
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in conducting polymers: applications for electrochemistry
Somayeh Tajika,
Hadi Beitollahi *b,
Fariba Garkani Nejadc,
Iran Sheikh Shoaiec,
Mohammad A. Khalilzadehd,
Mehdi Shahedi Asle,
Quyet Van Le*f,
Kaiqiang Zhangg,
Ho Won Jang*g and
Mohammadreza Shokouhimehr*g
*b,
Fariba Garkani Nejadc,
Iran Sheikh Shoaiec,
Mohammad A. Khalilzadehd,
Mehdi Shahedi Asle,
Quyet Van Le*f,
Kaiqiang Zhangg,
Ho Won Jang*g and
Mohammadreza Shokouhimehr*g
aResearch Center for Tropical and Infectious Diseases, Kerman University of Medical Sciences, Kerman, Iran
bEnvironment Department, Institute of Science and High Technology and Environmental Sciences, Graduate University of Advanced Technology, Kerman, Iran. E-mail: h.beitollahi@yahoo.com
cDepartment of Chemistry, Faculty of Science, Shahid Bahonar University of Kerman, Kerman 76175-133, Iran
dDepartment of Biomaterials, College of Natural Resources, North Carolina State University, Raleigh, USA
eMarine Additive Manufacturing Centre of Excellence (MAMCE), University of New Brunswick, Fredericton, NB E3B 5A1, Canada
fInstitute of Research and Development, Duy Tan University, Da Nang 550000, Vietnam. E-mail: levanquyet@dtu.edu.vn
gDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: mrsh2@snu.ac.kr; hwjang@snu.ac.kr
First published on 13th October 2020
Abstract
Scientists have categorized conductive polymers as materials having strongly reversible redox behavior and uncommon combined features of plastics and metal. Because of their multifunctional characteristics, e.g., simplistic synthesis, acceptable environmental stability, beneficial optical, electronic, and mechanical features, researchers have largely considered them for diverse applications. Therefore, their capability of catalyzing several electrode reactions has been introduced as one of their significant features. A thin layer of the conducting polymer deposited on the substrate electrode surface can augment the electrode process kinetics of several solution species. Such electrocatalytic procedures with modified conducting polymer electrodes can create beneficial utilization in diverse fields of applied electrochemistry. This review article explores typical recent applications of conductive polymers (2016–2020) as active electrode materials for energy storage applications, electrochemical sensing, and conversion fields such as electrochemical supercapacitors, lithium-ion batteries, fuel cells, and solar cells.
1. Introduction
It is well known that electrochemistry entails phenomena in which a chemical modification results from electric forces and, conversely, chemical processes generate electric forces. This field involves features and behaviors of the electrolytic conductors in solid or liquid form. Numerous such phenomena occur at the electrolytic and electronic conductor interface, wherein the electric charge passage is connected to a redox chemical reaction. This reaction rate may be followed as an electric current with increased sensitivity. The contacts make up the electrodes of galvanic cells that may be utilized to convert chemicals to electrical energy as batteries or generate chemical products via electrolysis.1,2The development of novel techniques for performing fast in situ analyses has been considered as a major challenge as they present high sensitivity and accuracy for the detection of diverse materials with various features in real samples. Experts in the field have demonstrated that electroanalytical procedures would be among the encouraging alternatives for the quantitative and qualitative analyses of replacing the traditional techniques. Electrochemical sensing mechanisms are accompanied by benefits like simplistic instruments, miniaturization, higher selectivity and sensitivity, simplified utilization, minor sample pretreatment, portability, and shorter analysis time.3–5 Put differently, electrodes are capable of sensing substances found in the host with no damages to the host system. Nonetheless, electrochemical sensors suffer from limitations such as the electrochemically active interference in the sample, poor stability, difficult electron transfer pathway, as well as decreased limits of detection (LOD) and sensitivity.6,7 Consequently, diverse electrochemical procedures are combined with various sensing electrodes to augment their LOD and sensitivity via the modification of the electrode materials.
The storage and conversion of energies contribute significantly to the conservation of the available energy and improvement of its usage because of the existence of multiple energy resources in nature. Notably, a shorter storage time of just a few hours would be crucial in majority of applications; nonetheless, a lengthier storage of few months is sometimes necessary. In this aspect, electrochemical energy storage (EES) technology has been introduced as an encouraging tool for the storage of electricity in small- and large-scale applications due to the flexibility, higher energy conversion efficiency, as well as easy maintenance.8,9
It is worth noting that the use of instruments for electrochemical energy storage and conversion enable the storage of surplus energy in the case of a greater electricity supply than demand, and its release in the case of a greater demand than supply. Such a situation leads to higher efficiency of the grid, higher reliability, as well as lower costs. On the one hand, the grid-scale energy storage needs electrochemical machines with suitable power and energy output features, longer cycle life, higher efficiency, and lower costs. Therefore, accelerated advancements in devising electroactive substances for EES tools offered numerous attractive organic mechanisms that are appropriate for higher power and energy uses.10–12
On the other hand, researchers have been greatly attracted by organic π-conjugated polymers since Lethebyin showed the electrochemical procurement and description of polyaniline (PANI) created by oxidizing aniline on a platinum electrode in dilute sulfuric acid for the first time.13 As such, the conductivity of polyacetylene has been examined and increased ten-fold by using iodine vapors. Polyacetylene instability in air has resulted in the discovery of diverse, novel conducting polymers (CPs) to be utilized in basic studies and industrial applications.14–16 Therefore, since Heeger, MacDiarmid, and Shirakawa were jointly granted the Nobel Prize in Chemistry in 2000 for their leading research on CPs, academic and industrial researchers have given significant consideration to these materials. Earlier studies on these materials have been about the true mechanism of polypyrrole (PPy) formation. Park's group intensively investigated CPs electrochemistry and showed the autocatalytic growth system of PANI with its growth kinetics on the surface of the electrode.17–21
Generally, CPs have been introduced as one of the classes of organic polymers with metallic or semiconductor features, such as magnetic, electrical, optical, and electronic properties, to retain the characteristics of the conventional organic polymers, including simpler synthesis, corrosion resistance, and lower costs. These materials may be either insulators or semiconductors in the neutral or undoped form, which may be converted to the doped form through the redox reaction to form delocalized charge carriers. However, an advantage of the CPs in comparison to numerous organic polymers has been their adjustable chemical structures that may be modified to alter the conductivity of the polymer. The CPs have charge mobility along with their p-electron backbones that handle their uncommon electronic features such as lower energy optical transition, better electrical conductivity, higher surface area, high electron affinity, and mechanical flexibility.22–28 The most effective parameters constructing the physical properties of CPs are conjugation length, degree of crystallinity, and intra- and inter-chain interactions.
One of the applicable methods for preparing CPs is the anodic oxidation of the appropriate monomers, e.g., thiophene, aniline, etc. From the viewpoint of molecular electrochemistry, the most relevant aspects for clarifying the mechanism of electropolymerization are the formation of oligomers, nucleation and growth steps, and the formation of the polymeric materials. Mechanistic details are important for optimizing the conditions of electropolymerization. They play important roles in determining the quality of the produced CPs. A central point of electrochemical research is the analysis of the “doping” process. Even in the earliest stages of research on CPs, there was no doubt that these processes were not comparable with the classic doping of inorganic semiconductors. They correspond to oxidation in the case of p-doping or reduction in the case of n-doping. Using the electrochemical terminology, this means that the doping process corresponds to redox reactions in the polymer matrix. It is very important to know the phenomenological details of such redox reactions, in which potential range charging occurs at the maximum level of oxidation before material degradation.29–31
On the other hand, the experts in the field have been considerably attracted by the CPs because of the respective potent applications in energy storage and conversion, electrochemical sensors, and so on. Therefore, this review deals with the new studies of the CPs as (i) electrodes for electrochemical sensors and (ii) electrochemical energy storage and conversion tools like the electrode substances for super-capacitors, fuel cells, lithium-ion batteries (LIBs), and solar cells.
2. Electrochemical features of the conducting polymers
In 1977, the organic polymer polyacetylene was discovered, which when doped with iodine exhibited a unique, high electronic conductivity for organic material. Since then, researchers have investigated conducting polymers because of their specific electrical features.16 Despite traditional organic polymers, these materials enjoy certain features like electrical conductivity, higher electron affinity, as well as redox activities.32These materials include the π-electron backbone controlling uncommon electronic features like the electrical conductivity, lower energy optical transition, higher electron affinity as well as lower ionization potential. Moreover, such an extended π-conjugated system of the conducting polymer possesses alternating single- and double-bonds in line with the polymer chain.33,34
It is worth mentioning that the conduction mechanism in these types of polymers is highly complicated because they exhibit conductivity through a range of approximately 15 orders of magnitude, and several of them contain diverse mechanisms in distinct regimes. Conducting polymers have shown greater electrical conductivity via multiple orders of magnitude. Moreover, CPs may be doped electrochemically, implying that CPs oxidation creates a p-doped state and CPs reduction leads to an n-doped state. Redox reactions may control the conductivity of CPs. Electrochemical preparation techniques may address the features, morphology, thickness, and conductivity of the polymer. In general, researchers have applied the terms soliton, polaron, and bipolar for explaining the electronic phenomena in such systems.35,36 The conductivity in the conducting polymers is affected by diverse parameters like the conjugation length, polaron length, overall length of the chain, as well as charge transfer to the near-by molecules.37 Such conditions could be justified by several models based on inter-soliton hopping, hopping between the localized state assisted by the lattice vibrations, intra-chain hopping of the bi-polarons, varied range hopping in 3 sizes, as well as the charging energy restricting the tunneling between the conducting domains.38
It is possible to functionalize CPs and monomers with distinctive materials for tailoring their features. Therefore, adding the substituents ameliorates the performance and physicochemical features of the major polymer chain such as the enhanced mechanical and electrical characteristics. For improving CPs features, it is possible to prepare the CPs composite from the strongly conductive nanomaterials, as well as catalysts.36
3. Chemical structure of CPs
The electronic properties of CPs differ from the inorganic crystalline semiconductors in two major areas; i.e., long-range order and their molecular nature. For a polymer to become conducting, it must contain overlapping π-molecular orbitals and a high degree of π-bond conjugation. This comprehensive π-conjugated system of the CPs has irregular single and double bonds all along the polymer chain (Fig. 1). When a highly conjugated polymer is in its neutral state, it is essentially an insulating material. It is only upon removal of a π-bond electron from the conjugated polymer backbone to form a radial cation defect (called a polaron) that the insulating polymer becomes conductive. Removal of π-bond electrons causes the remaining electrons in the π-orbitals to become delocalized along the length of the conjugated polymer backbone, thereby enabling the free movement of the polymer along the chain. The formation of the positively charged polymer backbone is accompanied by the incorporation of a dopant anion, which has the effect of electrostatically balancing the positive charges.39,40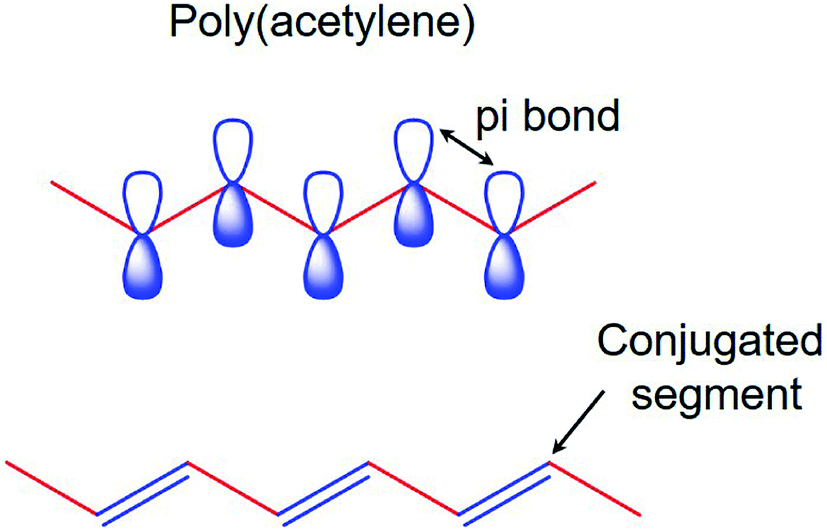 | ||
| Fig. 1 The π-conjugated system of conducting polymers. | ||
Some typical CPs include PPY, PANI, polycarbazole (PCZ), polythiophene (PTh) and its derivatives of poly(3,4-ethylene dioxythiophene) (PEDOT), and poly(3-hexylthiophene) (P3HT). These polymers contain the sp2 structure, which transports charge carriers (Fig. 2a). CPs have advantages such as controllable chemical/electrochemical properties and adjustable electrical conductivities from 10−11 to 105 S cm−1 upon doping (Fig. 2b). They are soft and extendable, similar to conventional polymers.41,42
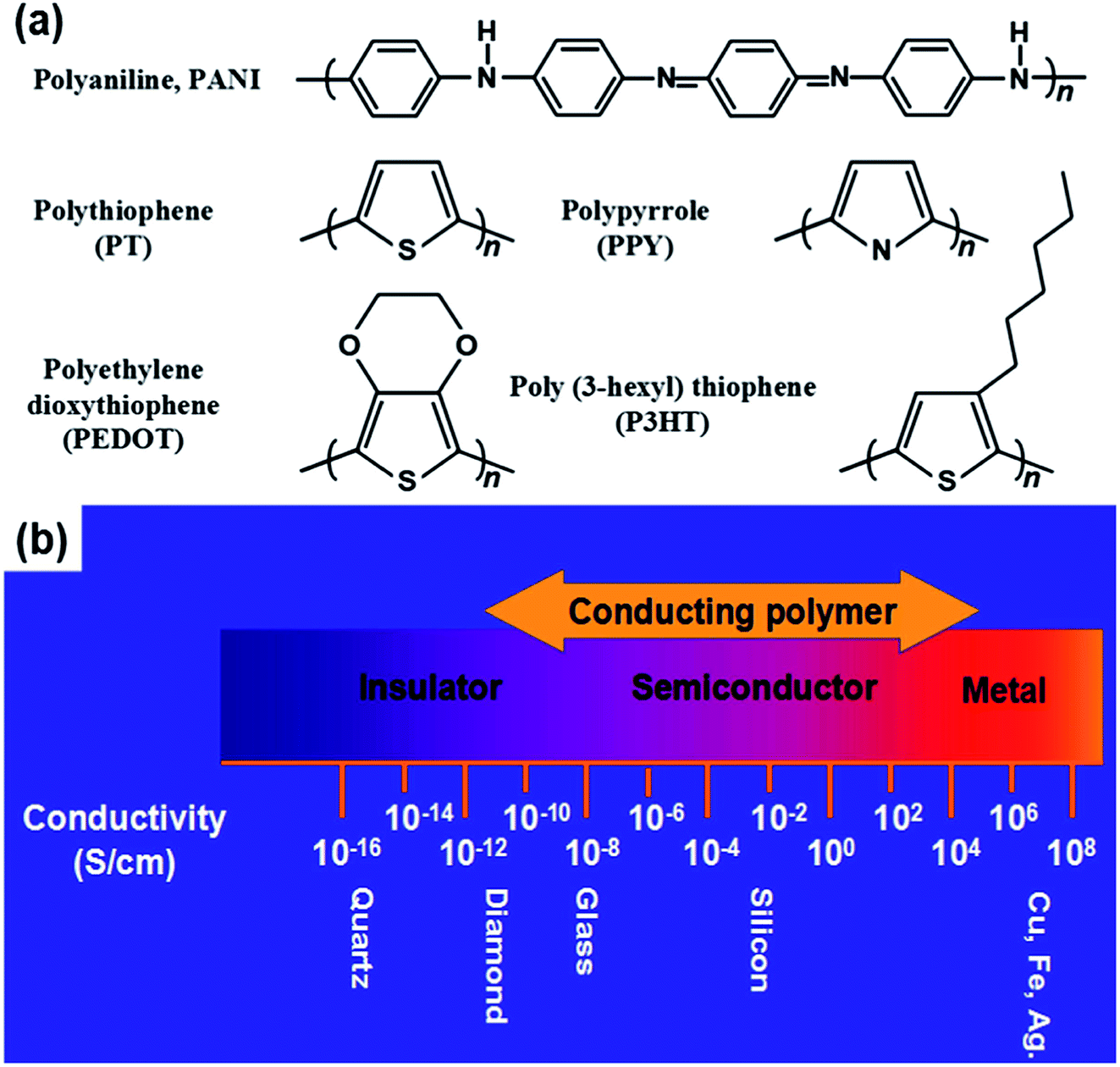 | ||
| Fig. 2 (a) Chemical structures of several CPs. (b) Electrical conductivity range of CPs. Reproduced with permission from ref. 42. Copyright 2020 Elsevier. | ||
The chemical structure of PPy consists of repeating units of the pyrrole monomer (a nitrogen-containing aromatic ring). The mechanism of pyrrole polymerization most likely involves oxidation, deprotonation, and crosslinking. The doping process of PPy is p-doping, where the polymer is oxidized and has a positive charge. One of the two leading theories is that pyrrole monomers are first oxidized to release a radical cation, followed by the coupling of two cations and the production of a bipyrrole. As oxidation continues, the chain continues to grow. The second theory advocates that a cation reacts with a neutral monomer. After oxidation and deprotonation, a dimer is formed, and the polymer chains keep growing by repeating this process. Eventually, a conduction band is developed by the delocalized electrons from the double bond in the backbone of PPy. PPy exhibits good electrical conductivity under physiological conditions due to its p-type conduction.43,44
PANI chains, resulting from the oxidative polymerization of aniline, are composed of two structural units, namely, the reduced [B–NH–B–NH], and the oxidized [B–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) QN−] repeat units, where B denotes the benzenoid and Q denotes a quinoid ring. The quinoid ring and benzenoid can transform into each other via the redox process. The protonated PANI chain is electrically conductive only when x
QN−] repeat units, where B denotes the benzenoid and Q denotes a quinoid ring. The quinoid ring and benzenoid can transform into each other via the redox process. The protonated PANI chain is electrically conductive only when x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y = 1:1 (i.e., benzenoid:quinoid = 3:1). PANI has a reversible acid/base doping process. PANI is conductive in acid form, while it is insulating in the emeraldine base form. The electrical conductivity of PANI increases with doping from the undoped insulating emeraldine base form (σ < 10−10 mho cm−1) to the fully doped, conducting emeraldine salt form (σ > 1 mho cm−1) by modulating its oxidative state through changing counter ions (dopant) and the degree of doping.45
:y = 1:1 (i.e., benzenoid:quinoid = 3:1). PANI has a reversible acid/base doping process. PANI is conductive in acid form, while it is insulating in the emeraldine base form. The electrical conductivity of PANI increases with doping from the undoped insulating emeraldine base form (σ < 10−10 mho cm−1) to the fully doped, conducting emeraldine salt form (σ > 1 mho cm−1) by modulating its oxidative state through changing counter ions (dopant) and the degree of doping.45
Carbazole is reported to form a conducting polymer with a conductivity of 0.1–1 S cm−1. It can be considered either as a derivative of pyrrole or aniline. PCZ has a bulky side chain of 4-[tris-(4-octyloxyphenyl)methyl]phenyl. Its big side chain prevents the π-stacking of polymers and reduces the interactions between polymers, which contributes to the high fluorescence efficiency, and also benefits the diffusion of explosives. Furthermore, the side chain moiety endows the polymer with good solubility and helps to form films with high quality by spin coating.46,47
Thiophene is an electron-rich aromatic ring, which can be oxidized to form p-doped highly conducting PThs. The electrochemical oxidation can result in well-adhering polymeric film formation. Moreover, the thickness of the polymer films can be controlled by changing the polymerization time during the electropolymerization process.48
PEDOT is a p-conjugated polymer that has received considerable interest due to its potential for a variety of applications. The rigid and linear conformation of PEDOT facilitates charge transport and favors its crystallization, resulting in favorable properties such as high charge/discharge capacities, fast response times, and high sensing capabilities. PEDOT is polymerized by either appropriate oxidants (chemically), or electrochemical current.49
The P3HT structure involves 2D sheets of π-stacked poly(thiophene) backbones separated by layers of n-hexyl side chains. It is thought that the conformation and packing of the polymer backbone plays a dominant role in the determination of its transport properties.50
4. Uses of the conducting polymers
4.1. Electrochemical sensing
According to the studies, electrochemical sensors and biosensors have been considered as electrochemical cells containing 3 or 2 electrodes. A characteristic 3-electrode system contains a reference, counter, and working electrode; the working electrode has a chemically stable solid conductive substance like gold, carbon, and platinum, the reference electrode generally contains the silver metal coated with a layer of silver chloride (Ag/AgCl), and a platinum wire is usually employed as an auxiliary electrode. On the contrary, a two-electrode system contains just the reference and working electrodes. Hence, it is possible to classify the electrochemical procedures into three key categories concerning the kinds of measurement, as follows:51–53 (i) current (voltammetric & amperometric), (ii) potential difference (potentiometry), and (iii) impedance (electrochemical impedance spectroscopy), out of which the bio-sensors based on the current measurements have a widespread application. On the one hand, the current measurement-based sensors have been described via the use of the potential to a working electrode against a reference electrode and the measurement of the current. This electric current results from the electrolysis because of the electrochemical oxidation or reduction at the working electrode surface. Nevertheless, this procedure is dependent on the mass transport rate of the molecules to the electrode, as well as the rate of electron transfer at the electrode surface.54,55It is notable that while developing the electroanalytical sensing mechanisms to determine different analytes, these sensing electrodes have been frequently modified with suitable substances for achieving the intended increase in the selectivity and sensitivity.56–59 Most electrocatalytic materials are based on noble or transition metal elements, either as pure electronic conducting metals or alloys, or as electronic semiconductors. Therefore, the substitution of precious metals with inexpensive organic compounds has been the subject of many active research efforts. For example, well-defined 2D covalent organic polymers and carbon organic frameworks act similar to CPs as they also include π-conjugated polymeric layers containing heterocycles and aromatic moieties with precisely controlled molecular structures. CPs can have intrinsic electrocatalytic properties toward certain redox reactions. It is also possible to use the inter-chain cavities in these materials to incorporate metals so that electrocatalysts can be generated. CPs have also been used to immobilize redox mediators, thus boosting the homogeneous electron transfer in the catalytic cycle. This approach is based on the efficient immobilization of biomolecules like proteins and enzymes through the very strong interactions with avidin via an affinity coupling with biotin. Therefore, their high conductivity and other properties discussed earlier make them suitable as catalysts for diverse redox reactions.60–62 According to the analyses, designing and constructing the CPs-modified electrodes is highly efficient in the electrochemical sensor technology.
In their research, Dechtrirat et al. procured a molecularly imprinted polymer (MIP) and a nanocomposite from the gold NPs (Au NP), and poly(3,4-ethylenedioxythiophene)/poly(styrene sulfonate) (PEDOT:PSS) was deposited on a screen-printed carbon electrode (SPCE). Fig. 3 presents the procedure for the construction of the electrochemical MIP sensor to detect nitrofurantoin (NFT). The one-pot concurrent in situ formation of the AuNPs and PEDOT:PSS was used to prepare the nanocomposite and inkjet-coat it on the SPCE. Then, this MIP film was coated on the modified SPCE using the co-electrodeposition of o-phenylenediamine and resorcinol in the presence of antibiotic NFT. Researchers also used DPV and showed the linearity of the potential of nearly 0.1 V versus Ag/AgCl in the 1 to 1000 nM NFT concentration range with 0.1 nM LOD at S/N = 3. Such a condition was 2 orders of magnitude less than that of the control MIP sensor with any nano-composite inter-layer, which reflected the advantageous impact of AuNP-PEDOT:PSS. Analyses showed the higher reproducibility of the electrode (relative standard deviation (RSD) of 3.1% for n = 6) and selectivity in comparison to the structurally associated molecules. Furthermore, it is possible to reuse it for at least 10 times, with stability for at least 45 days. Finally, researchers experienced its successful utilization for determining NFT in the (spiked) feed matrix with acceptable recovery.64
 | ||
| Fig. 3 The electrochemical MIP sensor for NFT determination: (a) the preparation of the electro-synthesized MIP on the nanocomposite modified SPCE. (b) Indirect voltammetric detection of NFT utilizing the K3[Fe(CN)6]/K4[Fe(CN)6] as the electrochemical probe. Reproduced with permission from ref. 64. Copyright 2018 Springer. | ||
Furthermore, the new sensitive electrochemical technique devised by Raj et al. has been used for simultaneously determining dopamine (DA) and 5-hydroxy-tryptamine (5-HT) with a graphene (GR) and poly4-amino-3-hydroxy-1-naphthalene-sulfonic acid-modified screen-printed carbon sensor. The researchers applied square wave voltammetry (SWV) and CV to study the electrochemical measurements, while EIS and field emission SEM were utilized for the characterization of the surface morphology of the modified sensor. It was found that this sensor simplified DA and 5-HT analysis in a concentration range between 0.05 and 100 μM and 0.05 to 150 μM with 2 nM and 3 nM LOD, respectively. It was further examined to determine 5-HT in the plasma specimens and confirm its selectivity toward 5-HT and DA in the presence of the usual metabolites in biological fluids. Consequently, Raj et al. substantially confirmed the analytical usability of this new sensor for simultaneously detecting 5-HT and DA in drug formulations, blood samples, and human urine.65
Another study conducted by Govindasamy et al. addressed the design and fabrication of an affordable modified electrode based on the sensitive detection of ascorbic acid (AA) by PANI and Ni (PANI–Ni) composites. They employed EIS, energy-dispersive X-ray spectroscopy, and electrochemical techniques for characterizing the composite formation. The PANI–Ni composite was utilized for modifying SPCE and then the final modified electrode was applied in the development of the AA sensor. This modified electrode showed very good electrocatalytic activities for oxidizing AA. In the next stage, researchers designed an amperometric sensor with the PANI–Ni composite that exhibited sensitive prompt signals for AA. Analyses showed a 2–1210 μM linear range, 0.4 μM LOD, and 0.479 μA μM cm−2 sensitivities; the researchers approved the acceptable reproducibility, stability, and iterability of their electrode. Finally, they confirmed the functional usability of the electrode in the samples of human urine.66
Talib et al. investigated the development of an electrochemical immuno-sensor based on poly(3,4-ethylene-dioxythiophene)/graphene oxide (PEDOT/GO)-modified SPCE to detect clenbuterol (CLB) (Fig. 4). Electropolymerization was used to modify the SPCE with the PEDOT/GO as a sensor platform, whereas electrochemical assay was implemented with the direct competitive format wherein free CLB and clenbuterol-horseradish peroxidase (CLB-HRP) in the solution would have competition for forming the binding with polyclonal anti-clenbuterol antibody (Ab) immobilized on the surface of the modified electrode. Researchers observed a linear standard CLB calibration curve with R2 of 0.9619 and 0.196 ng mL−1 LOD. They analyzed the milk sample and showed the ability of their immunosensor to detect CLB in real samples. Notably, their outputs could be compared with the enzyme-immunosorbent assay (ELISA).67
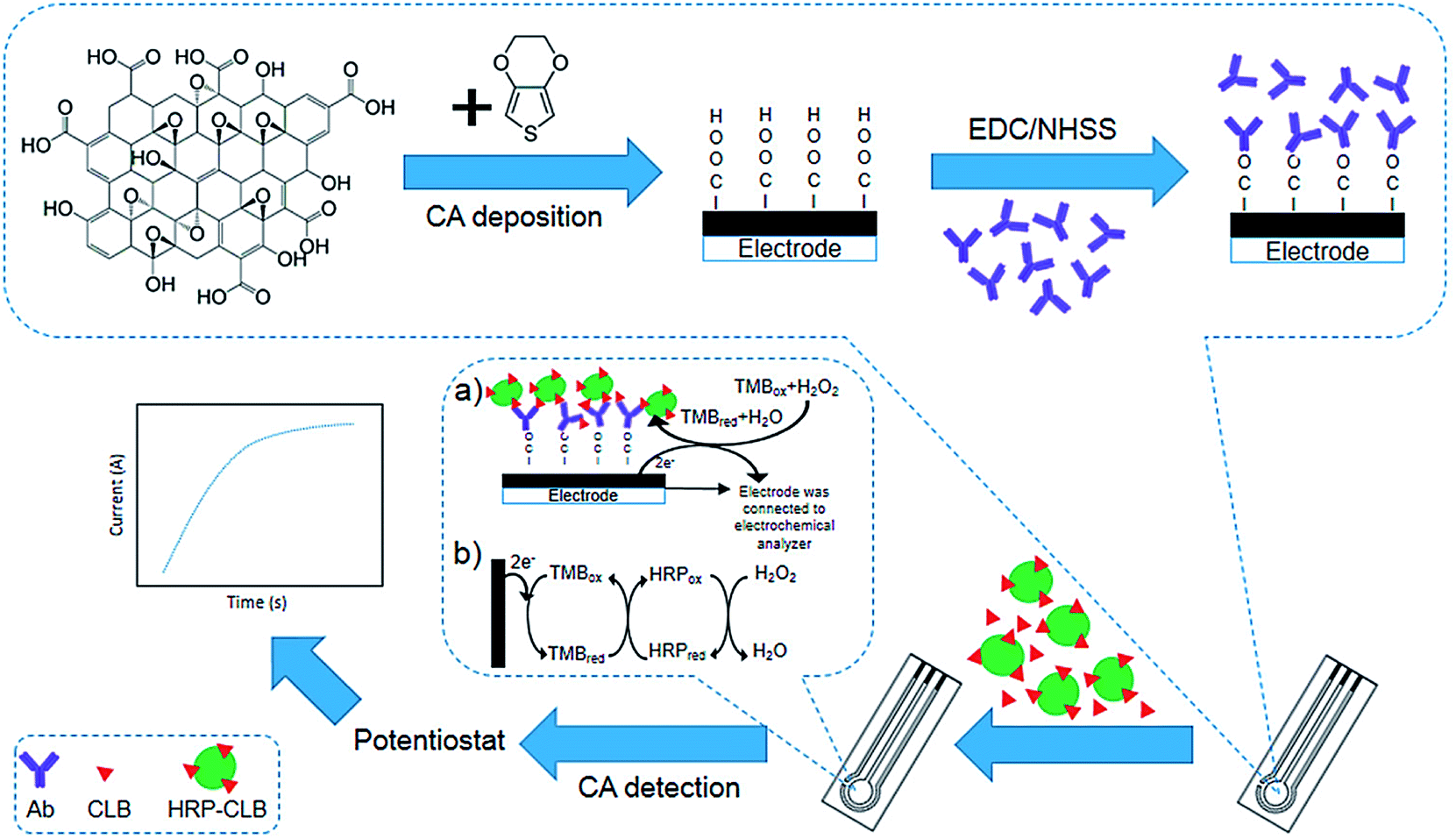 | ||
| Fig. 4 Construction of the clenbuterol hydrochloride (CLB) immunosensor. (a) The electrochemical immunosensor format was utilized to detect CLB. (b) The indirect electron transfer for the TMB redox is displayed by the complex TMB/HRP/H2O2 enzyme reaction on the modified SPCE to reduce the creation of the current. Reproduced with permission from ref. 67. Copyright 2018 MDPI. | ||
The investigation conducted by Sağlam et al. provided a new molecularly imprinted (MI) electrochemical sensor for selectively determining DA in the presence of an 80-fold excess of AA and UA. They used the Au NPs (Aunano)-decorated GCE coated with the poly(carbazole (Cz)-co-aniline (ANI)) copolymer film, incorporating DA as the template (DA imprinted-GC/P(Cz-co-ANI)-Aunano electrode, i.e., the DA-MIP-Aunano electrode. In the DA determination, the sensor electrode exhibited greater electroactivity for the analyte oxidation in 0.2 M phosphate buffer (PB) at pH 7. SWV was run into 10−4 to 10−5 M of DA and the oxidation peak potential was seen at 0.16 V. The limit of quantification (LOQ) and LOD were 6.7 × 10−6 M and 2.0 × 10−6 M, respectively. Furthermore, the binary and ternary synthetic mixtures, DA–AA, DA–UA–AA, and DA–UA, showed very good recovery for DA. Consequently, researchers observed the quantitative recovery of DA from the real samples of the bovine serum spiked with DA and detected it in the concentrated DA injection solution. They confirmed the validity of this new SWV in comparison to the earlier potentio-dynamic technique with a caffeic acid-modified-GC electrode.68
In their study, Deiminiat and Rounaghi used CV and proposed a method to measure ketamine using a one-step electropolymerization of the MIP with the polytyramine (pty), sol–gel, functionalized MWCNTs@Au NPs (fMWCNTs@AuNPs) nano-composite and ketamine on a pencil graphite electrode (PGE). They entrapped the f-MWCNTs@AuNPs nanocomposite in the polymer network to augment the rate of the electron transfer and their sensor sensitivity. After that, the chemical reduction technique was used to synthesize the f-MWCNTs@AuNPs nanocomposite. Electrochemical impedance spectroscopy (EIS), SWV, and CV were employed to examine the electrochemical behaviors of the modified electrode. Based on the optimal testing condition, a calibration curve of the MIP electrode was drawn and 2 dynamic linear ranges of 1.0–50.0 nM, and 50.0–1000.0 nM, and the LOD of 0.7 nM were observed for quantitatively measuring ketamine in the solution. These outputs showed very good stability, suitable iterability and reproducibility, and higher selectivity and sensitivity of this new sensor toward ketamine molecules. The researchers demonstrated the successful utilization of the electrochemically imprinted sensor to determine ketamine in biological samples with reasonable outputs.69
Another study dealt with the fabrication of a strongly sensitive voltammetric sensor for caffeine (CF) and aspirin (ASA) on a GCE modified with a composite film of MWCNT and poly(4-vinylpyridine) (P4VP). According to the CV of P4VP–MWCNT/GCE, the reaction of the electrode was one of the usual diffusion-limiting processes and the CF and ASA catalytic reactions were observed in 0.1 M PB (pH of 7.4). In order to detect ASA, a 30-fold enhancement was found in the anodic peak current (Ipa) at P4VP–MWCNT/GCE in comparison with the bare GCE. It was shown that at the optimal parameters, Ipa linearly depended on the CF (2.0–200 μM) and ASA (0.04–350 μM) concentrations. Moreover, the respective LODs of CF and ASA equaled 1.19 nM and 4.42 nM. They used DPV to determine CF and ASA in the presence of paracetamol (PCT) and revealed that it is possible to assay ASA in the biological samples without any interference from PCT and CF. Furthermore, this P4VP–MWCNT/GCE showed acceptable stability, recovery, and reproducibility in the biological and drug samples.70
The electropolymerization of a poly(sulfo-salicylic acid) film on a gold electrode modified by 2-mercapto-benzothiazole self-assembled monolayer and MCNTs (PSSA/CNTs/MBT/Au) was performed by Arvand and co-workers.71 Other researchers used their sensor to determine rutin (Fig. 5) and the electrocatalytic activities of the resultant sensor toward rutin oxidation were ascribed to the presence of the PSSA/MWCNTs nanocomposite. Based on the optimal conditions, a LOD of 1.8 nM and 2 linear calibration ranges from 0.01–0.8 to 0.8–10.0 μM were observed for the detection of rutin at the PSSA/CNTs/MBT/Au electrode. Consequently, the successful application of this electrode to determine rutin in the samples of red onion, red apple, orange, strawberry, salvia, and oat was achieved. This electrode presented several advantages, such as high repeatability, reproducibility, and great stability, with its probable use for the fast, sensitive, and selective detection of rutin.71 Table 1 presents some features of the conducting polymer-based sensors to determine diverse medicines.
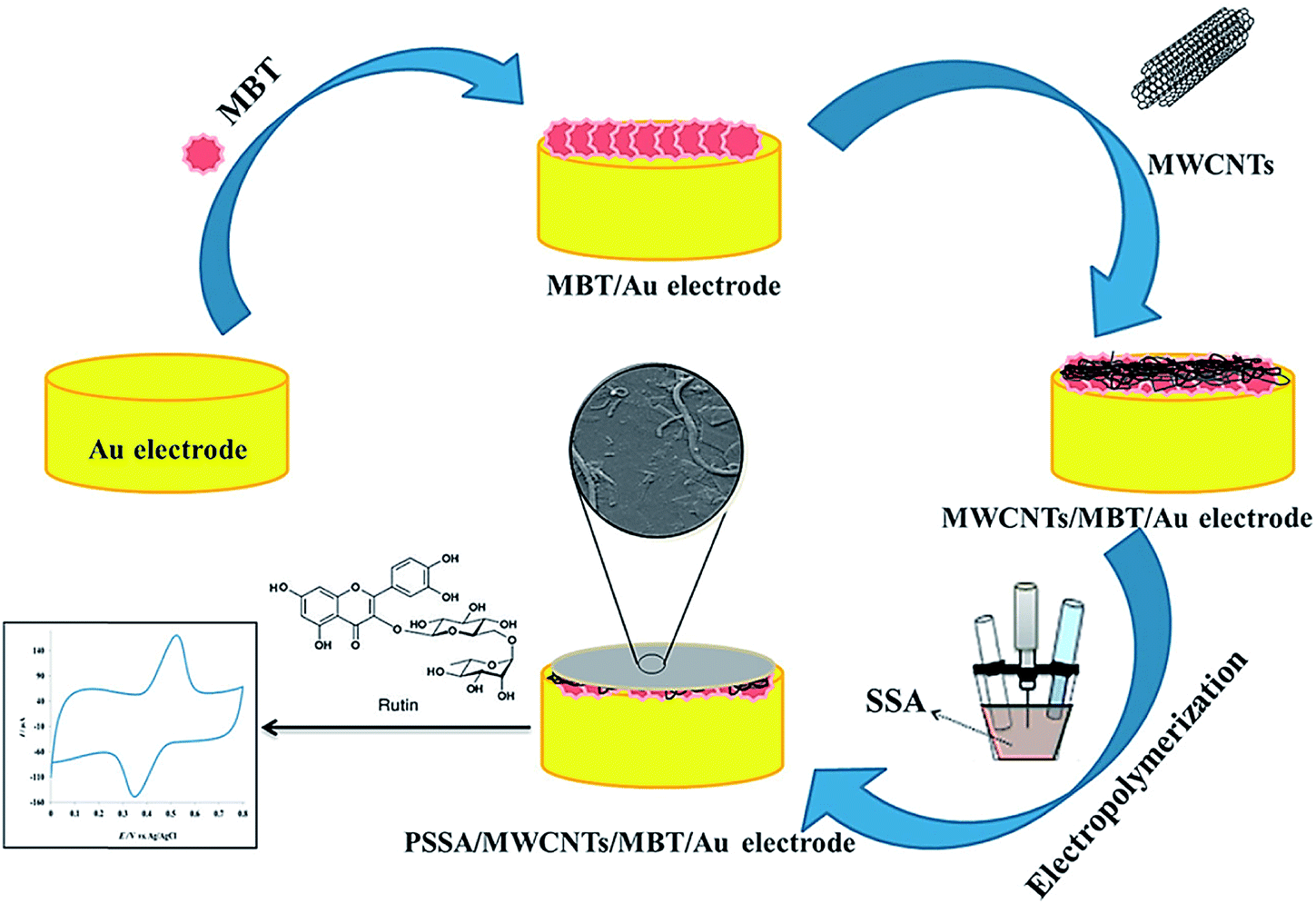 | ||
| Fig. 5 The preparation process of the PSSA/CNTs/MBT/Au-modified electrode for rutin sensing. Reproduced with permission from ref. 71. Copyright 2018 Elsevier. | ||
| Electrochemical sensor | Method | Analyte | Detection limit | Linear range | Ref. |
|---|---|---|---|---|---|
| MIP/GCE | DPV | Streptomycin | 0.5 nM | 2.0–1000.0 nM | 63 |
| MIP/AuNP-PEDOT:PSS/SPCE | DPV | Nitrofurantoin | 0.1 nM | 1.0–1000.0 nM | 64 |
| GR/p-AHNSA/SPCs | SWV | Dopamine | 2 nM | 0.05–100.0 μM | 65 |
| 5-Hydroxytryptamine | 3 nM | 0.05–150.0 μM | |||
| PANI–Ni/SPCE | Amperometric | Ascorbic acid | 0.62 μM | 2.0–1210.0 μM | 66 |
| SPCE/PEDOT/GO | Amperometric | Clenbuterol | 0.196 ng mL−1 | 5.0–150.0 ng mL−1 | 67 |
| DA imprinted-GC/P(Cz-co-ANI)-Aunano | SWV | Dopamine | 2.0 × 10−6 M | 10−4 to 10−5 M | 68 |
| Polytyramine/sol–gel/f-MWCNT@AuNPs MIP/PGE | SWV | Ketamine | 7 × 10−4 μM | 1.0 × 10−3 to 1.0 μM | 69 |
| P4VP–MWCNT/GCE | DPV | Aspirin | 4.42 nM | 0.04–350.0 μM | 70 |
| Caffeine | 1.19 nM | 2.0–200.0 μM | |||
| PSSA/MWCNTs/MBT/Au | DPV | Rutin | 1.8 nM | 0.01–10.0 μM | 71 |
Cui et al. designed a precise, sensitive, electrochemical sensing method for H2O2 levels, without oxygen interference, which has been considered as one of the prominent parameters in the environmental, clinical, and biological areas. They modified a boron-doped diamond (BDD) electrode using mesoporous platinum (MPrPt) via the electrodeposition of the Pt–Cu alloy and the anodic dissolution of Cu from the alloy and electropolymerization of the PPy (PPy3C:PPy = 4:1, molar ratio) co-polymer and a poly(pyrrole-3-carboxylic acid) (PPy3C). The analyses indicated the greater roughness factor of the resultant Pt and the more efficient surface area of the MPrPt/BDD, as well as the smaller resistance of the charge transfer in comparison to the ones in the Pt NPs-modified BDD electrode. The PPy3C–PPy/MPrPt/BDD electrode displayed a highly selective, sensitive, exact, reproducible, stable, and wider linear range of H2O2 response at the neutral pH based on ambient conditions with the same sensitivity and S/N ratio as the ones in control of nitrogen. Cui et al. revealed a 2 μM LOD for H2O2 with the linear range between 5 μM and 49 mM (4 orders of magnitude). It was found that MPrPt, PPy3C–PPy copolymer, and the BDD substrate had a synergic impact on the significant sensing function. The determination did not show any endogenous oxygen interferences of concern in the microanalysis, in vivo monitoring, and field utilizations.73
Harraz et al. demonstrated the synthesis of a new nanocomposite of α-Fe2O3/cross-linked PANI (α-Fe2O3/CPANI) through the simultaneous detection of the gelation and polymerization procedures. Therefore, researchers utilized the α-Fe2O3/CPANI nanocomposite as an effective hydrazine chemical sensor with greater selectivity and sensitivity. The electrochemical determination of hydrazine was performed at the α-Fe2O3/CPANI-modified GCE by CV and amperometry and then compared with the bare GCE or pure α-Fe2O3. The schematic representation of the synthesis of the α-Fe2O3/CPANI nanocomposite and the amperometric detection of hydrazine are shown in Fig. 6.
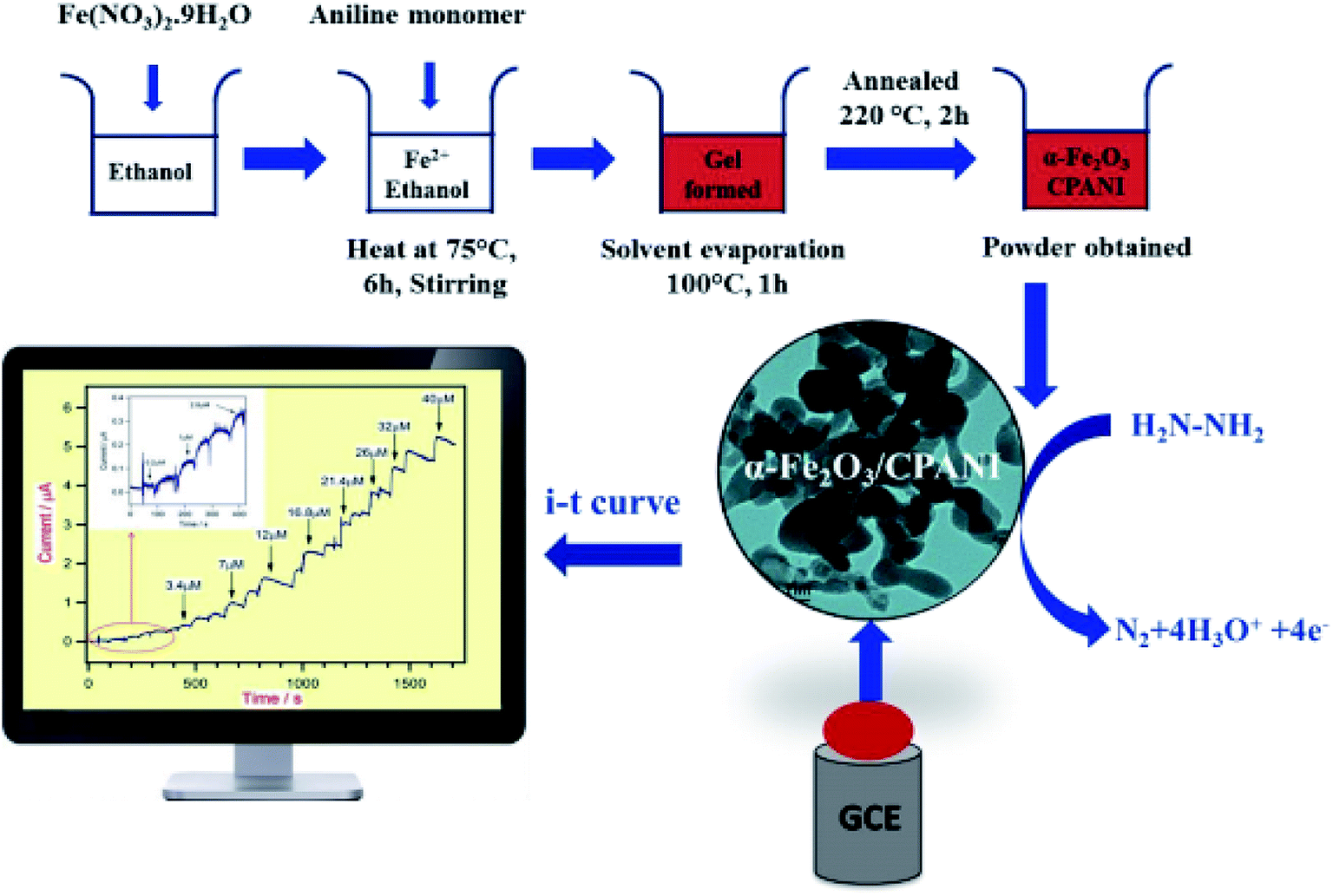 | ||
| Fig. 6 Schematic representation of the synthesis of the α-Fe2O3/CPANI-modified GCE, along with the amperometric detection of hydrazine. Reproduced with permission from ref. 74. Copyright 2016 Elsevier. | ||
On comparison to the pure α-Fe2O3 or bare GCE, the α-Fe2O3/CPANI nano-composite-modified GCE showed a more acceptable sensing function with the sensitivity of 1.93 μA μM−1 cm−2, 0.153 μM LOD at (S/N = 3), a wider linear range of hydrazine concentration between 0.2 μM and 40 μM, a correlation coefficient (R2) of 0.9983, and a response time of <12 s. This α-Fe2O3/CPANI-modified electrode exhibited electrochemical stability and higher selectivity as a trivial cross-sensitivity to the conventional bioactive interfering samples, and numerous metal ions have been reported.74
In another study, Liu et al. presented a poly(aniline-co-o-aminophenol)-modified GCE (PAOA/GCE) for aqueous nitrite detection. PAOA showed stable redox activities and conductivities in wider pH ranges than PANI, which made it appropriate for use as the electrode material in a neutral medium. Researchers used the electrochemical co-polymerization of aniline and o-aminophenol to prepare PAOA/GCE. FT-IR and SEM outputs confirmed PAOA formation and the electrode displayed a greater response towards nitrite oxidation in comparison to the PANI-modified GCE and the bare GCE. Furthermore, researchers determined the impacts of the rate of scan, temperature, and pH on the determination of nitrite. Consequently, it was possible to use PAOA/GCE in the wider pH-range between 2 and 8 and determine nitrite in a linear range between 5.0 × 10−6 and 2.0 × 10−3 M with 2 × 10−6 M LOD. Therefore, the electrode had very good stability, anti-interference abilities, and reproducibility, reflecting its attractiveness in the detection of aqueous nitrite in drinking water and ground water.75
Wang et al. presented a sensitive electroanalytical procedure for detecting trace amounts of Cu2+ by means of the phytate-functionalized PPy nanowires (PPyNWs)-modified GCE. Electrostatic absorption and ultrasonic mixing were used to prepare phytic acid/PPy (PA/PPy) NWs. The findings revealed that the PA/PPy NWs-modified GCEs and PPy NWs had electrochemical responses towards Cu2+. As a result of the synergistic impacts between PPy NWs and PA, the PA/PPy NWs-modified electrode displayed greater sensitivity as compared to that of the PPy NWs-modified electrode. Moreover, the PA/PPy NWs composite-functionalized electrodes had a suitable linear association with Cu2+ in the concentration range between 10 and 60 μg L−1 and LOD of 3.33 μg L−1 (S/N = 3). Consequently, the researchers utilized the electrochemical sensor for assaying Cu2+ in real water samples.76
Zuo et al. electrochemically determined Hg2+ at the trace level based on the poly(3,4-ethylenedioxythiophene) nanorods/graphene oxide nano-composite-modified GCE (PEDOT/GO/GCE). The researchers introduced this PEDOT/GO nano-composite using a simplified liquid–liquid interfacial polymerization method. Moreover, they used DPSV for determining low concentrations of Hg2+ on the PEDOT/GO/GCE. Then, they optimized the experimental condition like the accumulation time, deposition potential, and pH-values and based on the optimum conditions, the peak current was linearly related to the Hg2+ concentration in the 10.0 nM to 3.0 mM range. The analysis showed a 2.78 nM LOD at the signal to noise ratio of 3; ultimately, the outputs reflected the successful utilization for Hg2+ detection in the tap water samples.77
In one of the studies in the field, Selvan et al. in 2018 addressed the synthesis of the graphite electrodes coated with a CNT/asymmetrical N4 tetradentate Schiff base-ligand N,N′-bis(pyrrole-2-yl-methylene)-2-amino-benzylamine via condensing 2-amino-benzylamine. Then, pyrrole-2-carboxaldehyde was examined as one of the electrochemical sensors for simultaneously determining Hg(II) and Pb(II) in 0.1 M acetate buffer at the pH of 4.5 (Fig. 3). SWASV was used to investigate the electrochemical response features of the modified electrode toward Hg(II), as well as the Pb(II). A series of experiments have been accomplished for finding the best variables of deposition and stripping of the metal ions like deposition time, supporting electrolytes, and pH. Moreover, the linear calibration graph ranged between 3.3 and 66 nM Pb(II) ions and Hg(II) metal ions. The LOD was 0.36 nM for Hg(II) and 1.1 nM for Pb(II) (S/N = 3). Finally, researchers experienced the successful utilization of their sensor for determining Hg(II) and Pb(II) in real samples with acceptable recovery (Fig. 7).78
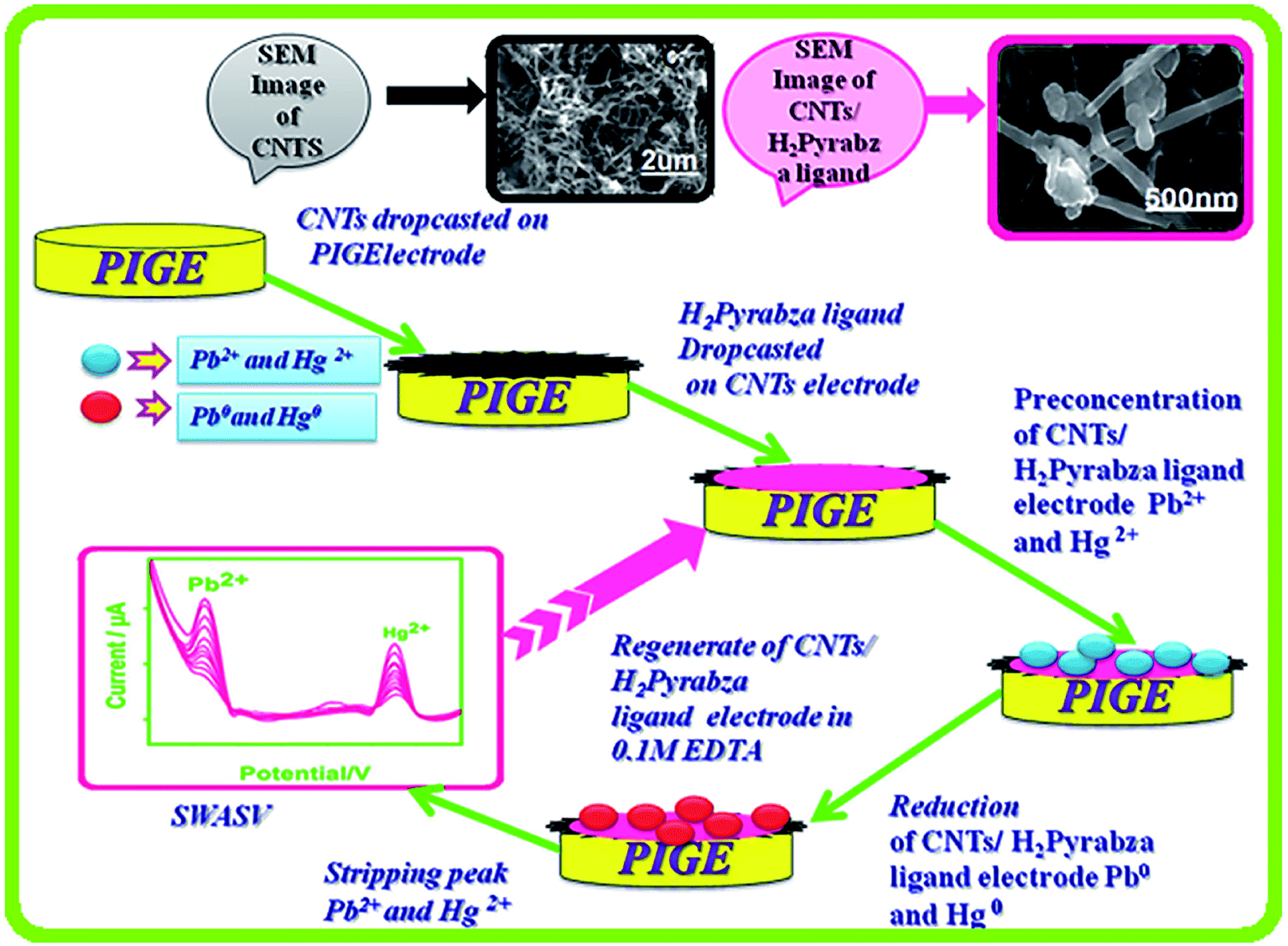 | ||
| Fig. 7 Fabrication procedure for the electrochemical sensor for the simultaneous determination of Hg(II) and Pb(II). Reproduced with permission from ref. 78. Copyright 2018 Elsevier. | ||
A strongly sensitive electrochemical aptasensor based on electropolymerized poly(pyrrolenitrilotriacetic) acid film and a novel aptamer functionalized with a penta-histidine peptide to quantify bisphenol A was designed by Kazane et al.79 The electrochemical signal of the [RuIII(NH3)6]3+ complex bound by the electrostatic interaction over the aptamer-modified electrode was used to estimate the surface coverage of the anti-bisphenol A aptamer of 1.84 × 10−10 mol cm−2. The binding of bisphenol A over the polymer film has been substantially described by the electrochemical techniques of SWV and EIS measurements. Therefore, the introduced label-free impedimetric aptasensor had a wide linear range from 10−11 to 10−6 M with 372 Ω sensitivity per unit of the log of concentration and a very good specificity towards the interfering parameters like 4,4′-dihydroxy-biphenyl and bisphenol P.
In their study, Sadriu et al. addressed the electrochemical synthesis of the isoproturon-imprinted PPy films over the GC electrodes in the ethanol and aqueous solution of pyrrole as a monomer, isoproturon as a template molecule, and LiClO4 as the supporting electrolyte (Fig. 4). CV and chrono-amperometry were used to run electro-polymerization. Moreover, the successful entrapment of the isoproturon template molecules was reported in PPy film, where they provided artificial recognition cavities. Following the electrochemical extraction of the template, the PPy film functioned as a MIP for selectively detecting isoproturon while the non-imprinted polymer (NIP) film that was constructed under similar conditions, except for the presence of isoproturon, had no interactions. CV was used to characterize the NIP and MIP films in the presence of the redox probes. EQCM (Electrochemical Quartz Crystal Microbalance) was used to estimate the polymer layer thickness and Faraday's law was used to calculate it. The analyses showed the isoproturon-selective determination even in the presence of carbamazepine and carbendazim interferents using the isoproturon-imprinted PPy films. In the next stage, the researchers utilized SWV to determine the LOD of 0.5 μg L−1 in milli Q water, whereas the LOD in the real water samples was 2.2 μg L−1 (Fig. 8).80
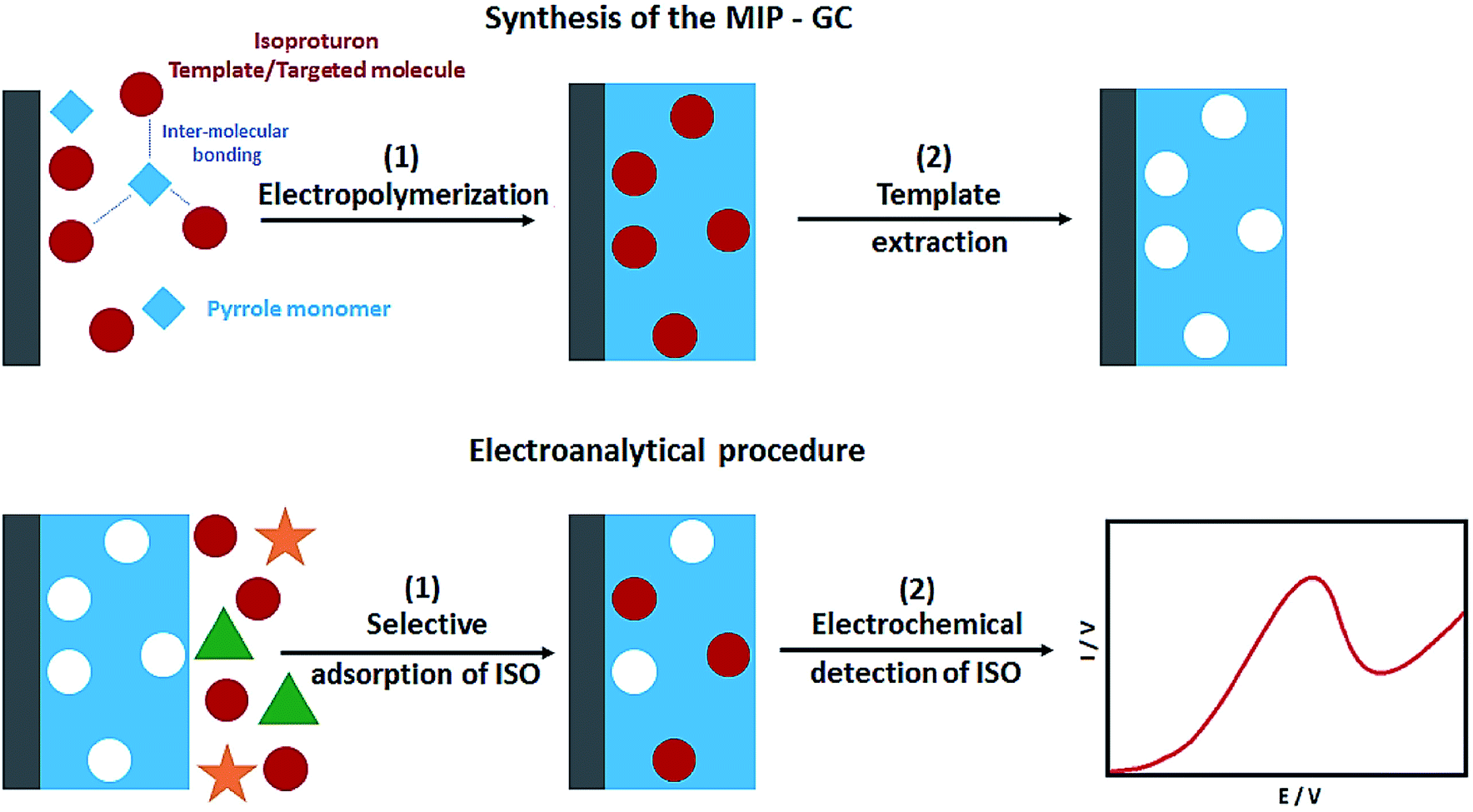 | ||
| Fig. 8 The process applied to the preparation of the imprinted PPy films on the GC substrate MIPGC and isoproturon electroanalysis. Reproduced with permission from ref. 80. Copyright 2020 Elsevier. | ||
Kaur et al. presented one of the nano-composites containing the conducting polymer (CP)–poly(3,4-ethylene-dioxythiophene) (PEDOT) and MWCNT with electrochemical deposition over the surface of the fluorine-doped tin oxide (FTO) to analyze malathion organophosphate (OP); –COOH functionalization of the MWCNTs was performed to covalently immobilize the enzyme acetylcholinesterase (AChE). FE-SEM, FTIR, and electrochemical investigations were used to procure the provided AChE/PEDOT-MWCNTs/FTO as well as PEDOT-MWCNTs/FTO bioelectrodes. Moreover, diverse optimization investigations were accomplished for distinct variables like the pH of 0.1 M phosphate buffer solution (PBS) (7.5), substrate concentration (0.3 mM), inhibition time (10 min), and AChE concentration (50 mU). Analyses showed a 1 fM LOD in the linear range between 1 fM and 1 μM so that it was possible to regenerate the inhibited AChE to 99% by 2-PAM. Consequently, the storage stability and re-usability of the procured bioelectrode were 30 days and 7 times, respectively. The recovery of the malathion from the spiked lettuce sample ranged from 96% to 98% on employing the designed bio-electrode.81 Table 2 presents some features of the conducting polymer-based sensors for determining diverse environmental contamination.
| Electrochemical Sensor | Method | Analyte | Detection limit | Linear range | Ref. |
|---|---|---|---|---|---|
| SOD/CNT/PEDOT/GCE | Amperometric | Superoxide | 1 μM | 20.0–3000.0 μM | 72 |
| PPy3C-PPy/MPrPt/BDD | Amperometric | H2O2 | 2 μM | 5.0 μM to 49.0 mM | 73 |
| α-Fe2O3/CPANI modified GCE | Amperometric | Hydrazine | 0.153 μM | 0.2–40.0 μM | 74 |
| PAOA/GCE | LSV | Nitrite | 2 × 10−6 M | 5.0 × 10−6 to 2.0 × 10−3 M | 75 |
| PA/PPy/GCE | DNPV | Cu2+ | 3.33 μg·L.−1 | 10–60 μg L−1 | 76 |
| PEDOT/GO/GCE | DPSV | Hg2+ | 2.78 nM | 10.0 nM to 3.0 mM | 77 |
| CNT/(H2pyrabza) ligand | SWASV | Pb(II) | 1.1 nM | 3.3–66.0 nM | 78 |
| Hg(II) | 0.36 nM | 3.3–66.0 nM | |||
| Aptamer-modified electrode | SWV | Bisphenol A | 10−11 mol L−1 | 10−11 to 10−6 mol L−1 | 79 |
| MIP-GC | SWV | Isoproturon | 0.5 μg L−1 | 2.5 × 10−9 to 5 × 10−6 M | 80 |
| AChE/PEDOT-MWCNTs/FTO | DPV | Malathion organophosphate | 1 fM | 1 fM to 1 μM | 81 |
Gu et al. devised a label-free electrochemical impedimetric immunosensor for monitoring melamine, which was substantially utilized to detect melamine in the spiked dairy samples. Researchers observed the co-electrodeposition of poly(pyrrole-co-pyrrole-1-propionic acid) and the rGO polymer layer (rGO–PPYPPA) on the electrode with a one-pot procedure and obtained a co-polymer layer with desirable electrochemical features. Then, they applied one of the competitive immunoassays to quantitatively analyze melamine. This rGO–PPYPPA copolymer ameliorated the conductivity of the sensor and created sufficient anchoring sites to bind antibodies and enhance immunobinding. Notably, certain immune interactions at the electrode surface, which restricted the transfer of the electrons of the redox probe Fe(CN)63−/4−, changed the electrochemical impedance. These alterations in the impedance were associated with the specific concentration of melamine. This immuno-sensor displayed wider detection ranges (10−11 to 10−2 M), lower LOD (1.2 × 10−11 M) and reasonable recovery (84.6 to 100.3%) in the real samples. The outputs provided a simplified flexible impedimetric immunosensing ground with greater potential for real-world applications.83
Consequently, Gursoy et al. prepared an appropriate co-polymer of poly-pyrrole and poly(3,4-ethylene-dioxythiophene) synthesized by the electropolymerization procedure and devised a novel lactose biosensor. Researchers observed the deposition of pyrrole and 3,4-ethylene-dioxythiophene monomers in the presence of sodium dodecyl-benzene sulphonic acid on a platinum disc electrode that was utilized as a working electrode. This sensor was based on the serial reaction of galactose oxidase and b-galactosidase immobilized on the co-polymer-modified platinum disc electrode. SEM, electrochemical analyses, and FTIR spectrometry were utilized to confirm the successful synthesis of the enzyme-immobilized copolymer. CV at 10.40 V was applied to determine the responses of the enzyme electrode to lactose. The bio-sensor response time was in the range between 8 and 10 s and the upper limit of the linear working segment was at a lactose concentration of 2.30 mM with 1.4 × 10−5 M LOD. Furthermore, the Michaelis–Menten constant equaled 0.65 mM of lactose. According to the outputs, this new biosensor had remarkable potential for determining the concentration of lactose in milk.84
Researchers modified a commercially available SPCE with the MWCNTs, AuNPs, as well as the poly-neutral red (PNR) film. Afterwards, the electrode was modified with the alcohol dehydrogenase (ADH) for use in the procurement process of the disposable amperometric biosensor to analyze ethanol. Alterations in the current intensity in the amperometric determination of ethanol have been measured to investigate the impacts of diverse modifications applied to SPCE on its electrochemical biosensor features towards ethanol. The alterations have been ascribed to the modifications of the conductivity and electrocatalytic activities of the electrodes. The researchers examined the bio-sensor responses for ethanol as a function of pH, working potential, and amounts of ADH and NAD+. The optimal values of the factors in the ethanol detection equaled 7.75 for pH, 150 units for the amounts of enzyme, +0.2 V for the working potential, and 7 mM for the amounts of coenzyme. Researchers also observed a sensitivity of 0.49 μA mM−1, a linear range of 320.2 to 1000 μM, and LOQ of 96.1 and 320.2 μM for the biosensor. Furthermore, they investigated the biosensor repeatability at +0.2 V with 400 μM ethanol solution. Consequently, 1.57% (n = 10) RSD was observed. According to the operational stability investigations, the initial amperometric responses of the biosensor to ethanol declined by 72.2% on day 7. As a result, according to the storage life examinations, the sensitivity of the biosensor diminished by 78.3% at the end of week 6. Next, researchers experimented this new biosensor to detect ethanol in the commercial alcoholic beverages so that the outputs were consistent with the results approved by the pertinent suppliers.85
A very simple and selective polymer film-modified electrode for vanillin determination in commercial food products was developed via electrochemical polymerization and overoxidation of pyrrole by Karabiberoğlu et al. The formation of both poly(pyrrole) and overoxidized poly(pyrrole) films on a glassy carbon electrode surface was characterized by electrochemical impedance spectroscopy, SEM, and X-ray photoelectron spectroscopy. Under the optimized conditions, the calibration graph was comprised of two linear segments of 0.032–1.50 μM and 3.0–150.0 μM with a detection limit of 0.012 μM. The selectivity of the modified electrode was examined in the presence of metals, inorganic ions, and organic substances. Moreover, the proposed method was successfully used for the assessment of vanillin contents in commercial food products with satisfactory results.86
In this regard, Mudila et al. used redox cyclic voltammetric analysis to explain the efficacious detection of urea, for which a modified PPy graphene oxide (PPY-GO, GO 20% w/w of PPY) nanocomposite electrode was designed. CV measurements indicated the efficient transfer of electrons in 0.1 M KOH electrolytic solution within the potential window range between 0 and 0.6 V. According to the analysis, the PPY-GO-modified electrode had a moderate electrocatalytic impact on the urea oxidation, which resulted in its detection in the electrolytic solution. Researchers utilized SWV in a concentration range of urea from 0.5–3.0 μM with 0.27 μM LOD to determine the current linear dependence versus the urea concentration. Finally, the nanocomposite film-modified electrode applied a synergistic impact like higher conductivity, a faster rate of electron transfer, and intrinsic catalytic abilities.88
Kausaite Minkstimiene et al. devised an amperometric glucose biosensor based on the graphite rod (GR) working electrode modified with the bio-composite containing the poly(pyrrole-2-carboxylic acid) (PCPy) particles and then the glucose oxidase (GOx) was examined. PCPy particles were synthesized using chemical oxidative polymerization with H2O2 as the initiator of the polymerization reaction, and were modified covalently with GOx (PCPy–GOx) following the activation of the carboxyl groups situated at the surface of the particles with a mixture of N-(3-dimethyl-aminopropyl)-N′ethylcarbodiimide hydrochloride (EDC) and N-hydroxy-succinimide (NHS). After that, the PCPy–GOx biocomposite was dispersed in a buffer solution that contained specific amounts of bovine serum albumin (BSA). The resultant biocomposite suspension was absorbed on the GR electrode surface, and then solvent airing and chemical cross-linking of the proteins were carried out with the glutaraldehyde vapor (GR/PCPy–GOx). According to the analyses, the current response of the GR/PCPy–GOx electrode to glucose gauged at +300 mV vs. Cl reference electrode was affected by the time interval of the synthesis of the PCPy particles, the pH of the GOx solution utilized for the modification of the PCPy particles, as well as the amounts of immobilized PCPy–GOx biocomposite. Moreover, the optimum pH of the buffer solution for activating the biosensor was 8.0. The LOD was 0.039 mmol L−1 with regard to the signal-to-noise ratio (S/N: 3), and this glucose biosensor was used to test human serum samples.89
Al-Sagur et al. published a measurable synthesis of the multifunctional conducting polyacrylic acid (PAA) hydrogel (MFH) integrated with rGO, lutetium phthalocyanine (LuPc2), and vinyl-substituted PANI (VS-PANI) as the powerful 3D matrix for glucose oxidase (GOx) immobilization (PAA-rGO/VS-PANI/LuPc2/GOx-MFH). Researchers then integrated multi-components like PAA with the rGO and VS-PANI via a free radical polymerization process with the use of the methylene bis-acrylamide as well as ammonium per-sulfate as the cross-linker and initiator. LuPc2 was doped to form the multifunctional hydrogel (PAArGO/VS-PANI/LuPc2-MFH). Fig. 9A and B respectively indicate the creation of the PAA-rGO/VS-PANI/LuPc2/GOx-MFH and SEM images of PAA-rGO/VS-PANI-MFH, PAA/VS-PANI-MFH. GOx was been immobilized in PAA-rGO/VS-PANI/LuPc2-MFH to fabricate the biosensor and utilized for the electrochemical determination of glucose. The PAA-rGO/VS-PANI/LuPc2/GOx-MFH biosensor showed a higher sensitivity of 15.31 μA mM−1 cm−2 for detecting glucose at concentrations ranging between 2 and 12 mM with LOD of 25 μM. Moreover, the PAA-rGO/VS-PANI/LuPc2-MFH bio-sensor displayed a faster response time (1 s) to the addition of glucose, which had a higher storage stability (three months). According to the real sample analysis, PAA-rGO/VS-PANI/LuPc2/GOx-MFH was efficiently utilized as the electrochemical biosensor in the clinical and industrial diagnosis.90
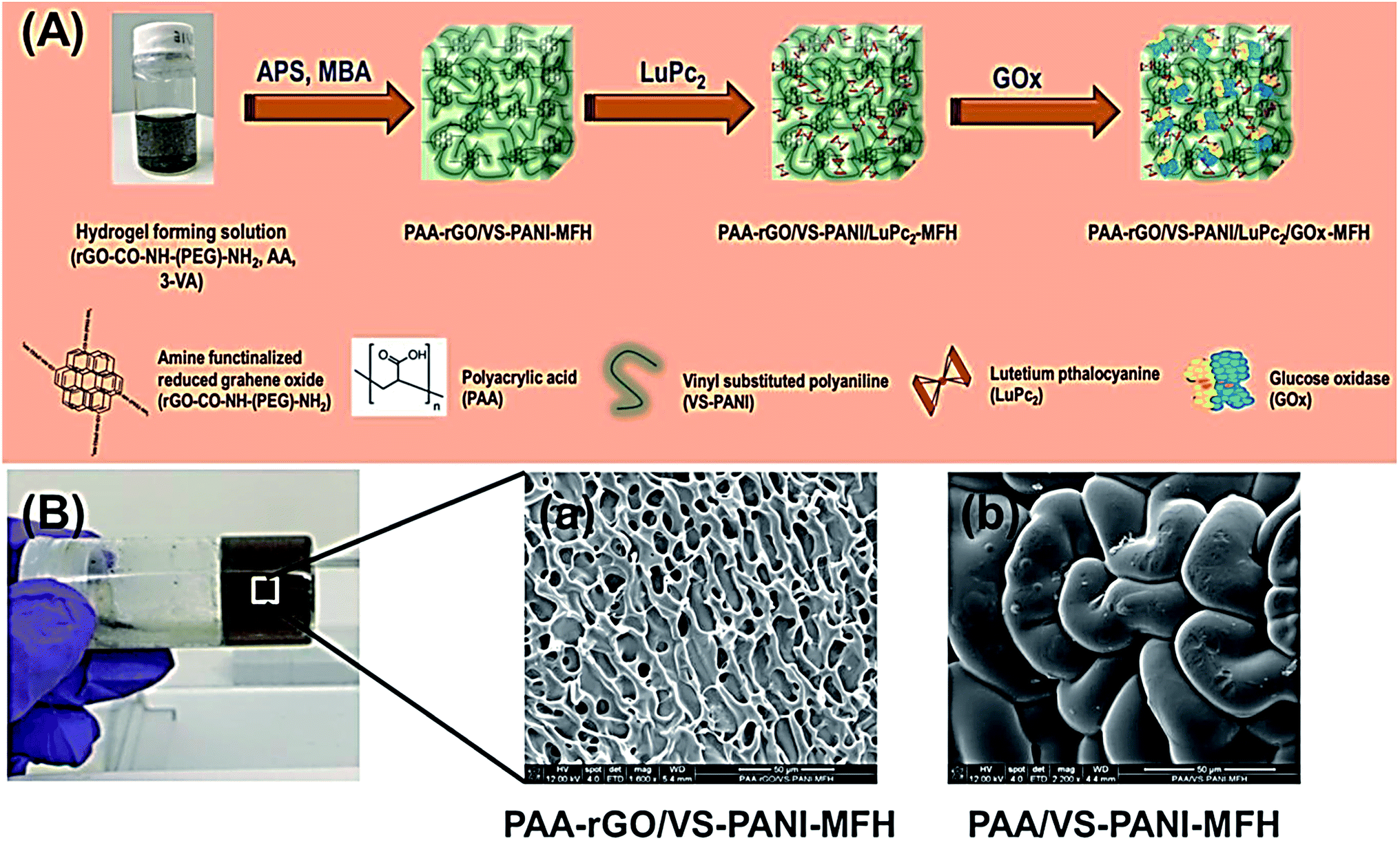 | ||
| Fig. 9 (A) The PAA-rGO/VS-PANI/LuPc2/GOx-MFH synthesis procedure. (B) SEM images of (a) PAA-rGO/VS-PANI-MFH, and (b) PAA/VS-PANI-MFH. Reproduced with permission from ref. 90. Copyright 2017 Elsevier. | ||
One of the novel selective and sensitive biosensors was presented by Shafaat et al. to determine cytochrome C as one of the biomarkers for cell apoptosis. This biosensor has been built based on the electrochemical entrapment of the cytochrome C aptamers in the CPs as the dopant anion on the SPCE surface. EIS, as one of the robust electrochemical techniques, was utilized as a detection technique. This aptasensor showed wider linear dynamic ranges for determining cytochrome C from 10 pM−1 nM; 3.29% RSD and 5 pM LOD were seen in the analyses. It also showed higher selectivity and acceptable stability for determining cytochrome C, even in the presence of the serum proteins (such as albumin, lysozyme, fibrinogen, immunoglobulin G) at the increased levels of concentration with success as one of the clinical detection tools for detecting cytochrome C.91
Serafín et al. provided a fast, sensitive method for determining the clinically related biomarker receptor tyrosine kinase AXL in the serum consisting of the amperometric disposable immunosensors. Therefore, they sandwiched the targeted protein between a particular capture antibody covalently immobilized on the SPCEs modified with the electro-polymerized poly(pyrrolepropionic acid) and a biotinylated detector antibody labeled with a streptavidin-horseradish peroxidase conjugate. Researchers also measured the amperometric response at −0.20 V versus the Ag pseudo-reference electrode of the SPCE by adding H2O2 in the presence of hydroquinone (HQ) as the mediator. Such an integrated immuno-sensing platform represented a lower LOD of 337 pg mL−1, acceptable selectivity as compared to the other non-target serum proteins and finally offered outputs with statistical compatibility with the ones observed in the use of a commercial ELISA kit. Hence, it could be said that the mentioned interesting characteristics, simplified operation and automation, and miniaturization of the pertinent equipment made this method an encouraging candidate for developing the tools for on-site clinical analyses.92
Kangkamano et al. devised a label-free electrochemical miRNA biosensor based on a pyrrolidinyl peptide nucleic acid (acpcPNA)/PPy/silver nanofoam (Ag NF)-modified electrode. This AgNF was electrodeposited as a redox indicator on the gold electrode that was functionalized with an electro-polymerized layer of PPy, which was considered as a conducting polymer, for the immobilization of the PNA probes. EIS was used to investigate the construction procedure of the method. Researchers utilized the biosensor for detecting miRNA-21 that was considered as a biomarker with abnormal expression in several kinds of cancer. They used CV and controlled the signals by changing the current of the Ag NF redox reaction prior to, and following, the hybridization. 2 PNA robe lengths were examined and the longer probe represented the more acceptable function. It was found that nucleotides overhanging on the electrode side had a greater impact on the signal than those on the solution side because of the higher insulation of the sensing surfaces. Based on the optimum conditions, the electrochemical signal was proportionate with the miRNA-21 concentration ranges between 0.20 fM and 1.0 nM, with a lower LOD of 0.20 fM. This biosensor exhibited higher specificity with the ability to differentiate between the complementary single- and double-base mismatched, and non-complementary targets. Analyses showed that 3 of 7 experimental plasma specimens had certain concentrations of 63 ± 4, 111 ± 4, and 164 ± 7 fM. Thus, this sensor exhibited an acceptable recovery of 81–119%. Consequently, the possible analyses of the biosensor without RNA extraction and or amplification showed the potential usefulness of the sensor for the prognosis as well as diagnosis of cancer in clinical applications.93
4.2. Energy storage and conversion
Experts in the field have considered energy as the most prominent universal issue because of the exhaustion of fossil fuels. Energy storage devices require electrode materials with good electronic conductivity to deliver maximum power. CPs are very attractive materials due to their broad range of conductivities, from semiconductor (10−11 to 10−3 S cm−1) to metal (10−1 to 106 S cm−1) behavior. Their charge storage can be enhanced by controlling the synthetic parameters (i.e., doping agent, temperature, pH, etc.) or by designing different morphologies, surface area, redox activity, etc. CPs have very high current density and are inexpensive when compared to carbonaceous electrode materials. Taking PANI as an example, a supercapacitor device based on PANI can exhibit a specific energy of 10 W h kg−1 with a slightly lower specific power of 2 kW kg−1, while a carbon-based supercapacitor device can only reach a specific power of 3–4 kW kg−1 and a specific energy of 3–5 W h kg−1. In particular, CPs are utilized in energy storage devices due to their high charge/discharge cycling rate, high conductivity, large surface area, high specific capacitance, and good redox reversibility.94,95 The conducting polymers generally have high specific capacitance values and can be desirable materials for developing the next generation of energy storage and conversion.Although experts in the field have found it necessary to the design of the nanostructured poly(3,4 ethylene-dioxythiophene) (PEDOT) with increased capacitance and mechanical flexibility to realize the high-performance supercapacitors, it is hard to make these types of electrodes with fast and affordable methods. Therefore, Wang et al. introduced a simplified, rapid, and flexible approach to making the mechanically powerful, strongly conductive, and versatile nanostructured PEDOT paper. The composite substance that could be fabricated in 30 min with the solution-based reactions exhibited a larger surface area (137 m2 g−1), lower sheet resistance (1.4 Ω cm−1), and higher active mass loading (7.3 mg cm−2). The symmetric PEDOT paper-based super-capacitors provided a higher specific electrode capacitance of 90 F g−1, 920 mF cm−2, and 54 F cm−3 and very good cycling stability (93% capacity retention after 15000 cycles at 30 mA cm−2) in 1.0 M H2SO4. The electrochemical function of the supercapacitors was retained at various bending angles, which showed the flexibility of the tools.99
He et al. demonstrated a simplified poly(o-methylaniline)-derived construction of the bifunctional microporous nitrogen-doped carbon micro-spheres (NCMSs) with increased electrocatalytic activities and stability for energy storage and the oxygen reduction reaction (ORR) in the supercapacitors. It was observed that when the pyrolysis temperature reached 900 °C, the dispersed NCMSs presented a higher surface area of 727.1 m2 g−1, acceptable total N-doping content, and a higher concentration of the quaternary N, exhibiting a more reasonable electrocatalytic activity for the ORR in comparison to the commercial Pt/C catalysts. The higher specific capacitance (414 F g−1) and very good durability showed their attractiveness for the advanced process of energy conversion and storage; therefore, their conducting polymer-derived method can offer novel ground for making heteroatom-doped carbon substances for energy device applications.100
Jena et al. addressed a new PANI derivative, poly(2-methyl thioaniline)-coated MWCNTs (PMTA@CNT) and the composite with graphene (PMTA@CNT/RGO) for use as the supercapacitor. The PMTA@CNT/RGO and PMTA@CNT electrodes exhibited considerably greater specific capacitances of 616 F g−1 and ∼522 F g−1 in 6 M KOH electrolyte at a current density of 1 A g−1. The obtained outputs were nearly 28 and 36% greater than the PANI@CNT/RGO and PANI@CNT electrodes procured by a synthesis procedure similar to the PMTA based electrode. It is worth mentioning that the PMTA-based composite electrode had a very good electrochemical function as compared with the PANI-based composite electrode. This could have been caused by the presence of an acceptable electron-donating group (–SCH3) in PMTA, which made the PMTA molecule electronically rich via the resonance effects that facilitated the transfer of the charges between the 2 components using the π-electrons of the quinoid rings in PMTA and benzenoid rings of the CNTs or graphene (Fig. 10). Hence, the PMTA composite electrode with acceptable capacitive behaviors encouraged its utilization for competing with the PANI composite-based electrode.101
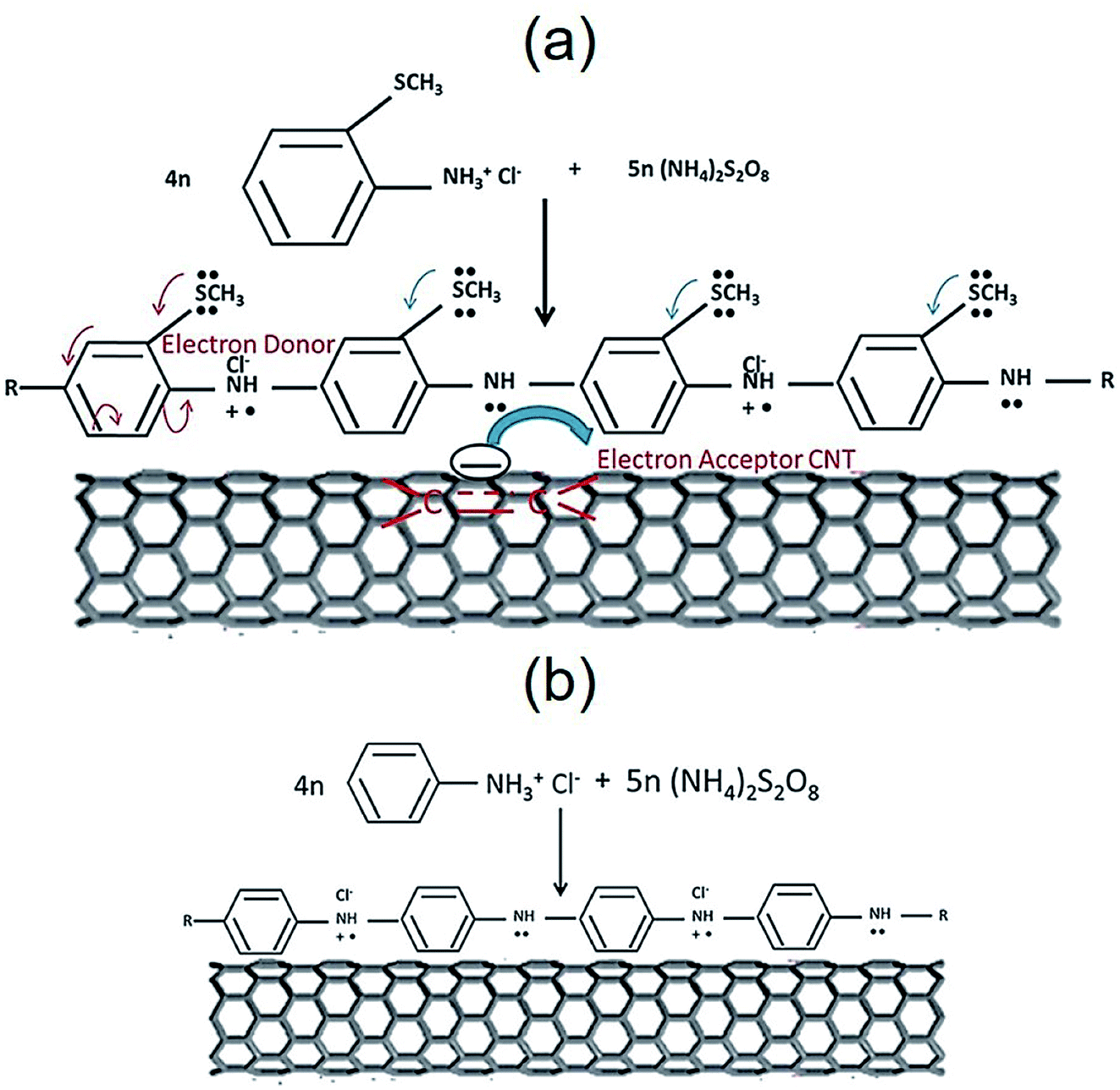 | ||
| Fig. 10 The electron conjugation process of the SCH3 group of (a) PMTA versus (b) PANI, accompanied by the traditional donor–acceptor state of the interaction (probable conduction mechanism). Reproduced with permission from ref. 101. Copyright 2017 Elsevier. | ||
Zhu et al. simply synthesized N-doped porous carbons (NPCs) with the nano-fiber-like structure by a one-step direct carbonization/KOH activation of the poly(aniline-co-p-phenylene-diamine) (P(ANI-co-PPDA)) at 600 to 900 °C in a N2 atmosphere. They showed that the greater heat treatment temperature enhanced the specific surface area (between 776 and 2022 m2 g−1), whereas it decreased the nitrogen content (from 6.96 to 1.18 wt%). The final NPCs (i.e., NPC-700) showed a higher surface area of 1513 m2 g−1, higher N content (6.43 wt%), and nanofiber-like morphology at 700 °C. The results indicated that in 6 M KOH electrolyte, the NPC-700 electrode had a capacitance of 316 F g−1 at 1.0 A g−1 and 167 F g−1 at 10.0 A g−1. The NPC-700 electrode exhibited an acceptable cycling stability with 81.6% capacitance retention after 5000 cycles at 1.0 A g−1. Finally, the simplified synthesis path and higher electrochemical functions of NPCs showed higher potential in the supercapacitor application.102
Ji et al. presented a quaternary electrolyte formulation for constructing a pristine carbon and PPy composite, which did not sacrifice electron or ion mobility. Lithium perchlorate (20 mM) was used as a supporting electrolyte and dopant, with sodium dodecyl-benzenesulfonate at a lower concentration of 1.43 mM as a surfactant, as well as pyrrole monomers and pristine carbon nanomaterials. Based on the analyses, the difference in the order of magnitude between the concentration of the dopant and surfactant ions allowed the as-deposited PPy to be dominantly doped by the small-sized and mobile perchlorate anions. Researchers succeeded in the preparation of the PPy composites with carbon black, CNTs, and electrochemically exfoliated graphene (EEG) with their newly fabricated quaternary electrolyte. Therefore, this PPy/EEG composite electrode exhibited 348.8 F g−1 specific capacitance with higher rate capability (190.7 F g−1 at 71 A g−1). Supercapacitor instruments based on the PPy/EEG composite electrodes exhibited higher rate behaviors of 500 mV s−1 and a longer cycle life of 5000 cycles.103
Gal et al. introduced the polyacetylene-based polyelectrolyte for application as quasi-solid-state dye-sensitized solar cells (DSSCs). Researchers used the phosphorus oxychloride activated polymerization of 2-ethynylpyridine to prepare the conjugated polyelectrolyte, and poly[2-ethynyl-N-(dichlorophosphino)pyridinium chloride] was utilized for the DSSC. The photoluminescence (PL) spectrum of the polymer solution demonstrated 2 maximal peaks of 471 and 489 nm relative to the photon energies of 2.64 eV and 2.54 eV. Poly[2-ethynyl-N-(dichloro-phosphino)pyridinium chloride] was used to fabricate the quasi-solid-state DSSCs with a SnO2:F/TiO2/D719 dye/solid-state electrolyte/Pt device, reflecting the highest power conversion efficiency (PCE) of 5.70%.106
Gopalan et al. reported the design and synthesis of a novel sulfonated PANI derivative consisting of thiol (−SH) groups (SPAN(SH)) for achieving the efficient dispersion of the AuNPs in SPAN(SH) (SPAN(SH)@GNP). They also assessed the photo-voltaic function of the poly(3-hexylthiophene-2,5-diyl): indene-C60 bisadduct (P3HT:ICBA) for elucidating the contribution of the SPAN(SH)@GNP NH (nanohybrid) buffer layer in the polymer solar cell (PSC) device architecture; ITO (indium tin oxide)/SPAN(SH) or SPAN(SH)@GNP(X%) NH/zinc oxide (ZnO)/Al (Fig. 11). According to the results, the PCE of the PSC was enhanced using the SPAN(SH)@GNP buffer layer. A buffer layer with 1.66 wt% of GNP based on the weight of SPAN(SH), (ITO/SPAN(SH)@GNP(1.66%)/P3HT:ICBA/ZnO/Al) highly improved the PCE (5.20%), which was significantly greater than the PCE (4.25%) of SPAN(SH) (ITO/SPAN(SH)/P3HT:ICBA/ZnO/Al). The very strong near field of the GNP was distributed laterally in the buffer layer and extended vertically into the adjacent P3HT:ICBA (active) layer, and provided a greater rate of exciton production in the active layer. Consequently, the synergistic impact of the energy level alignment, lower interfacial resistance, efficient hole extraction, and greater surface areas between the buffer/active layer significantly increased the PCE. Based on the outputs, designing the conducting polymer structure for proper interaction with PNP to achieve greater PCE is of great importance.107
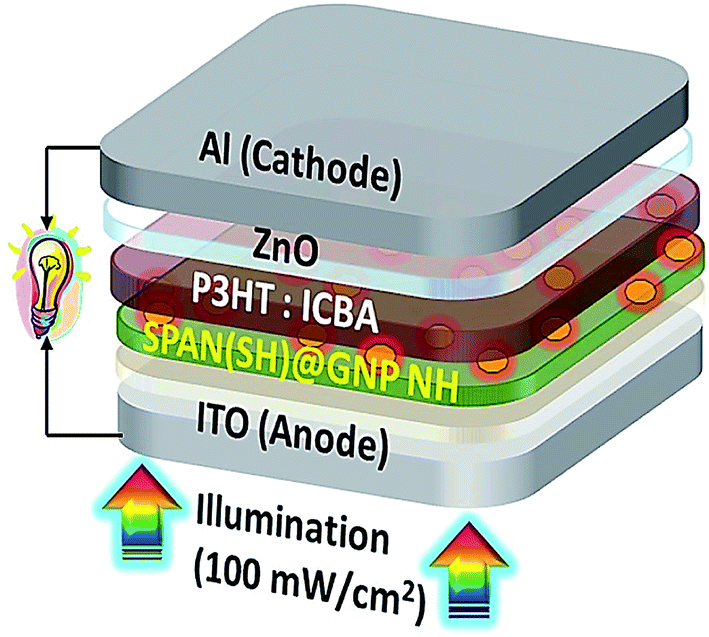 | ||
| Fig. 11 BHJ PSC with the tool architecture: ITO/SPAN(SH)@GNP(X%) NH/P3HT; ICBA/ZnO/Al. Reproduced with permission from ref. 107. Copyright 2018 Elsevier. | ||
Another study conducted by Alem et al. built poly(N-9′-heptadecanyl-2,7-carbazole-alt-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiadiazole (PCDTBT) based on the photovoltaic cells with the flexographic printing method, enabling the increased throughput-patterned active layers for deposition on the flexible substrates at the lowest cost. Such a condition has been met via the optimization of the flexographic plate pattern, solvent, drying process, and print speed. Researchers also incorporated halftone patterning, which is one of the popular processes in the graphics printing industry, and optimized the speed of printing; therefore, the homogeneity of the active layer print was considerably ameliorated. Future research on the appropriate solvents and drying conditions resulted in better uniformity and lower pin-hole formation. The usability of the flexographically-printed active layer was confirmed by making 1 cm2 photo-voltaic cells that displayed 3.5% efficiency, which could be compared with the other deposition methods. Hence, the flexography is appropriate for use as one of the construction techniques for bulk heterojunction organic photovoltaics.108
One of the device architectures for efficacious perovskite solar cells, which used poly(3-hexylthiophene) as one of the hole transport materials without any dopant, was introduced by Jung and co-workers. A thin layer of the broad-band gap halide perovskite was created on the top of the narrow bandgap light-absorbing layer via an in situ reaction of n-hexyl trimethyl ammonium bromide on the perovskite surface. The new instrument had a strong power conversion efficiency of 22.7% with ±0.51% hysteresis and exhibited suitable stability at 85% relative humidity without any encapsulation. In the case of encapsulation, it demonstrated a lengthier operational stability for 1370 h under 1 sun illumination at room temperature, which maintained 95% of the initial efficiency. Therefore, they extended our platform to the larger area modules (24.97 square centimeters) that have been constructed with a scalable bar-coating technique for poly(3-hexylthiophene) deposition and reached 16.0% PCE.109
It has been demonstrated that the chemical complexity of the conventional DSSC electrolyte required the development of greater selectivity and catalytic counter-electrode (CE) substances for the triiodide target molecule. For example, Sangiorgi et al. revealed that the molecularly imprinted PPy (MIP-PPy) could resolve the above problems. Various template molecules like L-2-aminopropionic acid (L-alanine) and 2-aminoacetic acid (glycine) have been investigated in the course of the electro-polymerization procedure for verifying MIP-PPy utilization as CE. Using the lower concentration of glycine resulted in an MIPPPy film that showed greater catalytic activity and electrochemical features on the triiodide reduction in comparison to the non-imprinted PPy (NIP-PPy). Gel-state DSSCs-based on the MIP substances have been procured and tested so that the optimized MIP-PPy CE with glycine as a template displayed nearly 20% enhancement in the power conversion efficiency and 50% reduction in the charge transfer resistance as compared to the cells based-on NIP-PPy. The obtained findings implied the possible enhancement of the PPy CE catalytic features with no addition of other substances or significant modifications in the generation procedure process; however, it enhanced the selectivity of the electrode.110
Researchers have introduced several CPs with positive effects for improving the rate capability and cycle life of the electrode materials due to their acceptable conductivity (between a few S cm−1 and 500 S cm−1), faster charge–discharge kinetics, as well as plastic features.113 Wang et al. procured a novel PTh-coated silicon composite anode substance with an in situ chemical oxidation polymerization procedure and utilized it as the anode in LIB. They described this substance structure with infrared spectroscopy, proving the occurrence of the oxidative polymerization of thiophene in the α-position. Poly-thiophene offered a more acceptable electric contact between the silicon particles; hence, the as-procured Si/PTh composite electrodes achieved more reasonable cycling performance in comparison to the bare Si anode. Finally, the specific capacity of the composite electrode retained 478 mA h g−1 following 50 cycles.114
Hua et al. designed a new electrodeposited hydrothermal technique for fabricating the high density β-Na0.33V2O5 nano-wire film on the metal current collector (Ti-foil). These researchers investigated the crystal structure, morphology, and impact of the deposition voltage, time, and temperature onto the procured specimens. Analyses showed a thickness of β-Na0.33V2O5 nano-wire film of ∼10.5 μm based on the experimental conditions of five hours, 90 °C temperature, and 2.5 V voltage. Coating β-Na0.33V2O5 nano-wire film with PPy and using it as a cathode of ILB revealed a higher reversible discharge capacity of 269.9 mA h g−1 at a current density of 50 mA g−1. On the contrary, this material cycling behavior has been highly more acceptable than the bare βNa0.33V2O5 nano-wire film. Finally, Hua et al. provided an interesting ground for exploring higher tap density cathode substances for LIB with greater electrochemical Functions.115
Another study conducted by Wang et al. designed a new 1D and co-axial PANI@tin dioxide@MWCNT (PANI@SnO2@MWCNT) composite as a conductive additive-free anode substance for the LIB. They initially fixed SnO2 NPs (5 nm) on the conductive MWCNT skeleton via self-assembly of the nanosized SnO2 particles over the MWCNT surface using the surfactant P123 and the in situ coating of a flexible layer of PANI with very good conductivity of the electron and lithium-ion (Fig. 12). According to their results, 1D and co-axial PANI@SnO2@MWCNT has been capable of the effective accommodation of the volume extension of the SnO2 NPs in the course of the lithiating and de-lithiating by wrapping a flexible coating layer of the PANI and buffer of 1D MWCNT. The synergistic action between MWCNT and PANI ameliorated the electronic and lithium ionic conductivity of the composite. The PANI@SnO2@MWCNT composite exhibited a lengthier cycling stability and more acceptable rate capability in comparison to the SnO2@ MWCNT composite, which exhibited a higher reversible charge/discharge specific capacity of 878/888 mA h g−1 at 0.2 A g−1 over 100 cycles, and 524/527 mA h g−1 at 1.0 A g−1 over 150 cycles, with no conductive additives in the electrode.116
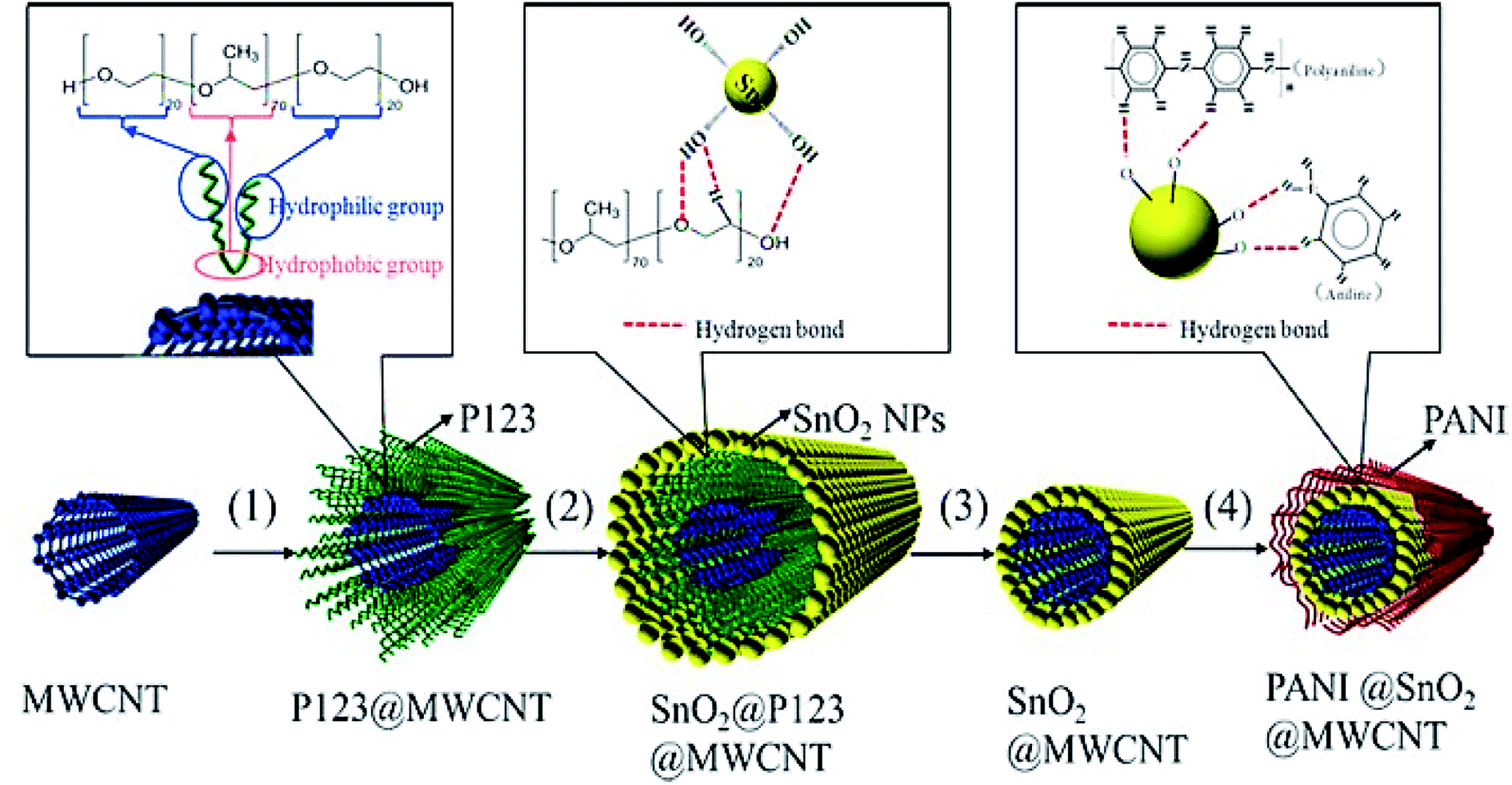 | ||
| Fig. 12 The preparation of PANI@SnO2@MWCNT; steps (1)–(4) represent the dissolution, self-assembly, heating, and in situ polymerization. Reproduced with permission from ref. 116. Copyright 2018 Elsevier. | ||
Experts in the field have considerably investigated ZnO as an electrode material in LIBs because of its high theoretical capacity. However, unsuitable electronic conductivity and numerous volumetric alterations in the course of the cycling restrict its industrial utilization. For example, Li et al. procured poly-pyrrole nanorings (PNRs) using the solution chemistry technique with pyrrole (Py) as raw material, ammonium persulfate (APS) as the oxidant, and cetyltrimethylammonium bromide (CTAB) as a surfactant. Moreover, zinc oxide NPs adsorbed on the PPy nanoring surface via a one-pot in situ sol–gel technique have been used to synthesize the ZnO/PNR composite. This composite showed a 3D interconnected network structure where the diameter of the PPy nanorings equals 80–100 nm and the average size of the ZnO nanocrystals with uniform distribution equals 10.49 nm. Therefore, a specific 3D conductive framework could establish suitable electronic contact between ZnO particles and buffer the volume variations in the course of the lithiation or delithiation procedures. The ZnO/PNR composite, as one of the electrode materials for LIBs, provides 1658 mA h g−1 first-cycle discharge capacity and 50.7% capacity retention over 150 cycles at 200 mA g−1, which reflects greater specific capacity and the significant stability of the cycle.117
Fard et al. built poly(pyrrole-co-aniline) (PPCA) hollow nanospheres (HN) as a catalyst support material using in situ emulsion polymerization. According to their study, a one-step and template-free strategy was introduced for the fabrication of the Pd NFs on a PPCA HN-coated GCE using a simplified electrochemical procedure. Moreover, CV, EIS, and chrono-amperometry towards the methanol oxidation as one of the model reactions in the alkaline medium have been used to evaluate the catalytic function of the Pd NFs/PPCA HN catalyst. The particular activities of Pd NFs/PPy (1.28 mA cm−2), Pd NFs/PPCA HN (1.79 mA cm−2), Pd NFs (0.78 mA cm−2), and Pd NFs/PANI (0.93 mA cm−2) showed that the PPCA-supported Pd NFs with higher surface area exhibited very good electrocatalytic activities in comparison to the other electrodes for the electro-oxidation reaction in alkaline medium. Such conditions could be the result of simpler charge transfer at the interface of the conductive copolymer, greater electrochemically available surface areas and electronic conductivity. Therefore, the proposed approach offers encouraging ground for direct methanol fuel cells.118
El-Moghny et al. recently devised new catalysts of the platinum NPs dispersed on PANi for formic acid electro-oxidation (FAO), which is a basic anodic reaction in the direct formic acid fuel cells (DFAFCs). Fig. 13 depicts the role of PANi in reducing CO poisoning and improving the catalytic activity of the Pt/PANi/GC electrode towards FAO. Therefore, the catalyst preparation scheme allowed the sequential electrodeposition of the fibril PANi and spherical PtNPs (ca. 65 nm in size) on the glassy carbon (GC) substrate and permitted the accurate control of the deposition sequence and loading. Moreover, incorporating PANi into the catalyst constituents could considerably (ca. 16 times) improve the catalytic activities of the catalyst toward FAO via shifting this mechanism toward a suitable dehydrogenation pathway and lessening the unfavorable toxic dehydration pathway. The researchers manipulated the deposition order and loading of diverse constituents of the catalyst to tune the catalytic efficacy. Multiple procedures have been utilized for confirming the substantial deposition of the catalyst and evaluating its composition, morphology, and crystal structure. Although PtNPs would be necessary for absorbing FA, PANi improved the PtNPs dispersion and mediated FAO to facilitate the charge transfer and mitigate the CO toxicity. Longitudinal continual (150 CVs) electrolysis testing demonstrated encouraging catalytic stability.126
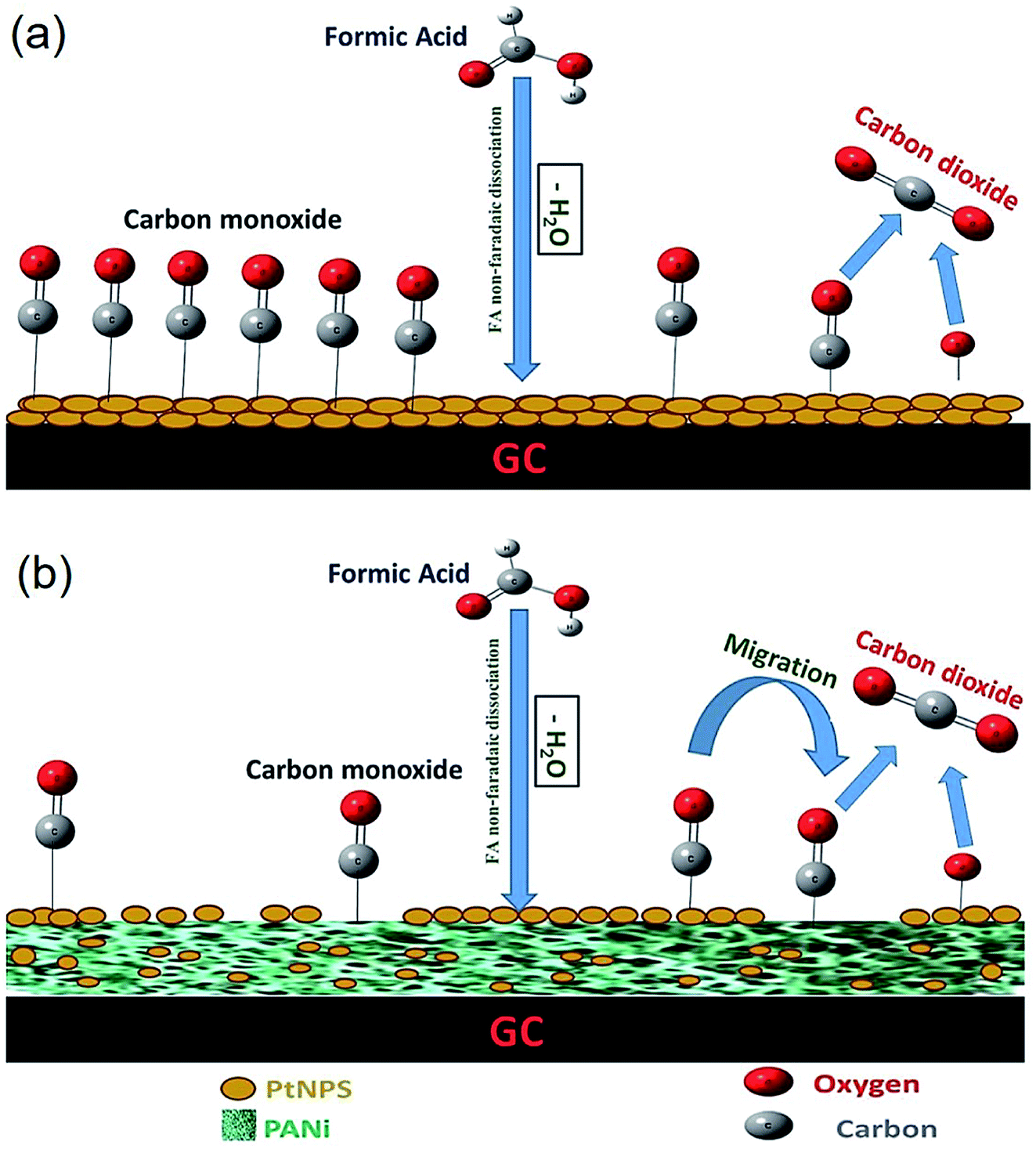 | ||
| Fig. 13 A representation of the poisoning dehydration pathway of FAO at the (a) Pt/GC and (b) Pt/PANi/GC electrodes. The amount of COads was lowered at the Pt/PANi/GC electrode, and PANi may further preferentially capture CO. Reproduced with permission from ref. 126. | ||
Another study conducted by Miglbauer et al. demonstrated the versatility of the conducting polymer poly(3,4-ethylene-dioxythiophene) (PEDOT) as an electrocatalyst for the peroxide fuel cells with no loss as a result of disproportionation. The researchers showed that PEDOT, as one of the cathodic catalysts to reduce peroxide to water, performed at a level on par with the most acceptable inorganic catalysts. Therefore, with the use of the PEDOT as the cathode and nickel as the anode material, open circuit potentials are possible in the range between 0.5 and 0.6 V with a power density of 0.20–0.30 mW cm−2. Thus, PEDOT functioning as the catalyst for the reduction of the hydrogen peroxide to water can be mechanistically understood. The resulting output would be a measurable hydrogen peroxide fuel cell cathode, used for the demonstration of the ability of the organic semiconducting substances as the electrocatalysts.127
Attar et al. were the first to demonstrate the electrocatalytic function of the PPy–copper oxide particles-modified carbon paste electrode (Cu2O/PPy/CPE) for the electrocatalytic oxidation of ethanol in alkaline media (Fig. 14). They procured the composite Cu2O/PPy with a simplified strategy containing the PPy film deposition on the CPE through a galvanostatic state, followed by the copper particles deposition at a fixed potential. Therefore, researchers applied CV and EIS for characterizing the electrochemical features of Cu2O/PPy/CPE and explaining the electro-oxidation system of ethanol. Attar et al. then examined and optimized the experimental variables affecting ethanol electro-oxidation. Based on the results, the electro-deposition of the copper particles on the PPy film augmented the catalytic activities toward ethanol oxidation, with the peak current density of 2.25 mA cm2 at 0.8 V vs. Ag/AgCl, which is 2.6 times greater than the peak current density observed in the PPy/CPE electrode. The saturation limit reached a value of 5 M. The acceptable catalytic activity, simplified procurement, and stability of the Cu2O/PPy composite indicate that it is a very good electrocatalyst for the oxidation of ethanol.128–131
 | ||
| Fig. 14 The preparation of Cu2O/PPy/CPE for ethanol electrocatalytic oxidation. Reproduced with permission from ref. 128. Copyright 2020 Elsevier. | ||
5. Conclusion
The electrochemical sensors and biosensors have been regarded as making a key contribution to diverse areas like medical diagnosis, medicine, environment, and food analyses. It has been revealed that the conducting polymers have an encouraging contribution to the generation of the latest electrochemical biosensors and sensors as a result of their distinct benefits in comparison to other substances. Notably, the recovery time, responsivity, higher selectivity and sensitivity, and reasonable stability contribute to the production of an acceptable sensor and its function. Generally, features of the charge transfer of the conducting polymers due to the existence of a p-conjugated mechanism resulted in the provision of an appropriate carrier for transferring electrons from the redox-active center to the electrode surface.As mentioned earlier, experts in the energy storage field are giving increasing consideration to electrical vehicles, grid-scale storage, and portable electronics for the optimal utilization of renewable energy resources like solar and wind energies. Therefore, studies on novel technology for the generation and storage of electrical energy should receive special attention. As the novel substances are progressively synthesized and prepared, a number of sustainable, eco-friendly energy conversion and storage mechanisms, such as in the super-capacitors, solar cells, LIB, as well as the fuel cells, would be strongly designed. As a result of their metal-like conductivity and reversible electrochemical doping/de-doping capability, conducting polymers have been regarded as encouraging substances for converting and storing energy. Molecular backbones usually consist of conjugated double bonds; moreover, the strongly conjugated polymer chains can be reversibly assigned the electrochemical features by a doping/de-doping procedure. Hence, via the modulation and control of the doping levels, their conductivity can be adjusted in various ranges between 10−10 and 104 S cm−1, which span the whole range from the insulators to the semi-conductors and conductors. Such conducting polymers maintain the benefits of conventional polymers like lower cost, simplified procurement, a suitable affinity for several other substances, and greater durability and flexibility. Finally, the CPs, particularly PPy PANI, and PEDOT, have adequate potential for use as the active electrode substances for electrochemical applications.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The financial support of the Future Material Discovery Program (2016M3D1A1027666) and the Basic Science Research Program (2017R1A2B3009135) through the National Research Foundation of Korea are appreciated. The supports provided by Kerman University of Medical Sciences, and Graduate University of Advanced Technology; Kerman, are appreciated. In addition, the supports from Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number of 104.05-2020.15 is also appreciated.References
- H. Gerischer, Principles of electrochemistry, The CRC handbook of solid state electrochemistry, 2019 Search PubMed.
- W. Schmickler and E. Santos, Interfacial electrochemistry, Springer Science & Business Media, 2010 Search PubMed.
- S. Tajik, H. Beitollahi, F. Garkani Nejad, K. Zhang, Q. V. Le, H. W. Jang, S. Y. Kim and M. Shokouhimehr, Sensors, 2020, 20, 3364 CrossRef CAS.
- H. Beitollahi, S. Tajik, Z. Dourandish, K. Zhang, Q. V. Le, H. W. Jang, S. Y. Kim and M. Shokouhimehr, Sensors, 2020, 20, 3256 CrossRef CAS.
- B. Nikahd and M. A. Khalilzadeh, J. Mol. Liq., 2016, 215, 253 CrossRef CAS.
- Z. C. Liu, D. S. Shin, M. Shokouhimehr, K. N. Lee, B. W. Yoo, Y. K. Kim and Y. S. Lee, Biosens. Bioelectron., 2007, 22, 2891 CrossRef CAS.
- K. Zhang, K. O. Kirlikovali, R. S. Varma, Z. Jin, H. W. Jang, O. K. Farha and M. Shokouhimehr, ACS Appl. Mater. Interfaces, 2020, 12, 27821 CrossRef CAS.
- Y. Shi, L. Peng, Y. Ding, Y. Zhao and G. Yu, Chem. Soc. Rev., 2015, 44, 6684 RSC.
- S. Tajik, H. Beitollahi, S. Z. Mohammadi, M. Azimzadeh, K. Zhang, Q. V. Le, Y. Yamauchi, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 30481 RSC.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291 CrossRef CAS.
- M. M. Farid, A. M. Khudhair, S. A. K. Razack and S. Al-Hallaj, Energy Convers. Manage., 2004, 45, 1597 CrossRef CAS.
- J. Kim, R. Kumar, A. J. Bandodkar and J. Wang, Adv. Electron. Mater., 2017, 3, 1600260 CrossRef.
- H. Letheby, J. Chem. Soc., 1862, 15, 161 RSC.
- N. Basescu, Z. X. Liu, D. Moses, A. J. Heeger, H. Naarmann and N. Theophilou, Nature, 1987, 327, 403 CrossRef CAS.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 16, 578 RSC.
- A. F. Diaz, J. I. Castillo, J. A. Logan and W. Y. Lee, J. Electroanal. Chem. Interfacial Electrochem., 1981, 129, 115 CrossRef CAS.
- Y. B. Shim and S. M. Park, Synth. Met., 1989, 29, 169 CrossRef.
- Y. B. Shim, M. S. Won and S. M. Park, J. Electrochem. Soc., 1990, 137, 538 CrossRef CAS.
- D. E. Stilwell and S. M. Park, J. Electrochem. Soc., 1988, 135, 2254 CrossRef CAS.
- D. E. Stilwell and S. M. Park, J. Electrochem. Soc., 1989, 136, 427 CrossRef CAS.
- S. Tajik, H. Beitollahi, M. R. Aflatoonian, B. Mohtat, B. Aflatoonian, I. Sheikh Shoaie, M. A. Khalilzadeh, M. Ziasistani, K. Zhang, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 15171 RSC.
- S. Tajik, H. Beitollahi, F. Garkani Nejad, M. Safaei, K. Zhang, Q. V. Le, R. S. Varma, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 21561 RSC.
- T. Yamamoto, H. Fukumoto and T. A. Koizumi, J. Inorg. Organomet. Polym., 2009, 19, 3 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309 CrossRef CAS.
- G. Wang, A. Morrin, M. Li, N. Liu and X. Luo, J. Mater. Chem. B, 2018, 6, 4173 RSC.
- T. O. Magu, A. U. Agobi, L. Hitler and P. M. A. Dass, J. Chem. Rev., 2019, 1, 19 CrossRef.
- M. R. Aflatoonian, S. Tajik, B. Mohtat, B. Aflatoonian, I. Sheikh Shoaie, H. Beitollahi, K. Zhang, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 13021 RSC.
- J. Simonet and J. Rault-Berthelot, Prog. Solid State Chem., 1991, 21, 1 CrossRef CAS.
- J. Huang, K. Wang and Z. Wei, J. Mater. Chem., 2010, 20, 1117 RSC.
- G. Inzelt, Conducting polymers: a new era in electrochemistry, Springer Science & Business Media, 2012 Search PubMed.
- N. Aydemir, J. Malmström and J. Travas-Sejdic, Phys. Chem. Chem. Phys., 2016, 18, 8264 RSC.
- D. G. Prajapati and B. Kandasubramanian, Macromol. Chem. Phys., 2019, 220, 1800561 CrossRef.
- S. Tajik, Z. Dourandish, K. Zhang, H. Beitollahi, Q. V. Le, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 15406 RSC.
- A. J. Heeger, Handbook of conducting polymers, ed T. A. Skotheim, New York, Marcel Dekker, 1986 Search PubMed.
- M. H. Naveen, N. G. Gurudatt and Y. B. Shim, Appl. Mater. Today, 2017, 9, 419 CrossRef.
- J. I. Kroschwitz, Electrical and Electronic Properties of Polymers, Wiley, New York, 1988 Search PubMed.
- M. Gerard, A. Chaubey and B. D. Malhotra, Biosens. Bioelectron., 2002, 17, 345 CrossRef CAS.
- T. O. Magu, A. U. Agobi, L. Hitler and P. M. Dass, J. Chem. Rev., 2019, 1, 19 CrossRef.
- A. Kausar and M. Siddiq, Polymer Science: Research Advances, Practical Applications and Educational Aspects, 2016, p. 177 Search PubMed.
- A. J. Heeger, J. Phys. Chem. B, 2001, 36, 8475 CrossRef.
- X. X. Wang, G. F. Yu, J. Zhang, M. Yu, S. Ramakrishna and Y. Z. Long, Prog. Mater. Sci., 2020, 100704 Search PubMed.
- P. Bocchetta, D. Frattini, M. Tagliente and F. Selleri, Curr. Nanosci., 2020, 16, 462 CrossRef CAS.
- Y. Liang and J. C. H. Goh, Bioelectricity, 2020, 2, 101 CrossRef.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, Sens. Actuators, B, 2015, 220, 534 CrossRef CAS.
- H. Nie, Y. Zhao, M. Zhang, Y. Ma, M. Baumgarten and K. Müllen, Chem. Commun., 2011, 47, 1234 RSC.
- R. Saraswathi, M. Gerard and B. D. Malhotra, J. Appl. Polym. Sci., 1999, 74, 145 CrossRef CAS.
- F. G. Zamani, H. Moulahoum, M. Ak, D. O. Demirkol and S. Timur, TrAC, Trends Anal. Chem., 2019, 118, 264 CrossRef.
- Z. Q. Feng, J. Wu, W. Cho, M. K. Leach, E. W. Franz, Y. I. Naim and D. Martin, Polymer, 2013, 54, 702 CrossRef CAS.
- M. Brinkmann and P. Rannou, Adv. Funct. Mater., 2007, 17, 101 CrossRef CAS.
- A. J. Bard, J. R. Faulkner, J. Leddy and C. G. Zoski, Electrochemical methods: fundamentals and applications. New York, Wiley, 1980 Search PubMed.
- K. Zhang, K. O. Kirlikovali, Q. V. Le, Z. Jin, R. S. Varma, H. W. Jang, O. K. Farha and M. Shokouhimehr, ACS Appl. Nano Mater., 2020, 3(5), 3964 CrossRef CAS.
- S. Kempahanumakkagari, A. Deep, K. H. Kim, S. K. Kailasa and H. O. Yoon, Biosens. Bioelectron., 2017, 95, 106 CrossRef CAS.
- M. D. Rahman, P. Kumar, D. S. Park and Y. B. Shim, Sensors, 2008, 8, 118 CrossRef CAS.
- H. Beitollahi, M. A. Khalilzadeh, S. Tajik, M. Safaei, K. Zhang, H. W. Jang and M. Shokouhimehr, ACS Omega, 2020, 5, 2049 CrossRef CAS.
- K. Zhang, T. H. Lee, J. H. Cha, R. S. Varma, J. W. Choi, H. W. Jang and M. Shokouhimehr, ACS Omega, 2019, 4, 21410 CrossRef CAS.
- S. A. Alqarni, M. A. Hussein, A. A. Ganash and A. Khan, Bio. Nano Sci., 2020, 1, 14 Search PubMed.
- M. Gerard, A. Chaubey and B. D. Malhotra, Biosens. Bioelectron., 2002, 17, 345 CrossRef CAS.
- S. Nambiar and J. T. Yeow, Biosens. Bioelectron., 2011, 26, 1825 CrossRef.
- M. Beley and P. C. Gros, Organometallics, 2014, 33, 4590 CrossRef CAS.
- R. M. Bullock, A. K. Das and A. M. Appel, Chem.–Eur. J., 2017, 23, 7626 CrossRef CAS.
- M. Ahlers, W. Müller, A. Reichert, H. Ringsdorf and J. Venzmer, Angew. Chem., Int. Ed., 1990, 29, 1269 CrossRef.
- Y. Wen, X. Liao, C. Deng, G. Liu, Q. Yan, L. Li and X. Wang, Microchim. Acta, 2017, 184, 935 CrossRef CAS.
- D. Dechtrirat, P. Yingyuad, P. Prajongtat, L. Chuenchom, C. Sriprachuabwong, A. Tuantranont and I. M. A. Tang, Microchim. Acta, 2018, 185, 261 CrossRef.
- M. Raj, P. Gupta, R. N. Goyal and Y. B. Shim, Sens. Actuators, B, 2017, 239, 993 CrossRef CAS.
- M. Govindasamy, V. Mani, S. M. Chen, A. Sathiyan, J. P. Merlin and G. Boopathy, Int. J. Electrochem. Sci., 2016, 11, e10814 Search PubMed.
- N. A. A. Talib, F. Salam and Y. Sulaiman, Sensors, 2018, 18, 4324 CrossRef.
- Ş. Sağlam, A. Arman, A. Arda, B. Ustamehmetoğlu, E. Sezer and R. Apak, Electroanal, 2019, 32, 964 CrossRef.
- B. Deiminiat and G. H. Rounaghi, Sens. Actuators, B, 2018, 259, 133 CrossRef CAS.
- H. Ghadimi, R. M. Tehrani, W. J. Basirun, N. J. Ab Aziz, N. Mohamed and S. Ab Ghani, J. Taiwan Inst. Chem. Eng., 2016, 65, 101 CrossRef CAS.
- M. Arvand, M. Farahpour and M. S. Ardaki, Talanta, 2018, 176, 92 CrossRef CAS.
- M. Braik, M. M. Barsan, C. Dridi, M. B. Ali and C. M. Brett, Sens. Actuators, B, 2016, 236, 574 CrossRef CAS.
- H. F. Cui, Y. F. Bai, W. W. Wu, X. He and J. H. Luong, J. Electroanal. Chem., 2016, 766, 52 CrossRef CAS.
- F. A. Harraz, A. A. Ismail, S. A. Al-Sayari, A. Al-Hajry and M. S. Al-Assiri, Sens. Actuators, B, 2016, 234, 573 CrossRef CAS.
- L. Liu, H. Cui, H. An, j. Zhai and y. Pan, Ionics, 2017, 23, 1517 CrossRef CAS.
- N. Wang, H. Dai, D. Wang, H. Ma and M. Lin, Mater. Sci. Eng., C, 2017, 76, 139 CrossRef CAS.
- Y. Zuo, J. Xu, X. Zhu, X. Duan, L. Lu, Y. Gao and Y. Yu, Synth. Met., 2016, 220, 14 CrossRef CAS.
- K. S. Selvan and S. S. Narayanan, J. Electroanal. Chem., 2018, 810, 176 CrossRef CAS.
- I. Kazane, K. Gorgy, C. Gondran, N. Spinelli, A. Zazoua, E. Defrancq and S. Cosnier, Anal. Chem., 2016, 88, 7268 CrossRef CAS.
- I. Sadriu, S. Bouden, J. Nicolle, F. I. Podvorica, V. Bertagna, C. Berho and C. Vautrin-Ul, Talanta, 2020, 207, 120222 CrossRef CAS.
- N. Kaur, H. Thakur and N. Prabhakar, J. Electroanal. Chem., 2016, 775, 121 CrossRef CAS.
- B. Xu, B. Zhang, l. Yang, F. Zhao and B. Zeng, Electrochim. Acta, 2017, 258, 1413 CrossRef CAS.
- Y. Gu, J. Wang, M. Pan, S. Li, G. Fang and S. Wang, Sens. Actuators, B, 2019, 283, 571 CrossRef CAS.
- O. Gursoy, S. Sen Gursoy, S. Cogal and G. Celik Cogal, Polym. Eng. Sci., 2018, 58, 839 CrossRef CAS.
- M. Bilgi and E. Ayranci, Sens. Actuators, B, 2016, 237, 849 CrossRef CAS.
- Ş. U. Karabiberoğlu and Ç. C. Koçak, Turk. J. Chem., 2018, 42, 291 Search PubMed.
- M. Dervisevic, E. Dervisevic, H. Azak, E. Cevik, M. Şenel and H. B. Yildiz, Sens. Actuators, B, 2016, 225, 181 CrossRef CAS.
- H. Mudila, P. Prasher, S. Rana, B. Khati and M. G. H. Zaidi, Carbon Lett., 2018, 26, 88 Search PubMed.
- A. Kausaite-Minkstimiene, L. Glumbokaite, A. Ramanaviciene, E. Dauksaite and A. Ramanavicius, Electroanal, 2018, 30, 1642 CrossRef.
- H. Al-Sagur, S. Komathi, M. A. Khan, A. G. Gurek and A. Hassan, Biosens. Bioelectron., 2017, 92, 638 CrossRef CAS.
- A. Shafaat, F. Faridbod and M. R. Ganjali, New J. Chem., 2018, 42, 6034 RSC.
- V. Serafín, R. M. Torrente-Rodríguez, M. Batlle, P. G. de Frutos, S. Campuzano, P. Yáñez-Sedeño and J. M. Pingarrón, Sens. Actuators, B, 2017, 240, 1251 CrossRef.
- T. Kangkamano, A. Numnuam, W. Limbut, P. Kanatharana, T. Vilaivan and P. Thavarungkul, Biosens. Bioelectron., 2018, 102, 217 CrossRef CAS.
- J. Yang, Y. Liu, S. Liu, L. Li, C. Zhang and T. Liu, Mater. Chem. Front., 2017, 1, 251 RSC.
- J. G. Ibanez, M. E. Rincón, S. Gutierrez-Granados, M. H. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731 CrossRef CAS.
- P. Sivaraman, A. P. Thakur and K. Shashidhara, Synth. Met., 2020, 259, 116255 CrossRef CAS.
- Y. Ge, R. Jalili, C. Wang, T. Zheng, Y. Chao and G. G. A. Wallace, Electrochim. Acta, 2017, 235, 348 CrossRef CAS.
- Y. Li, J. Niu, T. Xue, X. Duan, Q. Tian, Y. Wen and Z. Li, J. Electrochem. Soc., 2020, 167, 047512 CrossRef CAS.
- Z. Wang, P. Tammela, J. Huo, P. Zhang, M. Strømme and L. Nyholm, J. Mater. Chem. A, 2016, 4, 1714 RSC.
- Y. He, X. Han, Y. Du, B. Song, P. Xu and B. Zhang, ACS Appl. Mater. Interfaces., 2016, 8, 3601 CrossRef CAS.
- R. K. Jena, C. Y. Yue, M. M. Sk and K. A. Ghosh, Carbon, 2017, 115, 175 CrossRef CAS.
- D. Zhu, K. Cheng, Y. Wang, D. Sun, L. Gan, T. Chen and M. Liu, Electrochim. Acta, 2017, 224, 17 CrossRef CAS.
- S. Ji, J. Yang, J. Cao, X. Zhao, M. A. Mohammed, P. He and I. A. A. Kinloch, ACS Appl. Mater. Interfaces, 2020, 12, 13386 CrossRef.
- J. M. D'Arcy, H. D. Tran, V. C. Tung, A. K. Tucker-Schwartz, R. P. Wong, Y. Yang and R. B. Kaner, Proc. Natl. Acad. Sci., 2010, 107, 19673 CrossRef.
- C. Houarner-Rassin, E. Blart, P. Buvat and F. Odobel, Photochem. Photobiol. Sci., 2008, 7, 789 RSC.
- Y. S. Gal, S. H. Jin, S. Y. Shim and K. T. Lim, Mol. Cryst. Liq. Cryst., 2017, 654(1), 83 CrossRef CAS.
- S. A. Gopalan, A. I. Gopalan, A. Vinu, K. P. Lee and S. W. A. Kang, Sol. Energy Mater. Sol. Cells, 2018, 174, 112 CrossRef CAS.
- S. Alem, N. Graddage, J. Lu, T. Kololuoma, R. Movileanu and Y. Tao, Org. Electron., 2018, 52, 146 CrossRef CAS.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T. Y. Yang and J. Seo, Nature, 2019, 567, 511 CrossRef CAS.
- N. Sangiorgi, A. Sangiorgi, F. Tarterini and A. Sanson, Electrochim. Acta, 2019, 305, 322 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Electrochem. Commun., 2008, 10, 903 CrossRef.
- M. Tian and P. Wu, ACS Appl. Energy Mater., 2019, 2, 5066 CrossRef CAS.
- W. Zeng, L. Wang, X. Peng, T. Liu, Y. Jiang, F. Qin and Y. Zhou, Adv. Energy Mater., 2018, 8, 1702314 CrossRef.
- Q. T. Wang, R. R. Li, X. Z. Zhou, J. Li and Z. Q. Lei, J. Solid State Electrochem., 2016, 20, 1331 CrossRef CAS.
- K. Hua, X. Li, D. Fang, J. Yi, X. Wu, Z. Luo and S. Wang, Mater. Sci. Eng., B, 2018, 238, 26 CrossRef.
- M. S. Wang, Z. Q. Wang, Z. Chen, Z. L. Yang, Z. L. Tang, H. Y. Luo and W. Xu, Chem. Eng. J., 2018, 334, 162 CrossRef CAS.
- H. Li, S. Yang, Y. Zhao, T. Tan, X. Wang and Z. Bakenov, J. Nanomater., 2019, 4702849 CAS.
- L. A. Fard, R. Ojani, J. B. Raoof, E. N. Zare and M. M. Lakouraj, Appl. Surf. Sci., 2017, 401, 40 CrossRef CAS.
- R. Dillon, S. Srinivasan, A. S. Arico and V. Antonucci, J. Power Sources, 2004, 127, 112 CrossRef CAS.
- C. H. Choi, K. Chung, T. T. Nguyen and D. H. Kim, ACS Energy Lett., 2018, 3, 1415 CrossRef CAS.
- S. Tajik, H. Beitollahi, M. R. Aflatoonian, B. Aflatoonian, I. Sheikh Shoaie, M. A. Khalilzadeh, K. Zhang, Q. V. Le, H. W. Jang and M. Shokouhimehr, Microchem. J., 2020, 157, 104890 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu and S. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS.
- Z. Yang, H. Nie, X. Chen and S. Huang, J. Power Sources, 2013, 236, 238 CrossRef CAS.
- J. Peron, Z. Shi and S. Holdcroft, Energy Environ. Sci., 2011, 4, 1575 RSC.
- V. G. Khomenko, V. Z. Barsukov and A. S. Katashinskii, Electrochim. Acta, 2005, 50, 1675 CrossRef CAS.
- M. G. A. El-Moghny, H. H. Alalawy, A. M. Mohammad, A. A. Mazhar, M. S. El-Deab and B. E. El-Anadouli, Int. J. Hydrogen Energy, 2017, 42, 11166 CrossRef.
- E. Miglbauer, P. J. Wójcik and E. D. Głowacki, Chem. Commun., 2018, 54, 11873 RSC.
- A. El Attar, L. Oularbi, S. Chemchoub and M. El Rhazi, Int. J. Hydrogen Energy, 2020, 45, 8887 CrossRef CAS.
- K. Zhang, J. H. Cha, S. Y. Jeon, K. O. Kirlikovali, M. Ostadhassan, V. Rasouli, O. K. Farha, H. W. Jang, R. S. Varma and M. Shokouhimehr, Mol. Catal., 2020, 492, 110967 CrossRef CAS.
- K. Zhang, T. H. Lee, H. Noh, O. K. Farha, H. W. Jang, J. W. Choi and M. Shokouhimehr, Cryst. Growth Des., 2019, 19, 7385 CrossRef CAS.
- K. Zhang, K. O. Kirlikovali, J. M. Suh, J. W. Choi, H. W. Jang, R. S. Varma, O. K. Farha and M. Shokouhimehr, ACS Appl. Energy Mater., 2020, 3, 6019 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |