5.54 (2H, m, CHCH), 5.14–5.08 (2H, m, CHCH2), 3.67–3.57 (1H, m, CHCH3), 3.49 (2H, dd, J = 6.5, 1.2 Hz, CH2), 1.73 (3H, dq, J = 6.1, 1.4 Hz, CHCH3), 1.47 (3H, d, J = 7.0 Hz, CHCHCH3).:1). Analytical data were in accordance with literature values.26 1H NMR (400 MHz, CDCl3) δ 0.82 (3H, m, Ar-H), 7.62 (1H, s, Ar-H), 7.52–7.40 (2H, m, Ar-H), 7.34 (1H, dd, J = 8.5, 1.8 Hz, Ar-H), 6.12 (1H, dd, J = 17.3, 10.7 Hz, CHCH2), 5.43 (1H, septet, J = 7.3, 1.4 Hz, CHC(CH3)2), 5.15–5.10 (2H, m, CHCH2), 3.52 (2H, d, J = 7.3 Hz, CH2), 1.79 (6H, dd, J = 4.4, 1.4 Hz, CHC(CH3)2), 1.52 (6H, s, 2× CH3).:1 isomer ratio). Analytical data were in accordance with literature values.28 1H NMR (400 MHz, CDCl3) δ 7.83–7.78 (3H, m, Ar-H), 7.64 (1H, dd, J = 1.8, 0.8 Hz, Ar-H), 7.49–7.42 (2H, m, Ar-H), 7.35 (1H, dd, J = 8.4, 1.8 Hz, Ar-H), 6.09–6.02 (1H, m, CHCH2/CHCD2), 5.16 (0.5H, dd, J = 12.0, 2.0 Hz, CHCHH), 5.13 (0.5H, dd, J = 5.2, 2.0 Hz, CHCHH), 3.57 (1H, dd, J = 6.6, 0.7 Hz, CH2CHCD2); 13C NMR (100 MHz, CDCl3) δ 137.7 (2× C), 137.4 (CH), 137.2 (CH), 133.8 (2× C), 132.3 (2× C), 128.0 (2× CH), 127.8 (2× CH), 127.6 (2× CH), 127.5 (2× CH), 126.8 (2× CH), 126.1 (2× CH), 125.4 (2× CH), 116.2 (CH2), 115.7 (p, 1J(C-D) = 23.7 Hz, CD2), 40.4 (CH2), 39.8 (p, 1J(C-D) = 19.6 Hz, CD2); IR (thin film) 3053, 2923, 2113, 1689 cm−1; HRMS (EI) exact mass calculated for C13H10D2 [M]+ m/z 170.1059, found m/z 170.1080.:methanol, 98.5:1.5 then THF) afforded the title compound as a light pink solid (0.32 g, 90%).1H NMR (400 MHz, d6-DMSO) δ 7.39 (2H, d, J = 8.1 Hz, Ar-H), 7.25 (2H, d, J = 8.1 Hz, Ar-H), 4.32 (2H, d, J = 17.2 Hz, 2× NCHH), 4.11 (2H, d, J = 17.2 Hz, 2× NCHH), 3.64 (1H, q, J = 7.1 Hz, CHCH3), 2.49 (3H, s, NCH3), 1.40 (3H, s, CH3), 1.34 (9H, s, 3× CH3); 13C NMR (100 MHz, d6-DMSO) δ 174.3 (C), 173.0 (C), 169.3 (2× C), 141.6 (C), 132.5 (2× CH), 126.5 (2× CH), 79.9 (C), 61.8 (CH2), 47.5 (CH3), 45.5 (CH), 27.6 (3× CH3), 18.5 (CH3); IR (solid) 2978, 1724, 1713, 1367, 1150 cm−1; HRMS (ESI) exact mass calculated for C18H24BNNaO6 [M + Na]+ m/z 384.1589, found m/z 384.1575.CHH), 4.73–4.71 (1H, m, CCHH), 3.59 (1H, q, J = 7.2 Hz, CHCH3), 3.29 (2H, s, Ar-CH2C), 1.67 (3H, s, CCH3), 1.44 (3H, d, J = 7.2 Hz, CHCH3), 1.39 (9H, s, 3× CCH3); 13C NMR (100 MHz, CDCl3) δ 173.9 (C), 145.2 (C), 139.0 (C), 138.3 (C), 128.9 (2× CH), 127.3 (2× CH), 111.6 (CH2), 80.3 (C), 46.0 (CH), 44.1 (CH2), 27.8 (3× CH3), 21.9 (CH3), 18.3 (CH3); IR (thin film) 2978, 2934, 1728, 1452, 1148, 739 cm−1; HRMS (ESI) exact mass calculated for C17H24NaO2 [M + Na]+ m/z 283.1669, found m/z 283.1666.
CH2), 5.19 (1H, dq, J = 10.5, 1.7 Hz, CCHa), 5.17–5.15 (1H, m, CCHb), 3.59 (2H, d, J = 6.6 Hz, CH2).CH2), 5.10–5.05 (2H, m, CHCH2), 3.41 (2H, dd, J = 6.6, 1.5 Hz, CH2).CH2), 5.11–5.10 (1H, m, CHCHa), 5.09–5.05 (1H, m, CHCHb), 3.36 (2H, d, J = 6.7 Hz, CH2).CH2), 5.11–5.03 (2H, m, CHCH2), 3.34 (2H, d, J = 6.6 Hz, CH2).CH2), 5.13–5.06 (2H, m, CHCH2), 3.33 (2H, d, J = 6.6 Hz, CH2).CH2), 5.17 (1H, dq, J = 10.3, 1.4 Hz, CHCHa), 5.13 (1H, dq, J = 17.3, 1.6 Hz, CHCHb), 3.50 (2H, d, J = 6.6 Hz, CH2).CH2), 5.17–5.08 (2H, m, CHCH2), 3.61 (2H, d, J = 6.6 Hz, CH2).CH2), 5.42–5.41 (1H, m, CCHa), 5.16–5.04 (3H, m, CCHb, CHCH2), 3.27–3.25 (2H, m, CH2).CH2), 5.08–5.03 (2H, m, CHCH2), 4.86 (1H, s, OH), 3.32 (2H, d, J = 6.7 Hz, CH2).CH2), 5.16–5.10 (2H, m, CHCH2), 3.39 (2H, d, J = 6.5 Hz, CH2).CH2), 5.98–5.96 (1H, m, Ar-H), 5.10–5.02 (2H, m, CHCH2), 3.61 (2H, dd, J = 6.3, 1.2 Hz, CH2), 1.58 (9H, s, C(CH3)3).CH), 6.33 (dt, J = 15.8, 6.7 Hz, 1H, CH2CHCH), 4.96 (d, J = 8.4 Hz, 1H, NH), 4.58 (q, J = 6.8 Hz, 1H, CH), 3.72 (s, 3H, OCH3), 3.52 (d, J = 6.8 Hz, 2H, CH2), 3.06 (m, 2H, CH2), 1.41 (s, 9H, 3× CCH3). 13C NMR (101 MHz, CDCl3) δ 172.5 (C), 155.2 (C), 139.1 (C), 137.6 (C), 133.9 (C), 131.3 (CH), 129.6 (2× CH), 129.2 (CH), 129.0 (2× CH), 128.6 (2× CH), 127.3 (CH), 126.3 (2× CH), 80.1 (C), 54.6 (CH), 52.3 (CH3), 39.1 (CH2), 38.1 (CH2), 28.5 (3× CH3). IR (thin film): 1719, 1366, 1165, 692 cm−1. HRMS (ESI) exact mass calculated for C24H29NO4Na [M + Na]+ m/z 418.1989, found m/z 418.1979.CHa), 4.81–4.80 (1H, m, CCHb), 3.50 (2H, s, CH2), 1.72 (3H, s, CH3).CH), 6.45 (1H, dt, J = 15.8, 6.3 Hz, CH2CHCH), 3.73 (2H, d, J = 6.2 Hz, CH2CHCH).:1). Analytical data were in accordance with literature values.24 1H NMR (400 MHz, CDCl3) δ 7.82–7.77 (3H, m, Ar-H), 7.66 (1H, s, Ar-H), 7.62 (1H, s, Ar-H), 7.48–7.40 (2H, m, Ar-H), 7.39–7.32 (1H, m, Ar-H), 6.10 (1H, ddd, J = 16.9, 10.3, 6.4 Hz, CHCH2), 5.72–5.54 (2H, m, CHCH), 5.14–5.08 (2H, m, CHCH2), 3.67–3.57 (1H, m, CHCH3), 3.49 (2H, dd, J = 6.5, 1.2 Hz, CH2), 1.73 (3H, dq, J = 6.1, 1.4 Hz, CHCH3), 1.47 (3H, d, J = 7.0 Hz, CHCHCH3).:1). Analytical data were in accordance with literature values.26 1H NMR (400 MHz, CDCl3) δ 0.82 (3H, m, Ar-H), 7.62 (1H, s, Ar-H), 7.52–7.40 (2H, m, Ar-H), 7.34 (1H, dd, J = 8.5, 1.8 Hz, Ar-H), 6.12 (1H, dd, J = 17.3, 10.7 Hz, CHCH2), 5.43 (1H, septet, J = 7.3, 1.4 Hz, CHC(CH3)2), 5.15–5.10 (2H, m, CHCH2), 3.52 (2H, d, J = 7.3 Hz, CH2), 1.79 (6H, dd, J = 4.4, 1.4 Hz, CHC(CH3)2), 1.52 (6H, s, 2× CH3).:1 isomer ratio). Analytical data were in accordance with literature values.28 1H NMR (400 MHz, CDCl3) δ 7.83–7.78 (3H, m, Ar-H), 7.64 (1H, dd, J = 1.8, 0.8 Hz, Ar-H), 7.49–7.42 (2H, m, Ar-H), 7.35 (1H, dd, J = 8.4, 1.8 Hz, Ar-H), 6.09–6.02 (1H, m, CHCH2/CHCD2), 5.16 (0.5H, dd, J = 12.0, 2.0 Hz, CHCHH), 5.13 (0.5H, dd, J = 5.2, 2.0 Hz, CHCHH), 3.57 (1H, dd, J = 6.6, 0.7 Hz, CH2CHCD2); 13C NMR (100 MHz, CDCl3) δ 137.7 (2× C), 137.4 (CH), 137.2 (CH), 133.8 (2× C), 132.3 (2× C), 128.0 (2× CH), 127.8 (2× CH), 127.6 (2× CH), 127.5 (2× CH), 126.8 (2× CH), 126.1 (2× CH), 125.4 (2× CH), 116.2 (CH2), 115.7 (p, 1J(C-D) = 23.7 Hz, CD2), 40.4 (CH2), 39.8 (p, 1J(C-D) = 19.6 Hz, CD2); IR (thin film) 3053, 2923, 2113, 1689 cm−1; HRMS (EI) exact mass calculated for C13H10D2 [M]+ m/z 170.1059, found m/z 170.1080.:methanol, 98.5:1.5 then THF) afforded the title compound as a light pink solid (0.32 g, 90%).1H NMR (400 MHz, d6-DMSO) δ 7.39 (2H, d, J = 8.1 Hz, Ar-H), 7.25 (2H, d, J = 8.1 Hz, Ar-H), 4.32 (2H, d, J = 17.2 Hz, 2× NCHH), 4.11 (2H, d, J = 17.2 Hz, 2× NCHH), 3.64 (1H, q, J = 7.1 Hz, CHCH3), 2.49 (3H, s, NCH3), 1.40 (3H, s, CH3), 1.34 (9H, s, 3× CH3); 13C NMR (100 MHz, d6-DMSO) δ 174.3 (C), 173.0 (C), 169.3 (2× C), 141.6 (C), 132.5 (2× CH), 126.5 (2× CH), 79.9 (C), 61.8 (CH2), 47.5 (CH3), 45.5 (CH), 27.6 (3× CH3), 18.5 (CH3); IR (solid) 2978, 1724, 1713, 1367, 1150 cm−1; HRMS (ESI) exact mass calculated for C18H24BNNaO6 [M + Na]+ m/z 384.1589, found m/z 384.1575.CHH), 4.73–4.71 (1H, m, CCHH), 3.59 (1H, q, J = 7.2 Hz, CHCH3), 3.29 (2H, s, Ar-CH2C), 1.67 (3H, s, CCH3), 1.44 (3H, d, J = 7.2 Hz, CHCH3), 1.39 (9H, s, 3× CCH3); 13C NMR (100 MHz, CDCl3) δ 173.9 (C), 145.2 (C), 139.0 (C), 138.3 (C), 128.9 (2× CH), 127.3 (2× CH), 111.6 (CH2), 80.3 (C), 46.0 (CH), 44.1 (CH2), 27.8 (3× CH3), 21.9 (CH3), 18.3 (CH3); IR (thin film) 2978, 2934, 1728, 1452, 1148, 739 cm−1; HRMS (ESI) exact mass calculated for C17H24NaO2 [M + Na]+ m/z 283.1669, found m/z 283.1666.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the University of Glasgow and EPSRC to DP (DTA award ref: EP/K503058/1) is gratefully acknowledged. We also thank GlaxoSmithKline for samples from the New Methodology Expansion Set.References
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed; (b) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- (a) J. W. Lehmann, D. J. Blair and M. D. Burke, Nat. Rev. Chem., 2018, 2, 0115 CrossRef PubMed; (b) Y.-I. Jo, M. D. Burke and C.-H. Cheon, Org. Lett., 2019, 21, 4201–4204 CrossRef CAS PubMed; (c) A. W. Sun, S. Lackner and B. M. Stoltz, Trends Chem., 2019, 1, 630–643 CrossRef; (d) O. O. Grygorenko, D. M. Volochnyuk, S. V. Ryabukhin and D. B. Judd, Chem.–Eur. J., 2020, 26, 1196–1237 CrossRef CAS PubMed.
- (a) J. W. B. Fyfe, C. P. Seath and A. J. B. Watson, Angew. Chem., Int. Ed., 2014, 53, 12077–12080 CrossRef CAS PubMed; (b) J. W. B. Fyfe, E. Valverde, C. P. Seath, A. R. Kennedy, J. M. Redmond, N. A. Anderson and A. J. B. Watson, Chem.–Eur. J., 2015, 21, 8951–8964 CrossRef CAS PubMed.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. J. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- (a) J. Tsoung, K. Krämer, A. Zajdlik, C. Liébert and M. Lautens, J. Org. Chem., 2011, 76, 9031–9045 CrossRef CAS PubMed; (b) C. P. Seath, J. W. B. Fyfe, J. J. Molloy and A. J. B. Watson, Angew. Chem., Int. Ed., 2015, 54, 9976–9979 CrossRef CAS PubMed; (c) K. J. Broadley, M. G. P. Buffat, E. Burnell, R. H. Davies, X. Moreau, S. Snee and E. J. Thomas, Org. Biomol. Chem., 2016, 14, 2057–2089 RSC; (d) D. Phillips, J. F. M. Hewitt and D. J. France, ACS Omega, 2018, 3, 8451–8459 CrossRef CAS PubMed.
- (a) M. Moreno-Mañas, F. Pajuelo and R. J. Pleixats, J. Org. Chem., 1995, 60, 2396–2397 CrossRef; (b) E. Alacid and C. Nájera, Org. Lett., 2008, 10, 5011–5014 CrossRef CAS PubMed; (c) F. Chahdoura, C. Pradel and M. Gómez, Adv. Synth. Catal., 2013, 355, 3648–3660 CrossRef CAS.
- (a) PD 202091: L. J. Simons, B. W. Caprathe, M. Callahan, J. M. Graham, T. Kimura, Y. Lai, H. LeVine III, W. Lipinski, A. T. Sakkab, Y. Tasaki, L. C. Walker, T. Yasunaga, Y. Ye, N. Zhuang and C. E. Augelli-Szafran, Bioorg. Med. Chem. Lett., 2009, 19, 654–657 CrossRef CAS PubMed; (b) Plasmodium DHFR inhibitor: S. Kamchonwongpaisan, R. Quarrell, N. Charoensetakul, R. Ponsinet, T. Vilaivan, J. Vanichtanankul, B. Tarnchompoo, W. Sirawaraporn, G. Lowe and Y. Yuthavong, J. Med. Chem., 2004, 47, 673–680 CrossRef CAS PubMed; (c) Xenognosin A: J. C. Steffens, D. G. Lynn, V. S. Kamat and J. L. Riopel, Ann. Bot., 1982, 50, 1–7 CrossRef CAS.
- W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed.
- B. Wong, X. Linghu, J. J. Crawford, J. Drobnick, W. Lee and H. Zhang, Tetrahedron, 2014, 70, 1508–1515 CrossRef CAS.
- T. Hama, S. Ge and J. F. Hartwig, J. Org. Chem., 2013, 78, 8250–8266 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef CAS PubMed.
- S. E. Denmark and N. S. Werner, J. Am. Chem. Soc., 2008, 130, 16382–16393 CrossRef CAS PubMed.
- S. Lin, C.-X. Song, G.-X. Cai, W.-H. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 12901–12903 CrossRef CAS PubMed.
- H. Naka, K. Ito, M. Ueno, K. Kobayashi and Y. Kondo, New J. Chem., 2010, 34, 1700–1706 RSC.
- P. H. Lee, S.-Y. Sung and K. Lee, Org. Lett., 2001, 3, 3201–3204 CrossRef CAS PubMed.
- T. Itou, Y. Yoshimi, T. Morita, Y. Tokunaga and M. Hatanaka, Tetrahedron, 2009, 65, 263–269 CrossRef CAS.
- P. H. Lee, Y. Heo, D. Seomoon, S. Kim and K. Lee, Chem. Commun., 2005, 14, 1874–1876 RSC.
- M. Uchiyama, T. Furuyama, M. Kobayashi, Y. Matsumoto and K. Tanaka, J. Am. Chem. Soc., 2006, 128, 8404–8405 CrossRef CAS PubMed.
- M. Ochiai, E. Fujita, M. Arimoto and H. Yamaguchi, Chem. Pharm. Bull., 1982, 30, 3994–3999 CrossRef CAS.
- M. F. Semmelhack, A. Chlenov and D. M. Ho, J. Am. Chem. Soc., 2005, 127, 7759–7773 CrossRef CAS PubMed.
- G. Ortar, Tetrahedron Lett., 2003, 44, 4311–4314 CrossRef CAS.
- D. Seomoon and P. H. Lee, J. Org. Chem., 2008, 73, 1165–1168 CrossRef CAS PubMed.
- H. Tsukamoto, T. Uchiyama, T. Suzuki and Y. Kondo, Org. Biomol. Chem., 2008, 6, 3005–3013 RSC.
- K. Lee, J. Lee and P. H. Lee, J. Org. Chem., 2002, 67, 8265–8268 CrossRef CAS PubMed.
- I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 4983–4987 CrossRef CAS PubMed.
- K. D. Reichl, N. L. Dunn, N. J. Fastuca and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 5292–5295 CrossRef CAS PubMed.
- G. S. Coumbarides, M. Dingjan, J. Eames, A. Flinn and J. Northen, J. Labelled Compd. Radiopharm., 2006, 49, 903–914 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03338c |
| This journal is © The Royal Society of Chemistry 2020 |