
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of MOF-5 with mono-substituents: effects on drug delivery behavior†
Mengru Cai a,
Liuying Qina,
Longtai Youa,
Yu Yaoa,
Huimin Wua,
Zhiqin Zhanga,
Lu Zhang*c,
Xingbin Yin*a and
Jian Ni*ab
a,
Liuying Qina,
Longtai Youa,
Yu Yaoa,
Huimin Wua,
Zhiqin Zhanga,
Lu Zhang*c,
Xingbin Yin*a and
Jian Ni*ab
aSchool of Chinese Material Medica, Beijing University of Chinese Medicine, Beijing 102488, China. E-mail: yxbtcm@163.com; 602054@bucm.edu.cn
bBeijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, China
cDepartment of Biochemistry and Molecular Medicine, UC Davis NCI-designated Comprehensive Cancer Center, University of California Davis, Sacramento, CA, USA. E-mail: lluzhang@ucdavis.edu
First published on 7th October 2020
Abstract
Metal organic frameworks (MOFs) are widely used in drug carrier research due to their tunability. The properties of MOFs can be adjusted through incorporation of mono-substituents to obtain pharmaceutical carriers with excellent properties. In this study, different functional groups of –NH2, –CH3, –Br, –OH and –CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH are connected to MOF-5 to analyse the effect of mono-substituent incorporation on drug delivery properties. The resulting MOFs have similar structures, except for Br–MOF. The pore size of this series of MOFs ranges from 1.04 nm to 1.10 nm. Using oridonin (ORI) as a model drug, introduction of the functional groups appears to have a significant effect on the drug delivery performance of the MOFs. The IRMOFs can be ranked according to drug-loading capacity: MOF-5 > HO–MOF-5 > H3C–MOF-5 = Br–MOF-5 > H2N–MOF-5 > CH2CH–MOF-5. The ORI release from ORI @IRMOFs is explored at two different pH values: 7.4 and 5.5, and the ORI@IRMOFs are ranked according to the cumulative release percentage of ORI: ORI@MOF-5 > ORI@Br–MOF-5 > ORI@H3C–MOF-5 > ORI@H2N–MOF-5 > CH2CH–MOF-5 > ORI@ HO–MOF-5. In particular, the release behaviour of ORI@MOFs is described through a new model. The different drug delivery performance of MOFs may be due to the complex interactions between MOFs and ORI. In addition, the introduction of single substituents does not change the biocompatibility of MOFs. MTT in vitro experiments prove that this series of MOFs has low cytotoxicity. This study shows that the incorporation of single substituents can effectively adjust the drug delivery behaviour of MOFs, which is conducive to realization of personalized drug delivery modes. The introduction of active groups can also facilitate post-synthesis modification to achieve coupling of targeting groups. MOFs incorporated with single substituents perform favorably in terms of use as biomedical drug delivery alternative carriers in effective drug payload and flexible drug release.
CH are connected to MOF-5 to analyse the effect of mono-substituent incorporation on drug delivery properties. The resulting MOFs have similar structures, except for Br–MOF. The pore size of this series of MOFs ranges from 1.04 nm to 1.10 nm. Using oridonin (ORI) as a model drug, introduction of the functional groups appears to have a significant effect on the drug delivery performance of the MOFs. The IRMOFs can be ranked according to drug-loading capacity: MOF-5 > HO–MOF-5 > H3C–MOF-5 = Br–MOF-5 > H2N–MOF-5 > CH2CH–MOF-5. The ORI release from ORI @IRMOFs is explored at two different pH values: 7.4 and 5.5, and the ORI@IRMOFs are ranked according to the cumulative release percentage of ORI: ORI@MOF-5 > ORI@Br–MOF-5 > ORI@H3C–MOF-5 > ORI@H2N–MOF-5 > CH2CH–MOF-5 > ORI@ HO–MOF-5. In particular, the release behaviour of ORI@MOFs is described through a new model. The different drug delivery performance of MOFs may be due to the complex interactions between MOFs and ORI. In addition, the introduction of single substituents does not change the biocompatibility of MOFs. MTT in vitro experiments prove that this series of MOFs has low cytotoxicity. This study shows that the incorporation of single substituents can effectively adjust the drug delivery behaviour of MOFs, which is conducive to realization of personalized drug delivery modes. The introduction of active groups can also facilitate post-synthesis modification to achieve coupling of targeting groups. MOFs incorporated with single substituents perform favorably in terms of use as biomedical drug delivery alternative carriers in effective drug payload and flexible drug release.
Introduction
Improvements in drug delivery systems are necessary due to the inherent properties of drugs1 such as poor solubility,2 poor pharmacokinetics,3 poor biodistribution4 and adverse side effects.5 The use of nanotechnology to overcome such issues is constantly being updated.6 Many nanoparticles have been designed for such purposes, such as liposomes,7 quantum dots,8 graphene oxides,9 gold nanoparticles,10 mesoporous silica nanoparticles,11 carbon nanotubes12 and metal organic frameworks (MOFs).13 MOFs have a network structure comprising organic and inorganic units.14 The adjustable composition, structure, pore size and volume, easy functionalization and accessible metal sites of MOFs mean they have many advantages in the adsorption and release of biomolecules.15 Among these, tunable composition is advantageous because the linker is almost infinite, so that, theoretically, thousands of MOFs can be synthesized.16 Different MOFs can be obtained easily by changing the metal center or organic ligands. Adjustment of MOF structure can change the affinity between the drugs and MOFs, and can effectively control the release rate of drugs.17 Therefore, it is of great significance to study the influence of MOFs modified by different groups on drug-loading and drug-release behaviour. This is helpful to synthesize drug carriers with different characteristics and design suitable drug delivery systems according to requirements.Many MOFs have been reported as drug-loading materials over the past decade.18 An increasing number of functionalized MOFs have been synthesized and reported, to realize multifunctional drug delivery.19 Considerable efforts have also been made to improve the drug delivery capacity of MOFs.20 Many strategies have now been developed for efficient delivery of drugs, including surface adsorption, pore encapsulation, covalent binding and functional molecules as building blocks.21 Functionalizing the organic ligands of MOFs is an effective method to adjust the performance of MOFs. There have been systematic studies on the functionalization of organic ligands in many fields, such as gas storage,22 catalysis23 and adsorption.24 In addition, researchers have used functionalized MOFs to improve the performance of drug delivery. For example, Lázaro et al. used fumarate (fum) as an organic ligand to synthesize Zr-fum, which is an attractive alternative to UiO-66 in nanoscale drug delivery.25 Pander et al. synthesized NU-1000. The terminal hydroxyl groups in Zr6 nodes can facilitate the loading of carboxyl-containing drugs.26 However, the effect of functional groups with different properties and sizes on the delivery behaviour of MOFs has not been systematically studied.
In this contribution, we chose MOF-5 as the basic model compound because it has a simple cubic crystal structure and large porosity.27 We varied the chemical substituents at the 2-position of the phenylene unit in the MOF-5 framework using –CH3, –NH2, –Br, –OH and –CHCH2. These substituents were selected because the length of the ligand and the nature of the functional group varied, which can help to better understand drug delivery behaviour. Oridonin (ORI), a tetracyclic diterpenes isolated from Rabdosia rubescens, has been widely used in cancer therapy.28 It has a wide range of biological properties, including anti-tumor,29,30 anti-inflammatory31 and antibacterial effects,32 and prevention of liver fibrosis33 and other functions, which has attracted attention in biomedicine. However, its clinical application is limited due to its poor solubility and low bioavailability.34 At present, “donglingcao tablets” are mainly used for adjuvant treatment of various tumors. ORI preparation based on nanotechnology has thus attracted the attention of researchers, to improve the bioavailability of ORI.
Although there have been reports of drug delivery strategies based on functional group modification, there is still a lack of research on the impact of the introduction of different groups on the performance of MOFs as drug carriers. The present work systematically studies the effects of different sizes and polarities of functional-group-modified MOFs on drug delivery performance. Herein, the drug delivery behaviour (drug-loading capacity, drug-release rate and biological safety) of five modified MOF-5s are investigated experimentally. Five modified MOF-5s and MOF-5 are confirmed to have different drug delivery behaviours. The size and nature of the substituents have a great influence on drug delivery behaviour. Zn–O clusters, large conjugation of ligands and the presence of hydrogen bonds bring about multiple potential host–guest interactions between IRMOFs and ORI. Furthermore, the guest release process for the six MOFs is determined using four common models: a zero-order model, a first-order model, a Higuchi model and a Ritger–Peppas model. This study shows that the group-modified MOFs can have a significant impact on drug delivery. This research also provides a reference for the development of personalized drug delivery systems based on MOFs.
Experimental section
Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99.99%) and N,N-dimethylformamide (DMF, 99.5%) were obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Terephthalic acid (H2BDC) was provided by Tianjin Guangfu Fine Chemical Research Institute. 2-Hydroxymethyl terephthalic acid (98%), 2-bromoterephthalic acid (98%), 2-hydroxyterephthalic acid (98%) and 2-vinyl terephthalic acid (98%) were purchased from Chemsoon Co., Ltd. Triethylamine (TEA, AR) was got from Tianjin Fuchen Chemical Reagent Factory. Methanol (CH3OH, AR) were acquired from Beijing Chemical Factory. Oridonin (ORI, 98%) was supplied by Shanghai yuanye Bio-Technology Co., Ltd. High-glucose Dulbecco's Modified Eagle Medium (DMEM) was offered by Corning. Fetal bovine serum was available from Mediatech (International) Ltd. Dimethyl sulfoxide (DMSO) was taken from Tianjin Guangfu Fine Chemical Research Institute. PBS and penicillin–streptomycin mixture were provided by Solarbio. Fetal bovine serum was supplied by Mediatech (International) Ltd. 3-(4,5-Dimethylthiazol-2-yl)-2,5-dipheny-ltetrazolium bromide (MTT) was furnished from Beijing Biodee Biotechnology Co., Ltd.Synthesis of MOFs
MOF-5 was synthesized by direct addition of triethylamine which referred to the previous article and made some modifications.35,36 The synthesis method of functionalized MOF-5 with mono-substituent is similar to that of MOF-5 to ensure the consistency of the structure. And the modified MOFs were named NH2–MOF-5, CH3–MOF-5, Br–MOF-5, HO–MOF-5 and CH2CH–MOF-5. Next, the specific synthesis methods of the six MOFs are briefly introduced.
Synthesis of MOF-5
0.653 g Zn(NO3)2·6H2O and 0.135 g BDC was added to 20 ml DMF, and mixed by ultrasound. The mixed solution was placed on a magnetic stirrer. 1.1 ml of triethylamine was added to produce a white precipitate and stirred for 2 hours. The precipitate was collected by centrifugation and washed three times with DMF, then soaked in methanol for three days, and replaced with methanol every 24 hours. Finally, the product was dried in a vacuum for 12 hours (120 °C). At last, a white powder, MOF-5, was obtained.Synthesis of NH2–MOF-5
0.644 g Zn(NO3)2·6H2O and 0.144 g NH2–BDC was added to 20 ml DMF, and mixed by ultrasound. The mixed solution was placed on a magnetic stirrer. 1.1 ml of triethylamine was added and the mixture was stirred for 2 hours. After the same post-processing method as above, dry NH2–MOF-5 powder was obtained.Synthesis of CH3–MOF-5
0.654 g Zn(NO3)2·6H2O and 0.148 g CH3–BDC was added to 20 ml DMF, and mixed by ultrasound. The mixed solution was placed on a magnetic stirrer. 1.1 ml of triethylamine was added to produce a white precipitate and stirred for 2 hours. After solvent exchange and drying, CH3–MOF-5 powder is stored in a drying cabinet.Synthesis of Br–MOF-5
0.654 g Zn(NO3)2·6H2O and 0.201 g Br–BDC was dissolved in 20 ml DMF. 1.1 ml of triethylamine was added to produce a white precipitate and stirred for 2 hours. After solvent exchange and drying, Br–MOF-5 powder is stored in a drying cabinet.Synthesis of HO–MOF-5
0.654 g Zn(NO3)2·6H2O and 0.149 g HO–BDC was dissolved in 20 ml DMF. 1.1 ml of triethylamine was added and stirred for 2 hours. After solvent exchange and drying, HO–MOF-5 powder is stored in a drying cabinet.Synthesis of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH–MOF-5
CH–MOF-5
0.654 g Zn(NO3)2·6H2O and 0.158 g CH2CH–BDC was dissolved in 20 ml DMF. 1.1 ml of triethylamine was added and stirred for 2 hours. After solvent exchange and drying, CH2CH–MOF-5 powder is stored in a drying cabinet.
Characterization
The pore sizes of six MOFs were determined at 77 K by N2 adsorption on a Belsorp-max BET Sorptometer (Bel Japan Inc., Tokyo, Japan). The structural confirmation of the MOFs was achieved by Fourier Transform Infrared Spectrometer (FTIR). The sample surface was scanned in the 4000–400 cm−1 range. The purity of the powders was tested using X-ray powder diffraction (P-XRD). The analysis was performed using Cu-Kα (λ = 1.541 nm) radiation at 40 kV and 40 mA in the scan range of 2θ from 5 to 50° whose data was recorded in D/max-2550 X-ray diffractometer (Japan). Scanning electron microscopy (SEM) was performed to observe the surface morphology and particle size of the sample. Image came from a high-performance field emission scanning electron microscope Zeiss Merlin (Germany). In order to understand the thermal stability of the particles, the Thermogravimetric Analysis (TGA) is completed on the Synchronous Thermal Analyzer (Germany).Encapsulation of ORI
All drug-loading experiments were carried out by solvent adsorption method, and the supernatant concentration was measured by HPLC. By using the L9(34) orthogonal test table, three factors: the ratio of MOFs to drugs, the concentration of oridonin methanol solution, and the drug loading time are selected for the optimization of drug loading conditions.The drug loading was calculated using the formula below:
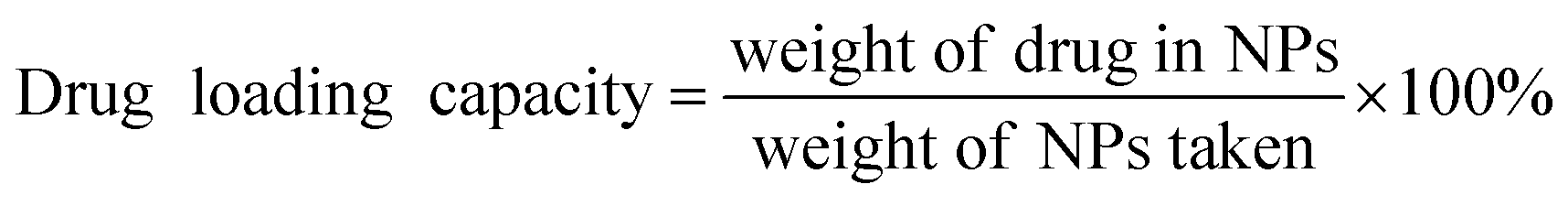
In addition, in order to study the physical state of the drug-loaded metal organic frameworks, SEM, TG and FTIR were performed.
In vitro drug release simulation
To simulate the release of ORI@MOFs, we measured the release of oridonin at pH 7.4 and 5.5 in vitro. Because the pH value of the tumor site is lower than normal tissue. Two pH values were selected to verify whether the drug delivery system is pH sensitive. And 5 mg of oridonin was placed in 40 ml of PBS at 37 °C with gentle shaking.37 1 ml of release medium was taken out at a certain time and 1.0 ml fresh PBS buffer is then refilled. Then the released-ORI content was measured by HPLC. Then the cumulative drug release of oridonin at each time point was calculated, and the drug release curve was plotted. In order to better understand the ORI release behavior of ORI@MOFs, four common models including the zero order model, first order model, Higuchi model and Riter–Peppas model was firstly chose to describe drug release process. Finally, we analyze the experimental data by computer to find the best release model for each nanoparticle, which can help predict the release of ORI at various times.Cell culture
HepG2 cells are supplied by Guangzhou Jeniobio Biotechnology. The cells were incubated using a DMEM added with 10% FBS and 1% penicillin–streptomycin solution (37 °C, 5% CO2). The medium is refreshed every three days.Cytotoxicity assay
The cytotoxicity of MOF-5, NH2–MOF-5, CH3–MOF-5, Br–MOF-5, HO–MOF-5 and CH2CH–MOF-5 was assessed by the MTT assays. The cells were cultured on 96 well plates at a density of 7000 cells per well. Cells are treated with different concentrations of IRMOF-n (5, 10, 15, 20, 25, 30, 35 μg ml−1) for 24 h. Following, the MTT solutions (5 mg ml−1, 20 μl) were added into each well and further incubated at 37 °C for 4 h in the dark. The culture supernatant was removed from all wells, DMSO (150 μl) was added to each well, and the plate was shaken for 10 minutes to solubilize the formazan crystals. The absorbance at 490 nm was measured with a microplate reader.
Results
Synthesis and structural characterization of MOFs
Six MOFs were successfully synthesized. The structures of all functional MOFs were characterized by powder X-ray diffraction (Fig. S1a†). The main peak positions of the five modified MOFs (diffraction peaks at 6.9° and 9.7°) were consistent with those of MOF-5, indicating they have the same topological structure. However, the peak intensity of each material was inconsistent, indicating there are still differences among the structures. For Br–MOF-5, the peaks were broad, indicating an amorphous structure, not a crystalline structure. The diffraction mode of Br–MOF-5 was poor, and the diffraction peaks were not obvious at 6.9° and 9.7°. This indicates that Br–MOF-5 may not have a topology similar to that of MOF-5. The powder X-ray diffraction results proved that MOFs with a certain topology can be obtained under the same conditions, although the quality and crystallinity of each material were different.Fig. S1b† represents the Fourier-transform infrared (FTIR) spectra of MOF-5, NH2–MOF-5, CH3–MOF-5, Br–MOF-5, HO–MOF-5 and CH2CH–MOF-5. The FTIR spectra of the six MOFs were generally similar, and the positions of the main peaks were consistent. This is because the six materials were similar in structure and composition, and only some of the groups were different. In the FTIR spectroscopy of the six MOFs, there was a clear broad absorption peak in the range 3500–3300 cm−1, showing the O–H stretching vibration peak. Peaks at 1580 cm−1 and 1690 cm−1 were caused by asymmetric stretching vibration of the C–O bond in terephthalic acid, while the peak at 1380 cm−1 was due to symmetric stretching vibration. The peak at 1300–400 cm−1 was the vibration peak of the frame structure of MOFs. The peak at 523 cm−1 was the absorption peak of the Zn–O bond, which indirectly characterizes the tetrahedral structure of the crystal. Compared with MOF-5, the FTIR spectrum of NH2–MOF-5 had two broad peaks in the range 3500–3300 cm−1, which are characteristic absorption peaks of primary amines. In the FTIR spectrum of Br–MOF-5, a double peak appeared at 600–500 cm−1, which is attributed to the bromine. In the FTIR spectrum of CH2CH–MOF-5, the absorption peak at 3100–3000 cm−1 was the C–H stretching vibration peak, contributed by –CCH2. The infrared spectra of CH3–MOF-5 and HO–MOF-5 were similar to that of MOF-5. According to the analysis, the characteristic peaks of methyl and hydroxyl groups existed in the framework of IRMOFs, so no new characteristic peaks appeared in the infrared spectra of these two materials. But for CH3–MOF-5, the peak intensity at 3000–2800 cm−1 increased, which proved the introduction of the methyl group. For HO–MOF-5, the peak intensity at 3500–3300 cm−1 proved the addition of hydroxyl groups. The FTIR spectra of the six MOFs thus confirmed the successful synthesis of the materials.
The pore size of the six MOFs was determined at 77 K by nitrogen adsorption. The pore size measurement results of the six MOFs are shown in Table 1. The difference in pore size between the materials is small, reducing the effect of the difference in pore size on the drug loading capacity.
| MOFs | Median pore width (nm) |
|---|---|
| MOF-5 | 1.0491 |
| NH2–MOF-5 | 1.0940 |
| CH3–MOF-5 | 1.0999 |
| Br–MOF-5 | 1.0558 |
| HO–MOF-5 | 1.0962 |
| CH2CH–MOF-5 |
1.0892 |
The SEM image was measured with a scanning electron microscope, and the results are shown in Fig. 1. The six IRMOFs are all irregularly round, with a relatively uniform size and a particle size below 100 nm.
 | ||
| Fig. 1 SEM image of ORI@MOF-5 (a), ORI@NH2–MOF-5 (b), ORI@CH3–MOF-5 (c), ORI@Br–MOF-5 (d), ORI@HO–MOF-5 (e) and ORI@CH2CH–MOF-5 (f). | ||
Thermal stability analysis of MOFs
It can be seen from the thermogravimetric curve in Fig. 2 that the six MOFs showed similar thermal stability (under N2 flow). The six MOFs are relatively stable within 450 °C. It can be seen that four MOFs are degraded at about 450 °C, including MOF-5, MOF-5–NH2, MOF-5–OH and MOF-5–CHCH2. MOF-5–CH3 and MOF-5–Br begin to degrade around 500 °C. The thermal stability of MOF-5–CH3 and MOF-5–Br is slightly better than that of MOF-5, MOF-5–NH2, MOF-5–OH and MOF-5–CHCH2. In general, six MOFs as drug carriers are thermally stable enough. However, different MOFs showed different weight loss processes during the heating process. Two weightlessness processes were observed in the thermogram of CH3–MOF-5, Br–MOF-5, HO–MOF-5 and CH2CH–MOF-5. The weight of the first stage is reduced (0–400 °C) by 15 wt%, which is due to the removal of solvent. The second weight loss process started at about 450 °C and could be attributed to the thermal decomposition of MOFs. For MOF-5 and NH2–MOF-5, there are three weightlessness events. The weight of the first stage is reduced (0–400 °C) by 15 wt%, which is due to the removal of water. The latter two stages are caused by the elimination of the solvent and the degradation of the metal frameworks, respectively. It shows that there is still a little residual solvent in MOF-5 and MOF-5–NH2. The six MOFs should be dried to remove residual water and solvent (DMF) before drug loading.
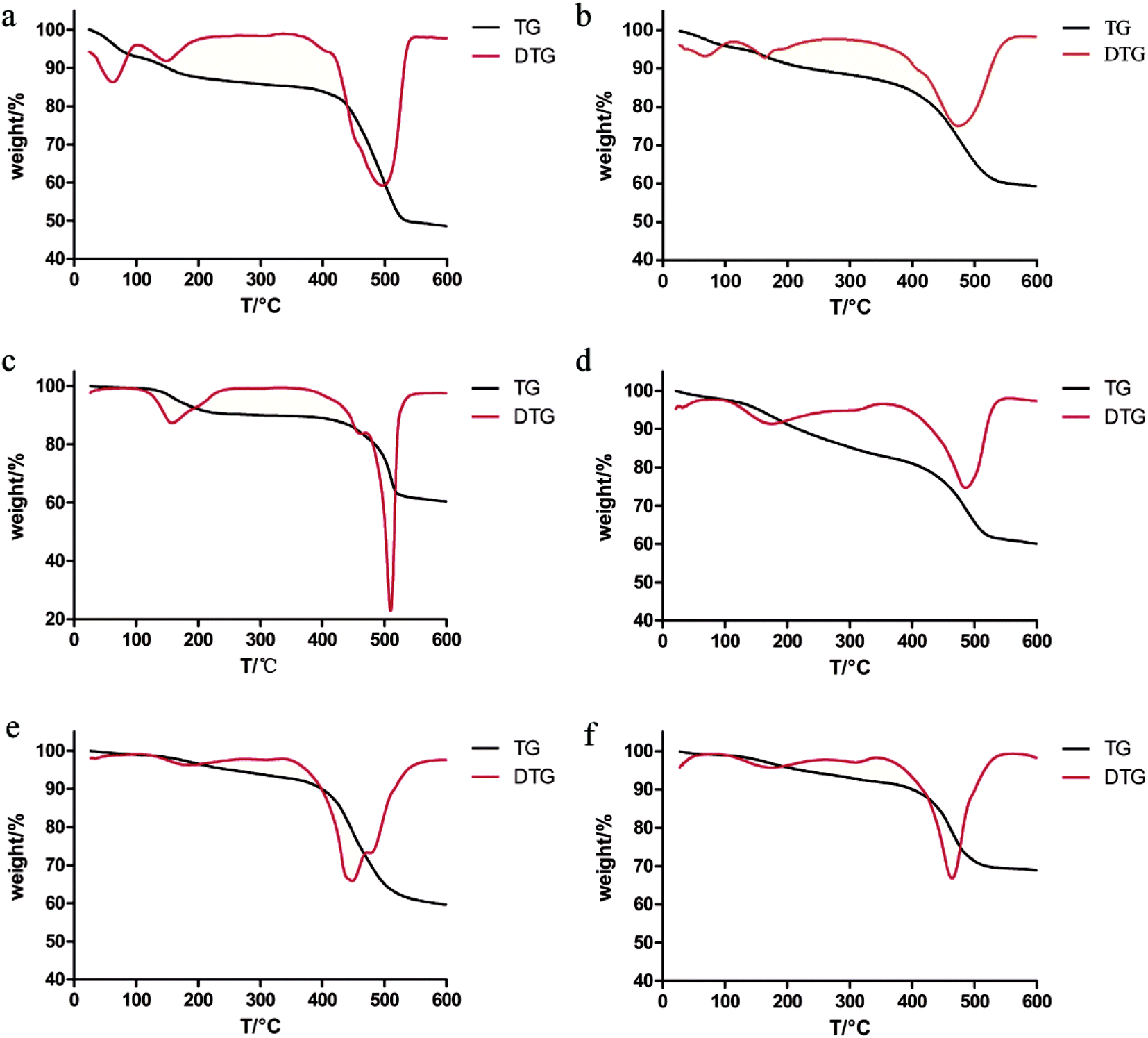 | ||
| Fig. 2 TG and DTG diagrams of MOF-5 (a), NH2–MOF-5 (b), CH3–MOF-5 (c), Br–MOF-5 (d), HO–MOF-5 (e) and CH2CH–MOF-5 (f). | ||
Drug loading and characterization of ORI@IRMOFs
The optimal drug-loading conditions were optimized through orthogonal experiments. For MOF-5, the drug-loading rate reached 50.61 wt% under optimized conditions of: MOF-5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ORI (1:3), magnetic stirring for 3 days and a concentration of ORI methanol solution of 15 mg ml−1. For NH2–MOF-5, the drug-loading rate reached 34.46 wt% under optimized conditions of: NH2–MOF-5:ORI (1:3), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For CH3–MOF-5, the drug-loading rate reached 38.99 wt% under optimized conditions of: CH3–MOF-5:ORI (1:3), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For Br–MOF-5, the drug-loading rate reached 39.97 wt% under optimized conditions of: Br–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For HO–MOF-5, the drug-loading rate reached 48.13 wt% under optimized conditions of: HO–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 10 mg ml−1. And for CH2CH–MOF-5, the drug-loading rate reached 27.37 wt% under optimized conditions of: HO–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 5 mg ml−1.
:ORI (1:3), magnetic stirring for 3 days and a concentration of ORI methanol solution of 15 mg ml−1. For NH2–MOF-5, the drug-loading rate reached 34.46 wt% under optimized conditions of: NH2–MOF-5:ORI (1:3), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For CH3–MOF-5, the drug-loading rate reached 38.99 wt% under optimized conditions of: CH3–MOF-5:ORI (1:3), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For Br–MOF-5, the drug-loading rate reached 39.97 wt% under optimized conditions of: Br–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 15 mg ml−1. For HO–MOF-5, the drug-loading rate reached 48.13 wt% under optimized conditions of: HO–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 10 mg ml−1. And for CH2CH–MOF-5, the drug-loading rate reached 27.37 wt% under optimized conditions of: HO–MOF-5:ORI (1:4), magnetic stirring for 5 days and a concentration of ORI methanol solution of 5 mg ml−1.
Although the six types of IRMOFs had similar pore sizes and structures, their properties changed and mediated host–guest interactions due to the functionalization of the organic ligands. Therefore, each IRMOF exhibited different drug-carrying capacity, as shown in Fig. 3. The drug-loading capacity of each MOF-5 with mono-substituent decreased compared to that of MOF-5. In addition, there were significant differences between each group, except for –CH3- and –Br-modified MOF-5. The IRMOFs can be ranked according to their drug-loading capacity: MOF-5 > HO–MOF-5 > H3C–MOF-5 = Br–MOF-5 > H2N–MOF-5 > CH2CH–MOF-5. The introduction of various groups has hindered the entry of ORI into the cavity of IRMOFs, and the effect of this spatial hindrance on the drug-loading capacity of IRMOFs is dominant. Thus, for CH2CH–MOF-5, the larger mono-substituted group CH2CH has stronger steric hindrance, resulting in the smallest drug-loading capacity. In addition, coordination binding, interactions among groups, hydrogen bond formation, π–π packing between the ORI and IRMOFs and electrostatic interactions all affect drug-loading capacity. HO–MOF-5 interacts with ORI (coordination binding, hydrogen bonding, π–π packing and electrostatic interaction) due to the presence of hydroxyl groups. This interaction can offset the steric hindrance of the group to a certain extent, so it has little effect on its drug-loading capacity. The methyl and halogen bromine atoms are non-polar groups, and they have only weak interaction with ORI, so the drug-loading capacity of these two modified groups will be lower than that of HO–MOF-5. The above results demonstrate the effect of substituent incorporation on the properties of MOFs, resulting in changes in drug-carrying capacity, thus benefiting the regulation of MOF drug-loading capacity.
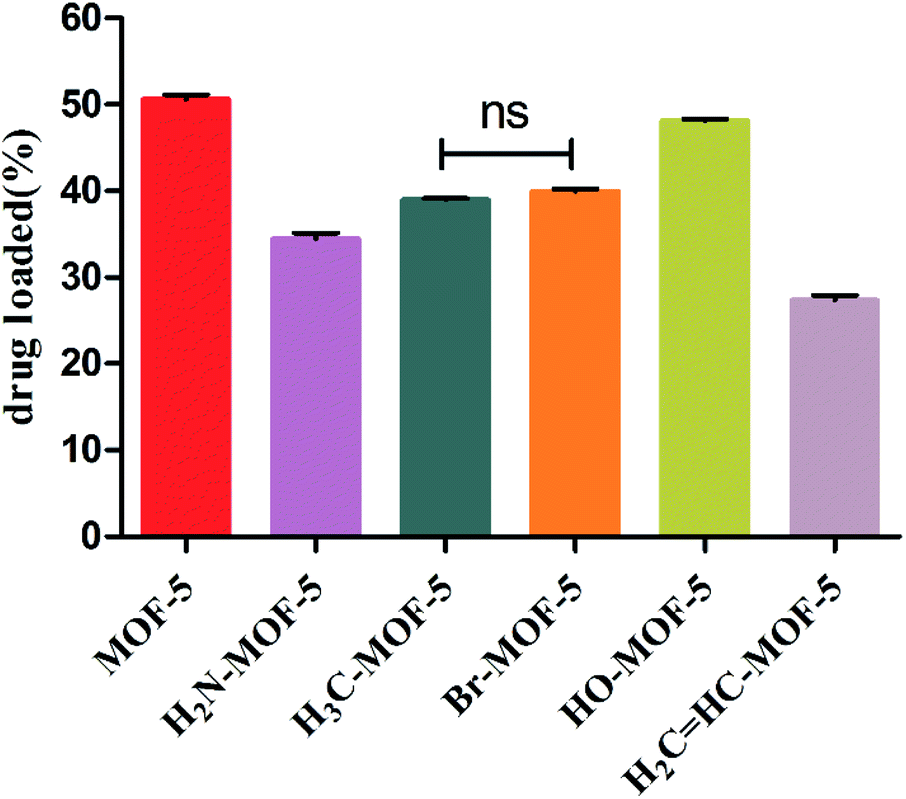 | ||
| Fig. 3 The drug loading capacity of MOF-5, NH2–MOF-5, CH3–MOF-5, Br–MOF-5, HO–MOF-5 and CH2CH–MOF-5. | ||
The chemical structures of the IRMOFs after ORI loading were examined using FTIR spectroscopy shown in Fig. S3.† The spectra of ORI@IRMOFs were generally similar to those of IRMOFs. In the spectra of ORI@IRMOFs, the characteristic peak of ORI completely disappeared after drug loading, indicating that ORI was completely adsorbed in the pores of IRMOFs. Subtle changes in the position and intensity of the absorption peak indicate there was a certain host–guest reaction between IRMOFs and ORI. In addition, SEM was used to characterize the morphology of ORI@IRMOFs (Fig. 4). After loading ORI, the morphology of IRMOFs did not change significantly. The ORI@IRMOFs were all spherical, and their average particle size was about 50 nm. Furthermore, Fig. 5 shows the thermal stability of six IRMOFs before and after drug loading. ORI@IRMOFs had higher thermal stability than ORI, but the thermal stability of ORI@IRMOFs is consistent with that of IRMOFs. This indicates that ORI completely enters the pores of IRMOFs, and IRMOFs have a certain protective effect on ORI. The ORI@IRMOFs could maintain a good structure up to 400 °C, and the skeleton began to collapse at 450 °C. There was a slight weight loss at 200 °C, which may be due to evaporation of water molecules and residual solvents.
 | ||
| Fig. 4 SEM image of ORI@MOF-5 (a), ORI@NH2–MOF-5 (b), ORI@CH3–MOF-5 (c), ORI@Br–MOF-5 (d), ORI@HO–MOF-5 (e) and ORI@CH2CH–MOF-5 (f). | ||
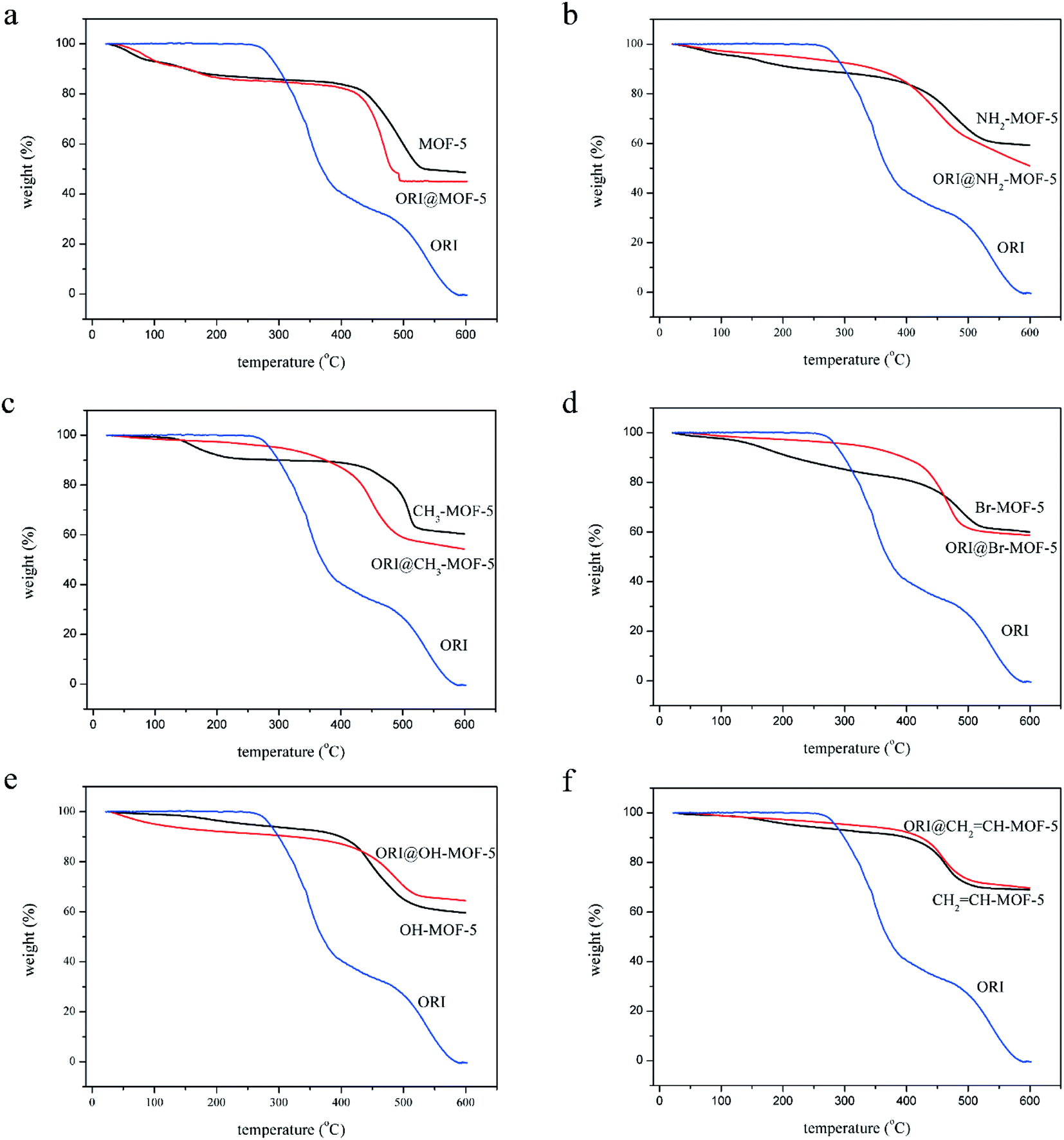 | ||
| Fig. 5 TG diagrams of ORI@MOF-5 (a), ORI@NH2–MOF-5 (b), ORI@CH3–MOF-5 (c), ORI@Br–MOF-5 (d), ORI@HO–MOF-5 (e) and ORI@CH2CH–MOF-5 (f). | ||
In vitro drug delivery
The ORI release from ORI@IRMOFs was explored at two different pH values: 7.4 and 5.5. As shown in Fig. 6a and c, the release patterns of the six drug-loaded materials are relatively similar, and the release process of ORI can be divided into three stages. In the first 5 hours, ORI was released rapidly, and the release rate of ORI slowed slightly in the next 7 hours. In the final stage, ORI is released at a very slow rate until the 48th hour. This is due to the complex structure of IRMOFs and ORI which leads to their various interactions. In order to better understand the behaviour of ORI release from ORI@MOFs, four commonly used models were selected to describe the drug release process including the zero order model, first order model, Higuchi model and Riter–Peppas model. The fitting results using different models were compared as shown in Table S1.† Comparing the fitting curves of the four simulation models with the experimental data, the fitting degree of the Riter–Peppas model is higher than other models. But none of them can provide a reasonable regression coefficient of the ORI drug molecule release profile. Since the guest–guest interaction and structural effects change with time, the shape of the release curve will be changed in the experiment, and the release equation should no longer satisfy the single term. Therefore, a new release kinetic model was established to observe the release behaviour of nanoparticles (Fig. S4†). It can be seen that the curve fitted by the new model is in good agreement with the experiment (R2 > 0.99). The fitting performance of the new model during drug release is better than other models, indicating that the model is more suitable for describing the release process of ORI molecules in IRMOFs system, and can be used to predict the drug release process of ORI-IRMOFs.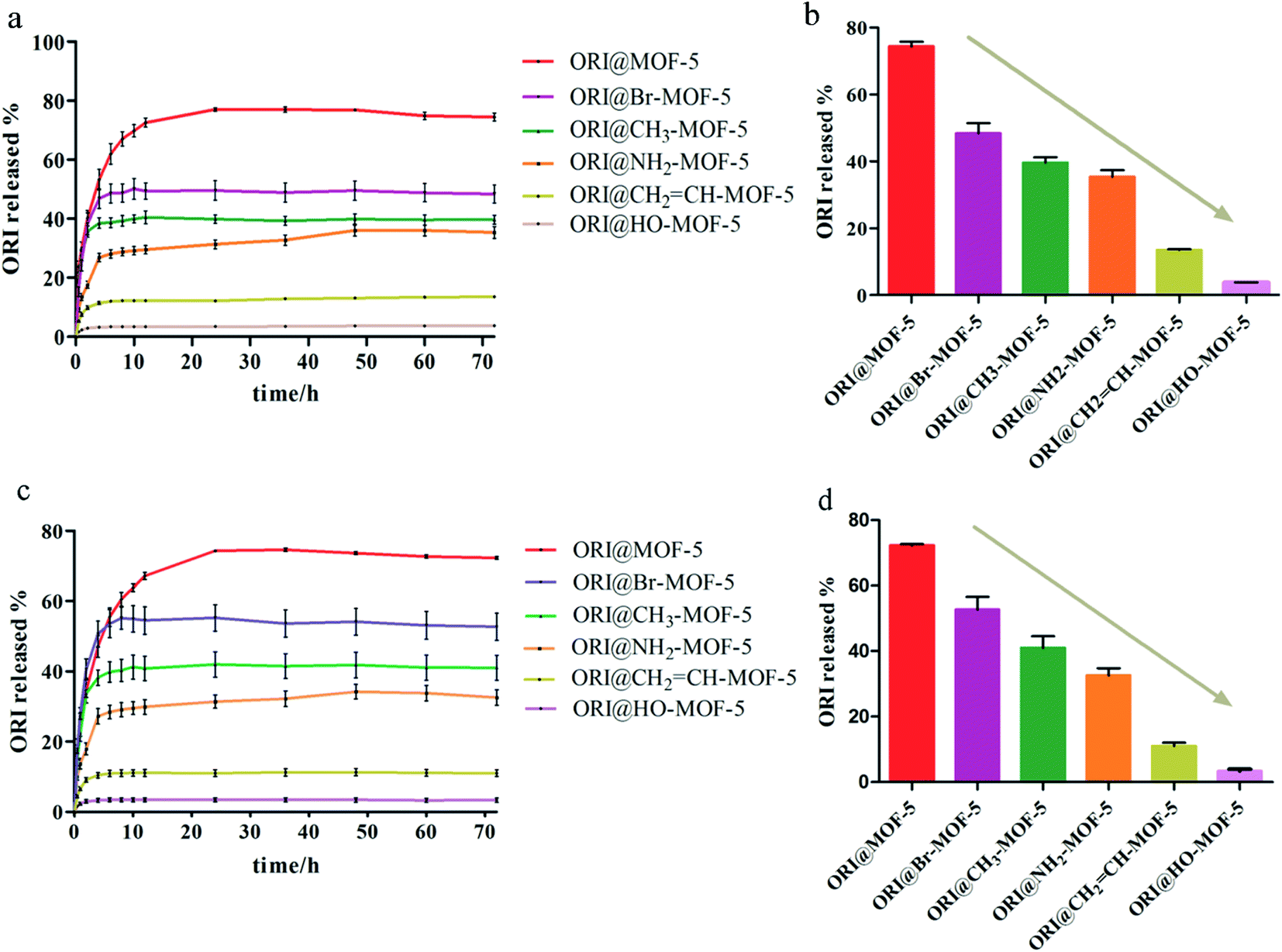 | ||
| Fig. 6 (a) The release curve of ORI@MOF-5, ORI@NH2–MOF-5, ORI@CH3–MOF-5, ORI@Br–MOF-5, ORI@HO–MOF-5 and ORI@CH2CH–MOF-5 under pH 5.5. (b) The total release of ORI under pH 5.5. (c) The release curve of ORI@MOF-5, ORI@NH2–MOF-5, ORI@CH3–MOF-5, ORI@Br–MOF-5, ORI@HO–MOF-5 and ORI@CH2CH–MOF-5 under pH 7.4. (d) The total release of ORI under pH 7.4. | ||
Obviously, there are close and numerous interactions between the ORI molecule and the host structure, which may be due to the existence of three types of drug molecules in the release profile. As shown in Fig. 7, free ORI at the edge of the frameworks is first released, and there is a weak interaction between ORI and the skeleton. This led to the burst release of the first phase. The free ORI in the centre of the pores is then released. Due to the molecular interaction between the drugs and the steric hindrance, they are released slowly in the second stage. The ORI attached to the frame is difficult to be released due to its hydrogen bond, π–π conjugation and binding with the frameworks. This forms the slowest ORI release rate in the third stage. Therefore, three processes of ORI release were formed.
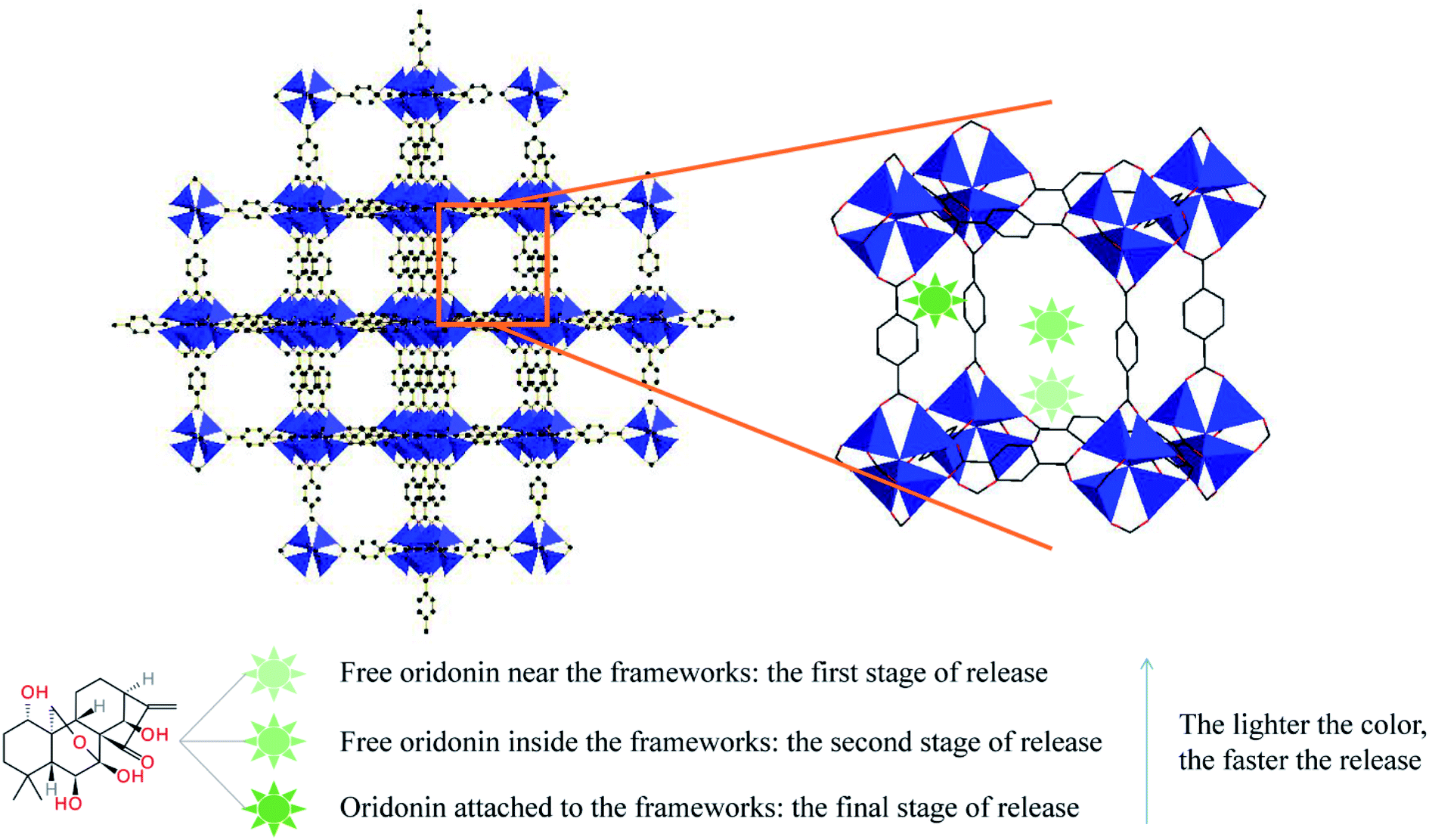 | ||
| Fig. 7 Schematic illustration of release process of ORI from MOF-5s through three states. | ||
Interestingly, the release rate and total amount of ORI varied under the same conditions. The release rate and total amount of each ORI-loaded MOF-5 with mono-substituent has decreased compared to MOF-5. And the cumulative release percentage of each group is significantly different from that of MOF-5. ORI@IRMOFs is ranked according to the cumulative release percentage of ORI: ORI@MOF-5 > ORI@Br–MOF-5 > ORI@H3C–MOF-5 > ORI@H2N–MOF-5 > CH2CH–MOF-5 > ORI@ HO–MOF-5. The release rate of IRMOFs is exactly the same as its drug loading capacity, except for HO–MOF-5. This is due to the functionalization of organic ligands, so that there is an interaction between IRMOFs and ORI, including steric hindrance, coordination binding, hydrogen bonding, π–π packing and electrostatic interaction. On the one hand, halogen bromine atoms, methyl groups and vinyl groups increase the hydrophobicity of IRMOFs, which prevents water from penetrating into the frame and slows the release of ORI.
On the other hand, the presence of these hydrophobic groups increases steric hindrance and prevents ORI from being expelled from the pores, and this hindrance is more powerful as the molecular weight of the functionalized group increases. The drug loading of HO–MOF-5 is higher than that of other functionalized IRMOFs, but the cumulative release rate is the lowest. This may be due to the complex interaction between HO–MOF-5 and ORI. Because of the existence of complex interactions (including coordination binding, hydrogen bonding, π–π packing and electrostatic interaction), HO–MOF-5 is easier to combine with ORI. And there is also the phenomenon of dead adsorption, which leads to ORI easy to enter the pores of HO–MOF-5, but it is difficult to discharge from the pores. The drug loading and release rate of NH2–MOF-5 are in the middle of all functionalized IRMOFs, that is, the blocking effect and affinity are in the middle of the six IRMOFs.
Safety evaluation of MOFs
Based on the above results, the drug-carrying capacity of IRMOFs can be adjusted by changing organic ligands. In addition, cytotoxicity and biocompatibility have validated the significance of IRMOFs as for the more stringent standard. The toxicities of MOF-5 with mono-substituent were evaluated by MTT assay. As depicted in Fig. 8, the cell viability of HepG2 was both above 90% at various concentrations, even if the concentration of IRMOFs was as high as 35 μg ml−1 indicating the negligible cytotoxicity of IRMOFs. These results suggest the biological compatibility of IRMOFs being used as new drug carriers.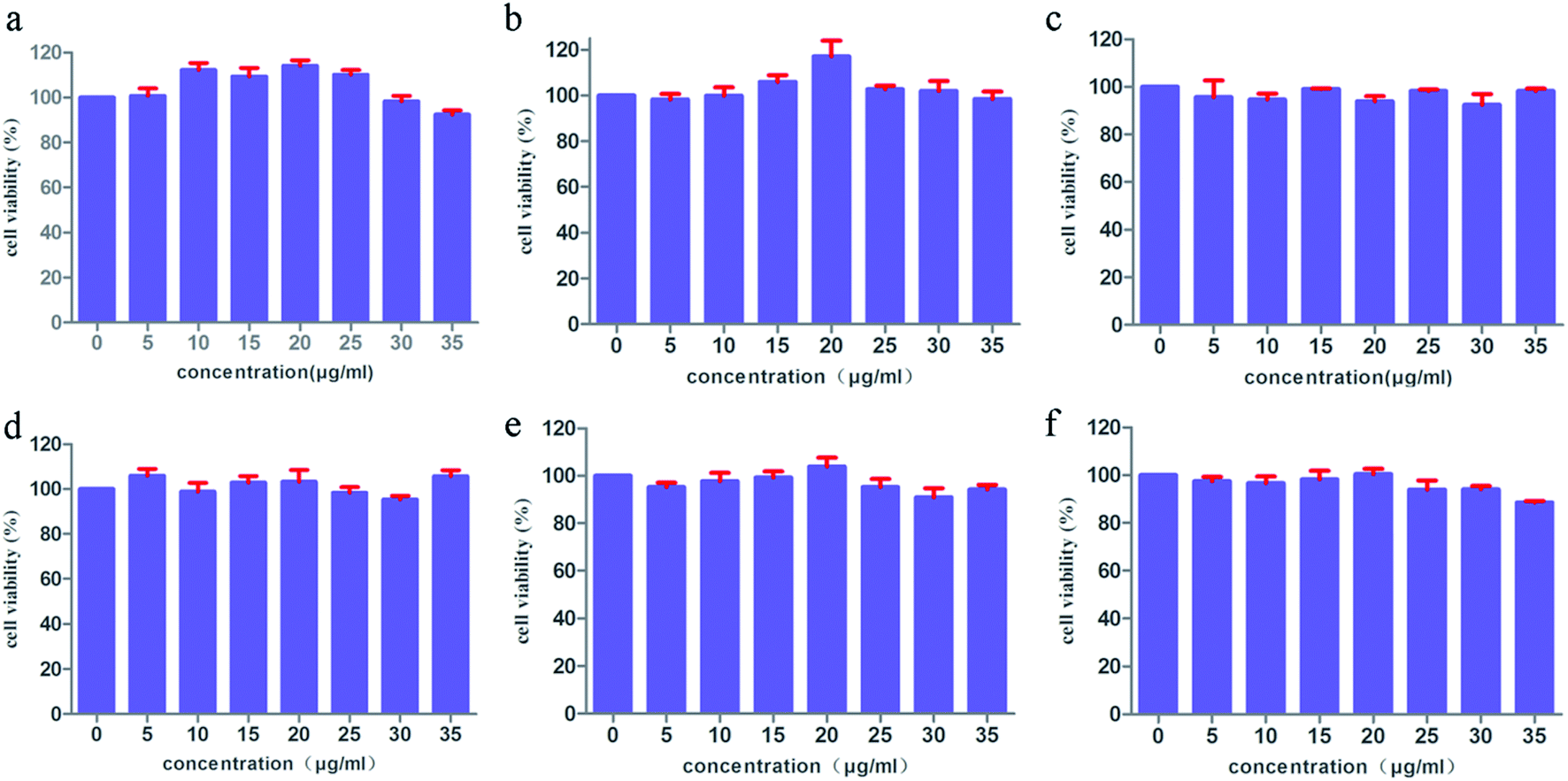 | ||
| Fig. 8 MTT assay data were presented as mean SD of viability% of three independent experiments: MOF-5 (a), NH2–MOF-5 (b), CH3–MOF-5 (c), Br–MOF-5 (d), HO–MOF-5 (e) and CH2CH–MOF-5 (f). | ||
Discussion
MOFs are a type of inorganic–organic hybrid porous crystalline material that is self-assembled by metal clusters or metal ions and multidentate bridging ligands under mild conditions.38 The diversity of metal ions and organic ligands enables the synthesis of an infinite number of MOFs. In order to investigate whether the presence of functional groups has an effect on the drug delivery capability of MOFs, the triethylamine direct addition method was used to introduce –CH3, –NH2, –Br, –OH and –CHCH2 to MOF-5. In addition to Br–MOF-5, –CH3, –NH2, –OH and –CHCH2 modified MOF-5s exhibit a typical MOF-5 topology, but the degree of crystal perfection is different. This may be due to the electron-withdrawing property of –Br leading to the weakening of the interaction between the Zn2+ metal center and the carboxyl group. The five IRMOFs have good thermal stability, suitable particle size and similar aperture, which are comparable to those of MOF-5. We used ORI as a model drug to study the effect of functionalization on the drug delivery ability of MOFs. It was found that the influence of each group on the drug delivery ability of MOFs is different. This may be due to the complex interactions among drugs and the MOFs. Increasing the length of the group will form steric hindrance, preventing the drug from entering and leaving the pores of the MOF material. This leads to a reduction in the drug-loading capacity and a slower release rate. Vinyl has the longest molecular chain, which means that the steric hindrance is the strongest, so the drug loading is the lowest, and drug release will be slower. The interactions between the drug and the group (coordination binding, hydrogen bonding, π–π packing and electrostatic interaction) will affect the loading and release of the drug. Hydroxyl and amino groups will increase the affinity among drugs and MOFs. As a result, the drug-loading capacity will be increased to a certain extent, and the drug release will be reduced accordingly.
In summary, the incorporation of substituents can modulate the drug delivery behavior of MOFs to design immediate-release or sustained-release drug delivery systems. This method is simple and easy to use, without any post-modification. This is consistent with the results of Jiang et al.39 When the substituent has a good affinity with the drug, it will increase the drug-loading capacity and also make it difficult to release the drug from the system. Therefore, a reversible interaction force between the drug and the substituent is necessary to increase the drug-carrying capacity without causing the drug to be difficult to release. As the length of the group increases, the drug-release rate slows down, which suggests that the ligand can be changed to control the drug-release rate.
Drug delivery systems usually need to be designed according to clinical needs. To reduce the dosing frequency, increase patient compliance or reduce the side effects of drugs, it is often necessary to design drugs as sustained-release preparations. Boi et al. designed and developed alginate microbeads for loading doxorubicin to control its release rate and reduce its toxicity.40 Meng et al. designed and developed a quaternized pectin–montmorillonite hybrid film to delay the release of 5-FU, which can improve the problem of dosing frequency due to short half-life.41 From our research, we found that the introduction of groups has a certain effect on the drug-release rate of MOF-based drug delivery systems. And as the length of the group increases, the sustained-release effect becomes more obvious. It is necessary to design drug delivery systems with different release rates according to the characteristics and needs of different drugs and diseases. Therefore, the development of MOF-based personalized drug delivery systems has useful research prospects.
Targeted drug delivery systems have received widespread attention in order to selectively deliver drugs to specific sites to improve the efficacy and reduce the toxicity and side effects of drugs.42 At present, the most-common active targeting strategy is to connect the targeting ligand to the drug delivery system, which requires the drug system to have an active group to graft the targeting ligand.43 Laha and colleagues linked folic acid (FA) to the amino group of IRMOF-3 to deliver curcumin to triple negative breast cancer cells.44 However, it was difficult to modify the FA to the surface of the MOFs without active groups. Therefore, it was necessary to modify the MOFs after synthesis, or to coat the MOFs with intermediates such as chitosan to facilitate the succession of targeting ligands. Gao et al. used the amino group of chitosan to connect with FA, and then wrapped ZIF-8 to achieve targeted drug delivery.45 This strategy of connecting target ligands and MOFs with intermediates can also achieve targeted drug delivery, but the process is cumbersome. Therefore, it is a good choice to modify the active substituents to MOFs for targeting ligand attachment.
ORI is a diterpene natural substance with a broad-spectrum anticancer effect. In this article, we studied the loading and release of ORI by a series of isostructural MOFs in vitro. There are many potential mechanisms for IRMOFs to load ORI, including pore encapsulation, surface adsorption and the interaction between ORI and IRMOFs. There are close and abundant interactions between ORI and IRMOFs, with possible interactions comprising coordination binding, π–π packing between the ORI and electron-rich frameworks of IRMOFs, hydrogen bonding, electrostatic interaction and space steric hindrance. The in vitro safety of six MOFs was investigated, and they were found to have good biocompatibility at the experimental dose. In the future, in vivo pharmacokinetics and pharmacodynamics of the drug delivery system will be studied in depth. In addition, drug delivery in vivo will be explored to investigate clinical application. There are certain limitations to studying the delivery rule of MOFs, using only ORI as a model drug. More model drugs are needed to investigate and analyze MOF delivery. It is hoped that our research will provide new ideas for the use of MOFs as drug carriers.
Conclusions
We have reported the synthesis of a series of isostructural Zn-based MOFs bearing mono-substituent functional groups of different lengths and properties. As drug carriers, the effects of the substituents on drug delivery behaviour were studied. The drug-loading and drug-release capabilities of the six drug delivery systems were compared, and it was found that the introduction of substituents had a significant effect on the drug delivery behaviour of MOFs. At the same time, MTT in vitro experiments further proved that MOF-5s with mono-substituents had negligible cytotoxicity and good biocompatibility. This study has introduced substituents into MOFs, and has also adjusted drug delivery behaviour and designed personalized drug delivery systems. It was also hoped to adjust the physical and chemical properties of MOFs. The incorporation of active groups can facilitate post-synthetic modification to meet the needs of targeted drug delivery. In general, such biocompatible drug delivery systems may become potential candidates for anti-tumor therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Beijing Natural Science Foundation (No. 7202121), the National Natural Science Foundation of China (No. 81703715), the Training Programme Foundation for the Beijing Municipal Excellent Talents (No. 2017000020124G295) and the Fundamental Research Funds for the Central Universities (No. 2020-JYB-ZDGG-046, No. 2020-JYB-XSCXCY-127).References
- A. Z. Wilczewska, K. Niemirowicz, K. H. Markiewicz and H. Car, Pharmacol. Rep., 2012, 64, 1020–1037 CrossRef CAS.
- X. Chen, S. W. Niu, D. H. Bremner, X. J. Zhang, H. M. Zhang, Y. Y. Zhang, S. D. Li and L. M. Zhu, Carbohydr. Polym., 2020, 247, 116672 CrossRef CAS.
- L. F. Huang, J. Huang, J. B. Huang, H. M. Xue, Z. Q. Liang, J. Wu and C. Chen, Biomater. Sci., 2020, 8, 2376–2393 RSC.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Expert Opin. Drug Delivery, 2007, 4, 621–633 CrossRef CAS.
- Y. Y. Zhong, X. S. Li, J. H. Chen, X. X. Wang, L. T. Wei, L. Q. Fang, A. Kumar, S. Z. Zhuang and J. Q. Liu, Dalton Trans., 2020, 49, 11045–11058 RSC.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
- X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- X. M. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. J. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS.
- Z. Liu, X. M. Sun, N. Nakayama-Ratchford and H. J. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Metal-Organic Frameworks: Opportunities for Catalysis, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- Y. J. Sun, L. W. Zheng, Y. Yang, X. Qian, T. Fu, X. W. Li, Z. Y. Yang, H. Yan, C. Cui and W. H. Tan, Nano-Micro Lett., 2020, 12, 184–212 CrossRef.
- C. B. He, D. M. Liu and W. B. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS.
- R. S. Forgan, Dalton Trans., 2019, 48, 9037–9042 RSC.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, e1707365 CrossRef.
- Y. J. Sun, L. W. Zheng, Y. Yang, X. Qian, T. Fu, X. W. Li, Z. Y. Yang, H. Yan, C. Cui and W. H. Tan, Nano-Micro Lett., 2020, 12, 184–212 CrossRef.
- Y. Yang, H. F. Yao, F. G. Xi and E. Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS.
- C. Yu, S. Bourrelly, C. Martineau, F. Saidi, E. Bloch, H. Lavrard, F. Taulelle, P. Horcajada, C. Serre, P. L. Llewellyn, E. Magnier and T. Devic, Dalton Trans., 2015, 44, 19687–19692 RSC.
- R. J. Marshall, Y. Kalinovskyy, S. L. Griffin, C. Wilson, B. A. Blight and R. S. Forgan, J. Am. Chem. Soc., 2017, 139, 6253–6260 CrossRef CAS.
- I. A. Lázaro, S. Haddad, J. M. Rodrigo-Munoz, R. J. Marshall, B. Sastre, V. del Pozo, D. Fairen-Jimenez and R. S. Forgan, ACS Appl. Mater. Interfaces, 2018, 10, 31146–31157 CrossRef.
- M. Pander, A. Zelichowska and W. Bury, Polyhedron, 2018, 156, 131–137 CrossRef CAS.
- Z. H. Mai and D. X. Liu, Cryst. Growth Des., 2019, 19, 7439–7462 CrossRef CAS.
- L. Wu, W. F. Wang, R. Liu, G. Wu and H. X. Chen, R. Soc. Open Sci., 2018, 5, 181378 CrossRef CAS.
- Z. Liu, L. Ouyang, H. Peng and W. Z. Zhang, Cell Proliferation, 2012, 45, 499–507 CrossRef CAS.
- J. M. Xu, E. A. Wold, Y. Ding, Q. Shen and J. Zhou, Molecules, 2018, 23, 474 CrossRef.
- A. P. Hu, J. M. Du, J. Y. Li and J. W. Liu, Inflammation Res., 2008, 57, 163–170 CrossRef CAS.
- J. Huang, L. J. Wu, S. Tashiro, S. Onodera and T. Ikejima, Acta Med. Okayama, 2005, 59, 261–270 CAS.
- Z. W. Yuan, P. Ouyang, K. X. Gu, T. Rehman, T. Y. Zhang, Z. Q. Yin, H. L. Fu, J. C. Lin, C. L. He, G. Shu, X. X. Liang, Z. X. Yuan, X. Song, L. X. Li, Y. F. Zou and L. Z. Yin, Pharm. Biol., 2019, 57, 710–716 CrossRef CAS.
- L. M. Kuo, C. Y. Kuo, C. Y. Lin, M. F. Hung, J. J. Shen and T. L. Hwang, Molecules, 2014, 19, 3327–3344 CrossRef.
- J. P. Li, S. J. Cheng, Q. Zhao, P. P. Long and J. X. Dong, Int. J. Hydrogen Energy, 2009, 34, 1377–1382 CrossRef CAS.
- S. X. Gao, N. Zhao, M. H. Shu and S. N. Che, Appl. Catal., A, 2010, 388, 196–201 CrossRef CAS.
- G. S. Chen, J. Y. Luo, M. R. Cai, L. Y. Qin, Y. B. Wang, L. L. Gao, P. Q. Huang, Y. C. Yu, Y. M. Ding, X. X. Dong, X. B. Yin and J. Ni, Molecules, 2019, 24, 3369 CrossRef CAS.
- J. Della Rocca, D. M. Liu and W. B. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS.
- K. Jiang, L. Zhang, Q. Hu, Q. Zhang, W. X. Lin, Y. J. Cui, Y. Yang and G. D. Qian, Chem.–Eur. J., 2017, 23, 10215–10221 CrossRef CAS.
- S. Boi, N. Rouatbi, E. Dellacasa, D. Di Lisa, P. Bianchini, O. Monticelli and L. Pastorino, Int. J. Biol. Macromol., 2020, 156, 454–461 CrossRef CAS.
- Y. J. Meng, S. Y. Wang, Z. W. Guo, M. M. Cheng, J. Li and D. Q. Li, Int. J. Biol. Macromol., 2020, 154, 413–420 CrossRef CAS.
- Y. Gao, J. J. Xie, H. J. Chen, S. E. Gu, R. L. Zhao, J. W. Shao and L. Jia, Biotechnol. Adv., 2014, 32, 761–777 CrossRef CAS.
- M. R. Cai, G. S. Chen, L. Y. Qin, C. H. Qu, X. V. Dong, J. Ni and X. B. Yin, Pharmaceutics, 2020, 12, 232 CrossRef CAS.
- D. Laha, K. Pal, A. R. Chowdhuri, P. K. Parida, S. K. Sahu, K. Jana and P. Karmakar, New J. Chem., 2019, 43, 217–229 RSC.
- X. C. Gao, X. Hai, H. Baigude, W. H. Guan and Z. L. Liu, Sci. Rep., 2016, 6, 37705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06106a |
| This journal is © The Royal Society of Chemistry 2020 |