
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dendritic, redox-responsive, supramolecular (Dr.S) system for lysis-triggered delivery for drug-resistant renal cancer
Yichu Yuan†
a,
Piaopiao Jin†b,
Yueming Wang†c,
Xinyu Zhaod,
Qida Hud,
Wangteng Wu d,
Jiwei Huang*c and
Nan Zhang*a
d,
Jiwei Huang*c and
Nan Zhang*a
aDepartment of Urology, Second Affiliated Hospital, Zhejiang University School of Medicine, No. 88 Jiefang Road, Hangzhou 310009, China. E-mail: nanzhang@zju.edu.cn; Tel: +86-571-87783550
bHealth Management Center, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
cDepartment of Urology, Renji Hospital, Shanghai Jiao Tong University School of Medicine, No. 160 Pujian Road, Shanghai 200127, China. E-mail: huangjiwei@renji.com; Tel: +86-21-68383716
dDepartment of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
First published on 13th October 2020
Abstract
Purpose: Aiming to improve the drug loading capacity of dendritic nanoparticles and enhance delivery efficacy in drug-resistant cancer, we developed and optimized a more advanced dendritic, redox-responsive, supramolecular (Dr.S) system for intravenous RAD001 administration. Materials and methods: The Dr.S system was engineered by linking 3rd generation polyamidoamine dendrimers (G3 PAMAM) with 8-arm polyethylene glycol (PEG) to encapsulate a molecular targeted agent RAD001. The drug-loading capacity was measured by ultraviolet-visible spectrophotometry. In vitro release behavior was determined with a two-compartment model, and the in vivo distribution pattern was tracked by Cy5.5 fluorescence. The therapeutic effect of Dr.S/RAD001 was evaluated in RAD001-resistant cancer cells and tumor-bearing nude mice, respectively. Results: The Dr.S system encapsulating RAD001 with a loading efficiency of 10.6% formed a core–shell structure, by shifting hydrophobic PAMAM/RAD001 components towards inner space and exposing the hydrophilic PEG on the surface. The Dr.S/RAD001 system could respond to a lysis-mimicking reduction stimulus, and functionally release cargoes to facilitate tumor accumulation and cellular internalization. These features contributed to the enhanced anti-tumor activity of RAD001 in renal cancers in vitro and in vivo. The Dr.S/RAD001 system also reversed acquired RAD001-resistance by a 60-fold increase in tumor accumulation of the therapeutics. Conclusion: The functional Dr.S/RAD001 system enables lysis-triggered release of RAD001 to achieve better tumor accumulation, which helps overcome acquired drug resistance in renal cancers.
1. Introduction
The majority of small molecule drugs are hydrophobic, and require modifications to be intravenously available.1–3 Otherwise the hydrophobic agents could only be administrated orally, which might limit their bioactivity and therapeutic effect. RAD001, with the generic name everolimus, is an orally administered derivative of rapamycin that inhibits the mammalian target of rapamycin (mTOR) Ser/Thr kinase.4 In metastatic renal cancer, RAD001 has been used for adjuvant targeted therapy, and showed prolonged progression-free survival.5 However, RAD001 did not increase overall survival, and meanwhile caused a high incidence of off-target related adverse events. Oral targeted agents, though administered more conveniently, lack tumor accumulation and bioactivity-associated efficacy in comparison to intravenous derivatives.6,7 Therefore, an intravenous formulation of RAD001 might overcome these disadvantages and increase the therapeutic efficacy.Nanotechnology brings possibility of intravenous administration of hydrophobic targeted agents, benefiting from enhanced solubility by polymer modification.6,8 We previously developed a dendritic supramolecular polymer for co-delivery of small chemotherapeutics and oligonucleotides.9 Briefly, the polymeric delivery system was synthesized from multi-arm polyethylene glycol (PEG) and low-generation dendrimers, to encapsulate doxorubicin by hydrophobic effect. A key defect in this design is the limited loading capacity due to space restraint of the low dendrimer generation. Meanwhile, development of drug resistance after long-term usage of targeted therapy is also a major problem.10 Previous studies have shown that polymeric modification on chemotherapeutics might help alleviate chemoresistance, possibly due to distinctive regulatory features of nanoparticle-mediated delivery, such as enhanced permeability and nanoassembly uptake.2,11–15
Herein, we report a more advanced dendritic, redox-responsive, supramolecular (Dr.S) system for intravenous RAD001 administration. We replaced the low-generation dendrimers with larger ones, and modified the nanoparticles with redox-responsive structures, aiming to improve loading capacity and delivery efficacy in drug-resistant cancer.
2. Materials and methods
2.1. Reagents
RAD001 was purchased from Selleck Chemicals (Houston, TX). Trimethylamine (Et3N), 1,4-butanediamine, cystamine dihydrochloride, dithiothreitol (DTT), hydroxyl N-hydroxylsuccinimide polyethylene glycol (NHS-PEG-OH, molecular weight [MW] = 2000 Da), eight-armed PEG (bPEG, MW = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), 1,1′-carbonyldiimidazole (CDI), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (Saint Louis, MO). Third-generation polyamidoamine dendrimers (G3 PAMAM) was purchased from Dendritech (Midland, MI). 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) was obtained from Beyotime Biotechnology (Haimen, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), 4-(dimethylamino)-pyridine (DMAP), and fluorescein isothiocyanate (FITC) were purchased from Aladdin Industrial (Shanghai, China). Dialysis membranes were obtained from Spectrum Laboratories (Rancho Dominguez, CA). Unmodified and fluorescence-tagged negative-control small interfering RNA (nc-siRNA, nc-siRNACy5.5+, and nc-siRNAFITC+) was purchased from Biomics Biotechnologies (Nantong, China).
000 Da), 1,1′-carbonyldiimidazole (CDI), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (Saint Louis, MO). Third-generation polyamidoamine dendrimers (G3 PAMAM) was purchased from Dendritech (Midland, MI). 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) was obtained from Beyotime Biotechnology (Haimen, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), 4-(dimethylamino)-pyridine (DMAP), and fluorescein isothiocyanate (FITC) were purchased from Aladdin Industrial (Shanghai, China). Dialysis membranes were obtained from Spectrum Laboratories (Rancho Dominguez, CA). Unmodified and fluorescence-tagged negative-control small interfering RNA (nc-siRNA, nc-siRNACy5.5+, and nc-siRNAFITC+) was purchased from Biomics Biotechnologies (Nantong, China).
2.2. Cells and mice
Human renal cell carcinoma ACHN cell line was purchased from American Type Culture Collection (Rockville, MD) and cultured in high glucose DMEM (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Sigma-Aldrich). Cells were grown under humidified air containing 5% CO2 at 37 °C, and were used within 3 months of thawing. RAD001-resistant ACHN cells (ACHN/R) were transformed from regular ACHN cells by induction using low-dose RAD001, starting from 0.1 μM and increasing stepwise to 5.0 μM eventually after 4 months of induction.The mice xenograft model was established in male BALB/c nude mice, 4–6 weeks of age and with a body weight of 18 ± 2 g, were purchased from HFK Bioscience (Beijing, China) and bred under standard pathogen-free conditions. Each mouse was injected subcutaneous in the subaxillary flank with 1 × 106 ACHN or ACHN/R cells subcutaneously injected with 100 μL of Matrigel (Becton Dickinson, Franklin Lakes, NJ). When the xenograft tumor volume reached 50–70 mm3, tumor-bearing mice were treated with the indicated regimen via tail-vein administration. All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and all procedures were approved by the Animal Care Committee of Zhejiang University School of Medicine.
2.3. Synthesis of bPEG-SS-PEG-PAMAM (Dr.S vector)
bPEG-SS-PEG was synthesized according to previous publications.16 bPEG-SS-PEG (2 g, 0.12 mmol) dissolved in dimethyl sulfoxide (DMSO, 10 mL) was added to a solution of CDI (2 g, 12 mmol) and Et3N (2 mL, 14.4 mmol) in DMSO (10 mL) at room temperature. The reaction mixture was stirred for 5 h. After that, the resulted product was precipitated in 1 L of ether/tetrahydrofuran solution (v/v = 4:1), and was redissolved in DMSO (10 mL), which was subsequently added dropwise to G3 PAMAM dendrimer in DMSO solution (10 mL) in the presence of Et3N (300 μL, 2.9 mmol). After being stirred overnight, the final product was purified by extensive dialysis against water using dialysis membrane (molecular weight cutoff [MWCO] = 14000) and was freeze-dried to yield the final product Dr.S vector as white solid (3.46 g, 64.7%).
2.4. Preparation of RAD001-loaded Dr.S (Dr.S/RAD001) system
RAD001 (15.0 mg) dissolved in DMSO (2 mL) and the Dr.S vector (40.0 mg) dissolved in water (10 mL) were mixed and stirred overnight. The solution was purified by dialysis against water using a dialysis membrane (MWCO = 14000) for 12 h. The purified solution was then passed through a 0.45 mm filter to remove the unloaded RAD001 and lyophilized to afford a white powder. The RAD001-embedded Dr.S was dissolved in water and mixed with nc-siRNA solution (amount according to indicated nitrogen:phosphate ratios [N:P ratios]) followed by 30 seconds of vortex to form the Dr.S/RAD001 nanoparticle system. Alternatively, the Dr.S/RAD001 could also be formed from Dr.S/nc-siRNA and RAD001. The mixture solution of the Dr.S vector and nc-siRNA was stirred for 30 min to form the Dr.S/nc-siRNA nanoparticles. The DMSO solution of RAD001 was added to the Dr.S/siRNA solution, and the mixture was stirred overnight, dialyzed, filtered, and freeze-dried to obtain the white powder.
2.5. Determination of loading capacity
The drug-loading content was measured by an ultraviolet-visible (UV-Vis) spectrophotometry (UV-2800, Hitachi, Tokyo, Japan). The Dr.S/RAD001 nanoparticles were dissolved in 20 mL of water and absorbance measured at λ = 279 nm. A calibration curve was established under identical conditions. The drug loading content was calculated using the following formula: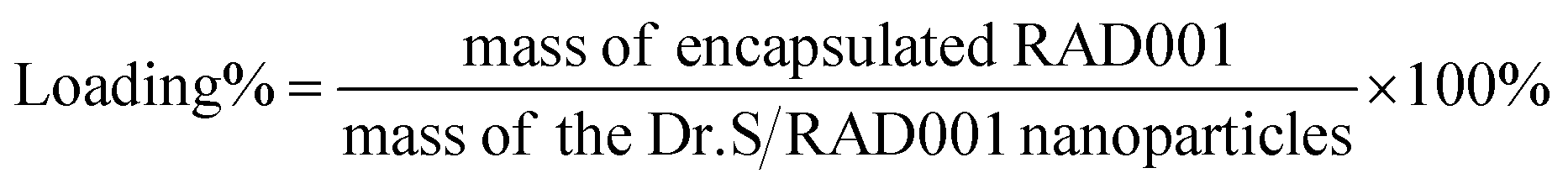
2.6. Physical characterization and measurement of time-dependent change after reduction stimulation
The particle size and ζ potential of the Dr.S/RAD001 system were analyzed at 25 °C by dynamic light scattering (DLS) on a Zetasizer 3000 (Malvern Instruments, Worcestershire, UK). The morphology of complexes was examined by transmission electron microscopy (TEM, HT-7700, Hitachi, Japan). The solution of the Dr.S/RAD001 system was then treated with 10 mM of DTT, a reduction agent. The particle size and ζ potential were measured by DLS at the indicated time to illustrate the time-dependent change patterns.2.7. Determination of the in vitro release behavior of the Dr.S/RAD001 system
The release pattern was determined in a two-compartment model. Firstly, 5.0 mg of Dr.S/RAD001 was dissolved in 10 mL of PBS and sealed in dialysis tubing (MWCO = 14000 Da). The solution was dialyzed against 40 mL of PBS in the absence or presence of 10 mM of DTT at 37 °C under stirring at 100 rpm on an orbital shaker. At defined time points, 1 mL of the medium was removed from the 40 mL solution out of the dialysis bag for high-performance liquid chromatography (HPLC) analysis. Each time 1 mL of PBS was added to the original system. RAD001 concentration in the acquired medium was measured by HPLC at a maximum absorption wavelength of 279 nm by a UV detector. The analytical column was a Hypersil-ODS C18 column (250 mm × 4.6 mm, 5 μm). The mobile phase was CH3CN:H2O (v/v = 7:3), with the flow rate of 1.0 mL min−1.
2.8. Characterization of pharmacokinetic and cellular internalization
In vivo distribution patterns were visualized by Cy5.5 fluorescence. Naked nc-siRNACy5.5+ or Dr.S/RAD001 with siRNACy5.5+ was intravenously administered into mice via tail-vein injection at an equivalent dose of 1.0 mg siRNACy5.5+ per kg mouse body weight. At the predetermined time points, the mice were anesthetized with 2.5% isoflurane and imaged using the Xenogen IVIS Lumina system (Caliper Life Sciences, Alameda, CA). The results were then analyzed using Living Image 3.1 software (Caliper Life Sciences, Alameda, CA). The cellular uptake of the Dr.S/RAD001 system were examined by confocal scanning laser microscopy (CLSM) in ACHN cells treated with the Dr.S/RAD001 system condemning nc-siRNAFITC+. The cells were seeded on 24-well plates and grown for 20 h. The nc-siRNAFITC+ was complexed with Dr.S/RAD001 at room temperature for 30 min before use. After cell uptake for 0, 6, 12, and 24 h, the cells were fixed with fresh 4% formaldehyde, then treated with DAPI for 10 min, and last stained with Alexa Fluor® 647 for 40 min. The images were acquired on a CLSM (Radiance 2100, Bio-Rad, Hercules, CA).2.9. Evaluation of antiproliferation effect by cytotoxicity assay
ACHN and ACHN/R cells were seeded in 96-well plates at 8000 cells per well in 200 μL of DMEM containing with 10% FBS for 18 h. Subsequently, the cells were incubated with culture medium containing Dr.S/RAD001, Dr.S and RAD001 with different concentrations for 72 h. Then, 100 μL of DMEM containing MTT stock solution (1 mg mL−1) was added to the wells. After incubation for another 4 h, each well was replaced with 100 μL of DMSO. The absorbance was measured at 570 nm using an ELISA plate reader (Model 550, Bio-Rad, Hercules, CA). The relative cell growth (%) related to control cells cultured in the media without the polymer was calculated by the following formula:where V% is the relative cell viability (%), Aexperimental is the absorbance of the wells culturing the treated cells, Ablank is the absorbance of the blank, and Acontrol is the absorbance of the wells culturing untreated cells. The dose-dependent viability patterns were then fitted by a non-linear regression approach using the following model:
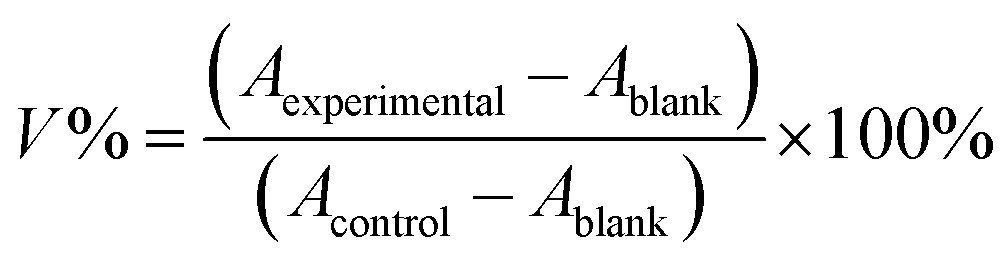
where IC50 is the half maximal inhibitory concentration, [RAD001] is the RAD001 dose, and a is a slope constant.
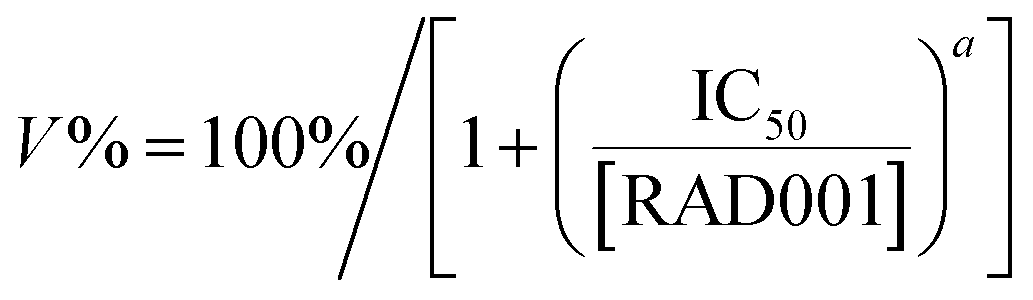
2.10. In vivo therapeutic study
For the tumor suppression study, the mice were randomly divided into groups (n = 3 per group). The mice bearing ACHN or ACHN/R tumors were treated with PBS, Dr.S, RAD001 (p.o., 2.5 mg kg−1), Dr.S/RAD001 (i.v. 2.5 mg kg−1 OSI-027 equivalent). Treatment (oral or intravenous administration) was performed every two days for 8 times. Tumor growth was monitored by measuring the perpendicular diameter of the tumor using calipers. Tumor volume (mm3) was calculated as:in which l and w indicate the length and width of the tumor. The tumor samples were collected for further immunoblotting, histology, immunohistochemistry studies.
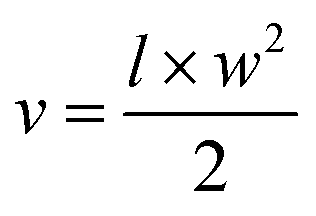
2.11. Determination of tumor accumulation in ACHN/R tumor
The treated mice were sacrificed at 24 h to collect tumor tissues, which were lysed in ice-cold radioimmunoprecipitation buffer supplemented with phosphatase inhibitor cocktails (Cell Signaling, Danvers, MA), centrifuged. The protein was then collected. Protein content and RAD001 concentration were detected by bicinchoninic acid protein assay and HPLC, respectively, which determined the RAD001 concentration normalized by the total protein content.2.12. Statistical analyses
Experiments were repeated at least in triplicate. Data are presented in term of mean ± s.d. Statistical analyses were performed using Prism 7 (GraphPad, La Jolla, CA) with the indicated analytical methods. Best-fit values and 95% confidential intervals (95% CIs) were calculated using indicated nonlinear regression. p < 0.05 was considered significant.3. Results
3.1. Synthesis and cargo loading of the Dr.S system
The G3 PAMAMs were successfully conjugated to a central 8-arm PEG scaffold (bPEG-SS-PEG) via redox-responsive disulfide linkages with a yield rate of 64.7%. The 1H NMR spectrum of the Dr.S vector demonstrated the characteristic peaks of PEG (–CH2CH2O–) at 3.67 ppm, disulfide bonds (–CH2SSCH2–) at 3.52–3.42 ppm, and PAMAM dendrimers at 3.45–2.32 ppm in the Dr.S vector (Fig. 1a). Inside the Dr.S vector, each multiarm PEG was functionalized with 8 PAMAMs according to stoichiometry of the integral values.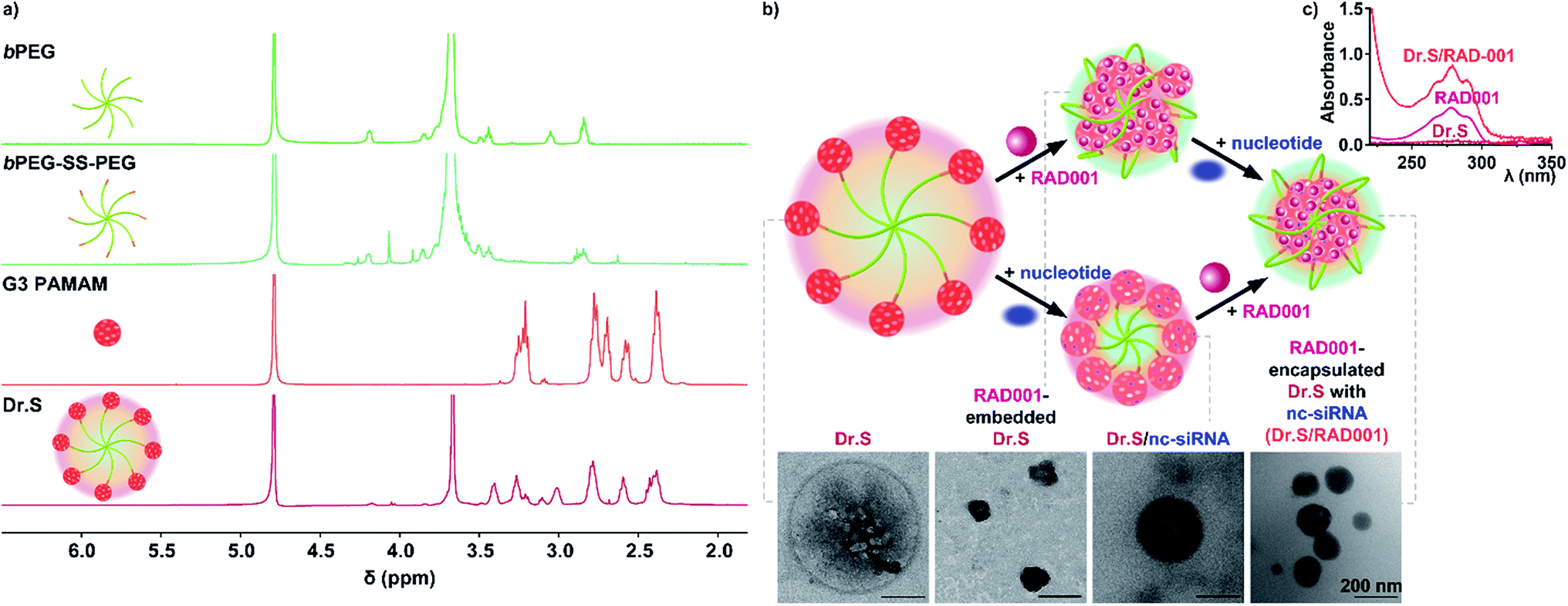 | ||
| Fig. 1 Characterization of the Dr.S system. (a) 1H NMR spectra of the Dr.S vector and its components, namely 8-arm polyethylene glycol (bPEG), 8-arm PEG with disulfide bond (bPEG-SS-PEG), 3rd generation polyamidoamine (G3 PAMAM). (b) RAD001 loading process by the Dr.S system with nucleotide condensation. The corresponding transmission electron microscopy (TEM) images were shown. The bar represents 200 nm. (c) Ultraviolet-visible spectra of the Dr.S vector, free RAD001, and the Dr.S/RAD001 system. | ||
Loading process were then achieved with the help of negative control siRNA (nc-siRNA), a scrambled version of siRNA sequence with no biological function, following encapsulation of the RAD001 cargos into the PAMAMs, while the more hydrophobic PAMAM/RAD001 shifted towards the inner space of the Dr.S vector, exposing the hydrophilic PEG chains to form a core–shell structure observed by TEM (Fig. 1b). The RAD001-loaded Dr.S (Dr.S/RAD001) system displayed a smooth and spherical appearance that would facilitate delivery efficacy. Alternatively, Dr.S/RAD001 could also be formed by nc-siRNA condensation followed by RAD001 encapsulation (Fig. 1b), indicating that RAD001 loading was a solubility–association interaction between cargos and the Dr.S vector. Loading capacity was then determined to be 10.6% by estimation of peak absorbance at the wavelength of 279 nm in ultraviolet-visible spectrometry (Fig. 1c). Therefore, Dr.S system was capable to load and encapsulate RAD001 for potent delivery.
3.2. Dr.S system mediates redox-responsive delivery
Functional delivery requires fundamental physical properties and additional responsive delivery features. In nucleic acid delivery complexes, the N:P ratio is defined as the ratio of amines on the carrier to the number of phosphates on the nucleotide backbone. The higher the N:P ratio is, the greater number of amine based vectors per nucleic acid complexed with.17 The drug-loaded Dr.S system shrank rapidly at N:P ratios lower than 5 (Fig. 2a), because the nucleic acid and cationic materials tend to form looser nanogels at a relatively low N:P ratio.18,19 Once the N:P ratio at which the nucleotides were completely condensed (N:P ratio = 5) was reached, the size of nanoparticles remained stably ranging from 169 nm to 186 nm (N:P ratio over 5), which facilitated its delivery in circulation along with the spherical shape (Fig. 2a). Meanwhile, the nanoparticle surface was equipped with positive charges, revealed by positive ζ-potential level persistently above 4 mV at the same N:P ratios (Fig. 2a). Therefore, the Dr.S system with N:P ratio of 20 was used for further investigation for stable and comparable physical properties as to nanoparticles with higher N:P ratios.
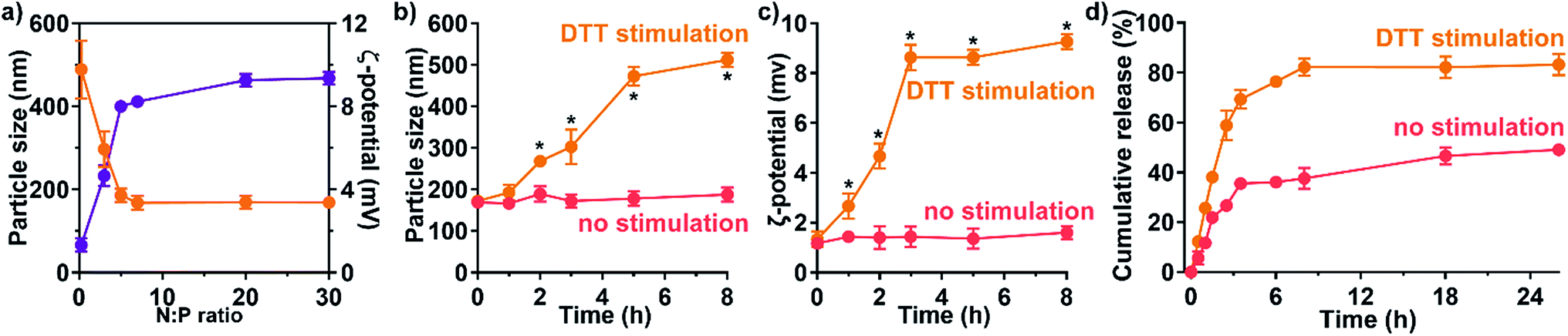 | ||
| Fig. 2 Redox-responsiveness of the Dr.S/RAD001 system. (a) The patterns of particle size (orange) and ζ-potential (purple) at different N:P ratios. (b and c) The time-dependent change of particle size (b) and ζ-potential (c) with DTT stimulation (yellow) or without any stimulation (red). *p < 0.05, vs. no stimulation, two-way ANOVA followed by post-hoc Bonferroni test. (d) The time-dependent release patterns of the Dr.S/RAD001 system with DTT stimulation (yellow) or without any stimulation (red). | ||
Functionalized redox-responsiveness by disulfide linkages was then verified by stimulation with reduction agent, DTT, resembling lysis-rich microenvironment. The physical properties, namely the particle size and the ζ-potential, were significantly altered after DTT stimulation within 2 h (Fig. 2b and c). At 8 h post stimulation, the size and the surface potential increased to 512 ± 16 nm (mean ± s.d.) and 9.27 ± 0.31 mV (s.d.), respectively. The change in size and surface charge indicated detachment process of PEG which featured increasing particle radius due to departure of PEG chains and positive ζ-potential after revealing the positive PAMAM core. We further investigated the stimuli-responsive release by high-performance liquid chromatography (HPLC) (Fig. 2d). As no stimulation was presented, only 45.6 (95% CI 42.8–48.5)% of the total loaded cargo could be freed from the Dr.S system. Under the condition of DTT stimulation, a first-order release pattern of free drug from the Dr.S/RAD001 system was demonstrated with a half-life (t1/2) of 1.6 (95% CI 1.5–1.8) h and a maximum cumulative release (Rmax) of 83.9 (95% CI 81.3–86.5)% by non-linear best-fit regression, which doubled the release amount triggered by reduction stimuli. Therefore, the Dr.S system was able to mediate redox-responsive delivery of RAD001.
3.3. Dr.S facilitates tumor retaining and cellular internalization
Accumulation in the tumor microenvironment is also an important parameter of delivery efficacy.6,20 The Dr.S/RAD001 system carrying nc-siRNA labelled with Cy5.5 (Dr.SCy5.5+/RAD001) via tail-vein injection mainly accumulated in the tumor site, while the naked nc-siRNACy5.5+ administration resulted in no tumor retaining, indicating better tumor retaining of the Dr.S system (Fig. 3a). Instead, naked nc-siRNACy5.5+ showed higher dose accumulation in the kidney than the Dr.SCy5.5+/RAD001 system, suggesting a quicker renal clearance without the Dr.S′ assistance. Dr.S system also showed enhanced cellular internalization ability (Fig. 3b). In 6 h post treatment with Dr.SFITC+/RAD001, the system had been distributed in the cytoplasm of the ACHN renal cellular carcinoma (RCC) cells. The internalized Dr.SFITC+/RAD001 system retained for the following 18 h, and still showed remarkable fluorescence intensity at 24 h post treatment. The Dr.S system therefore substantiated both tumoral and cellular retaining, possibly supported by enhanced permeability and retention (EPR) effect.21 | ||
| Fig. 3 Enhanced tumor retaining and cellular internalization. (a) Fluorescence distribution in mice treated with tail-vein injection of PBS, nc-siRNACy5.5+, or Dr.SCy5.5+/RAD001. The tumors and major organs were harvested to show fluorescence intensity. (b) The time-dependent internalization process of Dr.SFITC+/RAD001 (green) visualized by confocal microscopy. The cytoplasm was stained with Alexa Fluor 594 (red) and the nucleus was stained with DAPI (blue). | ||
3.4. Dr.S enhances anti-tumor ability of RAD001 in RCC
Dr.S/RAD001 increased antiproliferative effect of RAD001 in RCC cells (Fig. 4a). The cell viability assays showed significantly greater cytotoxicity of Dr.S/RAD001 with a half maximal inhibitory concentration (IC50) of 5.20 (95% CI 4.60–5.87) μM comparing to free RAD001 (13.9 [95% CI 12.4–15.6] μM) in ACHN cells (p < 0.001, F = 142.8; extra sum-of-squares F test). We further evaluated the therapeutic performance of the Dr.S/RAD001 system in mice based on cancer growth after tail-vein administration in C57BL/6 mice bearing ACHN tumors (Fig. 4b). In comparison with conventional oral treatment with RAD001, the Dr.S/RAD001 system administrated intravenously excelled in inhibiting tumor growth in vivo. Lower tumor growth rates were observed in the Dr.S/RAD001-treated mice than those in the control groups according to follow-up tumor volume measurements. The Dr.S delivery of RAD001 significantly facilitated inhibition of the final tumor volume by 56.4% (oral RAD001 vs. Dr.S/RAD001: 486 ± 41 mm3 vs. 212 ± 30 mm3; mean ± s.d., n = 3) (Fig. 4c). To investigate the underlying mechanism, we compared the expression of phosphorylated mTOR, the target of RAD001, by western blotting and immunohistochemistry (Fig. 4d and e). The results indicated that encapsulation of RAD001 with the Dr.S system could attenuated p-mTOR expression in the xenograft tumors in comparison to oral RAD001 treatment. Meanwhile, the tumor samples were analyzed with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays to reveal increased apoptotic cells in Dr.S/RAD001-treated tumors (Fig. 4e). We also observed increased apoptosis and necrosis in the Dr.S/RAD001 group (Fig. 4e). These results suggested a better inhibitory effect of the Dr.S/RAD001 system in vivo.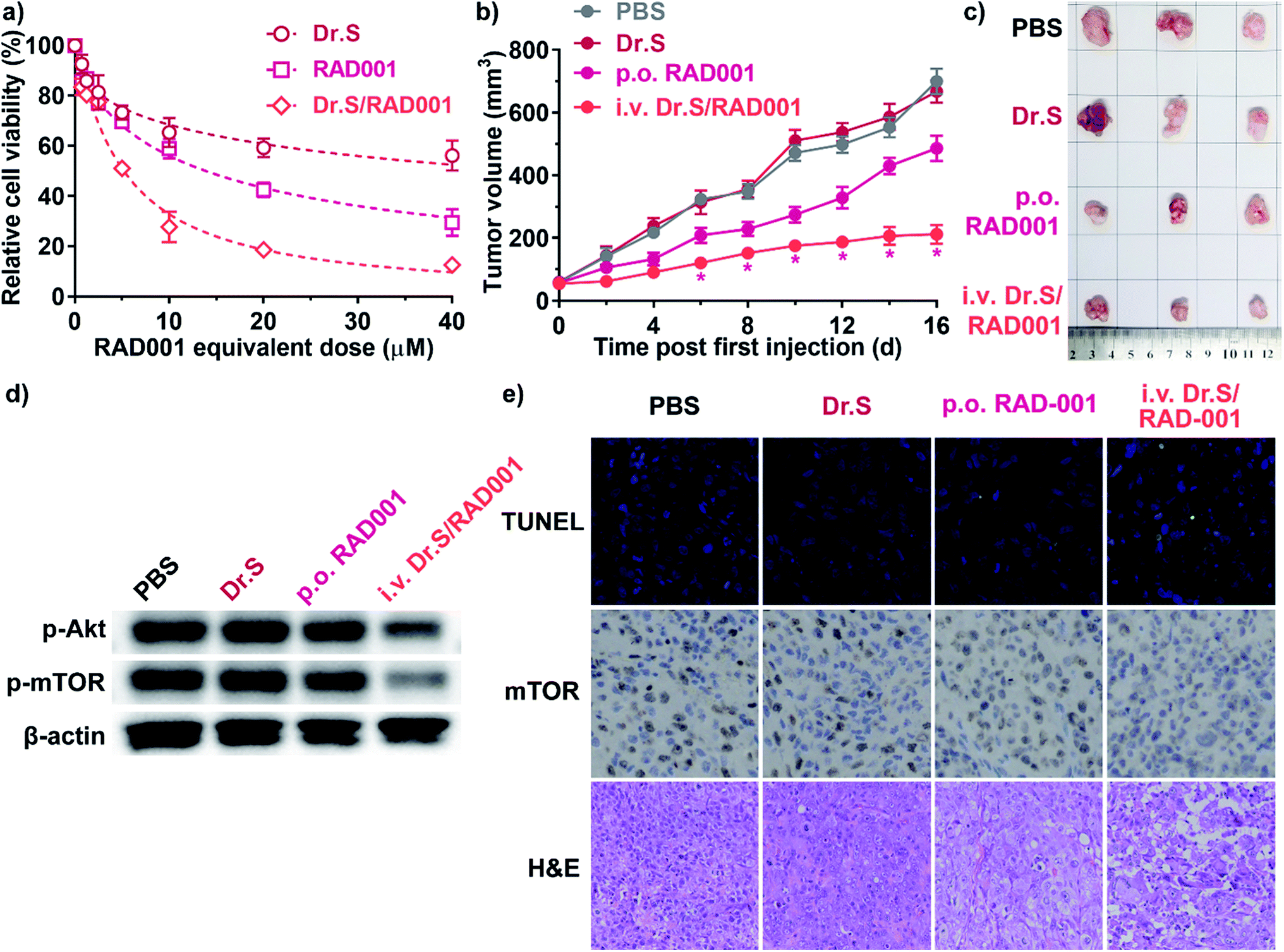 | ||
| Fig. 4 Therapeutic effect of the Dr.S/RAD001 system. (a) Dose-dependent antiproliferation of the Dr.S/RAD001 system (light red) in comparison with the Dr.S vector (dark red) and free RAD001 (rose). Best fit curves by non-linear regression models were shown in dashed lines. (b) Tumor growth patterns in mice treated with PBS (gray), the Dr.S vector (dark red), oral RAD001 (rose), and intravenous Dr.S/RAD001 (light red). *p < 0.05, vs. p.o. RAD001; two-way ANOVA followed by Bonferroni test. (c) The white-field photo of the xenograft tumors treated with PBS, the Dr.S vector, oral RAD001, and intravenous Dr.S/RAD001. (d) Attenuation of phosphorylated Akt (p-Akt) and mTOR (p-mTOR) expression by intravenous Dr.S/RAD001. (e) The intratumoral characterization of apoptotic cells (green) by the TUNEL assays and mTOR positive expression (dark brown), with histology analysis by H&E staining. | ||
3.5. Dr.S increased RAD001 sensitivity in drug-resistant RCC
Nanotechnologies were applied for overcoming drug-resistance in previous approaches by inducing better tumor penetration and enhanced tumor uptake.22,23 We therefore performed further studies on reversal of drug resistance by the Dr.S system. In vitro cell viability assays (Fig. 5a) determined that the Dr.S/RAD001 system efficiently suppressed proliferation of RAD001-resistant ACHN (ACHN/R) cells that free RAD001 showed little cytotoxicity (IC50 values, 43.9 [95% CI 32.2–66.3] μM with RAD001 vs. 5.54 [5.03–6.12] μM with Dr.S/RAD001; p < 0.001, extra sum-of-square test). Interestingly, the Dr.S/RAD001 system showed similar antiproliferative effect in both regular ACHN and resistant ACHN/R cells (p = 0.405, extra sum-of square test). We also compared the inhibitory effect of intravenous Dr.S/RAD001 system with oral RAD001 on RAD001-resistant xenograft tumors (Fig. 5b). Oral RAD001 unquestionably failed to reduce tumor growth rate and showed similar treatment effect as PBS-treated control group. Notably, the Dr.S/RAD001 significantly outperformed the oral RAD001 in treating drug-resistant xenograft tumors by volume reduction of 75.8% (Fig. 5c). A further analysis of intratumoral RAD001 dose by HPLC was then carried out to verify the possible mechanism of resistance reversal (Fig. 5d). The results revealed a substantially higher, namely 60-fold, dose of RAD001 in Dr.S/RAD001-treated tumors than those with oral RAD001 treatment.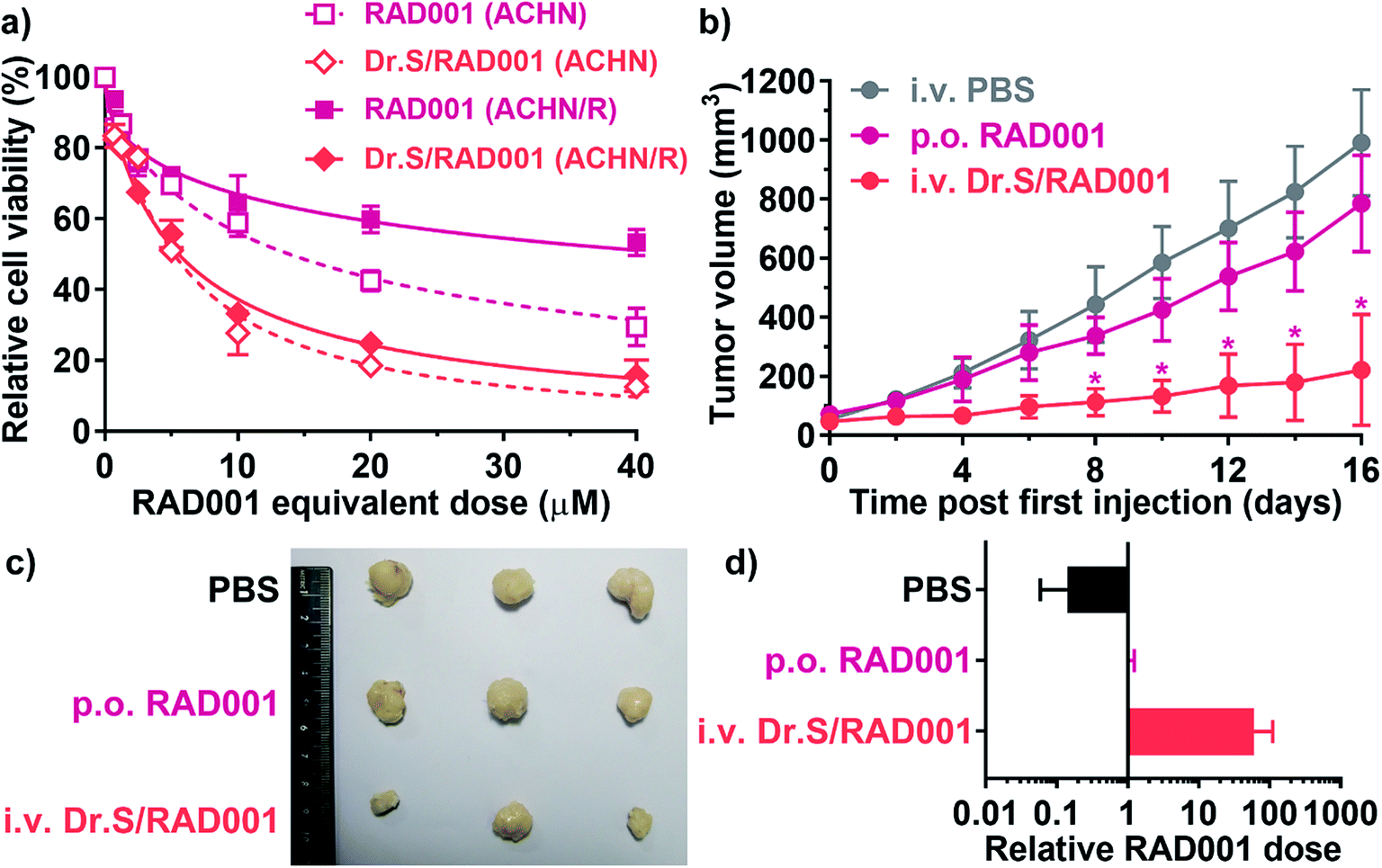 | ||
| Fig. 5 Therapeutic effect of the Dr.S/RAD001 system in RAD001-resistant RCC cells. (a) Dose-dependent antiproliferation of the Dr.S/RAD001 system (light red) in comparison with free RAD001 (rose) in regular (ACHN) and RAD001-resistant (ACHN/R) cells. Best fit curves by non-linear regression models were shown in dashed (ACHN) and solid (ACHN/R) lines. (b) Growth patterns of the xenograft RAD001-resistant tumors treated with PBS (gray), oral RAD001 (rose), and intravenous Dr.S/RAD001 (light red). *p < 0.05, vs. p.o. RAD001; two-way ANOVA followed by Bonferroni test. (c) The white-field photo of the drug-resistant xenograft tumors treated with PBS, oral RAD001, and intravenous Dr.S/RAD001. (d) Intratumoral RAD001 dose normalized to the control oral RAD001 group. | ||
4. Discussions
In the current study, we engineered a supramolecular Dr.S system with multiple 3rd generation dendrimers linked by a 8-arm PEG, which encapsulated molecular targeted therapeutic, RAD001, for redox-responsive delivery. The Dr.S/RAD001 system, triggered by reduction stimulus or lysis microenvironment, was able to functionally release the loaded RAD001 with a substantial enrichment in renal cancer. Even in renal cancer with acquired resistance, the system still worked properly to with enhanced tumor accumulation.The Dr.S system differs from our previous dendrimer-PEG design in several aspects.9 Firstly, the key dendrimer component in the Dr.S system, is a 3rd generation PAMAM, while the previous design used a 2nd generation dendrimer with different inner structures. The 3rd generation PAMAM is a commercially available dendrimer, which directly facilitates synthesis and lowers the production cost. Moreover, the 3rd generation PAMAM provides larger loading space in comparison to the 2nd generation dendrimer, leading to an increased loading capacity. Our results also showed that the Dr.S system using the 3rd generation PAMAM had little cytotoxicity, an important fundamental of a new nanomedicine. Secondly, our system was designed only for hydrophobic targeted therapeutics or other hydrophobic agents, as our previous approach was applied for co-delivery of doxorubicin and Bcl-2 siRNA. However, chemotherapeutics negatively regulate cancer metabolism, which might interfere with, sometimes inhibit, transfection of the co-delivered nucleotides.24 We therefore used the Dr.S system for drug delivery only. We adopted nc-siRNA in our nanoparticle system as a locker by complexing with G3 PAMAM to enhance encapsulation effect, via which the hydrophobic drugs loaded in the interior cavities could be locked tightly inside and avoid leakage in the complicated environment such as body circulation.
The Dr.S system also reverses RAD001 resistance in the renal cancer. Acquired resistance is a common situation in clinical settings after long time RAD001 use.25 Our preclinical approach using the Dr.S system indicated that nanoparticle encapsulation of the free RAD001 increased sensitivity in renal cancer, which is benefited from enhanced penetration and tumor accumulation of the intravenous nanoscale formulation. Other mechanisms might also contribute to the reversal of RAD001 resistance, such as regulation of intracellular proteins related to multidrug resistance (MDR) genes, and enhanced apoptosis.11 Another possible contributing factor is nanoparticle-induced cell cycle arrest, that increased cellular uptake and decrease drug efflux.26 Overall, application of the Dr.S nanosystem against RAD001 resistance is a feasible solution.
5. Conclusions
In conclusion, the redox-responsive Dr.S system enables lysis-triggered release of RAD001 to achieve tumor accumulation, which predominantly contributes to reversal of acquired resistance in renal cancers. Preclinical data shows Dr.S′ potential as a salvage therapy after development of RAD001 resistance. Future research should focus on further optimization and translational applications of the Dr.S system, to excel in pharmacodynamics and efficacy and to obviate adverse events.Funding
This work was supported financially by Zhejiang Provincial Natural Science Foundation (LY19H050006 and LY18H050002).Ethics approval
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Zhejiang University and approved by the Animal Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine.Availability of data and material
All data are included in the manuscript. Raw data would be available upon request.Authors' contributions
Yichu Yuan, Piaopiao Jin and Yueming Wang: Conceptualization, Methodology, Validation, Visualization, Formal analysis, Investigation, Writing-original draft. Xinyu Zhao: Conceptualization, Resources, Visualization. Qida Hu: Methodology, Visualization, Software, Validation, Investigation. Wangteng Wu: Methodology, Visualization, Software. Nan Zhang and Jiwei Huang: Methodology, Data curation, Validation, Investigation, Resources, Writing-review & editing, Funding acquisition.Conflicts of interest
The authors declare no potential conflicts of interest.References
- Q. D. Hu, H. Fan, Y. Ping, W. Q. Liang, G. P. Tang and J. Li, Chem. Commun., 2011, 47, 5572–5574 RSC
.
- J. Shen, Q. Wang, Q. Hu, Y. Li, G. Tang and P. K. Chu, Biomaterials, 2014, 35, 8621–8634 CrossRef CAS
- X. Zhang, H. Wang, Z. Ma and B. Wu, Expert Opin. Drug Metab. Toxicol., 2014, 10, 1691–1702 CrossRef CAS
- F. Janku, T. A. Yap and F. Meric-Bernstam, Nat. Rev. Clin. Oncol., 2018, 15, 273–291 CrossRef CAS
- R. J. Motzer, B. Escudier, S. Oudard, T. E. Hutson, C. Porta, S. Bracarda, V. Grunwald, J. A. Thompson, R. A. Figlin, N. Hollaender, G. Urbanowitz, W. J. Berg, A. Kay, D. Lebwohl, A. Ravaud and R.-S. Group, Lancet, 2008, 372, 449–456 CrossRef CAS
- M. O. Durymanov, A. A. Rosenkranz and A. S. Sobolev, Theranostics, 2015, 5, 1007–1020 CrossRef CAS
- M. C. Shoshan and S. Linder, Expert Opin. Drug Metab. Toxicol., 2008, 4, 273–280 CrossRef CAS
- P. van Hoogevest, X. Liu and A. Fahr, Expert Opin. Drug Delivery, 2011, 8, 1481–1500 CrossRef CAS
- K. Wang, Q. Hu, W. Zhu, M. Zhao, Y. Ping and G. Tang, Adv. Funct. Mater., 2015, 25, 3380–3392 CrossRef CAS
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS
- W. Jin, Q. Wang, M. Wu, Y. Li, G. Tang, Y. Ping and P. K. Chu, Biomaterials, 2017, 129, 83–97 CrossRef CAS
- H. Wang, Y. Li, M. Zhang, D. Wu, Y. Shen, G. Tang and Y. Ping, Adv. Healthcare Mater., 2017, 6, 1601293 CrossRef
- Q. Chen, Y. Yang, X. Lin, W. Ma, G. Chen, W. Li, X. Wang and Z. Yu, Chem. Commun., 2018, 54, 5369–5372 RSC
- C.-X. Yang, L. Xing, X. Chang, T.-J. Zhou, Y.-Y. Bi, Z.-Q. Yu, Z.-Q. Zhang and H.-L. Jiang, Mol. Pharm., 2020, 17, 1300–1309 CrossRef CAS
- Y. Yu, Q. Xu, S. He, H. Xiong, Q. Zhang, W. Xu, V. Ricotta, L. Bai, Q. Zhang, Z. Yu, J. Ding, H. Xiao and D. Zhou, Coord. Chem. Rev., 2019, 387, 154–179 CrossRef CAS
- Q. Hu, W. Wu, M. Wang, S. Shao, P. Jin, Q. Chen, H. Bai, X. Zhao, J. Huang, J. Wang, G. Tang and T. Liang, J. Controlled Release, 2020, 317, 67–77 CrossRef CAS
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS
- Y.-H. Zhang, Y. Chen, Y.-M. Zhang, Y. Yang, J.-T. Chen and Y. Liu, Sci. Rep., 2014, 4, 7471 CrossRef
- S. Mahajan and T. Tang, J. Phys. Chem. B, 2019, 123, 9629–9640 CrossRef CAS
- X. Jia, J. He, L. Shen, J. Chen, Z. Wei, X. Qin, D. Niu, Y. Li and J. Shi, Nano Lett., 2019, 19, 8690–8700 CrossRef CAS
- H. Nakamura, J. Fang and H. Maeda, Expert Opin. Drug Delivery, 2015, 12, 53–64 CrossRef CAS
- L. Deng, Z. Feng, H. Deng, Y. Jiang, K. Song, Y. Shi, S. Liu, J. Zhang, S. Bai, Z. Qin and A. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 31743–31754 CrossRef CAS
- Z. Mirza and S. Karim, Semin. Cancer Biol., 2019 DOI:10.1016/j.semcancer.2019.10.020
- W. F. Lai and M. C. Lin, PLoS One, 2015, 10, e0126367 CrossRef
- L. Hamieh, T. K. Choueiri, B. Ogorek, D. Khabibullin, D. Rosebrock, D. Livitz, A. Fay, J. C. Pignon, D. F. McDermott, N. Agarwal, W. Gao, S. Signoretti and D. J. Kwiatkowski, PLoS Genet., 2018, 14, e1007679 CrossRef
- J. Liu, T. Wei, J. Zhao, Y. Huang, H. Deng, A. Kumar, C. Wang, Z. Liang, X. Ma and X. J. Liang, Biomaterials, 2016, 91, 44–56 CrossRef CAS
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |