
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparation of iron(III), zinc(II) and lead(II) from a choline chloride–ethylene glycol deep eutectic solvent by solvent extraction
Stylianos Spathariotis†
 a,
Nand Peeters†b,
Karl S. Rydera,
Andrew P. Abbotta,
Koen Binnemansb and
Sofia Riaño*b
a,
Nand Peeters†b,
Karl S. Rydera,
Andrew P. Abbotta,
Koen Binnemansb and
Sofia Riaño*b
aSchool of Chemistry, University of Leicester, Leicester LE1 7RH, UK
bDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, P.O. Box 2404, B-3001 Heverlee, Belgium. E-mail: sofia.riano@kuleuven.be
First published on 8th September 2020
Abstract
Deep eutectic solvents (DESs) were used as alternatives to the aqueous phase in solvent extraction of iron(III), zinc(II) and lead(II). The selective extraction of iron(III) and zinc(II) was studied from a feed of ethaline (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of choline chloride:ethylene glycol) and lactiline (1:2 molar ratio of choline chloride:lactic acid), with the former DES being more selective. A commercial mixture of trialkylphosphine oxides (Cyanex 923, C923) diluted in an aliphatic diluent selectively extracted iron(III) from a feed containing also zinc(II) and lead(II). The subsequent separation of zinc(II) from lead(II) was carried out using the basic extractant Aliquat 336 (A336). The equilibration time and the extractant concentration were optimized for both systems. Iron(III) and zinc(II) were stripped using 1.2 mol L−1 oxalic acid and 0.5 mol L−1 aqueous ammonia, respectively. An efficient solvometallurgical flowsheet is proposed for the separation and recovery of iron(III), lead(II) and zinc(II) from ethaline using commercial extractants. Moreover, the process was upscaled in a countercurrent mixer-settler set-up resulting in successful separation and purification.
:2 molar ratio of choline chloride:ethylene glycol) and lactiline (1:2 molar ratio of choline chloride:lactic acid), with the former DES being more selective. A commercial mixture of trialkylphosphine oxides (Cyanex 923, C923) diluted in an aliphatic diluent selectively extracted iron(III) from a feed containing also zinc(II) and lead(II). The subsequent separation of zinc(II) from lead(II) was carried out using the basic extractant Aliquat 336 (A336). The equilibration time and the extractant concentration were optimized for both systems. Iron(III) and zinc(II) were stripped using 1.2 mol L−1 oxalic acid and 0.5 mol L−1 aqueous ammonia, respectively. An efficient solvometallurgical flowsheet is proposed for the separation and recovery of iron(III), lead(II) and zinc(II) from ethaline using commercial extractants. Moreover, the process was upscaled in a countercurrent mixer-settler set-up resulting in successful separation and purification.
Introduction
Primary production of metals generates large volumes of waste in the form of tailings and slags, which can contain significant metal concentrations. For instance, waste from zinc production in the form of jarosite contains approximately 40% iron, 9% zinc and 8% lead.1 Another example is the fayalite slag from primary copper production, which usually contains 40% iron, 0.8% copper, 0.25% lead and 3% zinc.2,3 Both wastes also contain other valuable metals such as scandium, cobalt and nickel, which prompted several industries and research groups to investigate their recovery.4,5 Furthermore, elements such as lead, copper and zinc are hazardous and it is important to remove them from the waste prior to disposal.1,2,6 Thus, reprocessing of secondary waste streams avoids the stockpiling of environmentally harmful elements and can cause the recovery of relatively valuable metals.Solvent extraction (SX) is the most commonly used technique in hydrometallurgy for the concentration and separation of metals.7 Hereby, a metal-rich aqueous phase is mixed with an immiscible organic phase that contains usually an extractant, a diluent and in some cases a phase modifier.8 During mixing, the metals are extracted to the organic phase based on their capability to form hydrophobic complexes with the extractant.9,10 Both phases are disengaged after mixing, resulting in the selective separation and purification of metals from the aqueous phase. Purification of the loaded organic phase by scrubbing is executed when co-extraction of non-desired solutes occurs. In the final stripping step, the loaded organic phase is contacted with an aqueous solution capable of stripping the desired metal resulting in a purified and concentrated aqueous metal phase. Then the recovery of the metal in its elemental state is usually achieved by electrowinning or precipitation.10,11
The separation and recovery of Fe(III), Pb(II) and Zn(II) from aqueous solutions, that mimic jarosite waste streams, has been broadly studied using SX. Reportedly, the extraction of Fe(III) and Zn(II) has been investigated by the extractant tri-n-butyl phosphate (TBP), resulting in more than 90% Zn(II) recovery.12 The separation of Pb(II) and Zn(II) from galena (PbS) was studied by using the extractants TBP and Cyanex 272 (C272) respectively, extracting 92% of Pb(II) by TBP and 95% Zn(II) by Cyanex 272 at equilibrium pH 3.0. Fe(III) impurities in these processes were removed by precipitation using an ammoniacal solution at pH 3.5.7 In general, the Zn(II) extraction from chloride media is performed by extractants such as Cyanex 923 (C923), Aliquat 336 (A336) or di-(2-ethylhexyl)phosphoric acid (D2EHPA).13–15
Recently, a new branch of extractive metallurgy has emerged as promising alternative to hydrometallurgy due to the increased selectivity, namely solvometallurgy. This branch replaces aqueous solutions by non-aqueous solvents such as molecular organic solvents, ionic liquids or deep-eutectic solvents (DESs). Thus solvents extraction is then not executed between aqueous and organic phases, but between non-aqueous and organic phases, named non-aqueous solvent extraction (non-aqueous SX). These non-aqueous solutions do not imply a completely anhydrous phase.16 DESs are evaluated as alternatives in both leaching and non-aqueous SX processes.8,17,18 DESs are mixtures formed by hydrogen bond acceptors and hydrogen bond donors that have a melting point that is lower than their individual components. DESs are usually easy to prepare from relatively inexpensive, biodegradable and recyclable compounds.19–23 Relatively little attention has been paid to the use of DESs as alternatives to aqueous phases in solvent extraction.21 Foreman achieved the extraction of transition metals using the quaternary ammonium extractant Aliquat 336 (A336) from a diluted system of 1:2 choline chloride:lactic acid.24 Riaño et al. studied the leaching and solvent extraction of B(III), Co(II) and Fe(III) from a non-aqueous feed of 1:2 choline chloride:lactic acid, indicating that DESs can act as aqueous alternatives to facilitate the extraction process.8
In this paper, DESs are employed to replace the aqueous phase in the solvent extraction process to purify and separate a mimicked jarosite waste stream containing Fe(III), Pb(II) and Zn(II). Ethaline and lactiline are mixtures of 1:2 molar ratio of choline chloride:ethylene glycol and choline chloride:lactic acid respectively. Overall ethaline and lactiline are relatively cheap and easy preparable DESs with relatively low viscosity.20,25 Chloride salts of Fe(III), Pb(II) and Zn(II) were dissolved in both DESs and the most efficient separation was achieved using non-aqueous solvent extraction by contacting the DES feed containing the metals with commercial extractants C923 and A336. Although the main goal is to evaluate DESs as non-aqueous phases to separate Fe(III), Pb(II) and Zn(II) in non-aqueous SX processes, some extraction mechanisms are proposed. Since DESs are not involved in the stripping processes, proposing stripping mechanisms was omitted. The metal recovery processes were up-scaled in countercurrent extraction cascades by using a small battery of mixer-settlers. Mutual solubilities of the two phase systems used in the mixer-settlers were determined after completion of the separation.
Experimental
Products
Choline chloride (99%), ethylene glycol (99.5%), anhydrous FeCl3 (99%), anhydrous AlCl3 (99%) and LaCl3·7H2O (99.99%) were purchased from Acros Organics (Geel, Belgium). ZnCl2 (98%) was purchased from Chemlab-Analytical (Zedelgem, Belgium). Ethanol (99.9%), PbCl2 (98%), methanol-d4 (99.8%), Aliquat 336 (A336, a mixture of quaternary ammonium chlorides, with 88.2–90.6% quaternary ammonium content) and anhydrous oxalic acid (99%) from Sigma-Aldrich (Diegem, Belgium). Cyanex® 923 (C923, a mixture of trialkylphosphine oxides), Cyanex® 272 (bis(2,4,4-trimethylpentyl)phosphinic acid) were obtained from Cytec Solvay Group (New Jersey, USA), TBP (tri-n-butyl phosphate) from Alfa Aesar (USA). The aliphatic and aromatic diluents Shell GTL GS190 (C10–C13 aliphatic hydrocarbon diluent) and Shellsol A150 (C9–C11 aromatic hydrocarbon diluent) were obtained from Shell (Rotterdam, The Netherlands), ammonia solution (25%), hydrochloric acid (37%) and lactic acid (88%) from VWR International (Leuven, Belgium). All the chemicals were used as received, without any further purification. Ultrapure water (18.2 MΩ cm) was obtained by a Merck Millipore (Overijse, Belgium) Reference A+ Milli-Q water purification system.Preparation of DES solutions
DESs were prepared by mixing choline chloride with ethylene glycol or lactic acid, both at a molar ratio of 1:2 at 60 °C, to make ethaline and lactiline respectively. The heavy phase (HP) (feed or heavy polar phase) was prepared by dissolving FeCl3, ZnCl2 and PbCl2 in the right amounts in the DES, to obtain the following concentrations: 2.80 g L−1 Fe(III), 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). The solution was stirred until it became transparent and homogeneous.
Extraction, scrubbing and stripping experiments
Extraction experiments were performed in 4 mL glass vials. The HP (DES phase containing the metals) and the LP (light phase or less polar phase) were mixed at 1:1 volume ratio at 2000 rpm, at temperature of 25 °C using a Nemus Life Turbo Thermo Shaker TMS-200 for 20 min, unless stated otherwise. After extraction, the vials were centrifuged at 5000 rpm for 20 s using a Thermo Scientific Heraeus Labofuge 200 centrifuge to assure complete phase disengagement. In the case of Zn(II) extraction from the DES feed with A336, the pre-equilibration of A336 was firstly done by mixing A336 with pure ethaline using a Burrell Wrist-Action Shaker at 450 rpm for 20 min. Samples were centrifuged in an Eppendorf Centrifuge 5804 at 4000 rpm for 1 min. Afterwards, the pre-equilibrated A336 phase was used for the Zn extraction, following the methodology described before. Extraction experiments in 40 mL tubes, containing 20 mL of each phase, were performed to get a homogenous large volume of the LP-loaded with metals-phase, in order to carry out the several subsequent scrubbing and stripping tests with the same starting LP phase, which were performed at 4 mL tubes.
Stripping of metals was carried out in 4 mL glass vials by contacting the loaded LP with the stripping phase containing the stripping agent following the same procedure as the extraction (mixing and centrifugation). Metal concentrations in the heavy DES and aqueous phases were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using an Optima 8300 spectrometer equipped with an axial (AX)/radial (RAD) dual plasma view, a GemTip Cross-Flow II nebulizer, a Scott double pass with inert Ryton spray chamber and a demountable one-piece Hybrid XLT ceramic torch with a 2.0 mm internal diameter sapphire injector. Dilutions were done with 2 vol% nitric acid solutions and all ICP-OES analysis were measured in triplicate. Samples were 1000 times diluted and scandium(III) was used as internal standard. A schematic overview of a batchwise non-aqueous extraction experiment is shown in Fig. 1.
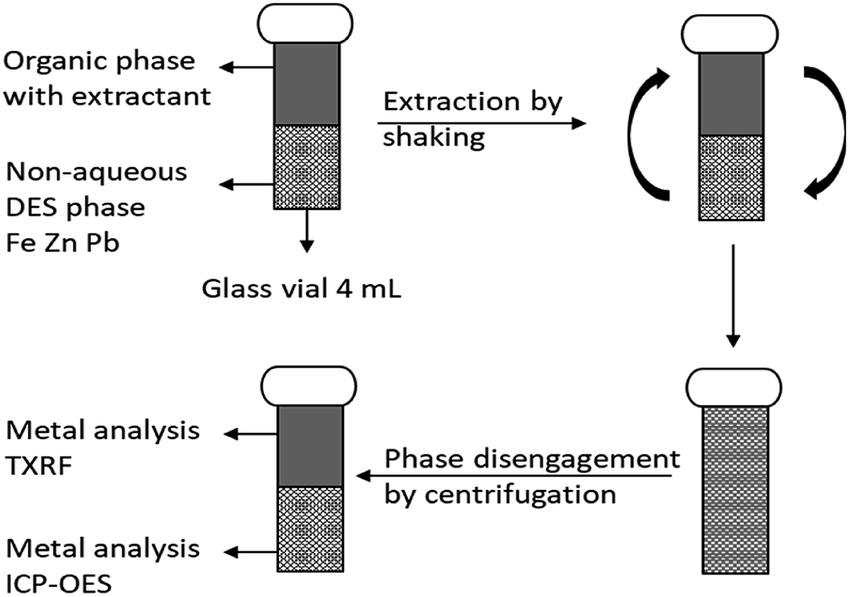 | ||
| Fig. 1 Schematic overview of a batchwise non-aqueous extraction experiment. Stripping is performed in the same way having the loaded with metals LP mixed with the stripping phased. | ||
In order to calculate the concentration of the metals in the LP, the concentration in the HP after extraction was subtracted from the initial:
| [M]LP = [M]HPi − [M]HP | (1) |
Percentage extraction, distribution ratio and separation factor
The percentage extraction (% E) is defined as the amount of metal extracted in the light phase [M]LP over the initial amount in the heavy phase [M]HPi:
 | (2) |
The distribution ratio (D) is defined as the concentration of metal extracted in the light phase [M]LP over the concentration left in the heavy phase at equilibrium [M]HP:
 | (3) |
The separation efficiency between two metals is correlated to the separation factor (α) which is the fraction of the distribution ratios of the two:
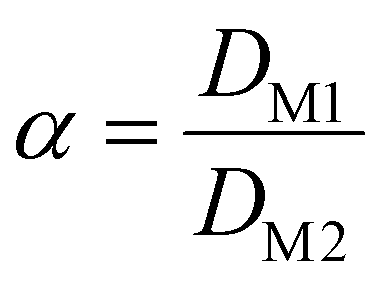 | (4) |
In a similar way, the percentage stripping or scrubbing (% S) is given by the metal concentration in the aqueous phase [M]aq after the stripping or scrubbing divided by its concentration before the extraction in the light phase [M]LP:
 | (5) |
Counter-current upscaling with mixer-settlers
The number of theoretical stages was estimated by constructing McCabe–Thiele diagrams for each process. This was done by varying the phase ratio (LP:HP) between 11:1 and 1:11, followed by plotting the determined metal concentrations in the corresponding LP and HP after phase disengagement. Afterwards, both extraction and stripping operations were continuously executed in Rousselet PTFE lab-scale mixer-settler units of universal type (Model UX 1.1), having a mixer volume of 35 mL, a settler volume of 143 mL and a settler area of 49 cm2. In each settler, one baffle and two PTFE coalescence plates were used to help the phase disengagement. Peristaltic pumps of the type ProMinent Beta 4 were used to pump the HP and LP phases via high density polyethylene plastic tubes. The extraction of Fe(III) and Zn(II), together with the Zn(II) stripping, were executed in two mixer-settler stages; the Fe(III) stripping was done in three stages. For all operations, the HP phase was the continuous phase, the flow rates of the HP and LP phases were 2.8 mL min−1 and 2.5 mL min−1 respectively, and the phase ratio LP:HP was kept at 1:1. A minor adjustment was done for the Zn(II) extraction by A336, whereby 20 wt% water was added to the HP phase to lower the viscosity and phase disengagement time. During the operation, samples (100 μL) of both the HP and LP phases were collected at the end of each settler after every 30 min of operation time. The HP phases were analyzed with ICP-OES as described earlier and the LP phases were analyzed using a total reflection X-ray fluorescence spectrometer (TXRF; Bruker S2 Picofox), equipped with a molybdenum X-ray source and operated at a voltage of 50 kV. The quartz glass sample carriers were first heated for half an hour at 60 °C in a hot air oven. Sample preparation was done by mixing 25 μL of loaded light phase together with 50 μL gallium(III) ICP standard and 925 μL ethanol. Analysis was done by adding 3 μL of this prepared sample on the preheated carriers followed by drying 30 min at the same temperature.
Mutual miscibility studies
Mutual miscibility experiments were done by mixing equal volumes of HP and LP for 60 min, followed by centrifuging at 3000 rpm for 10 minutes. The solubility of ethylene glycol (singlet, 3.46 ppm) and choline chloride (quartet, 4.00 ppm) in C923 (40 wt% diluted in aliphatic diluent) and A336 were determined with 1H NMR (Bruker Ascend 400 spectrometer, operating at 400 MHz) using 1,2-dichloroethane (singlet, 3.70 ppm) as internal standard. The solubility of A336 (singlet, 0.91 ppm) in ethaline (diluted with 20 wt% water) was determined by 1H NMR using 3-pentanone (triplet, 1.06 ppm) as internal standard. For all NMR measurements, deuterated methanol was used as solvent. The solubility of C923 (singlet, 55 ppm) (40 wt% diluted in aliphatic diluent) was determined by phosphor quantification with 31P NMR (Bruker Ascend 400 spectrometer, operating at 243 MHz) using TBP (singlet, −0.5 ppm) as internal standard. In order to include the metal concentration effect, chloride salts of Zn(II) and La(III) were dissolved in the DES to obtain a concentration of 1.60 g L−1 and 2.05 g L−1 respectively. All NMR spectra were analyzed using MestReNova software.Results and discussion
Solvent extraction of Fe(III) from Pb(II) and Zn(II)
The solvent extraction behavior of Fe(III), Pb(II) and Zn(II) was studied from two different DESs: lactiline (1:2 molar ratio of choline chloride:lactic acid) and ethaline (1:2 molar ratio of choline chloride:ethylene glycol). The studied LPs were TBP, C272 and C923 diluted in an aliphatic diluent (Shell GS190), at an initial extractant concentration of 30 wt% with the scope to optimize it further once the optimum phase was selected. The results are summarized in Table 1. The aliphatic diluent was chosen because it has low ecotoxicity and photochemical reactivity and is also biodegradable.16,26 Fe(III) exhibits a high affinity for C923 and is more easily and selectively extracted from ethaline than from lactiline. The percentage extraction of Zn(II) from ethaline was less than 20% for all tested extractants. These results differ from conventional aqueous solvent extraction, where C923 and C272 are known to be good Zn(II) extractants.7,13 This difference is very likely related to the coordination ability of the DES. ZnCl2 will coordinate with the available ligands in the DES, causing that the solubilized metal gets included in the DES framework. In this complex framework, ZnCl2 is most likely coordinated with the chloride anion of choline chloride to form negatively charged complexes and is surrounded by ethylene glycol molecules that coordinate with that negative charge via hydrogen bonding networks.27–29 The formation of this complex framework hinders the coordination of C923 and C272 with Zn(II) and hence lower the extraction efficiency. Furthermore, another explanation for the deviating C272 extraction behavior could be that the acidity of the DES is not in the preferred range. Ethaline has almost neutral pH (6.89) while lactiline (pH 3.1) is more acidic. Therefore, lactiline could favor Zn(II) extraction by C272.12,30–34
| Ex | DPb | DZn | DFe | % EPb | % EZn | % EFe | |
|---|---|---|---|---|---|---|---|
| a Shaking time: 60 min, 2000 rpm, 25 °C. Concentrations in the DES phase: 2.80 g L−1 Fe(III), 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). Concentration of extractants: 30 wt% each in aliphatic diluent. | |||||||
| Ethaline | TBP | 0.37 | 0.26 | 0.43 | 27.10 | 20.50 | 29.90 |
| C272 | 0.18 | 0.07 | 0.22 | 15.10 | 6.90 | 17.80 | |
| C923 | 0.00 | 0.04 | 20.44 | 0.00 | 4.30 | 95.30 | |
| Lactiline | TBP | 0.27 | 0.13 | 0.12 | 21.20 | 11.40 | 10.90 |
| C272 | 0.35 | 0.19 | 0.16 | 26.00 | 15.70 | 13.90 | |
| C923 | 0.27 | 0.09 | 0.78 | 21.30 | 8.40 | 43.70 | |
For the separation of Fe(III), Pb(II) and Zn(II) from ethaline, the best results for the selective Fe(III) extraction were obtained with C923. The co-extraction of Zn(II) was minimal and Pb(II) was not co-extracted. Extraction by TBP and C272 was less selective and lower for all three metals. Even lower extraction percentages were obtained when extracting from lactiline and none of the studied extractants allowed a selective separation of the metals.
The influence of the presence of multiple elements in the feed was investigated as co-extraction of different metals is known to influence metal distribution ratios in solvent extraction systems. A DES feed solution was prepared mimicking the composition of a real leachate of fayalite slag: 2.80 g L−1 (0.05 M) Fe(III), 0.41 g L−1 (0.002 M) Pb(II) and 1.96 g L−1 (0.03 M) Zn(II). The extraction behavior of C923 in an aliphatic diluent was also tested at equimolar concentrations (0.002 M): 0.11 g L−1 Fe(III), 0.41 g L−1 Pb(II) and 0.13 g L−1 Zn(II). This concentration was chosen because higher Pb(II) concentrations did not dissolve in ethaline. In both cases, C923 in aliphatic diluent was selective towards Fe(III) extraction from ethaline, with no observable extraction of Pb(II) and some co-extraction of Zn(II). The co-extraction of Zn(II) was higher in the feed with equimolar amounts of each metal (case (1) in Table 2), as the metal concentrations are lower and more free extractant molecules are available to allow co-extraction of impurity metals. At high metal concentrations in the feed, less free extractant molecules are present and thus the metal that has the highest affinity will be extracted preferentially and the co-extraction of less preferred metals will be suppressed. This type of loading effects can be exploited to increase the selectivity of solvent extraction systems.
| Fe/Pb/Zn (g L−1) | % EFe | % EPb | % EZn | |
|---|---|---|---|---|
| a Shaking time 20 min, 2000 rpm at 25 °C. Light phase: 30 wt% C923 in aliphatic diluent. | ||||
| 1 | 0.11/0.41/0.13 | 100.0 | 0.0 | 10.5 |
| 2 | 2.80/0.41/1.96 | 95.0 | 0.0 | 4.0 |
Furthermore, the influence of the concentration of the extractant on the extraction of Fe(III), Zn(II) and Pb(II) was investigated by varying the concentration of C923 in the diluent between 10 and 100 wt%. The highest selectivity was achieved at a C923 concentration of 40 wt%, which allowed 90% Fe(III) extraction in the LP phase with only 5% Zn(II) co-extraction (Fig. 2). Above this concentration, Zn(II) co-extraction increased and this caused a slight decrease on the percentage extraction of Fe(III). Pb(II) extraction remained insignificant at all cases. Therefore, this extracting phase and concentration (40% C923 in aliphatic diluent) will be used for the investigation of the associated extraction process.
 | ||
| Fig. 2 Effect of the C923 concentration in aliphatic diluent on the % E of Fe(III), Pb(II) and Zn. Concentrations in the HP phase: 2.80 g L−1 Fe(III), 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). Shaking speed 2000 rpm at 25 °C, equilibration time: 60 min. | ||
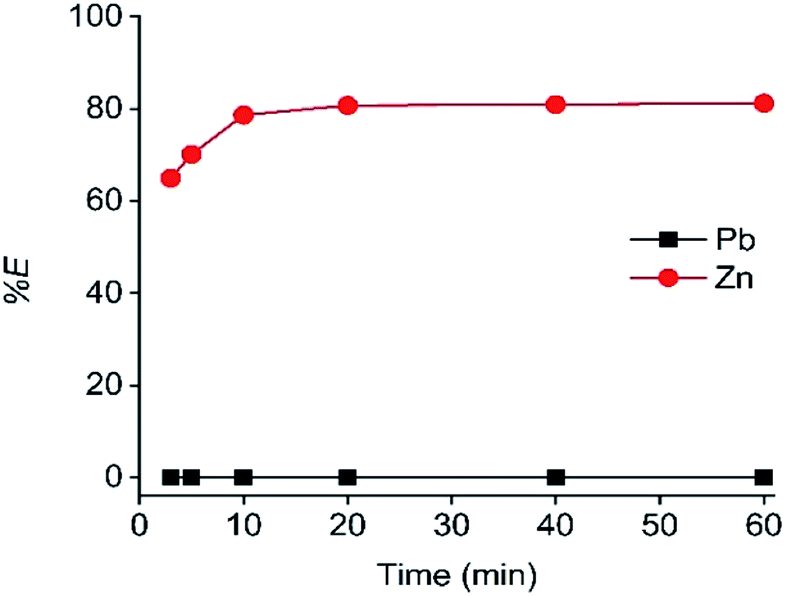
The contact time of the C923 extraction was also optimized (Fig. 3). The equilibrium was reached within 20 min of shaking, as a constant maximum Fe(III) extraction was achieved of 95%. Furthermore, Zn(II) co-extraction remained low and constant over time, reaching 3% at 20 min of equilibration.
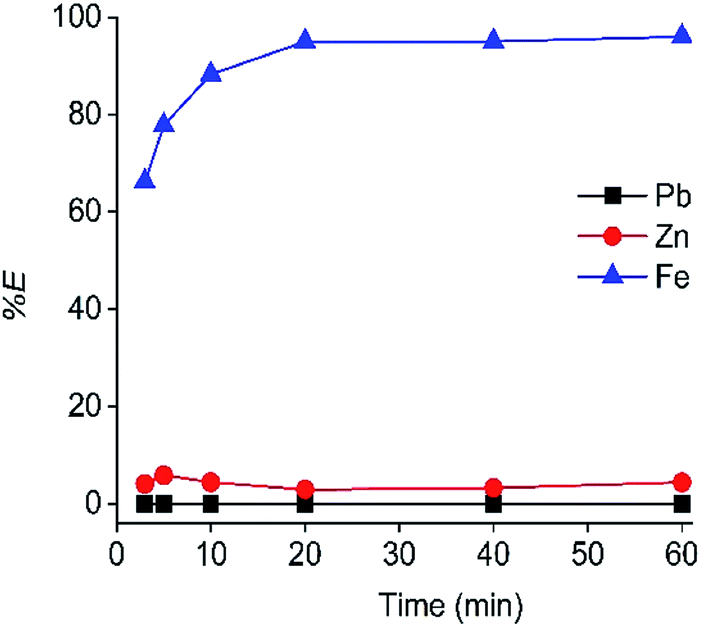 | ||
| Fig. 3 Effect of equilibration time on the extraction of Fe(III), Zn and Pb(II). Concentrations in the HP phase: 2.80 g L−1 Fe(III), 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). C923 concentration 40 wt% in aliphatic diluent. Shaking speed 2000 rpm at 25 °C. | ||
A possible mechanism for the extraction of Fe(III) by C923 is proposed. C923 is a solvating extractant formed by a mixture of four liquid trialkylphosphine oxides.35 Lloyd et al. confirmed the dominance of [FeCl4]− complexes in ethaline.36 Thus, it is most likely that these complexes accept a proton from ethylene glycol in the DES to form the neutral HFeCl4 species,37 which are then extracted by the solvating extractant C923.22,38,39 These assumptions are integrated in the proposed extraction mechanism (eqn (6)):22,37–42
| Fe(DES)3+ + H(DES)+ + 4Cl(DES)− + bC923(org) ⇆ [HFeCl4][C923]b(org) | (6) |
After the separation of Fe(III) from Pb(II) and Zn(II), recovery of Fe(III) was achieved by stripping the loaded LP phase with an aqueous solution. Several stripping agents were investigated and the results are summarized in Table 3. Among these, HCl and HNO3 have been used before for the stripping of Fe(III) and Zn(II) from C923 when extracting from chloride media,43,44 but in our case dilute solutions of HCl or HNO3 were insufficient for complete Fe(III) stripping. A 1.2 mol L−1 oxalic acid solution stripped a maximum amount of Fe(III) with insignificant co-stripping of Zn(II). From these stripping tests, it can be concluded that scrubbing of Zn(II) can be carried out with ammonia solution. Concentrations of NH3 below 0.1 mol L−1, caused the precipitation of Zn(II):
| Zn2+ + 2NH3 + 2H2O ⇌ Zn(OH)2(s) + 2NH4+ | (7) |
| Stripping agent | Concentration (mol L−1) | % SZn | % SFe |
|---|---|---|---|
| a Shaking time 20 min, 2000 rpm at 25 °C. Concentrations in LP phase: 2.66 g L−1 Fe(III) and 0.06 g L−1 Zn(II).b Below this concentration a precipitate was formed. | |||
| MilliQ | 0.0 | 29.3 | |
| HCl | 0.1 | 0.0 | 21.4 |
| HCl | 1.0 | 0.0 | 2.4 |
| HNO3 | 0.1 | 0.0 | 14.1 |
| HNO3 | 1.0 | 0.0 | 10.4 |
| Citric acid | 1.0 | 0.0 | 33.9 |
| NH3 | 0.1b | 26.3 | 30.9 |
| Oxalic acid | 0.1 | 0.0 | 7.0 |
| Oxalic acid | 1.2 | 2.4 | 89.0 |
Higher NH3 concentrations result in the formation of the soluble positively charged tetraammine zinc(II) complex, Zn(NH3)42+.
However, Zn(II) is only co-extracted in a relatively low quantity (i.e. 60 mg L−1). This would probably be reduced to a negligible amount when the extraction by C923 is executed in a multistage continuous counter-current process. Therefore, more detailed optimization of the Zn(II) scrubbing was omitted.
Separation of Pb(II) and Zn(II)
After complete Fe(III) extraction, the DES raffinate contained only Pb(II) and Zn(II). Different types of extractants and diluents were tested for the subsequent extraction of Zn(II) (Table 4). From previous experiments it was already known that C923, C272 and TBP were unsuitable extractants for Zn(II) from the HP (Table 1). Another type of extractant that was considered was Aliquat 336 (A336). n-Decanol was added as a modifier to enhance the miscibility between the A336 and the aliphatic diluent. The extraction of metals is also influenced by the physical properties of the diluent such as: density, viscosity, dielectric constant and miscibility. Subsequently, an aromatic diluent (Shellsol A150) was used because of its miscibility with A336 and its higher density. Furthermore, aromatic diluents improve the phase disengagement after mixing.45–47 Significant increase in Zn(II) extraction was observed for A336 diluted in the aromatic diluent, while Pb(II) extraction was below detection limit (Table 4).| Extractant | Diluent | DPb | DZn | % EPb | % EZn |
|---|---|---|---|---|---|
| a Shaking time: 60 min, 2000 rpm, 25 °C. Concentrations in the HP phase: 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). Concentrations of extractants: 30 wt% each.b 10 wt% n-decanol was added as phase modifier. | |||||
| TBP | Aliphatic | 0.00 | 0.06 | 0.00 | 5.40 |
| C923 | Aliphatic | 0.00 | 0.09 | 0.00 | 8.00 |
| C272 | Aliphatic | 0.00 | 0.00 | 0.00 | 0.00 |
| D2EHPA | Aliphatic | 0.12 | 0.05 | 10.30 | 5.00 |
| A336 | Aliphaticb | 0.00 | 0.32 | 0.00 | 24.50 |
| A336 | Aromatic | 0.00 | 0.56 | 0.00 | 36.00 |
| C272 | Aromatic | 0.00 | 0.07 | 0.00 | 6.90 |
The next studied parameters were the A336 concentration in the LP and the contact time. As shown in Fig. 4 and 5, undiluted A336 allowed the highest Zn(II) extraction efficiency and reached equilibrium after 20 min contact time. As reported, A336 can be suitable for Zn(II) extractions.48,49 To avoid miscibility issues and phase volume changes, A336 was pre-equilibrated with the DES before extraction. The dominance of [ZnCl4]2− complexes in ethaline has been reported in several publications.36,50–52 These complexes are described as being the extracted species during the extraction on Zn(II) by A336 in chloride media.14,48,49,53 Therefore, the Zn(II) extraction mechanism by A336 can be described as follows:53–55
| Zn(DES)2+ + 4Cl(DES)− + 2[A336][Cl](org) ⇌ [ZnCl4][A336]2(org) + 2Cl(DES)− | (8) |
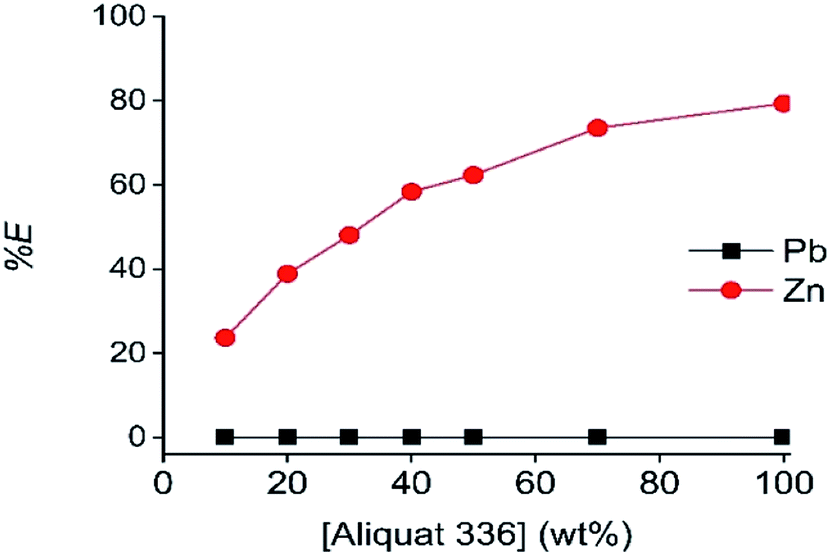 | ||
| Fig. 4 Effect of A336 concentration in aromatic diluent on the extraction of Pb(II) and Zn(II). Shaking speed 2000 rpm at 25 °C, equilibration time: 60 min. Concentrations in the HP phase: 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). | ||
 | ||
| Fig. 5 Effect of equilibration time on the Pb(II) and Zn(II) extraction by pre-saturated A336. Shaking speed is 2000 rpm at 25 °C. Concentrations in the HP phase: 1.96 g L−1 Zn(II) and 0.41 g L−1 Pb(II). | ||
Ammonia can form Zn(II) complexes, and it can be successfully applied as stripping agent (Table 5). With excess NH3, the Zn(OH)2 precipitate dissolves according to eqn (9). This critical point was determined at 0.1 mol L−1 NH3 where Zn(OH)2 is converted to the soluble tetraammine zinc(II) complex Zn(NH3)42+.14
| Zn(OH)2 + 4NH3 ⇌ [Zn(NH3)4]2+ + 2OH− | (9) |
| Stripping agent | Concentration (mol L−1) | % SZn |
|---|---|---|
| a Shaking time 20 min, 2000 rpm at 25 °C. Concentration in LP: 1.57 g L−1 Zn(II).b Below this concentration precipitation was formed. | ||
| MilliQ | 0.0 | |
| HCl | 0.1 | 0.0 |
| HCl | 1.0 | 0.0 |
| HNO3 | 0.1 | 0.0 |
| HNO3 | 1.0 | 0.0 |
| Oxalic acid | 0.1 | 0.0 |
| Oxalic acid | 1.0 | 0.0 |
| H2SO4 | 1.0 | 0.0 |
| Citric acid | 1.0 | 0.0 |
| NH3b | 0.1 | 6.7 |
| NH3 | 0.5 | 77.5 |
| NH3 | 1.0 | 74.3 |
| NH3 | 2.0 | 74.2 |
Lead(II) precipitation in ethaline
After several days of storage, a white precipitate was observed in the DES feed solution containing dissolved PbCl2, FeCl3 and ZnCl2. To determine its composition, the precipitate was filtered and washed with water and ethanol, followed by TXRF analysis. The obtained spectrum of the remaining solid confirmed the composition of PbCl2. The formation of this precipitate was evaluated as a function of time as shown in Fig. 6.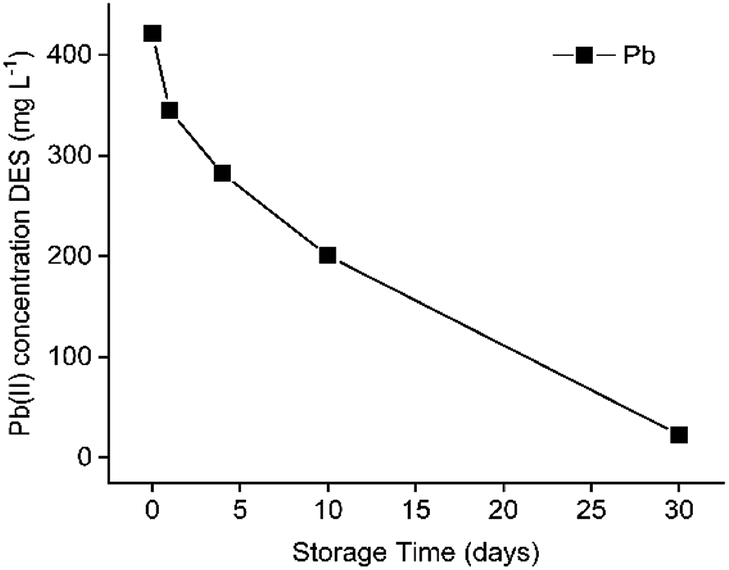 | ||
| Fig. 6 Decrease in Pb(II) concentration in ethaline with time after the dissolution of PbCl2 in ethaline. Initial concentration in HP: 0.41 g L−1 Pb(II). | ||
Fig. 6 shows that PbCl2 is unstable after being dissolved in the DES. It dissolves at 60 °C, but starts to precipitate after cooling. After 30 days, the precipitation is almost complete. In this way, Pb(II) is recoverable by isolating this precipitate, which enables the recycling of the DES. However, cementation or electrowinning is preferred because it accelerates the recovery process significantly.47 Since the emphasis of the work is on using DESs in non-aqueous SX processes to separate Fe(III), Pb(II) an Zn(II), further investigations on this precipitation tendency of Pb(II) were not executed.
Process scale-up using mixer-settlers
Based on the above results, a flowsheet for the separation and recovery of Fe(III), Pb(II) and Zn(II) is proposed (Fig. 7).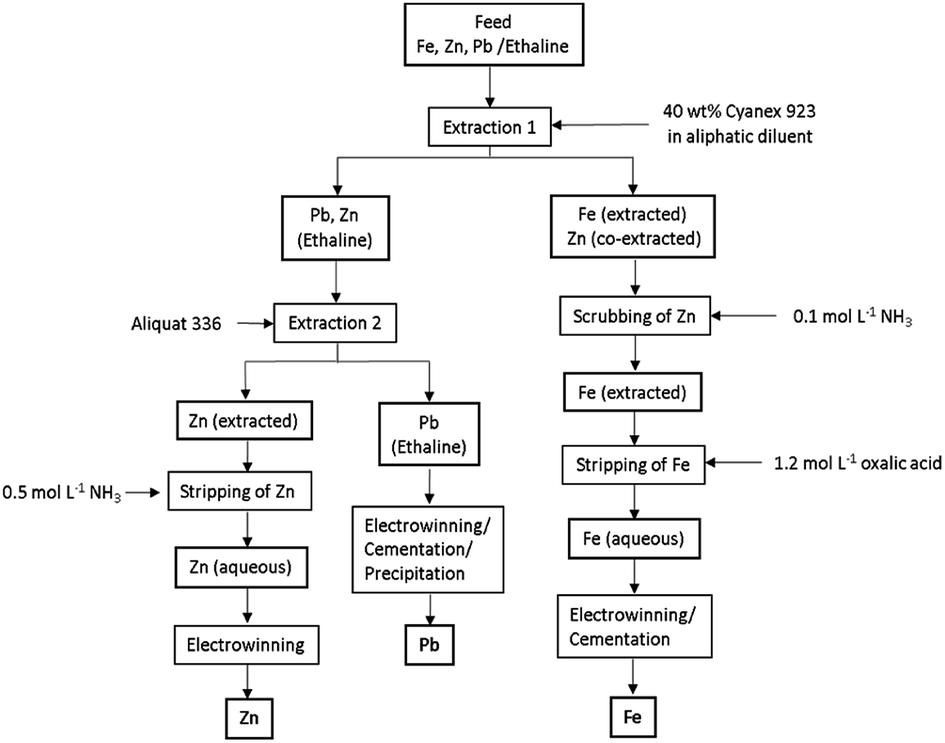 | ||
| Fig. 7 Proposed flowsheet for the separation and recovery of Fe(III), Zn(II) and Pb(II). | ||
This flowsheet was validated in a continuous counter-current circuit using mixer-settlers. Minor adjustments to the flowsheet were needed to obtain successful process validation. First, PbCl2 was not dissolved in the DES feed for mixer settler experiments. The reason for this is the Pb(II) precipitation tendency as described above and because the presence of solids in mixer-settlers is highly undesirable. Secondly, the Zn(II) extraction of the DES raffinate with pre-equilibrated A336 showed undesired features due to the relative high viscosity of both the HP and LP. This resulted in long phase disengagement times and lower Zn(II) extraction efficiency. Batch scale experiments proved that these problems were solved by adding 20 wt% water to the DES (HP) or by extracting at 40 °C. The former approach was chosen in order to reduce the process energy intensity. Stage numbers and phase ratios were first determined by constructing McCabe–Thiele diagrams. The vertical and horizontal solid lines in Fig. 8 represent the feed lines and operating lines respectively. Furthermore, the slope of the operating lines represents the used phase ratios, which are 1:1 for all operations. The theoretical number of stages are represented by the dashed lines, whereby one step is equal to one theoretical stage. Fig. 8 shows that two stages are required for the Fe(III) and Zn(II) extraction and the Zn(II) stripping, and three stages for the Fe(III) stripping. The phase ratio for all operations is 1:1.
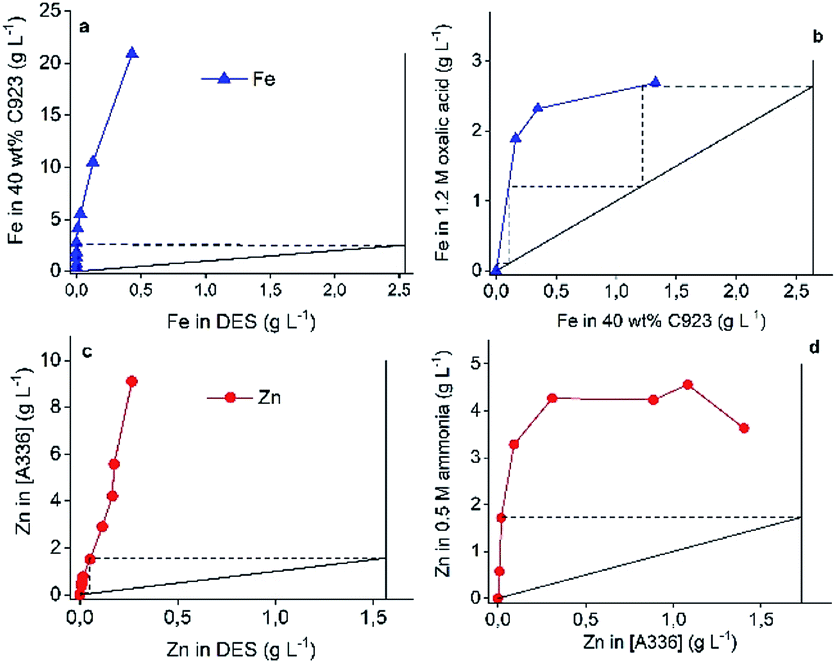 | ||
| Fig. 8 McCabe–Thiele diagrams for the DES Fe(III) extraction with 40 wt% C923 in aliphatic diluent (a), Fe(III) stripping with 1.2 mol L−1 oxalic acid (b), Zn(II) extraction in DES diluted with 20 wt% water by pre-equilibrated A336 (c) and Zn(II) stripping with stripping with 0.5 mol L−1 (d). DES feed contained 2.64 g L−1 Fe(III) and 1.96 g L−1 Zn(II). Shaking speed 2000 rpm, 3 h at 25 °C. | ||
The determined parameters were successfully used as input for the mixer-settler experiments, which are shown in Fig. 9. Each operation reached equilibrium after ca. one hour operation time, no formation of undesired features such as crud, third phase or precipitation were observed. Fig. 9 confirms the successful process up-scaling. Fe(III) was quantitatively extracted by 40 wt% C923 in aliphatic diluent and was subsequently completely stripped by 1.2 mol L−1 oxalic acid. The two stage counter-current extraction showed no co-extraction of Zn(II), as expected. Furthermore, the only Zn(II) remaining in the DES raffinate was diluted with 20 wt% water, hereafter completely extracted by pre-equilibrated A336 and quantitatively stripped with 0.5 mol L−1 ammonia solution.
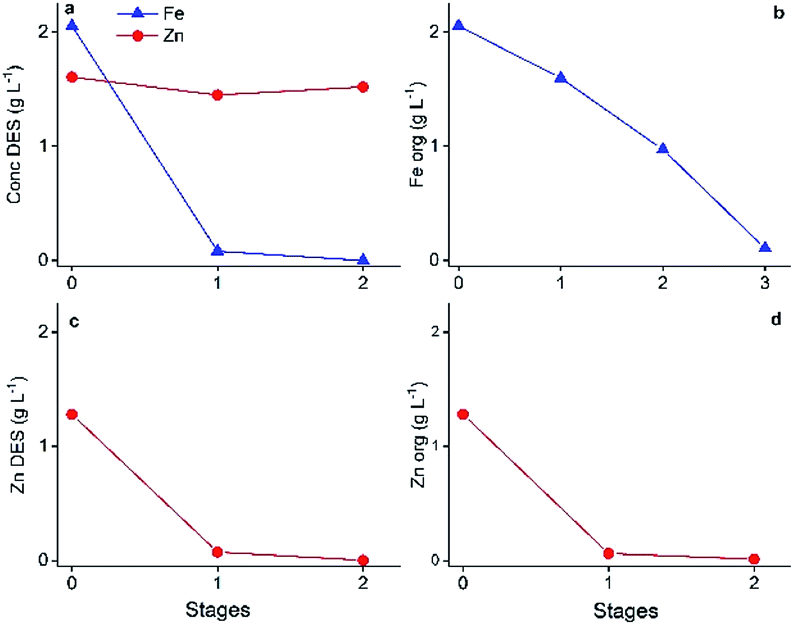 | ||
| Fig. 9 Evolution of the Fe(III) and Zn(II) concentrations during the mixer-settler experiments. Fe(III) extraction by 40 wt% C923 in aliphatic diluent (a). Fe(III) stripping by 1.2 mol L−1 oxalic acid (b). Zn(II) raffinate extraction by pre-equilibrated A336 (c) and Zn(II) stripping by 0.5 mol L−1 ammonia solution (d). All HP:LP ratios were kept 1:1, 850 rpm at room temperature. Initial DES feed contained 2.05 g L−1 Fe(III) and 1.60 g L−1 Zn(II). | ||
Mutual miscibility studies
Relatively high mutual miscibility causes considerable losses of the LP in HP and vice versa. This means that the lost amount of LP and/or HP should be added after each cycle in the process to compensate and ensure efficient extraction or stripping. Thus, mutual miscibility can lead to a cost-ineffective process. Therefore, the mutual miscibility of the two phase systems was studied using quantitative NMR techniques. The ethaline input in the mixer-settlers experiments contained 2.05 g L−1 Fe(III) and 1.60 g L−1 Zn(II). Fe(III) ions are ferromagnetic and are therefore not suitable for NMR measurements. In order to study the salt concentration effect on the mutual miscibility, alternative trivalent diamagnetic ions were tested. Al(III) and La(III) were tested and only La(III) was extracted by 40 wt% C923 in aliphatic diluent with an efficiency that is comparable with Fe(III) (84% E La(III) vs. 90% E Fe(III)). Therefore, La(III) was substituted in all Fe(III) containing systems for mutual miscibility studies.The solubility of pure A336 in ethaline (containing 1.28 g L−1 Zn(II), diluted with 20 wt% water) is 0.07 g L−1. C923 (40 wt% diluted in aliphatic diluent) is according to the obtained 31P NMR spectrum immiscible in undiluted ethaline (containing 2.05 g L−1 La(III) and 1.60 g L−1 Zn(II)) due to the absence of resonance peaks. Since the lowest detected concentration of TBP as internal standard is 0.50 g L−1, the C923 solubility is reported as lower as 0.50 g L−1. The choline chloride solubility is 0.35 g L−1 in C923 (40 wt% diluted in aliphatic diluent) and 1.50 g L−1 in undiluted A336. The ethylene glycol solubility is 56.11 g L−1 in C923 (40 wt% diluted in aliphatic diluent) and 203.94 g L−1 in undiluted A336. These latter values are very high and therefore undesirable from an industrial point of view. The solubility of ethylene glycol in C923 could be reduced by increasing the dilution of the extractant. For example, the solubility of ethylene glycol in 10 wt% C923 in aliphatic diluent was decreased to 19.75 g L−1 and choline chloride was observed to be completely immiscible as concluded from the absence of corresponding resonance peaks (lowest detected concentration 1,2-dichloroethane as internal standard is 0.01 g L−1). Fig. 1 confirms that 10 wt% C923 still ensured ca. 90% Fe(III) extraction, resulting in a more acceptable mutual miscibility and reduced consumption of the relative expensive C923. The solubility of ethylene glycol in A336 can be reduced by the same approach as well. Moreover, the 20 wt% water addition to the Zn(II) containing ethaline ensures acceptable extraction efficiencies at even diluted A336 conditions. For example, the Zn(II) extraction efficiency to 10 wt% A336 in aromatic diluent did not drop significantly (80% to ca. 78%) when ethaline was diluted with 20 wt% water, resulting in an ethylene glycol solubility in 10 wt% A336 (in aromatic diluent) of 14.50 g L−1 and immiscible choline chloride. Moreover, the solubility of 10 wt% A336 (in aromatic diluent) in ethaline (containing 1.28 g L−1 Zn(II), diluted with 20 wt% water) was further reduced from 0.07 g L−1 to 0.01 g L−1. According to the results, the used aliphatic and aromatic solvents were virtually completely immiscible with their corresponding contacted phases. A summary of the mutual miscibilities is given in Table 6. The mutual miscibility is often a major drawback in non-aqueous solvent extraction. Nevertheless, this section proves that the mutual miscibilities can be reduced to some extent by choosing a suitable diluent and by adding water. Furthermore, the water addition also enhances the Zn(II) extraction, improving the efficiency of the process.
| HP | LP | Solubility HP in LP (g L−1) | Solubility LP in HP (g L−1) |
|---|---|---|---|
| a Containing 2.05 g L−1 La(III) and 1.60 g L−1 Zn(II).b Containing 1.28 g L−1 Zn(II) and 20 wt% H2O.c Concentrations are based on the used internal standard concentration. | |||
| ChCl:EGa |
40 wt% C923 in aliphatic diluent | ChCl 0.35, EG 56.11 | C923 < 0.50c, aliphatic diluent <0.01c |
| ChCl:EGb |
Pure A336 | ChCl 1.50, EG 203.94 | A336 0.07 |
| ChCl:EGa |
10 wt% C923 in aliphatic diluent | ChCl < 0.01c, EG 19.75 | C923 < 0.50c, aliphatic diluent <0.01c |
| ChCl:EGb |
10 wt% A336 in aromatic diluent | ChCl < 0.01c, EG 14.50 | A336 0.01, aromatic diluent <0.01c |
Conclusions
C923 (40 wt% diluted in aliphatic diluent) could extract 95% of Fe(III) from an ethaline feed with minor co-extraction of Zn(II), while Pb(II) was not extracted. Subsequently, pre-equilibrated pure A336 could extract 80% of Zn(II) from the remaining ethaline raffinate with no Pb(II) co-extraction. Furthermore, Fe(III) and Zn(II) were stripped from their corresponding loaded organic phases by 1.2 mol L−1 oxalic acid and 0.5 mol L−1 ammonia respectively. For all SX operations, 20 minutes equilibration time at room temperature were the optimum conditions. The Pb(II) remaining in the ethaline, after Fe(III) and Zn(II) extraction, precipitated over time. An alternative and faster approach for Pb(II) recovery can be the addition of zinc metal to produce metallic lead via cementation. Separation and recovery of Fe(III) and Zn(II) were successfully achieved in a counter-current continuous circuit by using mixer-settlers. Only minor adjustments such as a 20 wt% water dilution of the ethaline raffinate, prior to the Zn(II) extraction with pre-equilibrated A336, were required. The mutual miscibility of choline chloride, C923, A336 in their corresponding contacted phases were reasonable, while the used aliphatic and aromatic solvents were not detectable. The miscibility of ethylene glycol in 40 wt% C923 in aliphatic diluent and in undiluted A336 were too high for industrial application. However, further dilution of both HPs and LPs reduced the miscibility of ethylene glycol, while extraction efficiencies were almost unaffected.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project has received funding from the European Union's EU Framework Programme for Research and Innovation Horizon 2020 under grant agreement no. 721385 (SOCRATES). This work reflects only the author's view, exempting the Community from any liability. Website: http://etn-socrates.eu/References
- M. Swain, K. K. Sahu and B. N. Roy, International Journal of Scientific and Technical Research in Engineering, 2016, 1, 36–46 Search PubMed , www.ijstre.com.
- Z. Peng, D. Gregurek, C. Wenzl and J. F. White, JOM, 2016, 68, 2313–2315 CrossRef.
- X. Wang, T. Van Gerven and B. Blanpain, in Element Recovery and Sustainability, ed. A. Hunt, RSC Green Chemistry, Cambridge, 22nd edn, 2013, pp. 29–58 Search PubMed.
- Y. Wang and C. Zhou, Hydrometallurgy, 2002, 63, 225–234 CrossRef CAS.
- G. Bulut, K. T. Perek, A. Gül, F. Arslan and G. Önal, Mining, Metall. Explor., 2007, 24, 13–18 CAS.
- M. Kerolli-Mustafa, L. Ćurković, H. Fajković and S. Rončević, Croat. Chem. Acta, 2015, 88, 189–196 CrossRef CAS.
- A. A. Baba and F. A. Adekola, J. King Saud Univ., Sci., 2013, 25, 297–305 CrossRef.
- S. Riaño, M. Petranikova, B. Onghena, T. Vander Hoogerstraete, D. Banerjee, M. R. S. Foreman, C. Ekberg and K. Binnemans, RSC Adv., 2017, 7, 32100–32113 RSC.
- J. Rydberg, Solvent Extraction Principles and Practice, Revised and Expanded, CRC Press, New York, 2004 Search PubMed.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- J. S. Morell and M. J. Jackson, Uranium Processing and Properties, Springer, New York, 2013 Search PubMed.
- S. I. El Dessouky, Y. A. El-Nadi, I. M. Ahmed, E. A. Saad and J. A. Daoud, Chem. Eng. Process., 2008, 47, 177–183 CrossRef CAS.
- S. Martı and F. Jose, J. Chem. Technol. Biotechnol., 2001, 302, 298–302 Search PubMed.
- B. Wassink, D. Dreisinger and J. Howard, Hydrometallurgy, 2000, 57, 235–252 CrossRef CAS.
- Z. Zhu, P. Yoko and C. Y. Cheng, Hydrometallurgy, 2017, 169, 213–218 CrossRef CAS.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2017, 3, 570–600 CrossRef.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345–386 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- K. Saji John, J. Saji, M. L. P. Reddy, T. R. Ramamohan and T. P. Rao, Hydrometallurgy, 1999, 51, 9–18 CrossRef CAS.
- V. Fischer, Prog. Chem., 2013, 25, 881–892 Search PubMed.
- M. R. S. Foreman, Cogent Chem., 2016, 2, 1–11 Search PubMed.
- A. R. Harifi-Mood and R. Buchner, J. Mol. Liq., 2017, 225, 689–695 CrossRef CAS.
- Shell, Solvents: GTL fluids and solvents, http://www.shell.com/business-customers/chemicals/our-products/solvents-gtl-solvents-and-fluids.html, accessed 1 May 2018 Search PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies and R. Rasheed, Inorg. Chem., 2004, 43, 3447–3452 CrossRef CAS.
- E. L. Smith, Trans. Inst. Met. Finish., 2013, 91, 241–248 CrossRef CAS.
- M. B. Mansur, REM, Rev. Esc. Minas, 2011, 64, 51–55 CrossRef.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- K. Sarangi, P. K. Parhi, E. Padhan, A. K. Palai, K. C. Nathsarma and K. H. Park, Sep. Purif. Technol., 2007, 55, 44–49 CrossRef CAS.
- A. P. Abbott, S. S. M. Alabdullah, A. Y. M. Al-Murshedi and K. S. Ryder, Faraday Discuss., 2018, 206, 365–377 RSC.
- A. Skulcova, A. Russ, M. Jablonsky and J. Sima, BioResources, 2018, 13, 5042–5051 CAS.
- E. Dziwinski and J. Szymanowski, Solvent Extr. Ion Exch., 1998, 16, 1515–1525 CrossRef CAS.
- D. Lloyd, T. Vainikka, M. Ronkainen and K. Kontturi, Electrochim. Acta, 2013, 109, 843–851 CrossRef CAS.
- A. Agrawal, S. Kumari and K. K. Sahu, J. Environ. Manage., 2011, 92, 3105–3111 CrossRef CAS.
- R. K. Mishra, P. C. Rout, K. Sarangi and K. C. Nathsarma, Hydrometallurgy, 2010, 104, 298–303 CrossRef CAS.
- A. Agrawal and K. K. Sahu, Miner. Process. Extr. Metall. Rev., 2010, 31, 121–134 CrossRef CAS.
- K. Larsson, C. Ekberg and A. Degaard-Jensen, Hydrometallurgy, 2012, 129–130, 35–42 CrossRef CAS.
- J. Rydberg, M. Cox, C. Musikas, G. Choppin and M. Dekker, J. Chem. Technol. Biotechnol., 2005, 80, 359–360 CrossRef.
- B. R. Reddy and D. N. Priya, J. Power Sources, 2006, 161, 1428–1434 CrossRef CAS.
- K. Larsson and K. Binnemans, J. Sustain. Metall., 2015, 1, 161–167 CrossRef.
- T. H. Nguyen, L. Wang and M. S. Lee, J. Korean Inst. Met. Mater., 2017, 55, 247–254 CrossRef CAS.
- N. K. Batchu and K. Binnemans, Hydrometallurgy, 2018, 177, 146–151 CrossRef CAS.
- J. Szymanowski, Hydroxyoximes and copper hydrometallurgy, CRC Press, Boca Raton, 1993 Search PubMed.
- F. Endres, D. MacFarlane and A. P. Abbott, Electrodeposition from ionic liquids, Wiley-VCH, Weinheim, 2008 Search PubMed.
- C. W. McDonald and T. Rhodes, Anal. Chem., 1974, 46, 300–301 CrossRef CAS.
- B. Pospiech and A. Chagnes, Sep. Sci. Technol., 2015, 50, 1302–1309 CrossRef CAS.
- M. J. Hartley, PhD thesis, University of Leceister, 2013.
- A. P. Abbott, J. C. Barron, G. Frisch, S. Gurman, K. S. Ryder and A. Fernando Silva, Phys. Chem. Chem. Phys., 2011, 13, 10224–10231 RSC.
- A. P. Abbott, J. C. Barron, G. Frisch, K. S. Ryder and A. F. Silva, Electrochim. Acta, 2011, 56, 5272–5279 CrossRef CAS.
- R. S. Juang, H. C. Kao and W. H. Wu, J. Membr. Sci., 2004, 228, 169–177 CrossRef CAS.
- P. Loyson, Solvent Extr. Ion Exch., 2000, 18, 25–39 CrossRef CAS.
- T. Sato, T. Shimomura, S. Murakami, T. Maeda and T. Nakamura, Hydrometallurgy, 1984, 12, 245–254 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |