
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved thermal and mechanical properties of bismaleimide nanocomposites via incorporation of a new allylated siloxane graphene oxide
Hao Jiang *,
Zhao Li,
Jiantuo Gan,
Lei Wang and
Yan Li
*,
Zhao Li,
Jiantuo Gan,
Lei Wang and
Yan Li
School of Materials Science and Engineering, Xi'an Shiyou University, Xi'an, 710065, P. R. China. E-mail: hjiang@xsyu.edu.cn
First published on 6th October 2020
Abstract
A thermosetting resin system based on bismaleimide (BMI) has been developed via copolymerization with 4,4′-diaminodiphenylsulfone in the presence of a newly synthesized graphene oxide, modified using allylated siloxane (AS-GO). The curing behavior of the AS-GO-containing resin system was evaluated using curing kinetics. The dispersibility of AS-GO in the resin was observed through polarizing optical microscopy (POM), which indicates that AS-GO has good dispersibility in the resin due to GO modified with allylated siloxane which has a good phase compatibility with BMI. The effect of AS-GO on the thermomechanical and mechanical properties of the cured modified resin was also studied. Results of thermogravimetric analysis indicated that the cured sample systems display a high char yield at lower concentrations of AS-GO (≤0.5 wt%) with an improved thermal stability. Using dynamic mechanical analysis, a marked increase in glass transition temperature (Tg) with increasing AS-GO content was observed. Mechanical property analyses revealed a possible effect of AS-GO as a toughener, and the results showed that an addition of 0.3% AS-GO maximized the toughness of the modified resin systems, which was confirmed by analysis of fracture surfaces.
1. Introduction
Bismaleimide (BMI) resins are widely used in the high-tech fields. Their applications are mainly based on their good thermal stability, great mechanical properties, good moisture, and radiation and corrosion resistance after the curing process.1–6 However, major drawbacks, including the inherent brittleness are caused by the high crosslink density and the aromatic nature of the network, limiting its further development in many fields and bringing some huge challenges for researchers. Efforts have been devoted to overcoming those problems by incorporating a microphase of dispersed elastomers or thermoplastic polymers,7–9 co-curing with other thermosetting materials,10–12 designing and synthesizing a new type of chain-extended BMI,13–15 and decreasing the crosslink density through Michael addition with active hydrogen-containing compounds.16–18 However, as far as the toughness is concerned, the mentioned-above methods are effective but usually result in accompanying shortcomings, such as reduction in modulus or poorer thermomechanical properties.19–21 Keeping in view of this issue, a new chemistry is needed to enhance the toughness of BMIs without compromising but rather improving the related physical and chemical properties. The concept of copolymerizing with nanomaterials is gaining popularity among the people working on modification of BMI resin, which will be desirable in many high-tech applications.It is well known that, due to its hexagonal honeycomb lattice structure composed of sp2 hybrid orbitals, graphene has peculiar and unique properties, such as superior tensile strength, high flexibility, mechanical property, good thermal conductivity, remarkable electron-transporting property and so on.22–30 In recent years, much attention has been paid to polymer nanocomposites reinforced with graphene. To our knowledge, graphene can be used as a new class of modifiers for resin matrixes, which results in the considerable improvements of new or modified properties.31–35 The oxidized form of grapheme (GO) is more compatible with the matrix and can drastic changes in the mechanical properties of the matrix.36 For high-tech composites for a desirable application, the thermomechanical properties are of prime importance. The functionalization of nanomaterial using polymers provides an efficient route to enhance the performance of BMI. Over the past few years, researchers have performed considerable research on the techniques that incorporate GO into BMI networks.37–42 However, it is rare to find modifications that can remarkably increase the compatibility between GO and BMI resin matrix while improving both the toughness and thermal stability of the cured products. Therefore, a related research is necessary. Due to the interfacial debonding between the BMI and graphene effects the composite properties, functionalization of GO could provide an effective method to solve the interfacial problem.
In the current work, a graphene oxide modified by allylated siloxane (AS) was designed and synthesized as a modifier of BMI. AS-GO was expected to be effective in toughening and enhancement of heat resistance due to its sufficient reactive sites of allyl group which can directly react with BMI to enter the cross-linked network and thermal conductive graphene structure. It was also expected that graphene oxide has better dispersion in the resin matrix due to surface modification. Therefore, AS-GO was characterized by XPS, FT-IR and XRD, the effects of AS-GO content on curing behavior, thermal stability and mechanical properties of the modified BMI system were studied and described in some detail.
2. Experimental
2.1 Raw materials
4,4′-Bismaleimide biphenyl methane was used as received, supplied by the Fengguang Chemical Co., Ltd. (Hubei, China). 4,4-Diaminodiphenyl sulfone (DDS) is chemically pure, supplied by Aldrich Chemical (USA). Diallyl bisphenol A (DABPA) was purchased from the Fuchen Chemical Co., Ltd (Tianjin, China). Graphite powder is industrial products, Jixihao New Energy Materials. 3-Glycidyl ether oxypropyl trimethoxysilane (silane coupler) and diallylamine (DAA) were purchased from Shanghai Aladdin Biotechnology. Sodium nitrate, hydrogen peroxide solution, concentrated sulfuric acid, concentrated hydrochloric acid and potassium permanganate were commercially supplied and were used as received.2.2 Characterization and measurements
DSC was performed with a MDSC2910 DSC instrument, the scanning range being from 25 °C to 350 °C with various heating rates under nitrogen. The dispersibility of AS-GO in the resin was examined using a polarized optical microscopy (POM) instrument. Fourier transform infrared (FTIR) spectra of the samples were obtained using a WQF-310 FTIR spectrometer supplied by Optical Instrument Factory (Beijing, China). X-ray photoelectron spectroscopy (XPS, Thermal Scientific K-Alpha XPS spectrometer) was used to investigate surface elemental composition. X-Ray Diffraction (XRD) patterns were collected using a D2 PHASER XRD instrument; the rotation velocity of the goniometer was 40 min−1. Thermo gravimetric analysis (TGA) of the samples was performed on a Perkin-Elmer Pyris 1 thermo gravimeter in inert atmosphere from 25 °C to 900 °C at a heating rate of 20 °C min−1. Dynamic mechanical analyzers (TA DMA 2980) of the samples were performed in three-point bending modes using a TA DMA 2980 instrument. The support span was 50 mm and the heating rate of 2 °C min−1 at an oscillation frequency of 1 Hz was selected. Scanning electron microscope (SEM, SM-6700F) was employed to examine the morphologies of the fracture surfaces of the cured resin. The mechanical properties of cured resins were determined according to GB/T 2567-2008.2.3 Preparation of specimens
A certain amount of graphene oxide, 80 ml ethanol, 20 ml glycol was firstly mixed and added into three-necked bottle. When the graphene oxide was dispersed evenly after ultrasonic treatment for a period of time, 25 mg of silane coupler mixed with DDA (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) through ethanol was added into the graphene oxide suspension. Then, ultrasonically mixed at 85 °C for 5 hours; in order to facilitate the removal of the sediment from the under layer, the reaction solution was allowed to stand after reaction. Next, carrying out vacuum freeze-drying after dialysis. Finally, the allylated siloxane modified graphene oxide (AS-GO) was obtained. The synthesis is schematically illustrated in Fig. 1. The BMI and DDS (molar ratio = 2:1) were mixed and heat treated to melt. Then, the DABPA was placed into the above mixture and mixed at 130 °C for 0.5 h. Finally, the AS-GO was added, until it was evenly dissolved in the system. The mixture was poured into a mold and degassed in a vacuum oven at 135 °C to ensure a complete removal of gas/air. Curing process was performed as followed: 150 °C/2 h + 180 °C/2 h + 210 °C/2 h + 240 °C/4 h.
:1) through ethanol was added into the graphene oxide suspension. Then, ultrasonically mixed at 85 °C for 5 hours; in order to facilitate the removal of the sediment from the under layer, the reaction solution was allowed to stand after reaction. Next, carrying out vacuum freeze-drying after dialysis. Finally, the allylated siloxane modified graphene oxide (AS-GO) was obtained. The synthesis is schematically illustrated in Fig. 1. The BMI and DDS (molar ratio = 2:1) were mixed and heat treated to melt. Then, the DABPA was placed into the above mixture and mixed at 130 °C for 0.5 h. Finally, the AS-GO was added, until it was evenly dissolved in the system. The mixture was poured into a mold and degassed in a vacuum oven at 135 °C to ensure a complete removal of gas/air. Curing process was performed as followed: 150 °C/2 h + 180 °C/2 h + 210 °C/2 h + 240 °C/4 h.
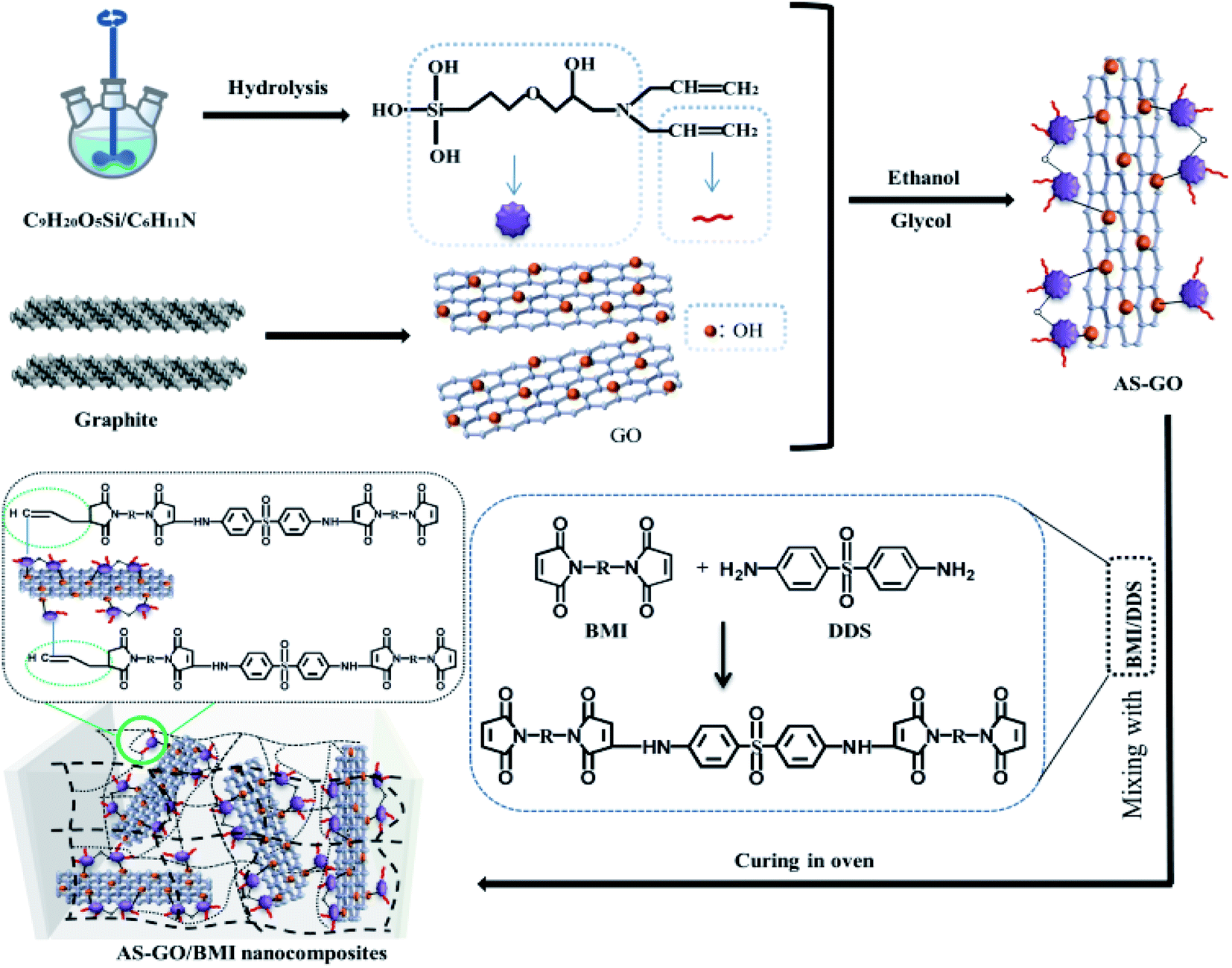 | ||
| Fig. 1 Schematic diagram for preparation of AS-GO/BMI nanocomposites. | ||
3. Results and discussion
3.1 Synthesis and characterization of AS-GO
The AS-GO was obtained by modification of GO by siloxanes containing allyl groups. Its structural information was investigated by FTIR spectra (Fig. 2), which also display the spectrum for the raw materials to confirm successful fabrication of AS-GO. The FTIR spectrum of DAA exhibits representative absorption peaks with the characteristic peaks appear at 3375 cm−1, which is related to nitrogen–hydrogen bond groups (N–H), while the peak at 920 cm−1 observed is assigned to the vibration bands of hydrocarbon groups (C–H).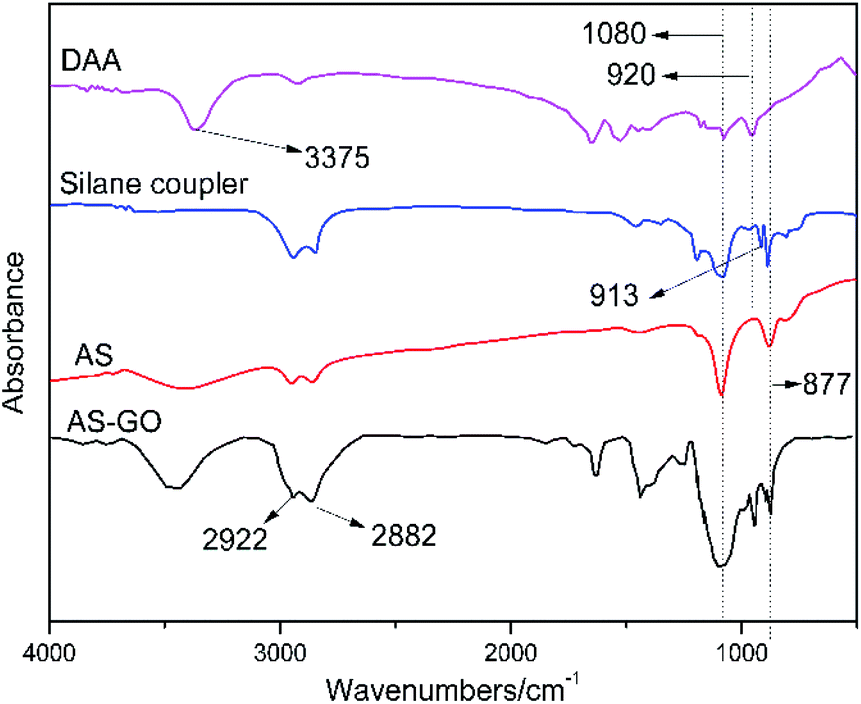 | ||
| Fig. 2 FTIR spectra of DAA, silane coupler, AS and AS-GO. | ||
For the silane coupler, the peak at 913 cm−1 was originated from epoxy group. For AS, the strong and broad peak at 3300–3500 cm−1 is associated to stretching vibration peak of the hydroxyl group. At the same time, no obvious peak at 3375 cm−1 is attributed to the epoxy group on the silane coupler is opened to form a hydroxyl group, hence the N–H bond disappears. The absorption peak at 920 cm−1 proved that AS was successfully synthesized, and there is no characteristic absorption peak of epoxy group at 913 cm−1, which also shows that the reaction is relatively complete.
As seen from the FTIR spectrum of AS-GO, the characteristic peaks appearing at 2882 cm−1 and 2922 cm−1 are related to methyl and methylene groups, the peak at 1080 cm−1 belongs to the Si–O–C stretching. In view the obvious absorption peaks at 877 cm−1 correspond to the C–Si stretching, revealing the allylated siloxane is successfully incorporated into the network of GO.
Fig. 3 displays the XPS information of GO and AS-GO nanomaterial respectively. This is applied to further analyze the bond state of the surface and the elementary composition. Compared with GO, the XPS spectrum of AS-GO present two characteristic peaks at Si 2s and Si 2p originating from AS, which further confirms the covalent functionalization of GO by AS. Similar phenomena have been reported in the literature.43,44 As can be seen from Fig. 3a that the C 1s peaks of GO are deconvoluted into six forms of carbon, including C–C (284.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.1 eV), C–OH (285.4 eV), C–O–C (286.3 eV), CO (287.1 eV), C(O)O (288.9 eV). The XPS spectrum of AS-GO (Fig. 3b) exhibits a new peak located at 283.7 eV is related to C–Si, and the C 1s signal of AS-GO exhibits a significant increase for C–C compared to GO, which indicate that AS molecules were grafted onto the GO surface. In addition, the C–OH content of AS-GO showed a significant decrease, which can be attributed to allylated siloxane reacts with the hydroxyl groups on the surface of GO resulting in a decrease in C–OH content.
C (284.1 eV), C–OH (285.4 eV), C–O–C (286.3 eV), CO (287.1 eV), C(O)O (288.9 eV). The XPS spectrum of AS-GO (Fig. 3b) exhibits a new peak located at 283.7 eV is related to C–Si, and the C 1s signal of AS-GO exhibits a significant increase for C–C compared to GO, which indicate that AS molecules were grafted onto the GO surface. In addition, the C–OH content of AS-GO showed a significant decrease, which can be attributed to allylated siloxane reacts with the hydroxyl groups on the surface of GO resulting in a decrease in C–OH content.
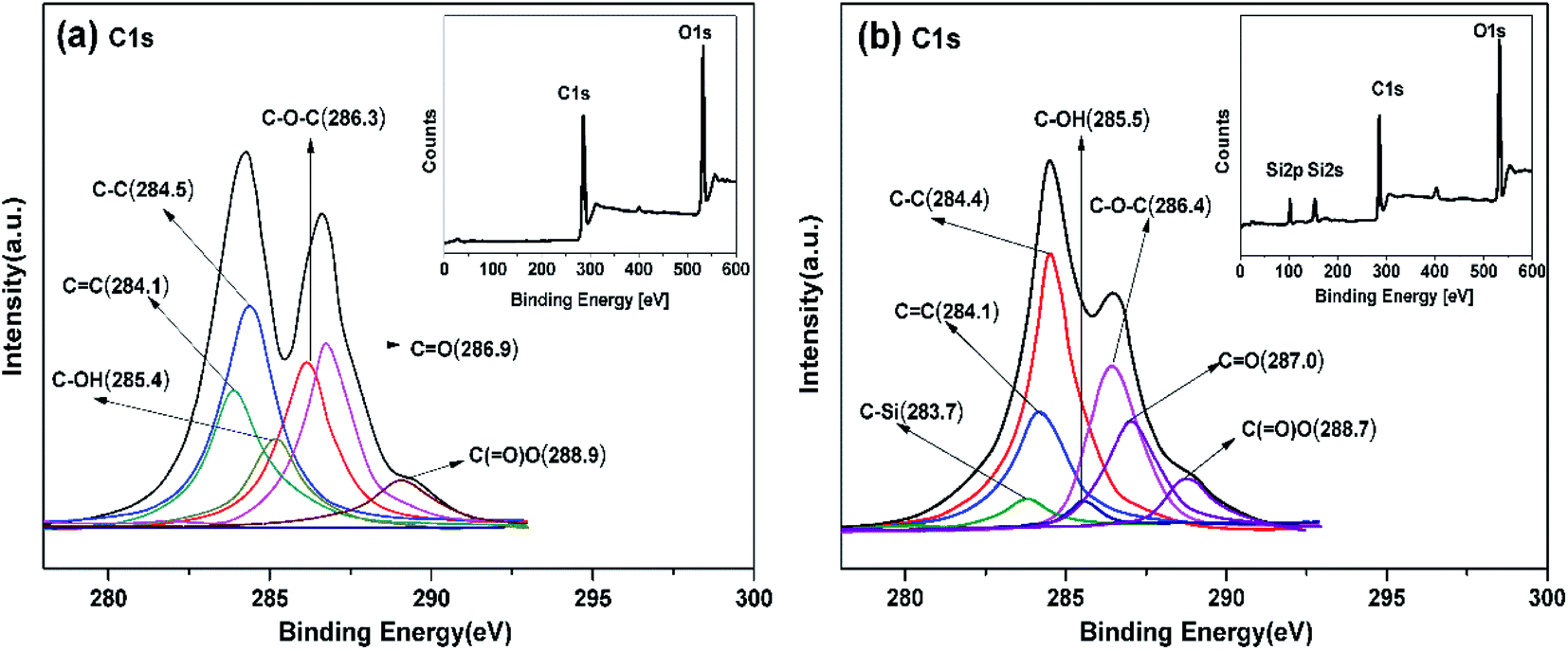 | ||
| Fig. 3 XPS spectra of GO (a) and AS-GO (b). | ||
The XRD patterns of GO and AS-GO are shown in Fig. 4. It can be observed that there is a strong diffraction peak at 2θ = 8.6°, corresponding to the characteristic diffraction peak of GO, according to the Bragg equation, lattice distance (d) is calculated to be 1.02 nm, which shows that the introduction of allylated siloxane expand the distance of graphene crystal planes. There is a strong diffraction peak at 2θ = 17.2°, corresponding to the characteristic diffraction peak of natural graphite, according to the Bragg equation, d = 0.52 nm, the diffraction peak position is similar to the characteristic diffraction peak of natural graphite, but the diffraction peak becomes wider. This shows that the GO crystal structure integrity is destroyed, and the degree of disorder increases after modification of surface with epoxy silane, the appearance of a strong diffraction peak at 2θ = 17.2° also indicates that some allyl-containing groups were reduced during modification of GO surface, which would facilitate dispersion in the resin. The XRD results confirm that GO is successfully reduced and combined with allylated siloxane.
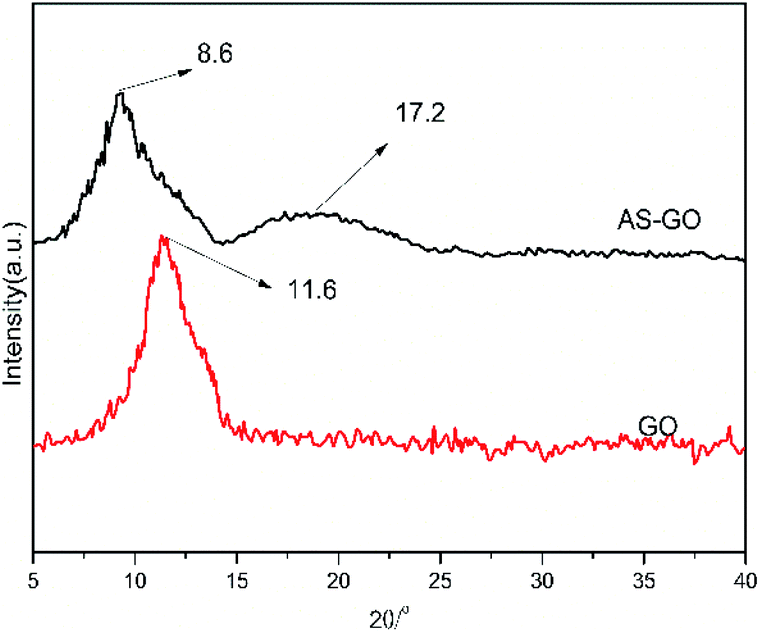 | ||
| Fig. 4 XRD spectra of AS-GO. | ||
3.2 Dispersion
To obtain excellent reinforcement effects on graphene/resin composite related materials, graphene must be uniformly dispersed in the resin matrix. In order to understand the dispersion of functional oxidation-modified graphene in the resin system, the modified resin system was characterized using a polarized microscope, as shown in Fig. 5. | ||
| Fig. 5 POM images of systems with AS-GO contents of (a) 0, (b) 1, (c) 3, and (d) 5 wt%. | ||
As can be seen from the Fig. 5, when the AS-GO content is low (0.1 wt% and 0.3 wt%), the overall dispersibility of AS-GO in the resin is better. This can be attributed to the presence of allyl groups on the surface of AS-GO, which can directly react with BMI to enter the cross-linking network, and increase the interaction force between the two phases, which can increase its compatibility, so that it can be relatively more evenly dispersed in the resin. Moreover, the molecular segments grafted on the surface of AS-GO increase the spacing of graphene sheets to prevent their agglomeration, and also can improve their dispersibility in the resin. The synergy of the two mechanisms makes As-GO and the resin matrix show good compatibility. However, when the content of AS-GO exceeds 0.3 wt%, AS-GO tends to reunite in the images (Fig. 5). This is because when the content of AS-GO is too much, the compatibility of AS-GO with the resin decreases due to the stacking effect, so a tendency to agglomerate occurs.
3.3 Curing behavior
The performance of the resin depends on the functional group, the degree of functional group reaction, and the specific characteristics of the cross-linked network structure. The curing reaction kinetics is an effective means of studying the polymer curing behavior. Taking 0.3 wt% AS-GO containing system as the research object, DSC analysis of different heating rate was carried out, as shown in Fig. 6a.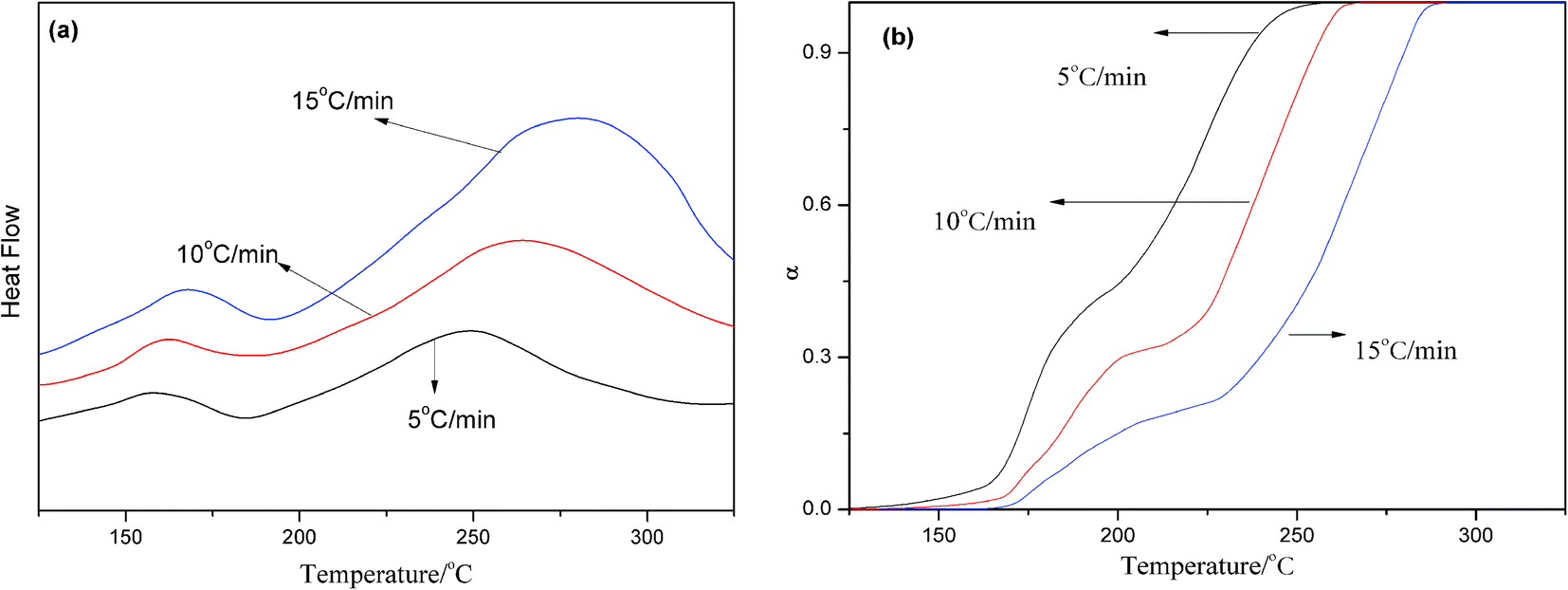 | ||
| Fig. 6 DSC curves (a) and variation in degree of cure (b) of 0.3 wt% AS-GO containing system with different heating rates. | ||
Using Flynn-wall-Ozawa (FWO) to find the apparent activation energy of the curing reaction can explore the relation between the apparent activation energy and the degree of curing α, thus studying the curing mechanism of the system. Fig. 6b shows the relationship between curing degree and temperature at different heating rates. It can be seen from the Fig. 6b that under the premise of a certain degree of curing, the higher the heating rate, the higher the curing temperature required. The curves do not cross, indicating that the curing kinetics are similar at different heating rates.
Generally speaking, in thermal analysis, the relation between reaction rate constant and temperature can be expressed using Arrhenius equation:45
 | (1) |
When calculating Ea, the relation between the heating rate and the reaction order can be expressed by an equation as follow:
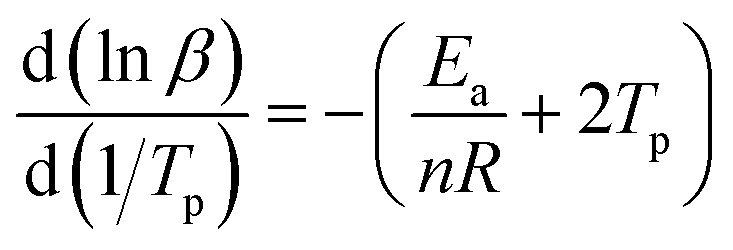 | (2) |
According to the FWO method, Ea can be obtained by calculating the slope of lnβ plotted against 1/Tp. The equation is shown in eqn (3):
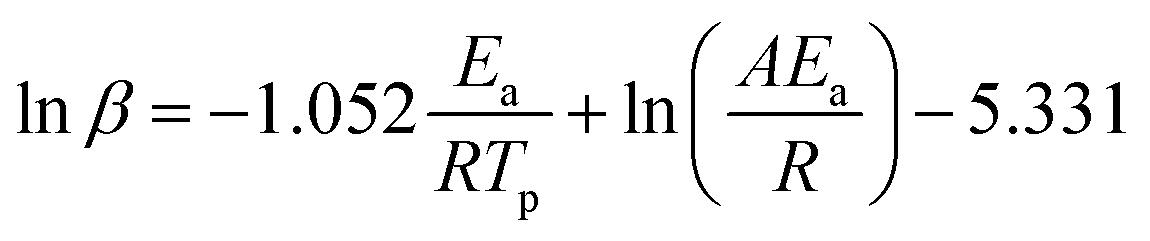 | (3) |
Fig. 7a is a straight line at multiple conversion rates obtained by the FWO method for the modified resin system. The apparent activation energy at each conversion rate is calculated from the slope as shown in Fig. 7b. The activation energy curve does not reveal the reaction mechanism, but its shape indicates the simplicity or complexity of the reaction, and it also provides kinetic control information and diffusion control mechanisms.
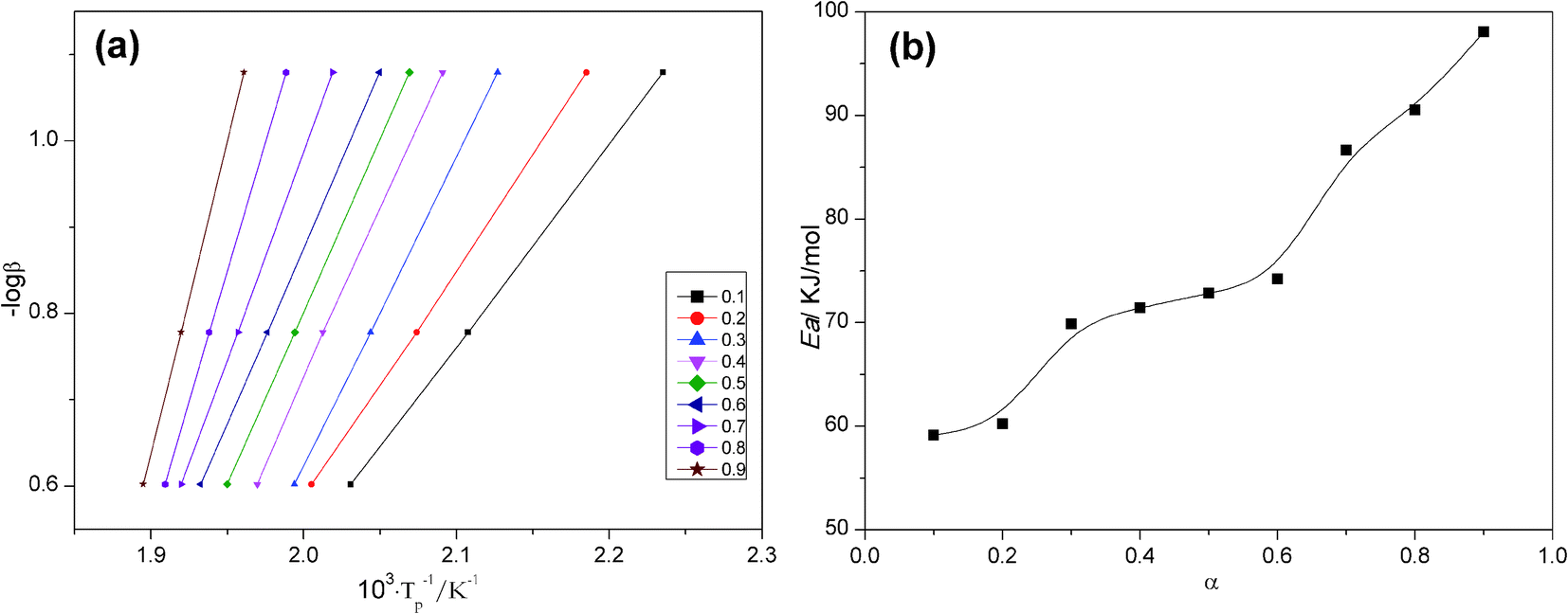 | ||
| Fig. 7 Activation energy; (a) Ozawa's plots for the curing of 0.3 wt% AS-GO containing system at different α and (b) correlation between Ea and α. | ||
As can be seen from Fig. 7b, for the AS-GO/BMI system, the non-isothermal DSC predicted that the reaction activation energy initially did not change much with the increase in the degree of curing. This can be attributed to the fact that the addition reaction dominates in the early stage of the reaction, and the activation energy has little effect on the degree of curing. When the curing degree of the system exceeds 20%, the reaction activation energy increases rapidly, which can be attributed to the self-polymerization reaction of the tertiary amine in the molecular chain of the system. After the curing degree reaches 30%, the moisture in the system decreases gradually, the activation energy of the system decreases, and the curing reaction is more likely to occur, so the activation energy of the system does not change much. With the gradual progress of the reaction, when the degree of curing exceeds 60%, the system enters a gel state and gradually forms a three-dimensional network structure. The relative molecular weight of the system tends to infinity, and the molecular movement becomes challenging. The concentration of active macromolecules gradually decreases, so the reaction becomes difficult, resulting in activation energy increases rapidly in the late stage of the reaction. At this stage, the system cannot maintain a certain equilibrium concentration of functional groups and the collision frequency between them. The reaction is changed from kinetic control to diffusion control, so the Ea value is increased.
To further verify the reaction mechanism, infrared spectroscopy was used to track and analyze the 0.3 wt% AS-GO-containing resin system at each stage of curing to determine the changes of groups under different curing degrees. The results are shown in Fig. 8. In the curing process of modified resin, the unsaturated double bond of maleimide undergoes diene addition with allyl group and then crosslinks. At the same time, it is accompanied by Diels–Alder reaction, homopolymerization and alternating copolymerization.
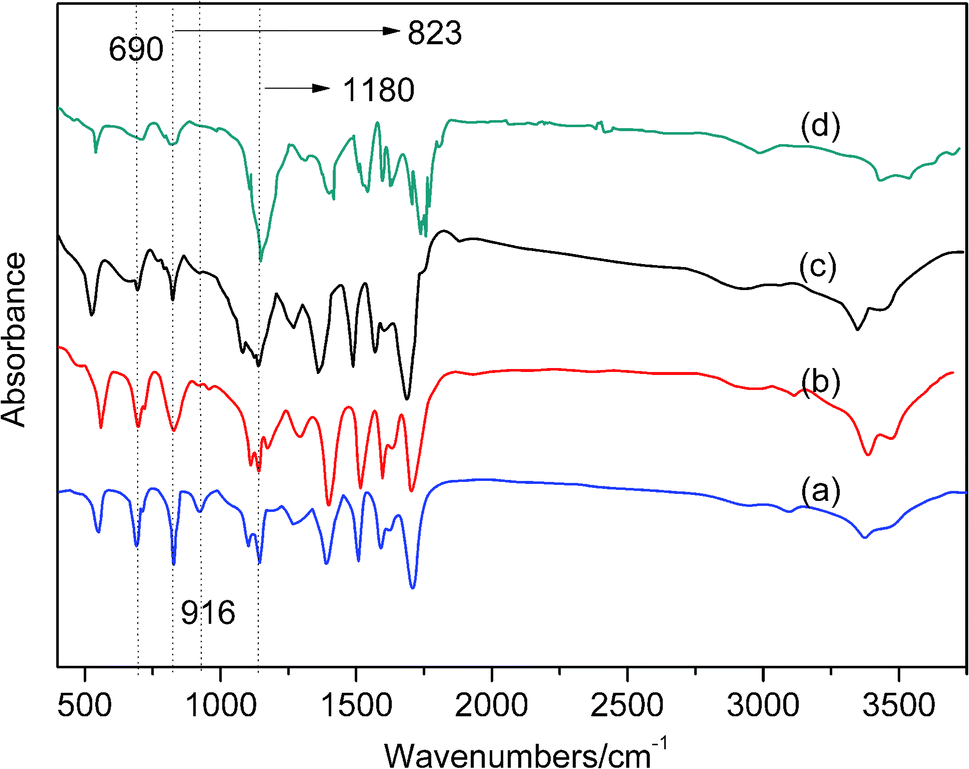 | ||
| Fig. 8 FTIR spectra of 0.3 wt% AS-GO containing system as a function of sequential cure conditions: (a–d) at sequential temperature of 150 °C, 180 °C, 210 °C, 240 °C respectively. | ||
As shown in Fig. 8, FTIR band at 690 cm−1 is assigned to the stretching vibration absorption peak of CC double bond in BMI, the peak characteristic of the maleimide double bonds are located at 690 cm−1 and 823 cm−1, the bands at 916 cm−1 correspond to allyl groups. It can be seen that, as the curing reaction proceeds, the maleimide double bonds and allyl groups decrease with reaction time. This can be attributed to the fact that, as the curing reaction proceeds, the CC double bond of BMI is open, and the diene addition reaction with the allyl compound and the Diels–Alder reaction gradually consume a large amount of maleimide double bond and olefin propyl double bond. In the later stage of the curing reaction, the steric hindrance effect also increases accordingly, and the collision between the active groups is hindered as the degree of crosslinking continues to increase, eventually, the curing reaction cannot fully proceed. Therefore, the stretching vibration absorption peak of CC double bond in BMI located at 690 cm−1 has not completely disappeared. The characteristic peak (1180 cm−1) of succinimide gradually strengthens from curve (a) to curve (d), which also shows that the crosslinking reaction of the system is gradually increasing. This also proves the rationality of the curing process.
3.4 Thermal stability analysis
TGA analysis was performed on the AS-GO/BMI cured resin, and the results are shown in Fig. 9. It can be seen that AS-GO has a certain effect on the initial decomposition temperature T0.05 of the modified system. With the increase of AS-GO content, the T0.05 of modified resin system showed an upward trend. The mass loss at the initial decomposition temperature is caused by the decomposition of the polymer network, since the heat resistance of graphene oxide is much higher than the matrix resin, and graphene oxide contains certain amino groups, carboxyl groups and other functional groups. Therefore, the matrix resin can form a stable composite structure with heat resistance and graphene oxide, so that the nanocomposite has better thermal performance.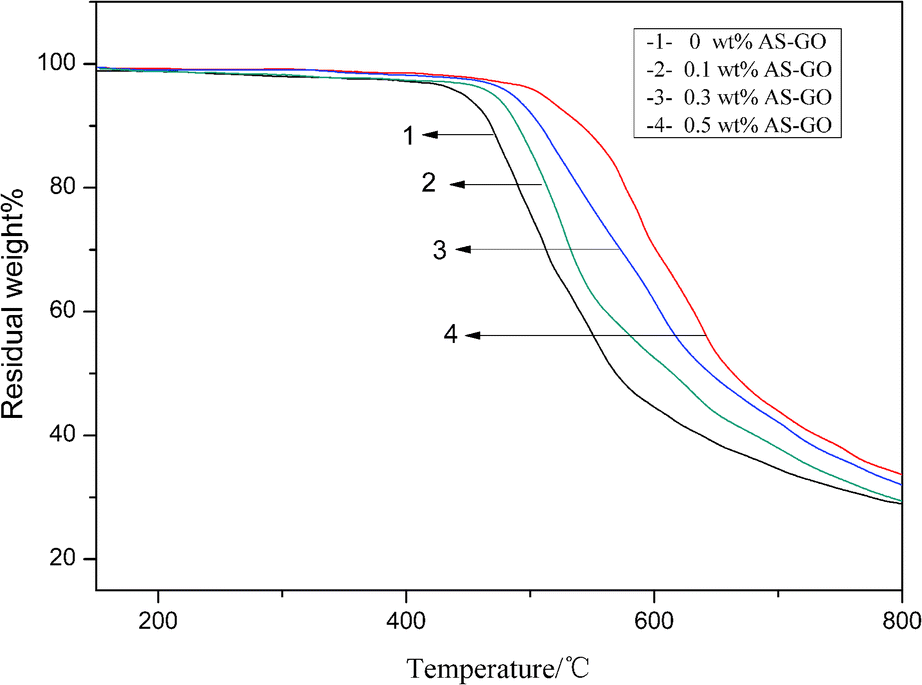 | ||
| Fig. 9 TGA curves of cured resin systems with different amount of AS-GO. | ||
As the temperature continues to increase, all systems follow a similar mass loss trend, but the system with a high AS-GO content has a slower loss rate. It can be explained that AS-GO has a certain improvement effect on the heat resistance of BMI. Until the temperature rises to 800 °C, the char yield of the modified cured systems are 29.1 wt%, 29.6 wt%, 32.2 wt%, 33.8 wt% respectively when the AS-GO loading is changed from 0 to 0.5 wt%. Generally speaking, allyl self-polymerizing aliphatic cross-linked network structure is relatively poor in heat resistance. When the temperature is high enough, the aliphatic cross-linked network structure formed by the self-polymerization of allyl groups in the unmodified system decomposes quickly, resulting in significant quality loss. Since the incorporation of AS-GO reduces the degree of allyl self-polymerization, the aliphatic cross-linked network structure is reduced, and at the same time, more aliphatic cross-linked network structures with better heat resistance are produced. The introduction of graphene oxide with higher heat resistance makes the crosslink density of the whole system increase, so the heat resistance is also improved. Therefore, compared with the unmodified system, the AS-GO containing system has a slower decomposition rate and higher char yield at high temperature.
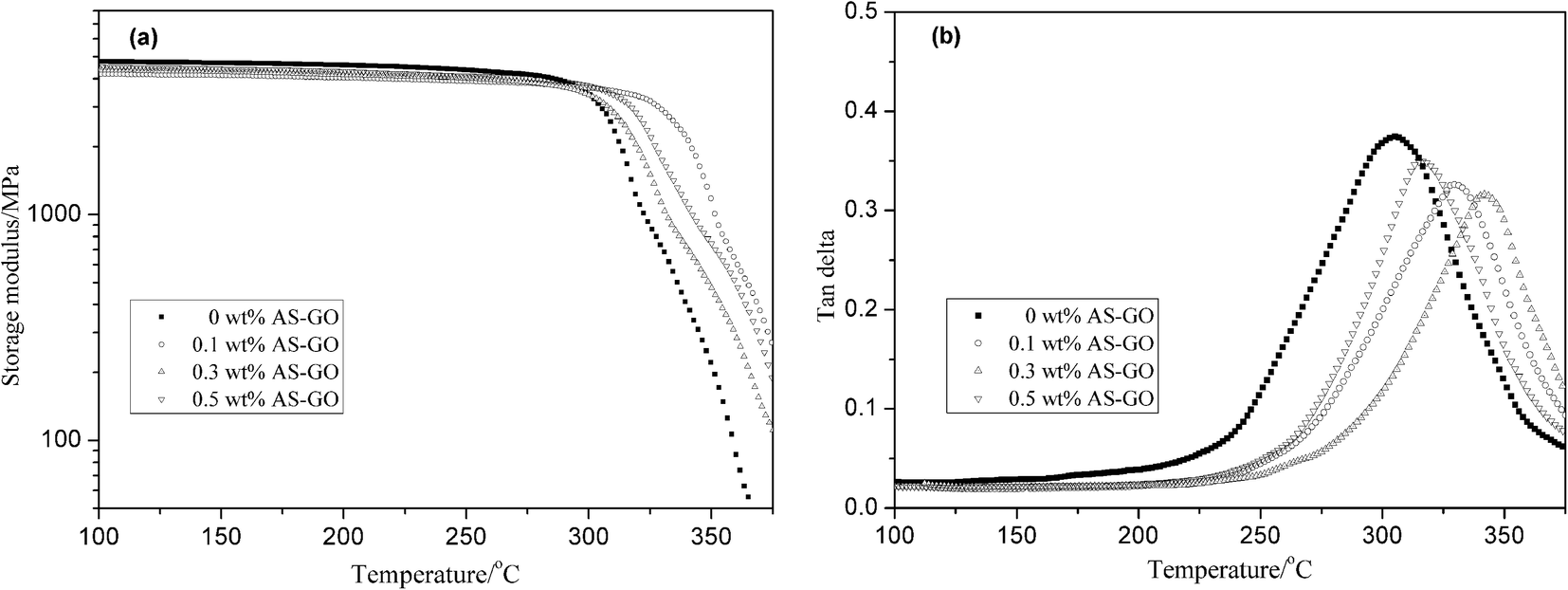
DMA measurements were performed using the AS-GO-containing cured resin systems and the storage modulus and loss tangent tanδ are presented in Fig. 10a and b respectively. It can be seen from Fig. 10a that the storage modulus of all cured systems does not change much in the range of room temperature to 300 °C (the glassy region). The storage modulus of the resin system is mainly related to the secondary relaxation of the molecular segment at this stage. The kinetic energy of the molecule cannot overcome the barrier of rotation in the main chain of the polymer, and cannot excite the movement of the segment. This also shows that all modified resins have better heat resistance. When the temperature continues to rise, the storage modulus decreases rapidly. This can be attributed to the fact that the energy obtained from the thermal motion of molecules at high temperature overcomes the barrier of rotation in the main chain of the polymer, the movement of the chain segment is excited, and the slack part of the resin system chain is activated. As can be seen from Fig. 10a, the maximum decrease rate of storage modulus of different content systems is inversely related to the content of AS-GO. As the content of AS-GO increases, the bulk density of the modified resin curing network increases, the mobility of the polymer chain is restricted, obstructed of the secondary movement of the molecular network chain becomes smaller. Therefore, the storage modulus of the cured network is relatively higher.
 | ||
| Fig. 10 Storage modulus (a) and tan(delta) (b) of cured resin systems. | ||
The glass transition temperature (Tg) of the thermosetting resin represents its maximum service temperature, and it is an important physical parameter. It is also an important indicator to evaluate its heat resistance. Tg can be directly expressed by the mechanical loss factor tanδ curve.
As shown in Fig. 10b, the Tg of the modified system with different contents of AS-GO is 305 °C/0 wt% AS-GO, 316 °C/0.1 wt% AS-GO, 329 °C/0.3 wt% AS-GO, 342 °C/0.5 wt% AS-GO respectively. The Tg of the modified resin system increased with the increase of AS-GO content. This can be attributed to AS-GO that can be connected to the resin network structure through a covalent bond to form a body structure while providing a heat-resistant structure, as a result, the crosslink density of the cured network increases, reflected in the higher values of Tg.
3.5 Mechanical properties
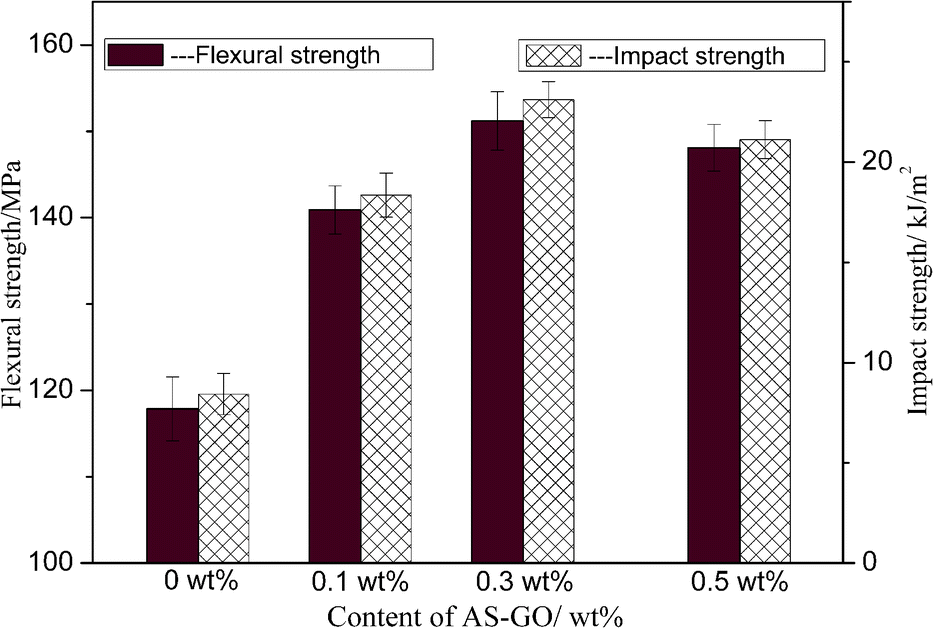
Strength is a measurement of the ability of a material to resist external damage and characterizes the material's stress limit. Different forms of destructive force have different strength indicators, it is important in practical applications. Impact strength is a measure of the ability of a material to resist impact damage under high-speed impact, flexural strength is a measure of the maximum stress that a material can withstand when it ruptures under bending load or reaches a specified deflection.Fig. 11 shows the effect of different AS-GO content on mechanical properties of the cured BMI system. It can be obtained that the AS-GO has a significant effect on the performance of the cured resin. Impact strength increase and reach a maximum value (23.12 kJ m−2) at 0.3 wt% and then decrease when the amount of AS-GO is varied from 0 to 0.5 wt%, which represents a 174% improvement. The flexural strength and impact strength showed roughly the same increasing trend, the maximum flexural strength reaches 151 MPa with a 29% increase as compared with that of the neat resin.
 | ||
| Fig. 11 Effect of AS-GO on the mechanical properties of cured resin system GO. | ||
When the amount of AS-GO is less than 0.3 wt%, AS-GO can be well dispersed in the resin matrix. At this stage, AS-GO reacts with the resin system and becomes part of the entire cross-linked macromolecular structure. It can be considered that AS-GO acts as a chain extender (as shown in Fig. 1). With the introduction of AS-GO, the modified resin system formed a large number of cavity structures, and the distance between molecular chains increased relatively. Meanwhile, the modified system also formed a large number of interpenetrating network structures, which increased the cross-linking density of the system, hence difficultly to make the molecular chain segment slip relatively. Thus the performance of the system can be improved due to the transmission and dispersion of stress when the cured resin is under the action of external forces.
When the amount of AS-GO is more than 0.3 wt%, the cross-linking density of cured resin can be greatly improved which leads to the increasing rigidity of the molecular chain. Therefore, the toughness decreases instead. On the other hand, excess AS-GO makes its dispersion become difficult and affects the phase structure of the system, which will form some defects in the resin matrix, resulting in a decrease in the performance of the cured resin. The effect of AS-GO content on the modified resin can also be further confirmed from the change of the fracture morphology of the cured resin of the system.
Fig. 12 is the SEM image of the fracture surface of the cured system with different AS-GO content. It can be noticed that the fracture surface of the unmodified system (Fig. 12a) is relatively smooth, almost no ridges can be seen, and it shows less gullies and less stress dispersion, which is a typical brittle fracture characteristic. As expected, it can be seen from Fig. 12b–d that, with addition of AS-GO, cross-section is undulating, rough and presents continuous gullies and tree branches, there are many restrictions between cracks, the damage is not carried out in a plane, and the direction of fracture also tends to be scattered. In addition, it can be found that the AS-GO was entangled with the BMI (as indicated by the red arrows in Fig. 12c) to form the interphase. The tearing of the sample at the edge of the interphase invalidated the composites. These gullies and micro-cracks indicate that the resin casting body produced micro-plastic deformation during the fracture process. The direction of crack propagation is changed, and the energy dissipation path of the resin casting body is increased, indicating the improvements in toughness. However, at higher AS-GO loading (0.5 wt%), a decreasing trend in toughness of cured resin systems is reflected in relatively few stripes, a smoother surface and strip chasm being observed, which can be attributed to a higher proportion of unreacted AS-GO accumulation agglomeration (as indicated by the red arrows in Fig. 12d) has an adverse effect on toughness. All these observations agree well with the impact strength values measured.
 | ||
| Fig. 12 Effect of AS-GO content on the morphology of fractured surface with AS-GO contents; (a) 0, (b) 0.1 wt%, (c) 0.3 wt%, (d) 0.5 wt%. | ||
4. Conclusion
In this work, the graphene oxide modified by allylated siloxane (AS-GO) has been synthesized and used to prepare a thermosetting resin with BMI resin. Various formulations containing 0 to 0.5 wt% AS-GO were made, and characterized for thermal and mechanical properties of modified resin. Characterizations including XPS, FT-IR and XRD results demonstrated that the allylated siloxane molecule was successfully linked onto the GO surfaces. The curing behavior and reactions of the AS-GO-containing resin system were evaluated using the curing kinetics method and FTIR under sequential cure conditions. Due to the chemical interaction between the BMI and the functional groups of AS-GO surfaces, the thermal and mechanical properties of the AS-GO/BMI nanocomposites were significantly improved. TGA results showed that modified BMI resin systems displayed the higher char yield at various formulations containing of AS-GO as compared to that neat resin. Using DMA, Tg was found to increase with increasing AS-GO loadings, which can be attributed to AS-GO can be connected to the resin network structure through a covalent bond to form a body structure while providing a heat-resistant structure. As expected, a suitable amount of AS-GO (0.3 wt%) led to a higher packing density of mesogenic networks, hence maximized the impact and flexural strengths of the modified which increases 174% and 29%, respectively.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors wish to thank for financial support from the Provincial Superiority Discipline of Materials Science and Engineering of Xi'an Shiyou University (ys37020203). Research fundings from the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 51702257); the National Science Foundation of Shaanxi Province (No. 2019JQ-286) and Scientific Research Program of Shaanxi Education Department (No. 19JK0660) are greatly acknowledged by the authors.References
- M. Liu, Y. Duan, Y. Wang and Y. Zhao, Mater. Des., 2014, 53, 466–474 CrossRef CAS.
- R. J. Iredale, C. Ward and I. Hamerton, Prog. Polym. Sci., 2017, 69, 1–21 CrossRef CAS.
- D. Zhuo, A. Gu, G. Liang, J. T. Hu, L. Yuan and X. Chen, J. Mater. Chem., 2011, 21, 6584–6594 RSC.
- M. H. Haque, P. Upadhyaya and S. Roy, Compos. Struct., 2014, 108, 57–64 CrossRef.
- M. Shibata, N. Tetramoto, A. Imada, M. Neda and S. Sugimoto, React. Funct. Polym., 2013, 73, 1086–1095 CrossRef CAS.
- M. Arslan, B. Kiskan, E. C. Cengiz, R. Demir-Cakan and Y. Yagci, Eur. Polym. J., 2016, 80, 70–77 CrossRef CAS.
- C. P. Vázquez, C. Joly-Duhamel and B. Boutevin, Macromol. Chem. Phys., 2013, 214, 1621–1628 CrossRef.
- X. Xiong, R. Ren and P. Chen, J. Appl. Polym. Sci., 2013, 130, 1084–1091 CrossRef.
- D. Zhuo, A. Gu, G. Liang, J. T. Hu, L. Cao and L. Yuan, Polym. Degrad. Stab., 2011, 96, 505–514 CrossRef CAS.
- I. Gouzman, N. Atar, E. Grossman, R. Verker, A. Bolker, M. Pokrass, S. Sultan, O. Sinwani, A. Wagner, T. Lück and C. Seifarth, Adv. Mater. Technol., 2019, 4, 1900368 CrossRef CAS.
- Q. Zhang, H. Wei, Y. Liu, J. Leng and S. Du, RSC Adv., 2016, 6, 10233–10241 RSC.
- M. Shibata and E. Miyazawa, Polym. Bull., 2017, 74, 1949–1963 CrossRef CAS.
- X. Xiong, P. Chen, Q. Yu, N. Zhu, B. Wang, J. Zhang and J. Li, Polym. Int., 2010, 59, 1665–1672 CrossRef CAS.
- Z. Ding, L. Yuan, Q. Guan, A. Gu and G. Liang, Polymer, 2018, 147, 170–182 CrossRef CAS.
- X. Xiong, X. Ma, P. Chen, L. Zhou, R. Ren and S. Liu, React. Funct. Polym., 2018, 129, 29–37 CrossRef CAS.
- F. E. Yu, S. C. Jiang, H. Y. Lee, J. H. Ho, J. M. Hsu and C. S. Chern, J. Polym. Res., 2015, 22, 187 CrossRef.
- H. L. Su, J. M. Hsu, J. P. Pan, T. H. Wang, F. E. Yu and C. S. Chern, Polym. Eng. Sci., 2011, 51, 1188–1197 CrossRef CAS.
- Q. T. Pham, J. M. Hsu, J. P. Pan, T. H. Wang and C. S. Chern, Polym. Int., 2013, 62, 1045–1052 CAS.
- C. Liu, Y. Dong, Y. Lin, H. Yan, W. Zhang, Y. Bao and J. Ma, Composites, Part B, 2019, 165, 491–499 CrossRef CAS.
- J. Zhou, Y. Li, N. Li and X. Hao, J. Compos. Mater., 2017, 51, 2585–2595 CrossRef CAS.
- K. Krishnadevi, V. Selvaraj and D. Prasanna, RSC Adv., 2015, 5, 913–921 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- L. Chen, G. Shi, J. Shen, B. Peng, B. Zhang, Y. Wang, F. Bian, J. Wang, D. Li, Z. Qian and G. Xu, Nature, 2017, 550, 380–383 CrossRef CAS.
- K. K. De-Silva, H. H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS.
- L. Minati, G. Speranza, V. Micheli, M. Dalla Serra and M. Clamer, Dalton Trans., 2020, 49, 3333–3340 RSC.
- N. Palaniappan, I. S. Cole and A. E. Kuznetsov, RSC Adv., 2020, 10, 11426–11434 RSC.
- G. Achagri, Y. Essamlali, O. Amadine, M. Majdoub, A. Chakir and M. Zahouily, RSC Adv., 2020, 10, 24941–24950 RSC.
- P. P. Brisebois and M. Siaj, J. Mater. Chem. C, 2020, 8, 1517–1547 RSC.
- Y. Wang, S. Li, H. Yang and J. Luo, RSC Adv., 2020, 10, 15328–15345 RSC.
- A. T. Smith, A. M. LaChance, S. Zeng, B. Liu and L. Sun, Nano Mater. Sci., 2019, 1, 31–47 CrossRef.
- J. Zhang, Z. Zhang, Y. Jiao, H. Yang, Y. Li, J. Zhang and P. Gao, J. Power Sources, 2019, 419, 99–105 CrossRef CAS.
- M. Huskić, S. Bolka, A. Vesel, M. Mozetič, A. Anžlovar, A. Vizintin and E. Žagar, Eur. Polym. J., 2018, 101, 211–217 CrossRef.
- F. Fang, P. Song, S. Ran, Z. Guo, H. Wang and Z. Fang, Compos. Commun., 2018, 10, 97–102 CrossRef.
- F. Fang, S. Ran, Z. Fang, P. Song and H. Wang, Composites, Part B, 2019, 165, 406–416 CrossRef CAS.
- W. Xu, B. Zhang, X. Wang, G. Wang and D. Ding, J. Hazard. Mater., 2018, 343, 364–375 CrossRef CAS.
- J. S. Jayan, A. Saritha, B. D. Deeraj and K. Joseph, Polym. Eng. Sci., 2020, 60, 773–781 CrossRef CAS.
- W. Li, M. Wang, Y. Yue, W. Ji and R. Ren, RSC Adv., 2016, 6, 54410–54417 RSC.
- C. Liu, H. Yan, Q. Lv, S. Li and S. Niu, Carbon, 2016, 102, 145–153 CrossRef CAS.
- Z. Wang, W. Wu, M. H. Wagner, L. Zhang and S. Bard, Polym. Degrad. Stab., 2016, 128, 209–216 CrossRef CAS.
- Z. Chen, H. Yan, T. Liu and S. Niu, Compos. Sci. Technol., 2016, 125, 47–54 CrossRef CAS.
- Z. Chen, H. Yan, Q. Lyu, S. Niu and C. Tang, Composites, Part A, 2017, 101, 98–107 CrossRef CAS.
- Y. Jiang, S. Yan, Y. Chen and S. Li, J. Adhes. Sci. Technol., 2019, 33, 1974–1988 CrossRef CAS.
- Y. J. Wan, L. X. Gong, L. Tang, L. B. Wu and J. X. Jiang, Composites, Part A, 2014, 64, 79–89 CrossRef CAS.
- X. Wang, W. Xing, P. Zhang, L. Song, H. Yang and Y. Hu, Compos. Sci. Technol., 2012, 72, 737–743 CrossRef CAS.
- H. Jiang, R. Wang, S. Farhan, D. Zhang and S. Zheng, Polym. Int., 2016, 65, 430–438 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |