
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting the photochemical synthesis of [FeFe]-hydrogenase mimics: reaction optimization, mechanistic study and electrochemical behaviour†
Sergio Aguado a,
Luis Casarrubios*a,
Carmen Ramírez de Arellanob and
Miguel A. Sierra*a
a,
Luis Casarrubios*a,
Carmen Ramírez de Arellanob and
Miguel A. Sierra*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Center for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad Complutense, 28040-Madrid, Spain. E-mail: sierraor@ucm.es; lcasarru@ucm.es
bDepartamento de Química Orgánica, Center for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad de Valencia, 46100-Valencia, Spain
First published on 12th August 2020
Abstract
The photoreaction of [(μ-S)2Fe2(CO)6] and alkenes or alkynes has been optimized to readily obtain functionalized [FeFe]-hydrogenase mimics. Irradiation under low CO pressure in THF produces the corresponding photo-adducts in good/acceptable (alkenes/alkynes) yields, with retention of the starting olefin stereochemistry. DFT-calculations provide plausible reaction pathways in both, singlet and triplet states. The DFT-calculation based in the singlet state is energetically more favorable. The electrochemical behavior of the synthesized compounds is also presented, including studies in acidic media. The electrochemical properties of the products vary in the presence of a double bond (cycloaddition of [(μ-S)2Fe2(CO)6] to alkynes), respect to a single bond (cycloaddition to alkenes).
Introduction
One of the challenges of chemistry in the 21st century is to develop viable methods to produce hydrogen.1 Hydrogenases are metalloenzymes able to generate molecular hydrogen by reducing protons in anaerobic media.2 The use of hydrogenase bio-inspired analogues is a promising option for the production of hydrogen. Current research in this field is mainly focused on two different approaches. The first one uses whole organisms, inorganic hybrids or supported enzymatic systems.3 The second approach uses mimics of hydrogenases, namely synthetic small molecules that when coupled to other reagents and catalysts are able to produce hydrogen.4Fragment I is the basic motif of the hydrogen production moiety of a [FeFe]-hydrogenase (Fig. 1). Much synthetic research in this field targeted the preparation of mimics with a simplified structural motif II similar to I.4b A second group of [FeFe]-H2ase synthetic models has the [(μ-SR)2Fe2(CO)6] motif as the essential core (III in Fig. 1).4a This second group of mimics has been less studied in the photocatalytic production of hydrogen.4c
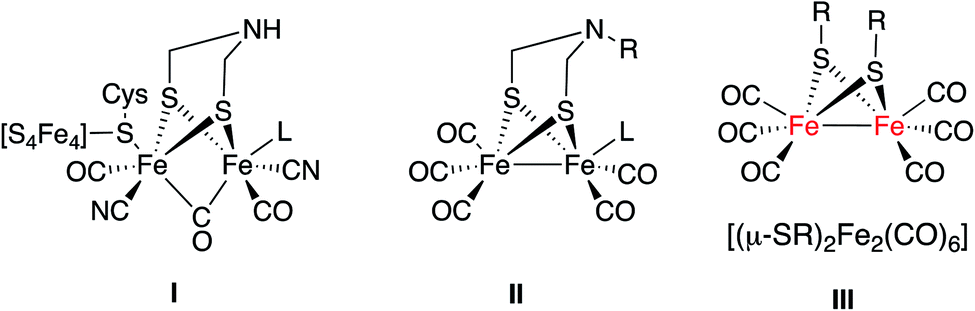 | ||
| Fig. 1 Schematic representation of [FeFe]-H2ase active site (I) and their synthetic mimics (II and III). | ||
The preparation of type III mimics is achieved by thermal reaction of either Fe2(CO)9 or Fe3(CO)12 and sulphides or disulphides to yield compounds having structure 1 (Scheme 1).4b This approach is versatile and provides access to sophisticated structures. However, again the reaction conditions are not tolerated by several classes of substrates. Additionally, the precursors of the sulphides or disulphides are not always easy to access.5
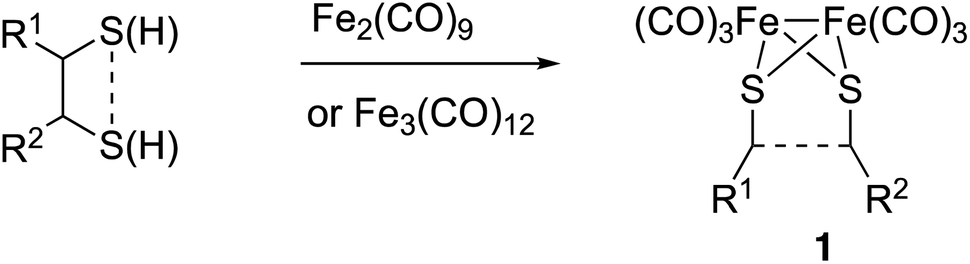 | ||
| Scheme 1 Synthesis of [FeFe]-H2ase synthetic models. | ||
An alternative and potentially useful approach to introduce the [(μ-S)2Fe2(CO)6] 2 moiety into substrates not compatible with the conditions used by standard approaches would be the photocycloaddition of [(μ-S)2Fe2(CO)6] and alkenes or alkynes. The photochemical reaction of 2 and simple unfunctionalized substrates has been previously reported. However, yields of photoreactions are usually very poor. Thus, irradiation of [(μ-S)2Fe2(CO)6] 2 and simple olefins6 including 1- and 2-pentene 3a and 3b yielded the corresponding photoadducts 4a and 4b in 6.9% and 8.9% yield, respectively (Scheme 2).
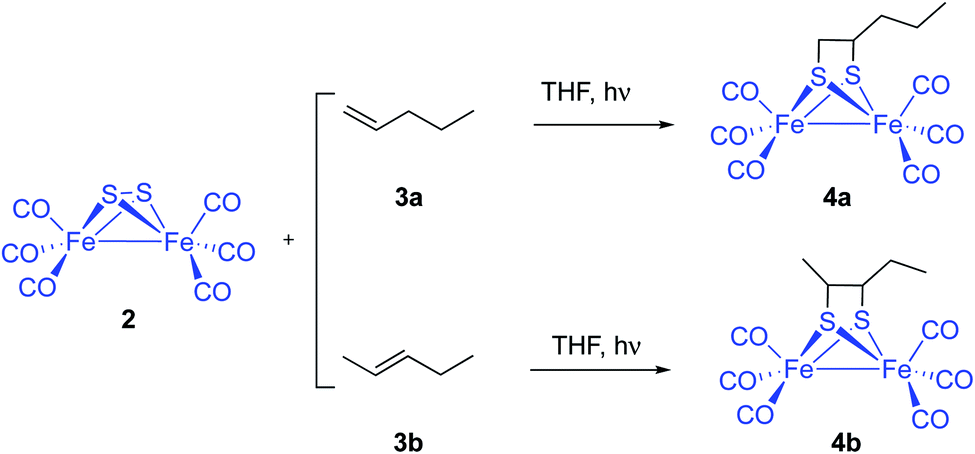 | ||
| Scheme 2 Photocycloaddition of 2 with 1- and 2-pentene 3a and 3b. | ||
Similar low yields were obtained with both, acyclic6 and cyclic7 dienes. The only exceptions are ethylene8 and p-benzoquinone,9 that produce the corresponding photoadducts in 65% and 53% yields, respectively. Finally, several C60[S2Fe2(CO)6]n (n = 1–6) and C70[S2Fe2(CO)6]n (n = 1–4) mixtures were obtained from [(μ-S)2Fe2(CO)6] and C60 and C70 fullerenes. The C60[S2Fe2(CO)6] adduct was separated from the mixture with a 52% yield based on recovered C60, while C70[S2Fe2(CO)6] adduct was obtained with a 63% based on recovered C70.10 The mechanism of these photoreactions remains unexplored. The mechanisms and synthetic applications for organometallic compounds photochemistry are intrinsically different from their all-carbon counterparts, being a subject of general interest.11
Despite the reported low yields for the photocycloaddition of [(μ-S)2Fe2(CO)6] and alkenes/alkynes, this reaction may be a good alternative to include this [FeFe] moiety into substrates incompatible with the reaction condition used by other synthetic approaches to these classes of compounds. We report herein a useful optimized approach to incorporate the [(μ-S)2Fe2(CO)6] into smooth reaction conditions to different classes of substrates, as well as a proposal for the reaction mechanism using DFT calculations.
Results and discussion
Complex [(μ-S)2Fe2(CO)6] 2 and 1-hexene were used to tune up the reaction conditions. Light source and solvent were first investigated. Thus, irradiation of equimolar amounts of [(μ-S)2Fe2(CO)6] and 1-hexene in anhydrous THF using 4 × 60 W blue light LEDs did not produce any reaction product. The reagents were recovered unaltered after 72 hours of irradiation. The use of medium pressure Hg-lamps (Pyrex filter and Pyrex well) produced the desired photoadduct 4c in 31% (400 W) and 47% (125 W) isolated yields. A 6.9% yield for the reaction of 1-pentene and [(μ-S)2Fe2(CO)6], using a high-pressure Hg-lamp and quartz glassware, was previously reported. Thus, filtering the UV component of the irradiation source clearly increases the reaction yield. This yield improvement is probably due to a smaller decomposition of the diiron complexes by the CO-ligands photo-removing effect (see below). Other solvents like MeCN (14%), benzene (18%), and Et2O (11%) produced lower isolated yields of the adduct 4c (Scheme 3).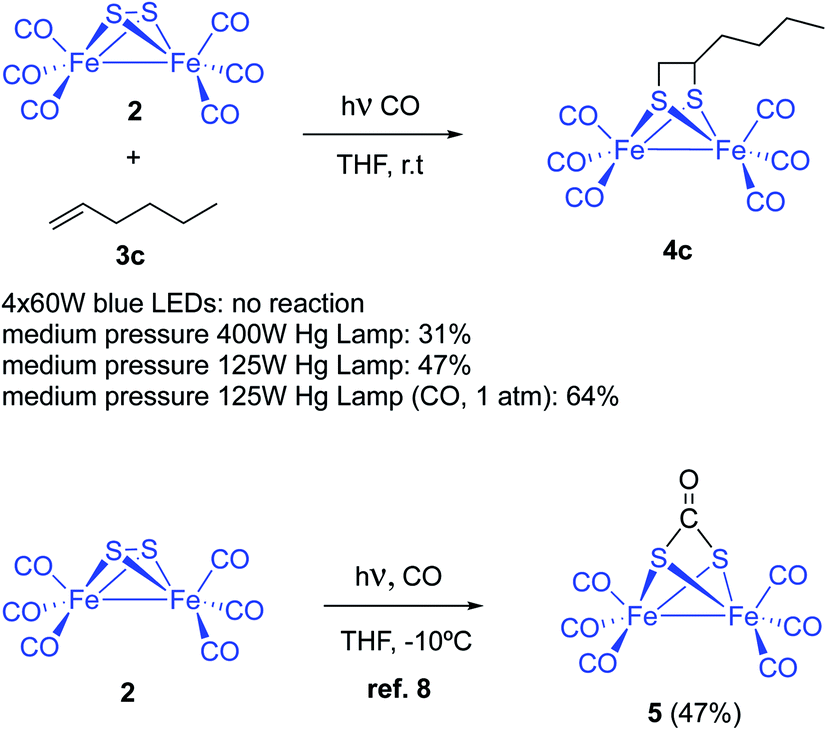 | ||
| Scheme 3 Photochemical reaction between [(μ-S)2Fe2(CO)6] 2 and olefins or alkynes 3. Initial optimizations. | ||
Dependence of yields with the choice of solvent pointed to a competitive light-induced CO dissociation leading to, either decomposition or tetrameric species.12 Thus, THF would fill iron coordination vacants avoiding or retarding competitive undesired reactions. This hypothesis would imply a yield increment under CO-atmosphere. However, it has been reported that complex [(μ-S)2Fe2(CO)6] reacts with CO to form the CO adduct 5 with a 47% yield (Scheme 3).8 Nevertheless, the reaction of [(μ-S)2Fe2(CO)6] 2 and 1-hexene was repeated under 1 atm (14 psi) of CO and, compound 4c was obtained with a 64% isolated yield. The reaction crude material was cleaner and decomposition of the starting diiron complex 2 was not observed. Therefore, it is clear that CO atmosphere hampers the photo-extrusion of CO and thence the decomposition of the [(μ-S)2Fe2(CO)6], increasing the reaction yields. However, the use of higher pressures of CO (40 psi) resulted in lower yields of the desired product. Competitive CO insertion to produce 5 might be the cause of these lower yields.8
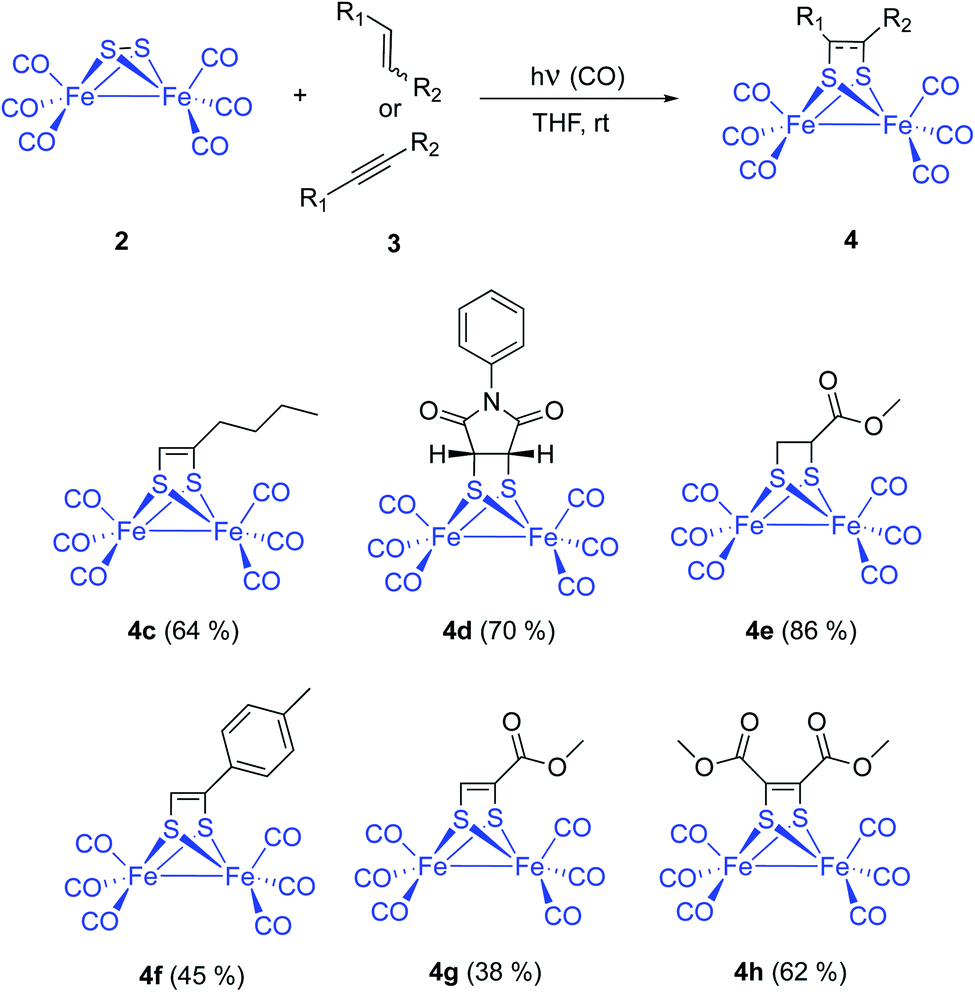 | ||
| Scheme 4 Photochemical reaction of [(μ-S)2Fe2(CO)6] with olefins and alkynes. Substrate scope. | ||
Fine tuning of the reaction conditions of this photocycloaddition allows a yield increment from the described 6–9% up to 65% in the case of simple aliphatic olefins. Functionalized alkenes like N-phenylmaleimide 3d and methyl acrylate 3e were reacted with [(μ-S)2Fe2(CO)6] 2 to form the corresponding photoadducts 4d and 4e with 70% and 86% isolated yields, respectively. These yields were achieved with THF as the choice solvent under 1 atm of CO and a 125 W medium pressure Hg-lamp (Pyrex filter and Pyrex well) (Scheme 4).
Series of both, terminal and disubstituted alkynes were next tested as starting substrates. p-Tolyl acetylene 3f formed the corresponding adduct 4f in 45% yield, while methyl propyolate 3g and dimethyl acetylendicarboxylate 3h yielded the corresponding adducts 4g (38%) and 4h (62%). Although functionalized alkynes formed the corresponding cycloadducts in lower yields than those obtained for alkenes, they could be used as substrates for the cycloaddition process. Therefore, the method is general and tolerates a variety of functional groups (vide infra).13
Photoadducts derived from the reaction of 2 with olefins could be formed either as cis- or trans-isomers in the newly formed metallacycle. The symmetry of our molecules avoids the assignation of the cis–trans stereochemistry by conventional NMR techniques. Crystals of compound 4d suitable for X-ray diffraction were grown from a DCM/hexane solution. The X-ray structure determination of 4d unambiguously confirms the cis arrangement of the fused bicyclic system (Fig. 2). Molecular structure of 4d shows a [(μ-SR)2Fe2(CO)6] complex with a butterfly structure for the [2Fe–2S] cluster. Both iron atoms adopt a distorted square-pyramidal geometry. The Fe–Fe bond length (2.4966(3) Å) lies in the range found for similar ethylenedithiolate-hexacarbonyl-di-iron structures14 (2.454–2.546 Å). Fe–Fe bond length in compound 4d is shorter than in metalloenzymes Hydrogenase DdI (ca. 2.55 Å) or CpI (ca. 2.62 Å).15 The dithiolate bridging ligand and both iron atoms form two fused five-membered metallocycles with the nitrogen substituent N-Ph group bending towards the Fe(1) atom. This conformation implies short intramolecular distances between the nitrogen atom N(1) and the closest carbonyl group C(23)–O(23) [N(1)⋯C(23) 3.221(2) Å; N(1)⋯O(23) 3.609(7) Å]. An intramolecular C–H⋯OC–Fe interaction is observed [C(12)⋯O(23) 3.310(2) Å; H(12)⋯O(23) 2.45 Å; C(12)–H(12)⋯O(23) 150.7°]. This interaction lies within the expected parameters for an intramolecular C–H⋯OC–Fe hydrogen bond with the oxygen of a terminal carbonyl group acting as a hydrogen bond acceptor (i.e. mean values C⋯O 3.50 Å; H⋯O 2.64 Å; C–H⋯O 138.0°).16 Interactions between N-arene group and the closest carbonyl group has been previously described to produce an enlargement on the C–Fe–Fe angle for the implicated carbonyl group in azadithiolates diirion structures.5,17 Thus, in compound 4d the C(23)–Fe(1)–Fe(2) angle is 5.25(5)° larger than the C(26)–Fe(2)–Fe(1) angle.
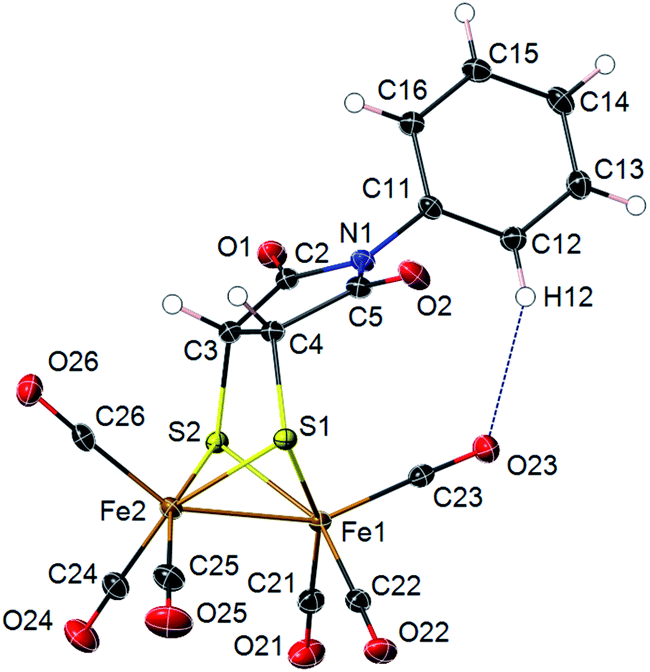 | ||
| Fig. 2 X-ray thermal ellipsoid plot for compound 4d (50% probability level) showing a C–H⋯OC intramolecular interaction. Selected bond lengths (Å) and angles (°): Fe(1)–Fe(2) 2.4966(3), Fe(1)–C(21) 1.7983(15), Fe(1)–C(22) 1.7990(15), Fe(1)–C(23) 1.8102(15), Fe(1)–S(1) 2.2428(4), Fe(1)–S(2) 2.2473(4), Fe(2)–C(24) 1.7995(16), Fe(2)–C(25) 1.8004(16), Fe(2)–C(26) 1.8077(15), Fe(2)–S(1) 2.2521(4), Fe(2)–S(2) 2.2513(4), S(1)–C(4) 1.8289(4), S(2)–C(3) 1.8434(4), C(3)–C(4) 1.5203(18), S(1)–Fe(1)–S(2) 81.488(14), S(1)–Fe(2)–S(2) 81.197(13), Fe(1)–S(1)–Fe(2) 67.478(12), Fe(1)–S(2)–Fe(2) 67.416(12), C(31)–N(1)–C(2) 119.64(10), C(31), C(12)⋯O(23) 3.310(2) Å; H(12)⋯O(23) 2.45 Å; C(12)–H(12)⋯O(23) 150.7°. | ||
Substrates having electroactive moieties were next tested. Methyl trans-2-ferrocenylacrylate 3i reacts with 2 and the corresponding photo-adduct 4i is isolated in 43% yield. The trans stereochemistry of the starting ferrocene derived olefin 3i is again maintained in the final adduct (δ = 4.13 and 3.16 ppm, d, J = 6.3 Hz for both C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –S groups). To confirm that the stereochemistry of the starting material is retained in the photocycloaddition, NOE experiments were performed for complex 4i on a 500 MHz NMR spectrometer. Irradiation of the signal at 3.16 ppm, corresponding to the CH–CO proton, showed a main NOE effect with the proton at 3.98 ppm (substituted Cp ring). This observed NOE effect points to a trans relative disposition of the CO2Me and the Fc moieties which is in good agreement with the concerted proposed calculated mechanism (see below).
–S groups). To confirm that the stereochemistry of the starting material is retained in the photocycloaddition, NOE experiments were performed for complex 4i on a 500 MHz NMR spectrometer. Irradiation of the signal at 3.16 ppm, corresponding to the CH–CO proton, showed a main NOE effect with the proton at 3.98 ppm (substituted Cp ring). This observed NOE effect points to a trans relative disposition of the CO2Me and the Fc moieties which is in good agreement with the concerted proposed calculated mechanism (see below).
Nucleotide 3j was next tested. In this case the product incorporating the [(μ-S)2Fe2(CO)6] moiety was obtained with a 24% isolated yield. Despite the high functionalization of 3j, no by-products were obtained, and unaltered starting materials could be recovered (Scheme 5).
 | ||
| Scheme 5 Photochemical reaction between [(μ-S)2Fe2(CO)6] and olefins or alkynes. Compatibility of the process with functionalized substrates. Compound 4i was a racemic material, one single enantiomer is represented for simplicity. | ||
The possibility of achieving a double photocycloaddition to obtain tetrametallic systems is also addressed. N,N′-(1,4-phenylene)dimaleimide 3k reacts with [(μ-S)2Fe2(CO)6], with no further reaction progress observed (tlc) after 15 hours of irradiation. From the crude reaction mixture, tetrametallic complex 4k was obtained with a 26% isolated yield. An analogous reaction was carried out with ferrocene complex 3l. A mixture of pentametallic complex 4m (single diastereomer, 52%) and trimetallic complex 4l (35%) was obtained. It is worthy to note that the four stereogenic centers of complex 4m are formed in a totally stereoselective way maintaining the configuration of the starting olefins (Scheme 6). A high degree of diastereoselectivity has also been achieved in this reaction. An analogous result was obtained from the bis-allyl derivative of hydroquinone 3o which lead to a mixture of dimetallic 4o and tetrametallic complex 4p in 55% and 20% isolated yields, respectively. Although the analysis of the crude mixtures of 4p showed a single product, there are no reasons to believe that a complete stereoselectivity was achieved in this case. Probably, 4p is a mixture of diastereomers but the chiral centres are well-separated and the differences in their NMR data may be null. Finally, the bis-propargyl derivative of hydroquinone 3n was not able to form the tetrametallic derivative, while bimetallic derivative 4n was obtained with just a 17% yield (Scheme 6). This is in good agreement with the observed lower reactivity of alkynes.
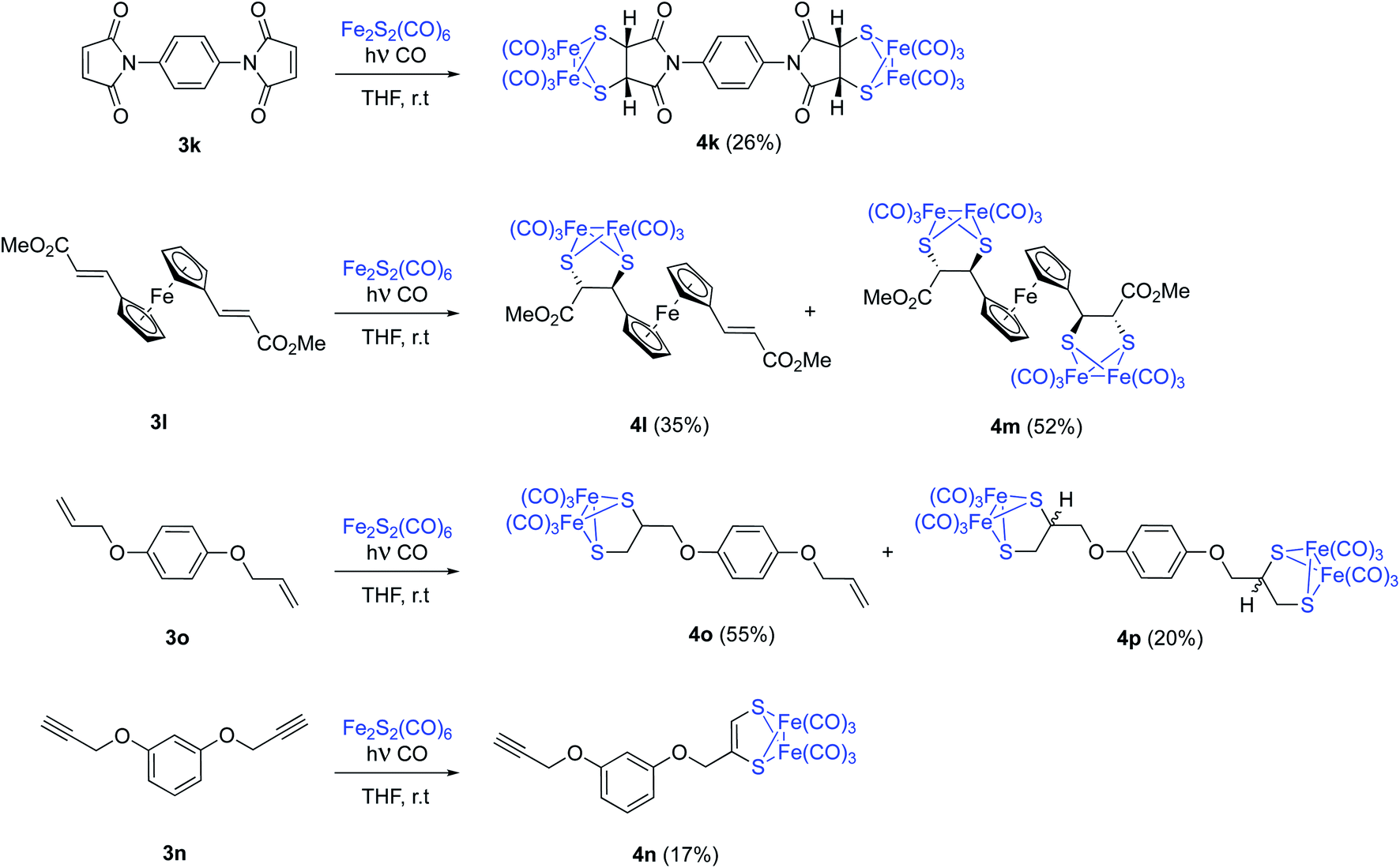 | ||
| Scheme 6 Photochemical reaction between [(μ-S)2Fe2(CO)6] and olefins or alkynes. Synthesis of polymetallic systems. Compounds 4l, 4m were racemic mixtures. Only one enantiomer is depicted for clarity. | ||
Mechanistic studies
According to a previous theoretical study,18 irradiation of the starting diiron complex 2 with UV-light would generate two butterfly isomers or a rhombus isomer by breaking one or both of the Fe–Fe and S–S bonds (see Fig. 4 in ref. 18). This study concludes that photochemical reactions of complex 2 should proceed through Fe–Fe butterfly biradical Fe2(CO)6S2 intermediates (Fig. 3).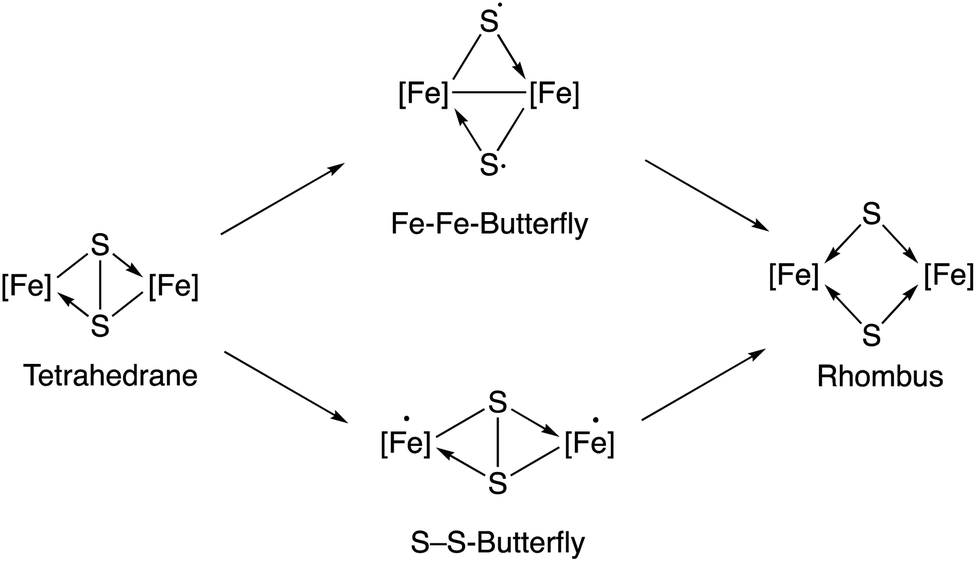 | ||
| Fig. 3 Species implied in the photolysis of complex 2 according to Bruce King et al.18 | ||
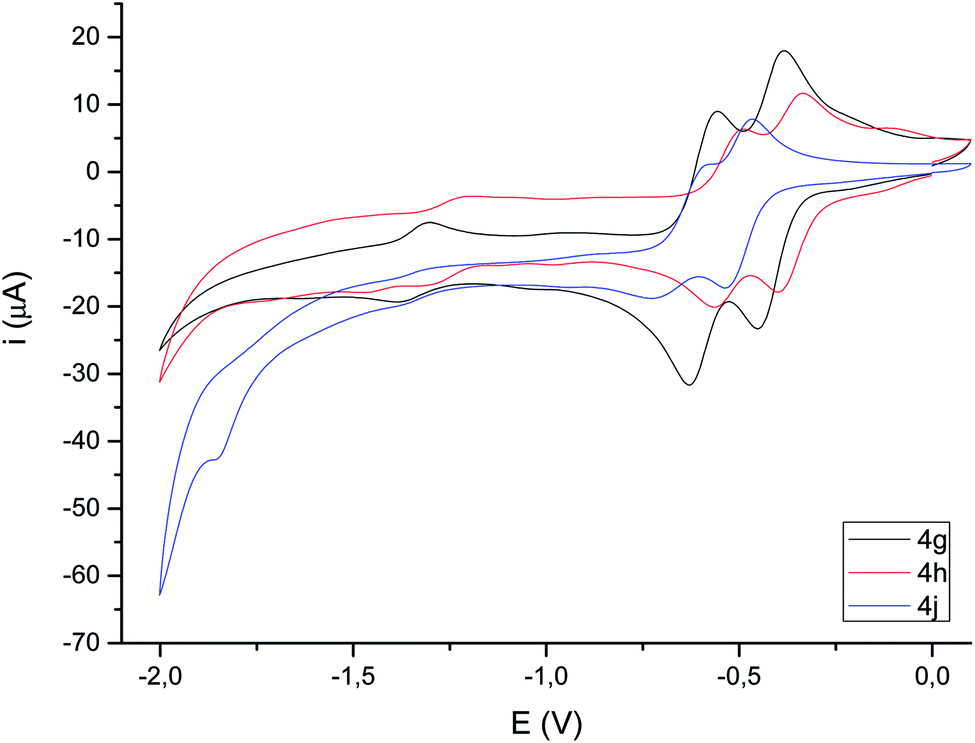 | ||
| Fig. 4 Cyclic voltammograms (focused on reduction) of selected compounds 4c,d (10−3 M in CH3CN), 10−1 M [N(nBu)4]PF6, counter-electrode: Pt; working electrode: glassy carbon; reference electrode: Ag/AgCl; scan rate: 100 mV s−1; values given in V. For full voltammograms see the ESI.† | ||
However, optimization of the Fe–Fe butterfly using unrestricted uBP86 functional together with the command guess(mix, always) or restricted BP86 yielded the same energy minimum. Careful examination of the spin densities and bond distances in the output files did not match a biradical species in any case. Optimization of the reaction pathway was calculated for the cycloaddition between starting complex 2 and both, methyl propiolate 3g (Scheme 7) and methyl acrylate 3e (Scheme 8).
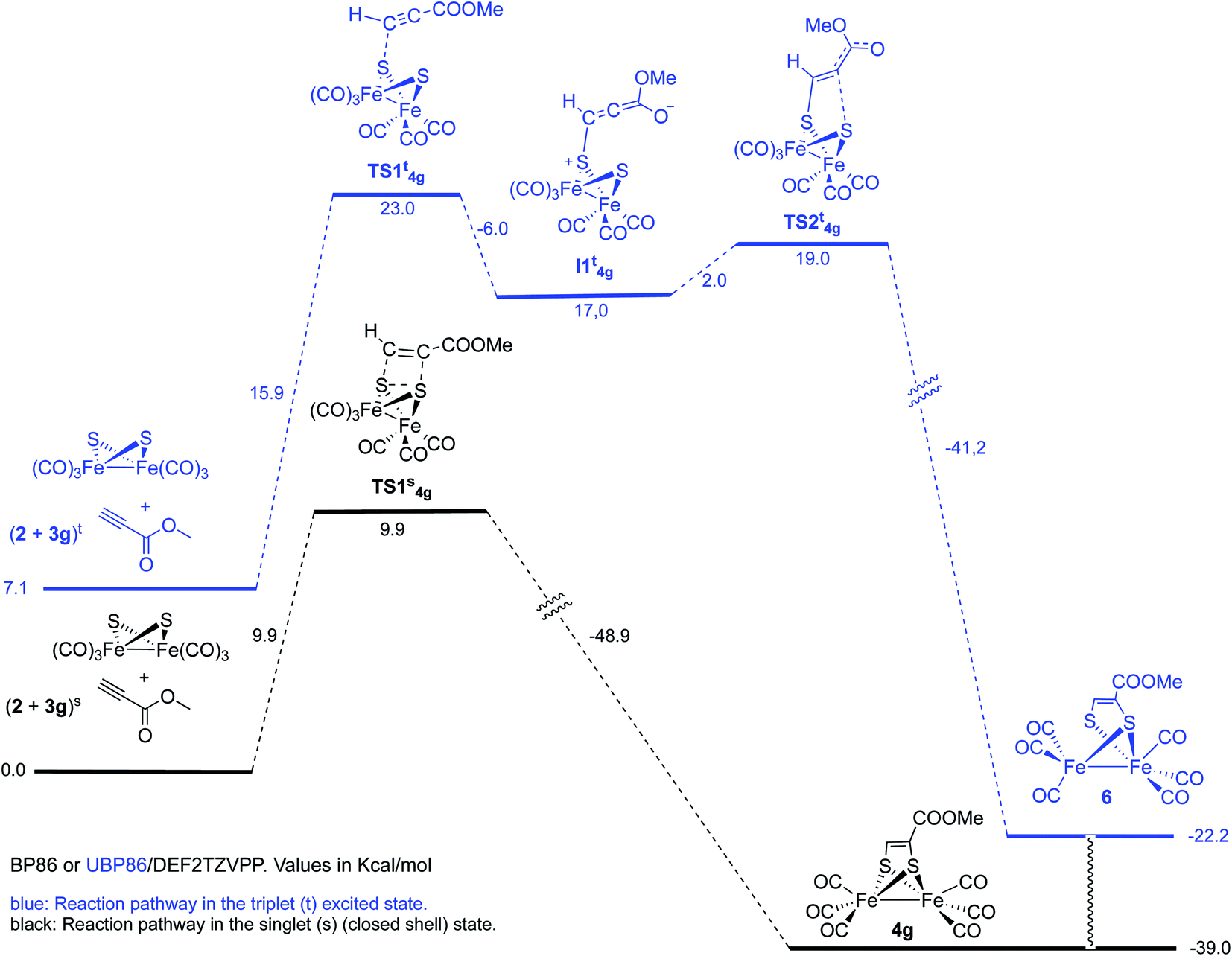 | ||
| Scheme 7 Reaction of complex 2 with methyl propiolate 3g. | ||
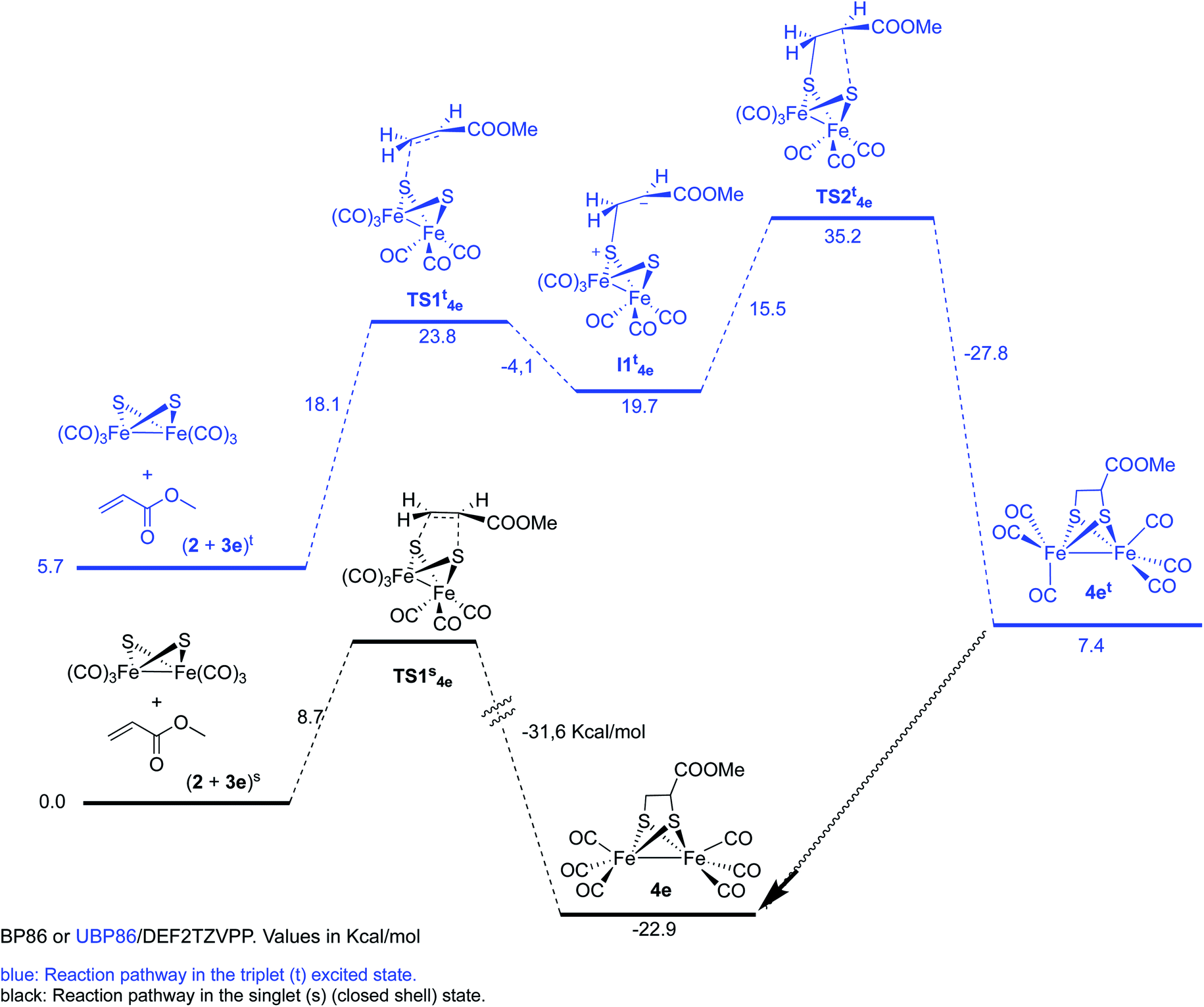 | ||
| Scheme 8 Reaction of complex 2 with methyl acrylate 3e. | ||
We first tested the possibility of reacting methyl propiolate with the FeFe-butterfly intermediate in the singlet state. In order to contemplate the possibility of biradicals implied in the process, broken spin symmetry [UBP86 + guess(mix, always)] was compared to restricted RBP86. Both calculations for the first TS of the pathway converged to the same minimum which should, in principle, discard a triplet diradical reaction pathway. Restricted singlet-state calculations for a concerted cycloaddition reaction are shown in Scheme 7. An alternative pathway involving triplet excited states was also contemplated. This process should involve a stepwise cycloaddition having a ΔΔG‡ = 30.1 kcal mol−1 in the rate-determining step, which makes the process less probable than the concerted pathway (ΔΔG‡ = 9.9 kcal mol−1). Moreover, the calculated final product 6 for this alternative mechanism has a structure different to the experimentally isolated complex 4g. These species, lacking one Fe–S bond were not observed in any of the experiments carried out in this work.19
The reaction of complex 2 and methyl acrylate 3e was also calculated in the singlet and triplet spin states. Results for the singlet state are similar to those obtained for the methyl propiolate. A concerted reaction pathway with a low activation barrier (ΔΔG‡ = 8.7 kcal mol−1) drives the reaction to the formation of the experimentally isolated product 4e. Unrestricted UBP86 singlet state was also tested and again it converged to the same energy minimum obtained with restricted BP86 one. While the triplet initial state of the reagents was found to be only 5.7 kcal mol−1 over the singlet, the two steps process was found to have an overall ΔΔG‡ of 35.2 kcal mol−1 which makes this process unfavorable when compared to the singlet concerted cycloaddition mechanism (Scheme 8). Therefore, we can safely conclude that the photoreaction of [(μ-S)2Fe2(CO)6] with alkenes and alkynes is a concerted process, which additionally accounts for the observed retention of the stereochemistry of the starting olefins into the obtained final products.20
Electrochemistry
Cyclic voltammograms of compounds 4 were recorded in CH3CN solution vs. Ag/AgCl (3 M). The reduction of the compounds clearly depends on the presence of a double bond in the bimetallic cycle. Thus, while alkene-derived products (for example 4c–e) show a quasi-reversible reduction wave at about −1.44 V to −1.61 V, assigned to a one-electron [FeIFeI]/[Fe0FeI] process,21 the analogous alkyne-derived compounds (for example 4f–h) show two quasi-reversible reduction waves about −0.92 V to −1.03 V and −1.08 V to −1.20 V (Fig. 4 and Table 1). Alkyne-derived compounds present a double bond within the metallacycle that is not present for alkene-derived compounds.| Epc1 | Epa1 (ΔE) | E(1) | Epc2 | Epa2 (ΔE) | E(2) | Epc3 | Epa3 (ΔE) | E(3) | |
|---|---|---|---|---|---|---|---|---|---|
| a Data (V) obtained from 10−3 M acetonitrile solutions, containing 0.1 M [N(nBu)4]PF6 as supporting electrolyte at 20 °C. Potentials are relative to Ag/AgCl. | |||||||||
| 4c | −1.15 | −0.96 (0.19) | −1.06 | −1.59 | — | — | — | — | — |
| 4d | −0.93 | −0.84 (0.09) | −0.89 | −1.74 | −1.64 (0.10) | — | — | — | — |
| 4e | −1.05 | −0.89 (0.17) | −0.97 | −1.45 | — | — | — | — | — |
| 4f | −0.70 | −0.60 (0.09) | −0.65 | −1.47 | — | — | — | — | — |
| 4g | −0.46 | −0.39 (0.07) | −0.42 | −0.63 | −0.56 (0.07) | −0.60 | −1.39 | −1.30 (0.09) | −1.34 |
| 4h | −0.40 | −0.34 (0.07) | −0.37 | −0.57 | −0.49 (0.08) | −0.53 | −1.31 | — | — |
| 4i | −1.08 | −0.87 (0.21) | −0.97 | −1.56 | — | — | — | — | — |
| 4j | −0.54 | −0.47 (0.07) | −0.50 | −0.72 | −0.59 (0.13) | −0.65 | −1.86 | — | — |
| 4k | −1.11 | −0.82 (0.29) | −0.96 | −1.71 | — | — | — | — | — |
| 4l | −1.06 | −0.91 (0.15) | −0.99 | −1.83 | — | — | — | — | — |
| 4m | — | — | — | — | — | — | — | — | — |
| 4n | −0.77 | −0.60 (0.18) | −0.68 | −1.62 | — | — | — | — | — |
| 4o | −1.10 | −0.91 (0.18) | −1.00 | −1.60 | — | — | — | — | — |
| 4p | −1.10 | −0.99 (0.12) | −1.04 | −1.50 | — | — | — | — | — |
Compounds derived from alkenes behave like the analogous derivatives having [(μ-SR)2Fe2(CO)6] structures.21 However, compounds derived from alkynes show a strongly displaced anodic wave (even lower than those derivatives of type II in Fig. 1), together with the new reversible wave (see Fig. 5 for comparison). A similar behaviour has been reported for complex 4h.22 DFT calculations (BP86/Def2tzvpp/SCRF, CPCM-MeCN) were performed for further understanding this anodic displacement and the electrochemistry of these complexes. The LUMO in complex 4e is clearly centered in the [FeFe] moiety, while the LUMO of complex 4g having a double bond has a strong component in the organic moiety of the metallacycle (Fig. 6). Therefore, the strong anodic displacement caused by the presence of one double bond in the metallacycle may be explained by the reception of the electron by the organic moiety. Contrary to the complexes having one double bond those complexes having a saturated moiety receipt the electron into the metallic moiety, which accounts for a reduction potential in the −1.44 to −1.61 range. This situation is maintained in the radical-anions 4e˙−, and 4g˙−. The LUMO orbital of 4e˙− is still localized across the [FeFe] fragment while radical anion 4g˙− has the LUMO located in the organic moiety, which is easily reducible (still with strong anodic displacements, giving lower reduction potentials than their saturated congeners, due to the presence of the metals).23 This proposal nicely explain the “strong effect of the dithiolene and (in their case) tetrachloro-biphenyl dithiolate groups on the level of the LUMO” reported by Gloaguen and Schollhammer.22
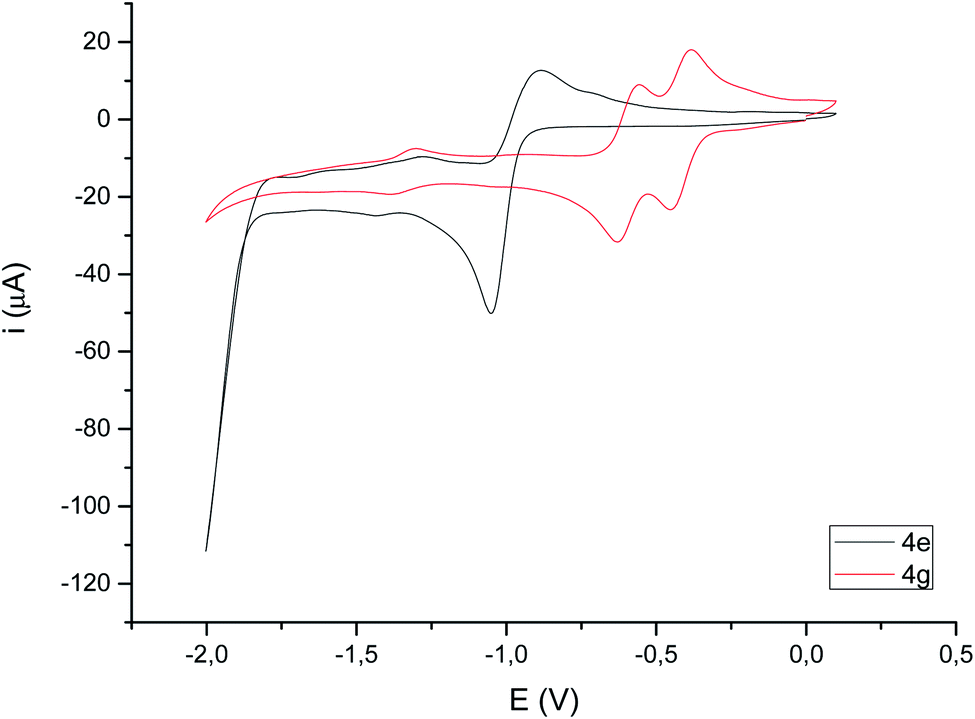 | ||
| Fig. 5 Cyclic voltammograms (focused on reduction) of compounds 4e and 4g (10−3 M in CH3CN), 10−1 M [N(nBu)4]PF6, counter-electrode: Pt; working electrode: glassy carbon; reference electrode: Ag/AgCl; scan rate: 100 mV s−1; values given in V. | ||
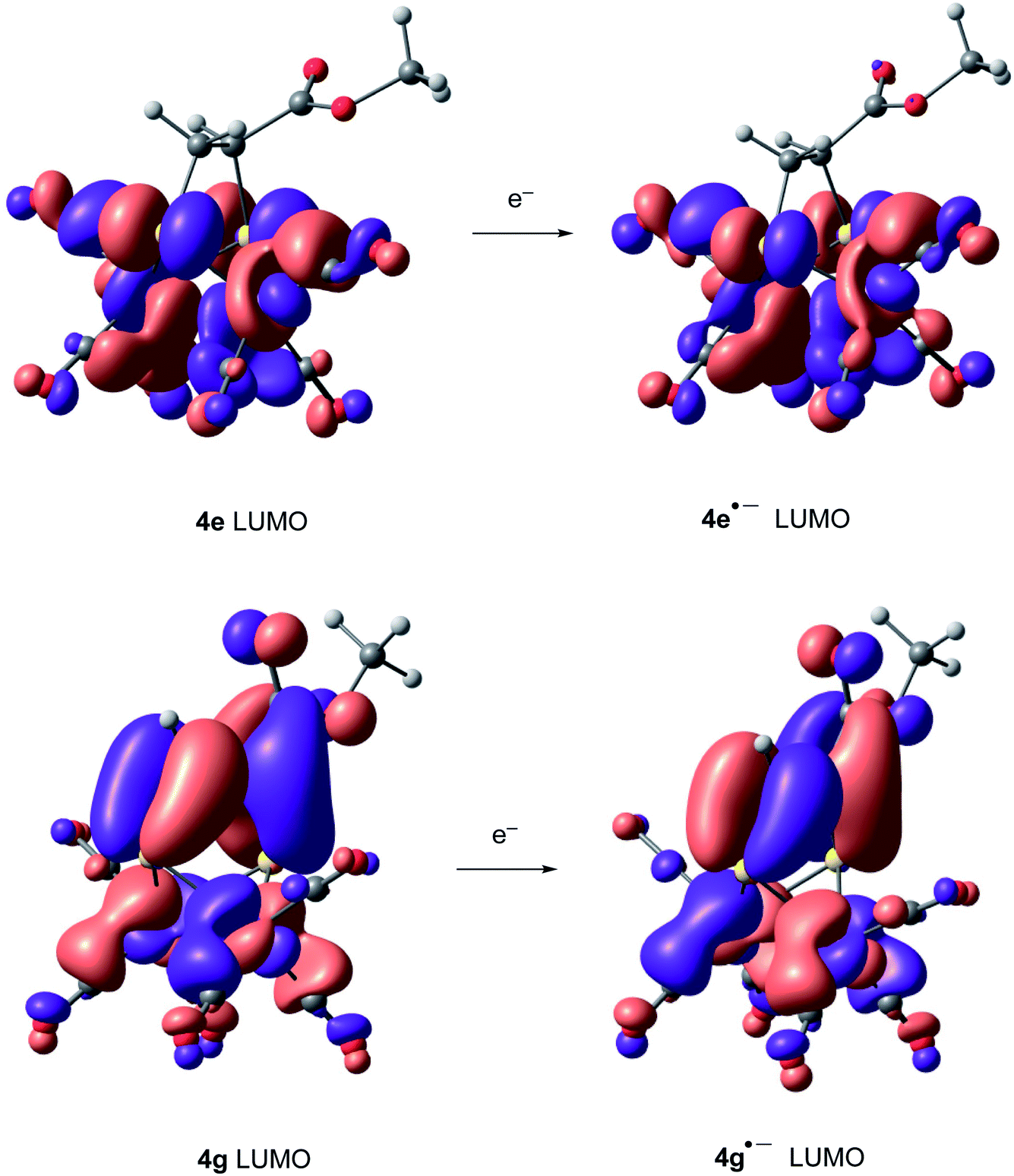 | ||
| Fig. 6 LUMO's of complexes 4e, 4e˙−, 4g and 4g˙−. | ||
The electrochemical behavior in acidic media of complexes 4e and 4g (as representative examples of complexes having either a saturated or double bond in the bridge joining the sulfur atoms) was next studied. None of these complexes showed electrocatalytic behavior in their first reduction wave in the presence of increasing amounts of acetic acid (pKa ∼ 22.3 in MeCN)24 (up to 20 eq., see Fig. 7), while a peak appears around −1.80 V which increases its intensity with the concentration of acid. These results are fully consistent with those reported in the literature for related compounds.25 It should be noted that the first reduction wave at −0.42 V for compound 4g (the one attributed to the reduction of the double bond) remains quasi-reversible, while the second reduction wave at −0.60 V losses its quasi-reversibility in the presence of AcOH as previously reported.22
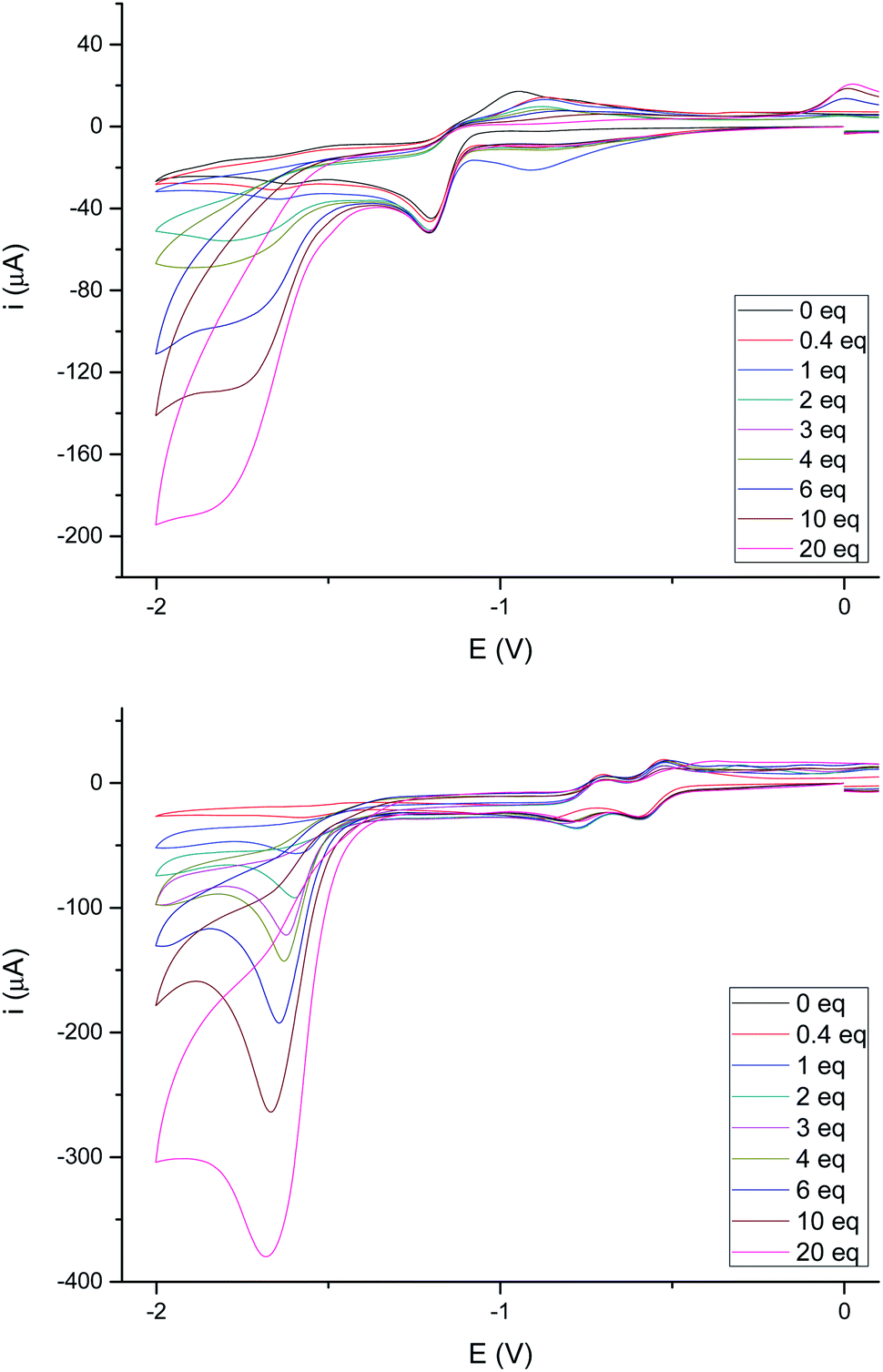 | ||
| Fig. 7 (Top) Cyclic voltammograms of 4e with added HOAc (0–20 eq.). Data (V) obtained from 10−3 M acetonitrile solutions, containing 0.1 M [N(nBu)4]PF6 as supporting electrolyte at 20 °C. Potentials are relative to Ag/AgCl. (Bottom) Cyclic voltammograms of 4g with added HOAc (0–20 eq.). Data (V) obtained from 10−3 M acetonitrile solutions, containing 0.1 M [N(nBu)4]PF6 as supporting electrolyte at 20 °C. Potentials are relative to Ag/AgCl. | ||
The behavior of complexes 4e and 4g towards a stronger acid (CF3COOH, pKa ∼ 12.6 in MeCN)24 was next studied. Fig. 8 shows the behavior of these complexes upon increasing additions of CF3COOH. The reduction of 4e (Fig. 8, top) becomes irreversible upon addition of less than 1 eq. of CF3COOH, as reported in the literature for related compounds.26 However, the intensity of the reduction wave at −0.97 V steadily increases with the concentration of acid and slightly shifts towards more negative values (up to 20 eq. of added CF3COOH). Therefore, the species generated in the electrochemical reduction are able to catalyze the proton reduction.26a In addition, reduction of protons is also observed around −1.60 V, even at low acid concentrations (<0.5 eq.).
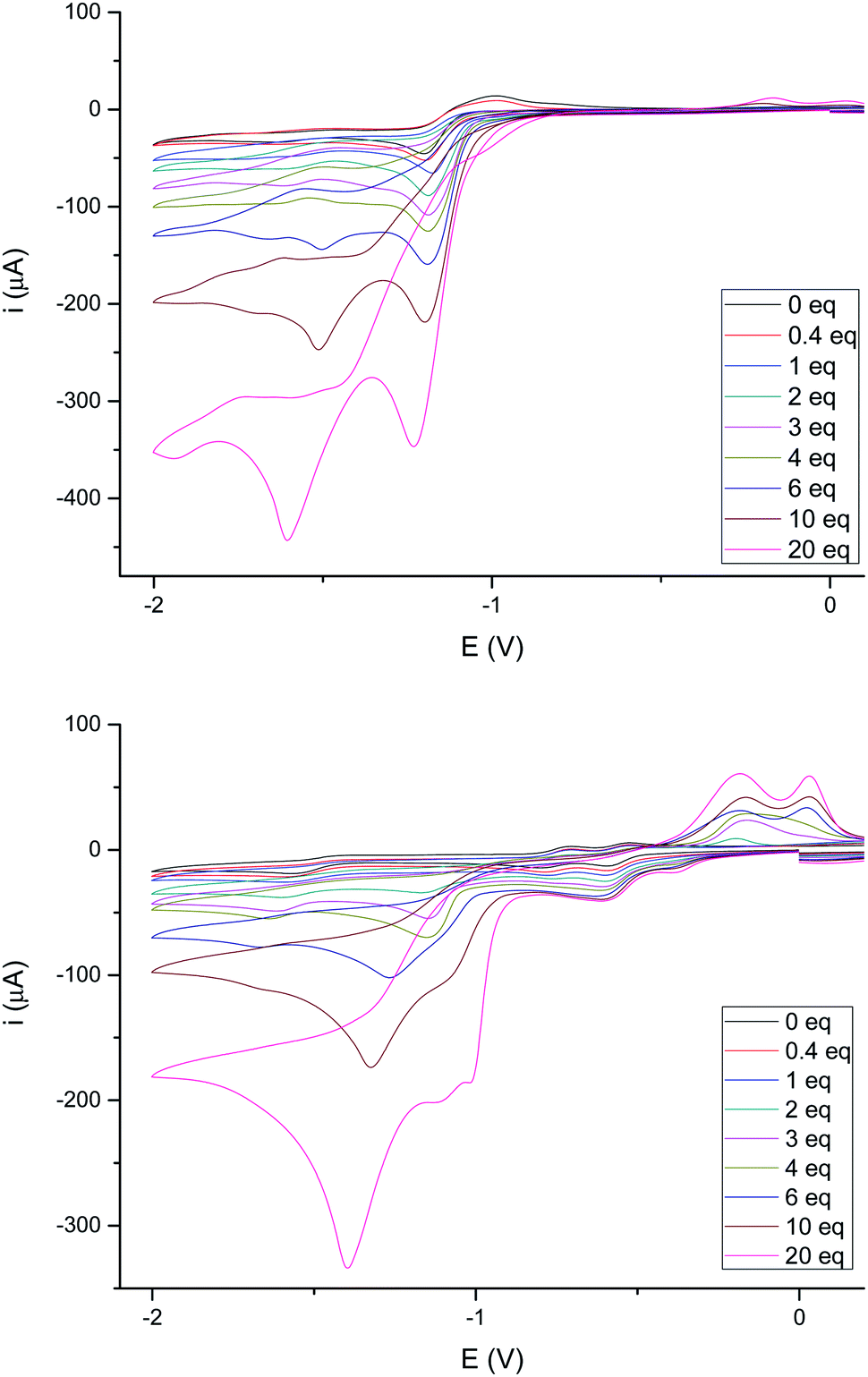 | ||
| Fig. 8 (Top) Cyclic voltammograms of 4e with added TFA (0–20 eq. of H+). Data (V) obtained from 10−3 M acetonitrile solutions, containing 0.1 M [N(nBu)4]PF6 as supporting electrolyte at 20 °C. Potentials are relative to Ag/AgCl. (Bottom) Cyclic voltammograms of 4g with added TFA (0–20 eq. of H+). Data (V) obtained from 10−3 M acetonitrile solutions, containing 0.1 M [N(nBu)4]PF6 as supporting electrolyte at 20 °C. Potentials are relative to Ag/AgCl. | ||
Complex 4g (Fig. 8, bottom) behaves differently. In this case, both waves (−0.42 V and −0.60 V) increase their intensity with the acid concentration until the ratio 4g/acid exceeds 4 equivalents. This supports the participation of the double bond in the first reduction event, generating species that are able to catalyze the reduction of protons.
Conclusions
A smooth and efficient photochemical method to prepare functionalized [FeFe]-hydrogenase mimics has been developed. Irradiation of [(μ-S)2Fe2(CO)6] and alkene/alkynes under medium-low CO pressures produce the corresponding photoadducts in good (alkenes) or acceptable yields (alkynes). The formation of photoadducts derived from alkenes occurs with retention of the stereochemistry of the starting olefin, as demonstrated by NOE measurements and X-ray diffraction. The process is compatible with substrates having ferrocene moieties, as well as functional groups like imides and esters. The photocycloaddition occurs through a concerted reaction pathway as demonstrated by extensive DFT-calculations. The stereochemistry of these reactions is compatible with the computed pathway. Alternative reaction pathways involving triplet states are considerable higher in energy, and, for alkynes, predict the formation of products that have not been detected experimentally.Photoadducts formed from alkynes present a double bond within the metallacycle, that strongly affect the electrochemistry of these compounds. Thus, in the presence of this double bond two strongly anodic displaced quasi-reversible reduction waves appear. These reduction events are compatible with the one electron17 reduction of the double bond “conjugated” with the [FeFe]-moiety. This one electron reduction, forms a radical-anion with a formal [Fe0FeI] state, which facilitate the second reduction to form the [Fe0Fe0] state.
The electrochemistry of complexes 4 in the presence of acids reveals a different behaviour between complexes having a double bond in the dithiametallacycle and those lacking this insaturation. Thus, complexes lacking the insaturation in the metallacycle behave like the analogous products reported in the literature. For this compounds, in the presence of soft acids (AcOH) the species derived from the quasi-reversible reduction wave around −0.97 V are electrocatalytically inactive, and a new electrocatalytically active band appears at −1.80 V. Complexes 4 having a double bond in the metallacycle behave similarly towards soft acids. However, in the presence of strong acids (CF3COOH) the species formed upon reduction in the wave around −0.97 V are able to reduce protons. For these unsaturated complexes, a new reduction wave appears around −1.60 V that is also catalytically active. Therefore, for complexes having a double bond, both waves (−0.42 V and −0.60 V) become catalytically active, showing the participation of dithiolene ligand in its structure.
Further work to apply these smooth methodologies to prepare more sophisticated [FeFe]-mimics, together with post functionalization of the photoadducts, is now underway in our laboratories.
Experimental section
General
Flame-dried glassware was used for moisture-sensitive reactions, and anhydrous solvents were taken from a Pure Solvent PS-MD-5 apparatus. Silica gel (Merck: 230–400 mesh) was used as stationary phase for purification of crude reaction mixtures by flash column chromatography. NMR spectra were recorded at 25 °C in DMSO-d6 or CDCl3 on a 300 and 500 MHz spectrometers. IR spectra were taken on a MIR (8000–400 cm−1) spectrometer using the attenuated total reflectance (ATR) technique. HRMS experiments were recorded on an Agilent 6500 accurate mass apparatus with a Q-TOF analyzer. Cyclic voltammograms were recorded using a Metrohm Autolab Potentiostat model PGSTAT302N with a glassy carbon working electrode, Ag/AgCl 3 M as reference and a Pt wire counter electrode. All the measurements were performed under Ar, at room temperature from CH3CN solutions containing 0.1 M [NnBu4]PF6 as supporting electrolyte, with analyte concentrations of 1 mM (scan rate 0.1 V s−1). When needed, an ultrasound bath was used to promote solubilization in those samples were a suspension was initially obtained.Computational details
Theoretical calculations have been performed using the Gaussian 09-D.01 software package27 at the BP86/Def2tzvpp28 level for all atoms. A SCRF, CPCM29 solvent model for THF was also used. Compounds 4e, 4g and their corresponding radical anions were also calculated using MeCN as solvent in order to match the conditions used in the electrochemical experiments An ultrafine-grid was used as integration grid for all the calculations as implemented in the G09 software suite.General procedure for the synthesis of [FeFe]-hydrogenase mimics
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) yielded pure 4c (159 mg, 64%) as a dark red solid. 1H NMR (300 MHz, CDCl3) δ 0,90 (t, 3H, J = 6.9 Hz, CH3), 1.25–1.57 (m, 6H, 3× CH2), 1.80 (dd, 1H, J = 12.3, 4.8 Hz, CH), 2.57–2.73 (m, 2H, CH2). 13C NMR (75 MHz, CDCl3) δ 208.7, 54.6, 42.2, 36.9, 31.8, 22.6, 14.0. IR (film): ν 2962, 2931, 2863, 2074, 2029, 1976 cm−1. Anal. calcd for C12H12Fe2O6S2: C, 33.67; H, 2.83; S, 14.98. Found C, 33.59; H, 2.91; S, 15.21.:3) yielded pure 4d (211 mg, 70%) as an orange solid. 1H NMR (300 MHz, CDCl3) δ 3.92 (s, 2H, 2× CH), 7.25–7.34 (m, 2H, Ar), 7.38–7.51 (m, 3H, Ar). 13C NMR (75 MHz, CDCl3) δ 206.5, 169.9, 130.7, 129.1, 129.0, 125.7, 54.3. IR (film): ν 2925, 2854, 2083, 2043, 1998, 1717, 1374, 1184 cm−1. ESI-HRMS m/z calcd for C16H7Fe2NNaO8S2 [M + Na]+ 539.82047; found 539.82269.:2) yielded pure 4e (95 mg, 86%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 2.57–2.53 (m, 2H, CH2), 3.28–3.32 (m, 1H, CH), 3.77 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 207.8, 170.2, 53.3, 52.8, 38.8. IR (film): ν 2078, 2034, 1983, 1737 cm−1. Anal. calcd for C10H6Fe2O8S2: C, 27.94; H, 1.41; S, 14.91. Found C, 28.30; H, 1.77; S, 14.85.:2) yielded pure 4f (120 mg, 45%) as a red-orange solid. 1H NMR (300 MHz, CDCl3) δ 2.31 (s, 1H, CH3), 6.35 (s, 1H, CHS), 7.09 (d, 2H, J = 8.2 Hz, Ar), 7.21 (d, 2H, J = 8.2 Hz, Ar). 13C NMR (75 MHz, CDCl3) δ 207.7, 161.8, 140.1, 133.1, 130.7, 129.4, 125.2, 21.5. IR (film): ν 2074, 2029, 1976 cm−1. Anal. calcd for C15H8Fe2O6S2: C, 39.16; H, 1.75; S, 13.94. Found C, 39.04; H, 1.88; S, 13.75.:1) yielded pure 4g (95 mg, 38%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.68 (s, 3H, CH3), 7.37 (s, 1H, CH). 13C NMR (75 MHz, CDCl3) δ 206.2, 160.8, 157.6, 153.1, 52.2. IR (film): ν 2080, 2041, 1994, 1719, 1253 cm−1. ESI-HRMS m/z calcd for C10H4Fe2NaO8S2 [M + Na]+ 450.79390; found 450.79282.:2) yielded pure 4h (176 mg, 62%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.72 (s, 6H, 2× CH3). 13C NMR (75 MHz, CDCl3) δ 206.9, 162.3, 155.5, 53.3. IR (film): ν 2086, 2050, 2008, 1728, 1255 cm−1. ESI-HRMS m/z calcd for C12H7Fe2O10S2 [M + H]+ 486.81744; found 486.81522.:2) yielded pure 4i (155 mg, 43%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.16 (d, 1H, J = 6.3 Hz, CHCO), 3.86 (s, 3H, OCH3), 3.98 (bs, 1H, Cp), 4.08 (s, 5H, Cp), 4.13 (d, 1H, J = 6.3 Hz, CH–Cp), 4.21–4.28 (m, 3H, Cp). 13C NMR (300 MHz, CDCl3) δ 208.0, 170.9, 84.9, 69.5, 69.1, 68.9, 68.7, 65.4, 56.9, 56.0, 53.40. IR (film): ν 2075, 2033, 1980, 1735, 1266 cm−1. ESI-HRMS m/z calcd for C20H15Fe3O8S2 [M + H]+ 614.82522; found 614.82740.:6) yielded pure 4j (59 mg, 24%) as a red solid. 1H NMR (500 MHz, CDCl3) δ 2.07 (s, 3H, CH3), 2.10 (s, 3H, CH3), 2.15 (s, 3H, CH3), 4.34–4.46 (m, 3H, 3× CH–O), 5.63 (t, 1H, J = 5.2 Hz, CH), 5.92 (t, 1H, J = 5.2 Hz, CH), 6.20 (d, 1H, J = 5.2 Hz, CH), 8.15 (s, 1H, Ar), 8.46 (s, 1H,
:2) yielded pure 4c (159 mg, 64%) as a dark red solid. 1H NMR (300 MHz, CDCl3) δ 0,90 (t, 3H, J = 6.9 Hz, CH3), 1.25–1.57 (m, 6H, 3× CH2), 1.80 (dd, 1H, J = 12.3, 4.8 Hz, CH), 2.57–2.73 (m, 2H, CH2). 13C NMR (75 MHz, CDCl3) δ 208.7, 54.6, 42.2, 36.9, 31.8, 22.6, 14.0. IR (film): ν 2962, 2931, 2863, 2074, 2029, 1976 cm−1. Anal. calcd for C12H12Fe2O6S2: C, 33.67; H, 2.83; S, 14.98. Found C, 33.59; H, 2.91; S, 15.21.:3) yielded pure 4d (211 mg, 70%) as an orange solid. 1H NMR (300 MHz, CDCl3) δ 3.92 (s, 2H, 2× CH), 7.25–7.34 (m, 2H, Ar), 7.38–7.51 (m, 3H, Ar). 13C NMR (75 MHz, CDCl3) δ 206.5, 169.9, 130.7, 129.1, 129.0, 125.7, 54.3. IR (film): ν 2925, 2854, 2083, 2043, 1998, 1717, 1374, 1184 cm−1. ESI-HRMS m/z calcd for C16H7Fe2NNaO8S2 [M + Na]+ 539.82047; found 539.82269.:2) yielded pure 4e (95 mg, 86%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 2.57–2.53 (m, 2H, CH2), 3.28–3.32 (m, 1H, CH), 3.77 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 207.8, 170.2, 53.3, 52.8, 38.8. IR (film): ν 2078, 2034, 1983, 1737 cm−1. Anal. calcd for C10H6Fe2O8S2: C, 27.94; H, 1.41; S, 14.91. Found C, 28.30; H, 1.77; S, 14.85.:2) yielded pure 4f (120 mg, 45%) as a red-orange solid. 1H NMR (300 MHz, CDCl3) δ 2.31 (s, 1H, CH3), 6.35 (s, 1H, CHS), 7.09 (d, 2H, J = 8.2 Hz, Ar), 7.21 (d, 2H, J = 8.2 Hz, Ar). 13C NMR (75 MHz, CDCl3) δ 207.7, 161.8, 140.1, 133.1, 130.7, 129.4, 125.2, 21.5. IR (film): ν 2074, 2029, 1976 cm−1. Anal. calcd for C15H8Fe2O6S2: C, 39.16; H, 1.75; S, 13.94. Found C, 39.04; H, 1.88; S, 13.75.:1) yielded pure 4g (95 mg, 38%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.68 (s, 3H, CH3), 7.37 (s, 1H, CH). 13C NMR (75 MHz, CDCl3) δ 206.2, 160.8, 157.6, 153.1, 52.2. IR (film): ν 2080, 2041, 1994, 1719, 1253 cm−1. ESI-HRMS m/z calcd for C10H4Fe2NaO8S2 [M + Na]+ 450.79390; found 450.79282.:2) yielded pure 4h (176 mg, 62%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.72 (s, 6H, 2× CH3). 13C NMR (75 MHz, CDCl3) δ 206.9, 162.3, 155.5, 53.3. IR (film): ν 2086, 2050, 2008, 1728, 1255 cm−1. ESI-HRMS m/z calcd for C12H7Fe2O10S2 [M + H]+ 486.81744; found 486.81522.:2) yielded pure 4i (155 mg, 43%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 3.16 (d, 1H, J = 6.3 Hz, CHCO), 3.86 (s, 3H, OCH3), 3.98 (bs, 1H, Cp), 4.08 (s, 5H, Cp), 4.13 (d, 1H, J = 6.3 Hz, CH–Cp), 4.21–4.28 (m, 3H, Cp). 13C NMR (300 MHz, CDCl3) δ 208.0, 170.9, 84.9, 69.5, 69.1, 68.9, 68.7, 65.4, 56.9, 56.0, 53.40. IR (film): ν 2075, 2033, 1980, 1735, 1266 cm−1. ESI-HRMS m/z calcd for C20H15Fe3O8S2 [M + H]+ 614.82522; found 614.82740.:6) yielded pure 4j (59 mg, 24%) as a red solid. 1H NMR (500 MHz, CDCl3) δ 2.07 (s, 3H, CH3), 2.10 (s, 3H, CH3), 2.15 (s, 3H, CH3), 4.34–4.46 (m, 3H, 3× CH–O), 5.63 (t, 1H, J = 5.2 Hz, CH), 5.92 (t, 1H, J = 5.2 Hz, CH), 6.20 (d, 1H, J = 5.2 Hz, CH), 8.15 (s, 1H, Ar), 8.46 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 8.97 (s, 1H, Ar). 13C NMR (125 MHz, CDCl3) δ 207.4, 170.4, 169.7, 169.5, 157.5, 154,7, 152.6, 150.9, 148.9, 143.3, 130.2, 86.7, 80.6, 73.2, 70.6, 63.0, 20.9, 20.7, 20.5. IR (film): ν 2079, 2042, 2000, 1751, 1577, 1224 cm−1. ESI-HRMS m/z calcd for C24H19Fe2N4O13S2 [M + H]+ 746.90844; found 746.91145.:2) yielded pure 4k (107 mg, 26%) as a reddish solid. 1H NMR (300 MHz, DMSO-d6) δ 4.48 (s, 4H, CH), 7.39 (s, 4H, Ar), 13C NMR (75 MHz, CDCl3) δ 182.0, 170.4, 130.6, 126.4, 54.2. IR (film): ν 2080, 2039, 2008, 1979, 1781, 1708 cm−1. ESI-HRMS m/z calcd for C26H12Fe4N3O16S4 [M + NH4]+ 973.64953; found 973.64842.:3) yielded 153 mg (35%) of pure complex 4l. Pure complex 4m (154 mg, 52%) was also isolated.
CH), 8.97 (s, 1H, Ar). 13C NMR (125 MHz, CDCl3) δ 207.4, 170.4, 169.7, 169.5, 157.5, 154,7, 152.6, 150.9, 148.9, 143.3, 130.2, 86.7, 80.6, 73.2, 70.6, 63.0, 20.9, 20.7, 20.5. IR (film): ν 2079, 2042, 2000, 1751, 1577, 1224 cm−1. ESI-HRMS m/z calcd for C24H19Fe2N4O13S2 [M + H]+ 746.90844; found 746.91145.:2) yielded pure 4k (107 mg, 26%) as a reddish solid. 1H NMR (300 MHz, DMSO-d6) δ 4.48 (s, 4H, CH), 7.39 (s, 4H, Ar), 13C NMR (75 MHz, CDCl3) δ 182.0, 170.4, 130.6, 126.4, 54.2. IR (film): ν 2080, 2039, 2008, 1979, 1781, 1708 cm−1. ESI-HRMS m/z calcd for C26H12Fe4N3O16S4 [M + NH4]+ 973.64953; found 973.64842.:3) yielded 153 mg (35%) of pure complex 4l. Pure complex 4m (154 mg, 52%) was also isolated.
Complex 4l (35%). 1H NMR (300 MHz, CDCl3) δ 3.04 (d, 1H, J = 6.1 Hz, CHCO), 3.77 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.95 (s, 1H, Cp), 4.03 (d, 1H, J = 6.1 Hz, CHS), 4.14–4.49 (m, 7H, Cp), 6.03 (d, 1H, J = 15.8 Hz, CH), 7.49 (d, 1H, J = 15.8 Hz, CH). 13C NMR (300 MHz, CDCl3) δ 207.9, 170.6, 167.4, 144.6, 115.9, 86.5, 79.8, 72.0, 71.9, 71.4, 70.7, 69.7, 69.4, 67.0, 56.5, 55.9, 53.5, 51.6. IR (film): ν 2075, 2034, 1976, 1723(sh), 1701, 1629, 1435, 1264, 1195, 1157 cm−1. ESI-HRMS m/z calcd for C24H19Fe3O10S2 [M + H]+ 698.84638; found 698.84799.
Complex 4m (52%). 1H NMR (300 MHz, CDCl3) δ 3.09 (d, 1H, J = 6 Hz, CHCO), 3.11 (d, 1H, J = 6 Hz, CHCO), 3.86 (s, 3H, OCH3), 3.87–3.91 (m, 2H, Cp), 3.89 (s, 3H, OCH3), 4.03–4.08 (m, 3H, CHS, Cp), 4.12–4.20 (m, 5H, Cp). 13C NMR (300 MHz, CDCl3) δ 208.5, 170.8, 170.8, 86.1, 86.0, 70.7, 70.4, 70.1, 70.1, 69.7, 69.5, 66.5, 65.6, 56.2, 56.1, 55.8, 53.6, 53.5. IR (film): ν 2075, 2034, 1976, 1701, 1629, 1434, 1306, 1263, 1194 cm−1. ESI-HRMS m/z calcd for C30H18Fe5NaO16S4 [M + Na]+ 1064.61211; found 1064.60748.
:1) yielded pure 4n (40 mg, 17%) as a red solid. 1H NMR (300 MHz, CDCl3) δ 2.53 (t, 1H, J = 2.4 Hz, ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 4.21 (d, 2H, J = 1.9 Hz, OCH2), 4.66 (d, 2H, J = 2.4 Hz, C–CH2O), 6.23 (t, 1H, J = 1.9 Hz, CH), 6.37–6.41 (m, 2H, Ar), 6.59–6.62 (m, 1H, Ar), 7.18 (dd, 1H, J = 9.2, 7.4 Hz, Ar). 13C NMR (75 MHz, CDCl3) δ 207.5, 158.9, 158.7, 158.6, 158.3, 138.8, 130.2, 108.1, 107.6, 102.5, 75.8, 66.6, 56.0. IR (film): ν 2074, 2030, 1970, 1886, 1502, 1208 cm−1. Anal. calcd for C18H10Fe2O8S2: C, 40.79; H, 1.90; S, 12.10. Found, C, 40.43; H, 2.14; S, 12.54.:1) yielded 77 mg (55%) of pure 4o and 46 mg (20%) of pure 4p as red solids.
CH), 4.21 (d, 2H, J = 1.9 Hz, OCH2), 4.66 (d, 2H, J = 2.4 Hz, C–CH2O), 6.23 (t, 1H, J = 1.9 Hz, CH), 6.37–6.41 (m, 2H, Ar), 6.59–6.62 (m, 1H, Ar), 7.18 (dd, 1H, J = 9.2, 7.4 Hz, Ar). 13C NMR (75 MHz, CDCl3) δ 207.5, 158.9, 158.7, 158.6, 158.3, 138.8, 130.2, 108.1, 107.6, 102.5, 75.8, 66.6, 56.0. IR (film): ν 2074, 2030, 1970, 1886, 1502, 1208 cm−1. Anal. calcd for C18H10Fe2O8S2: C, 40.79; H, 1.90; S, 12.10. Found, C, 40.43; H, 2.14; S, 12.54.:1) yielded 77 mg (55%) of pure 4o and 46 mg (20%) of pure 4p as red solids.
Complex 4o (55%). 1H NMR (300 MHz, CDCl3) δ 2.10 (dd, 1H, J = 13.2, 5.5 Hz, CH2S), 2.75 (dd, 1H, J = 13.2, 7.4 Hz, C
2S), 2.98–3.06 (m, 1H, CHS), 3.79 (dd, 1H, J = 9.7, 7.0 Hz, SCHC2), 3.97 (dd, 1H, J = 9.7, 5.8 Hz, SCHC2), 4.49 (bd, 2H, J = 5.4 Hz, C2CH), 5.27 (d, 1H, J = 10.5 Hz, C2), 5.39 (d, 1H, J = 16.1 Hz, C2), 6.04 (ddt, 1H, J = 16.1, 10.5, 5.4 Hz, CH), 6.74–6.91 (m, 4H, Ar). 13C NMR (75 MHz, CDCl3) δ 208.4, 153.5, 152.3, 133.50, 117.8, 115.9, 115.7, 70.8, 69.6, 52.7, 39.1. IR (film): ν 2074, 2029, 1969, 1885, 1502, 1208 cm−1. Anal. calcd for C18H14Fe2O8S2: C, 40.48; H, 2.64; S, 12.00. Found, C, 40.31; H, 2.74; S, 12.20.
Complex 4p (20%). 1H NMR (300 MHz, CDCl3) δ 2.10 (dd, 2H, J = 13.1, 5.6 Hz, C
2S), 2.75 (dd, 2H, J = 13.1, 7.5 Hz, C2S), 2.97–3.06 (m, 2H, CHS), 3.79 (dd, 2H, J = 9.7, 7.0 Hz, OC2), 3.96 (dd, 2H, J = 9.7, 5.6 Hz, OCH2), 6.81 (s, 4H, Ar). 13C NMR (75 MHz, CDCl3) δ 208.3, 152.9, 115.8, 70.7, 52.6, 39.1. IR (film): ν 2075, 2032, 1985, 1506, 1223 cm−1. Anal. calcd for C24H14Fe4O14S4: C, 32.83; H, 1.61; S, 14.61. Found, C, 33.11; H, 1.97; S, 15.10.
Crystal data for compound 4d
C16H7Fe2NO8S2, M = 517.05, monoclinic, a = 13.23734(18), b = 7.41493(10), c = 18.8284(2) Å, β = 92.6332(12)°, V = 1846.13 Å3, space group P2(1)/c, Z = 4, T = 120(2) K, λ = 0.71073 Å, Dcalcd = 1.860 g cm−3, μ = 1.844 cm−1, 42161 reflections measured, 6112 unique (Rint = 0.0427), dark red tablets obtained by CH2Cl2/n-hexane diffusion, crystal structure solved by dual-space methods with all non-hydrogen atoms refined anisotropically on F2 using the programs SHELXT30 and SHELXL-2018,31 hydrogen atoms were included using a riding model, GOF = 1.028, R (Fo, I > 2σ(I)) = 0.0270, Rw (Fo2, all data) = 0.0610.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Support for this work under grants CTQ2016-77555-C2-1-R and CTQ2016-81797-REDC (Programa Redes Consolider) from the AEI (Spain) is gratefully acknowledged. MAS thanks the Fundación Ramón Areces for a grant from the XVIII Concurso Nacional de Ayudas a la Investigación en Ciencias de la Vida y de la Materia (CIVP18A3938).References
- (a) Hydrogen as a Fuel: Learning from Nature, ed. R. Cammack, M. Frey and R. Robson, Taylor&Francis, 2001 Search PubMed; (b) Compendium of Hydrogen Energy Volume 1: Hydrogen Production and Purification, ed. V. Subramani, A. Basile and T. N. Veziroğlu, Elsevier, 2015 Search PubMed; (c) I. Dincer and C. Zamfirescu, Sustainable Hydrogen Production, Elsevier, 2016 Search PubMed; (d) Compendium of Hydrogen Energy Volume 4: Hydrogen Use, Safety and the Hydrogen Economy, ed. M. Ball, A. Basile and T. N. Veziroğlu, Elsevier, 2016 Search PubMed; (e) J. Corredor, M. J. Rivero, C. M. Rangel, F. Gloaguen and I. Ortiz, J. Chem. Technol. Biotechnol., 2019, 94, 3049–3063 CrossRef CAS.
- (a) W. Lubitz, H. Ogata, O. Rüdiger and W. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed; (b) P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- See, for example: (a) E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed; (b) K. A. S. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed; (c) K. A. Brown, B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed; (d) Y. Honda, H. Hagiwara, S. Ida and T. Ishihara, Angew. Chem. Int. Ed., 2016, 55, 8045–8048 (Angew. Chem., 2016, 128, 8177–8180) CrossRef CAS PubMed.
- Revisions: (a) L. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef PubMed; (b) Y. Li and T. B. Rauchfuss, Chem. Rev., 2016, 116, 7043–7077 CrossRef CAS PubMed; (c) F. Gloaguen, Inorg. Chem., 2016, 55, 390–398 CrossRef CAS PubMedRepresentative examples: (d) H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed; (e) Y. Na, J. Pan, M. Wang and L. Sun, Inorg. Chem., 2007, 46, 3813–3815 CrossRef CAS PubMed; (f) S. Gao, J. Fan, S. Sun, X. Peng, X. Zhao and J. Hou, Dalton Trans., 2008, 2128–2135 RSC; (g) P. Li, M. Wang, L. Chen, J. Liu, Z. Zhao and L. Sun, Dalton Trans., 2009, 1919–1926 RSC; (h) U.-P. Apfel, D. Troegel, Y. Halpin, S. Tschierlei, U. Uhlemann, H. Görls, M. Schmitt, J. Popp, P. Dunne, M. Venkatesan, M. Coey, M. Rudolph, J. G. Vos, R. Tacke and W. Weigand, Inorg. Chem., 2010, 49, 10117–10132 CrossRef CAS PubMed; (i) J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed; (j) D. Zheng, M. Wang, L. Chen, N. Wang and L. Sun, Inorg. Chem., 2014, 53, 1555–1561 CrossRef CAS PubMed; (k) T. Yu, Y. Zeng, J. Chen, Y.-Y. Li, G. Yang and Y. Li, Angew. Chem. Int. Ed., 2013, 52, 5631–5635 (Angew. Chem., 2013, 125, 5741–5745) CrossRef CAS PubMed.
- An alternative route to incorporate moieties II and III in substrates incompatible with the standard synthetic approaches to these compounds is the use of Fe2[(μ-SCH2)2(NC6H4N3)](CO)6 reagents through a Cu-catalyzed alkyne-azide cycloaddition. See, A. D. Merinero, A. Collado, L. Casarrubios, M. Gómez-Gallego, C. Ramírez de Arellano, A. Caballero, F. Zapata and M. A. Sierra, Inorg. Chem., 2019, 58, 16267–16278 CrossRef CAS PubMed.
- A. Kramer and I.-P. Lorenz, J. Organomet. Chem., 1990, 388, 187–193 CrossRef CAS.
- A. Kramer, R. Lingnau, I.-P. Lorenz and H. A. Mayer, Chem. Ber., 1990, 123, 1821–1826 CrossRef CAS.
- J. Messelhäuser, K. U. Gutensohn, I.-P. Lorenz and W. Hiller, J. Organomet. Chem., 1987, 371, 377–388 CrossRef.
- R. D. Adams and S. Miao, Inorg. Chem., 2004, 43, 8414–8426 CrossRef CAS PubMed.
- M. D. Westmeyer, T. B. Rauchfuss and A. K. Verma, Inorg. Chem., 1996, 35, 7140–7147 CrossRef CAS PubMed.
- Selected examples from these laboratorios (a) I. Fernández, F. P. Cossío and M. A. Sierra, Acc. Chem. Res., 2011, 44, 479–490 CrossRef PubMed; (b) I. Fernández and M. A. Sierra, Top. Heterocycl. Chem., 2013, 30, 65–84 Search PubMed; (c) A. Santiago, M. A. Gómez-Gallego, C. Ramírez de Arellano and M. A. Sierra, Chem. Commun., 2013, 49, 1112–1115 RSC; (d) G. M. Chu, I. Fernández and M. A. Sierra, Chem.–Eur. J., 2013, 19, 5899–908 CrossRef CAS PubMed; (e) J. G. Muntaner, L. Casarrubios and M. A. Sierra, Org. Biomol. Chem., 2014, 11, 286–297 RSC; (f) A. R. Rivero, I. Fernández and M. A. Sierra, Chem.–Eur. J., 2014, 20, 1359–1366 CrossRef CAS PubMed; (g) G. M. Chu, A. Guerrero-Martínez, I. Fernández and M. A. Sierra, Chem.–Eur. J., 2014, 20, 1367–1375 CrossRef CAS PubMed; (h) B. Eguillor, M. A. Esteruelas, I. Fernández, M. Gómez-Gallego, M. Martín-Ortiz, M. Oliván and M. A. Sierra, Organometallics, 2015, 34, 1898–1910 CrossRef CAS; (i) G. M. Chu, I. Fernández, A. Guerrero-Martínez, C. Ramírez de Arellano and M. A. Sierra, Eur. J. Inorg. Chem., 2016, 844–852 CrossRef CAS; (j) G. M. Chu, I. Fernández, A. Guerrero-Martínez, C. Ramírez de Arellano and M. A. Sierra, Inorg. Chem., 2016, 55, 2737–2747 CrossRef CAS PubMed.
- L. E. Bogan, D. A. Lesch and T. B. Rauchfuss, J. Organomet. Chem., 1983, 250, 429–438 CrossRef CAS.
- Good yields for the synthesis of alkyne adducts similar to 4 have been reported by the reaction of the lithium salts of terminal alkynes with [(μ-S)2Fe2(CO)] and subsequent addition of acid in: (a) D. Seyferth, G. G. Womack and L. C. Song, Organometallics, 1983, 2, 776–779 CrossRef CAS; (b) D. Seyferth and G. B. Womack, Organometallics, 1986, 5, 2360–2370 CrossRef CAS.
- 91 structures found in CSD-5.4 October 2019; C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., 2016, B72, 171–191 Search PubMed.
- (a) Y. Nicolet, A. L. Lacey, X. Vernéde, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed; (b) J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- D. Braga, F. Grepioni, K. Biradha, V. R. Pedireddi and G. R. Desiraju, J. Am. Chem. Soc., 1995, 117, 3156–3166 CrossRef CAS.
- (a) T. Liu, M. Wang, Z. Shi, H. Cui, W. Dong, J. Chen, B. Åkermark and L. Sun, Chem.–Eur. J., 2004, 10, 4474–4479 CrossRef CAS PubMed; (b) S. Gao, J. Fan, S. Sun, X. Peng, X. Zhao and J. Hou, Dalton Trans., 2008, 10, 2128–2135 RSC.
- I. Silaghi-Dumitrescu, T. E. Bitterwolf and R. B. King, J. Am. Chem. Soc., 2006, 128, 5342–5343 CrossRef CAS PubMed.
- We can hypothesize that decomposition of these products may be responsible for the lower yields experimentally observed when using alkynes instead of olefins as starting materials. However, decomposition was never observed under our reaction conditions and unreacted alkyne was recovered in all cases.
- The triplet state stepwise mechanism in Scheme 8 occurs through a zwitterionic intermediate that may rotate leading to partial or complete loss of the stereochemistry of the starting olefin. The torquoselectivity of reactions involving these species has been profusely studied. For examples from these laboratories, see: F. P. Cossío, A. Arrieta and M. A. Sierra, Acc. Chem. Res., 2008, 41, 925–936 CrossRef PubMed.
- G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS PubMed.
- The electrochemistry of complex 4h was reported previously: K. Charreteur, M. Kdider, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Inorg. Chem., 2010, 49, 2496–2501 CrossRef CAS PubMed . These authors reported that this complex was unstable in MeCN/NnBu4PF6 and electrochemical data were not given in this solvent. In our hands complex 4h was stable and electrochemistry could be measured without incidences. The reduction potentials measured in dichloromethane parallels our findings in MeCN (see text).
- For a similar behavior, see: (a) M. G. Avello, M. C. de la Torre, M. A. Sierra, H. Gornitzka and C. Hemmert, Chem.–Eur. J., 2019, 25, 13344–13353 CrossRef CAS PubMed; (b) S.-H. Wu, J.-J. Shen, J. Yao and Y.-W. Zhong, Chem.–Asian J., 2013, 8, 138–147 CrossRef CAS PubMed , and the pertinent references therein.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS PubMed.
- (a) J. F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2004, 566, 241–247 CrossRef CAS; (b) J. F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2006, 595, 47–52 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision, D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A Gen. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (c) F. Weigend, Phys. Chem. Chem. Phys., 2008, 8, 1057–1065 RSC.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1988, 102, 1995–2001 CrossRef; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXT– Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The graphics for the electrochemical measurements of complexes 4, the cartesian coordinates of species 4e, 4e˙−, 4g and 4g˙− as well as a copy of the IR and NMR spectra for all the new compounds prepared through this work. CCDC 1960548 contains the supplementary crystallographic data for compound 4d. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06002j |
| This journal is © The Royal Society of Chemistry 2020 |