
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cellulose hydrolysis using ionic liquids and inorganic acids under dilute conditions: morphological comparison of nanocellulose†‡
Jacobs H. Jordan ,
Michael W. Easson* and
Brian D. Condon
,
Michael W. Easson* and
Brian D. Condon
USDA, Agricultural Research Service, Southern Regional Research Center, 1100 Robert E. Lee Blvd, New Orleans, LA 70124, USA. E-mail: michael.easson@usda.gov
First published on 28th October 2020
Abstract
The preparation of cellulose nanocrystals (CNCs) from cellulose extracted from cotton gin motes (CGM) using an ionic liquid (1-butyl-3-methylimidazolium chloride, [BMIm]Cl) under dilute conditions is reported. The concurrent process involves minimal swelling of cellulose with an ionic liquid and hydrolysis of the cellulose initiated by the addition of either phosphoric (H3PO4), hydrochloric (HCl), or sulfuric (H2SO4) acid. The obtained nanocrystals had similar physical properties (e.g. crystallinity) to the counterparts prepared under conventional conditions and exhibited superior thermal properties for sulfate CNCs. Additionally, the obtained CNCs had low surface functionalization, yet were colloidally stable for >90 days, which is a desirable trait for post-functionalization of CNCs. This process represents a general strategy utilizing dilute ionic liquids in the preparation of nanocellulose under mildly acidic conditions.
Introduction
Ionic liquids (ILs) are regarded as a “green solvent” and possess favourable properties; they have a low melting point (<100 °C), are recoverable, and are readily tailored to specific applications.1,2 ILs have increasingly been applied in processing lignocellulose biomass.3–6 For example, an IL (1-butyl-3-methylimidizolium chloride, [BMIm]Cl) was used to dissolve cellulose with the aid of a solid acid support.5 In another example, Xia et al.7 screened a number of ionic liquids and deep eutectic solvents (DES) for suspension pre-treatment of cellulose biomass. The rate of enzymatic hydrolysis was affected using neat, 1.0, or 2.0 M ILs, and aqueous ILs (1.0 or 2.0 M) were found to be an effective suspension pre-treatment.The advent of IL use in biomass processing has more recently increased with the use of acidic ionic liquids.8 Thus, ILs have also been utilized for the hydrolysis of cellulose to produce cellulose nanocrystals (CNCs) and nanofibers (CNFs) from various lignocellulose sources.9 This is in contrast to traditional methods to prepare CNCs from the hydrolysis of cellulose using strong minerals acids: hydrochloric acid (HCl), sulfuric acid (H2SO4), and phosphoric acid (H3PO4).10–13 Mao et al. described the use of mildly acidic ionic liquids for the production of CNCs; they compared the case of the neat protic ionic liquid (PIL) obtained from BMIm and hydrogen sulfate ([BMIm]HSO4) and dilute IL premixed with H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14.3, mol mol−1).14 Yields were considerably improved with the use of the dilute IL [BMIm]HSO4 (ca. 48 vs. 90%, respectively). In contrast, Yang et al. used neat ILs as a pretreatment for the dissolution and regeneration of cellulose prior to H2SO4 hydrolysis or TEMPO oxidation; the resulting CNCs were obtained in low yield and had very low (<25%) crystallinity.15
:14.3, mol mol−1).14 Yields were considerably improved with the use of the dilute IL [BMIm]HSO4 (ca. 48 vs. 90%, respectively). In contrast, Yang et al. used neat ILs as a pretreatment for the dissolution and regeneration of cellulose prior to H2SO4 hydrolysis or TEMPO oxidation; the resulting CNCs were obtained in low yield and had very low (<25%) crystallinity.15
Lazko et al. prepared thermally stable CNCs via a two-step swelling/hydrolysis method using the neat ionic liquid [BMIm]Cl for 1 h follow by hydrolysis for 2, 5 or 16 h upon the addition of H2SO4 (4, 2 or 1 wt%, respectively).16 This method was improved with a Brønsted acid IL 1-(4-sulfobutyl)-3-methylimidazolium hydrogen sulfate ([SBMIm]HSO4) that was effectively used for the production of CNCs. Firstly, cellulose fibres were swollen in anhydrous [BMIm]Cl and then hydrolysed by the subsequent addition of [SBMIm]HSO4 at rather low concentrations (<4%).17 A more recent alternative used a combination of swelling in [BMIm]Cl (45 °C, 2 h) followed by 2-fold addition of oxalic acid (70 wt%) to initiate hydrolysis; optimum conditions were 7 h and 90 °C.18
In another two-step procedure,19 cellulose from bleached hard and softwood pulps and microcrystalline cellulose (MCC) from cotton linters were swollen in neat [BMIm]HSO4 for 24 h and then diluted with water to catalyse the formation of CNCs; reaction conditions were 12 h, ≥100 °C, and H2O:IL ratio of 1:4. This resulted in CNCs with low sulfur content and high crystallinity (77–82%).19 Treatment of microcrystalline cellulose (MCC) from cotton linters with neat [BMIm]HSO4 for 1.5 at T ≥ 90 °C similarly resulted in highly crystalline (>90%) CNCs.20
Iskak et al. investigated the effect of hydrolysis temperature and time on CNC production using neat [BMIm]Cl;21 they showed increasing temperature and time resulted in more crystalline CNCs with higher CNC yield and improved aspect ratio. In another instance, an ultrasonic pre-treatment in combination with an IL was employed to allow extraction of CNCs using milder acid hydrolysis (20–23 wt% H2SO4); the sulfuric acid was added directly to the suspension in a 2-step, tandem, 1-pot process.22 CNCs with improved thermal stability and re-dispersibility were prepared by using the ionic liquid [BMIm]BF4 by a simple rotary evaporator procedure.23 The PIL [BMIm]HSO4 was used to prepare CNCs (80 °C, 3 h); esterified CNCs with long chain fatty acids from methyl laurate were also prepared using a binary mixture of [BMIm]HSO4 and [BMIm]BF4 in conjunction with a lipase via a lipase-catalysed transesterification.24
In addition to the preparation of nanocellulose, ILs have been used as a solvent and reagent for preparation of functionalized CNCs. Tetrabutylammonium acetate (TBAA) was shown to dissolve cellulose in the presence of dimethylsulfoxide (DMSO) or crown ethers without pretreatment;25 subsequently TBAA used in combination with DMSO or N,N-dimethylacetamide (DMA) and acetic anhydride to produce hydrophobic, acetylated CNCs in a one-pot process, which were added as a support in a PLA matrix.26 Partially acetylated CNCs were prepared from successive rounds of pulping/processing of extractive free wood with the IL 1-ethyl-3-methylimidazolium acetate [EMIm]OAc at 60 °C for 2 h.27
Different extraction protocols lead to differing properties such as length, crystallinity, aspect ratio, and amount and type of surface functional groups. For example, hydrolysis with HCl gave hydroxyl CNCs that were selected as a toughening agent in epoxy adhesives28 and a two-step oxidation/reduction strategy afforded sterically stabilized neutral CNC suspensions.29 In contrast, the use of [BMIm]Cl and oxalic acid promoted the formation of CNCs with the carboxylate moiety; the CNCs were characterized with high crystallinity (80%) and both longer (>300 nm) and wider (∼37 nm) than H2SO4 hydrolysed CNCs.18 The production of nanocellulose with acid blends resulted in different morphologies and allowed fine-tuning of the number of surface charge groups and zeta (ζ) potential.11 Sulfate-modified CNCs were used to stabilised oil-in-water Pickering emulsions, and it was found that the amount of CNCs required for stabilisation was reduced for CNC suspension with a lowered ζ-potential (i.e. with a lower surface charge density),30 and similarly, sulfate CNCs were modified with an amino terminated polystyrene to render amphiphilic CNCs for enhanced emulsion stabilisation.31 CNCs derived from cotton had a greater aspect ratio compared to wood CNCs, which was found to have a beneficial impact on the toughening effect of polyvinyl alcohol composites.32
Other examples have sought to reduce the costs associated with the use of the (usually recoverable) IL. For example, a custom PIL was prepared by neutralization of ethanolamine and sulfuric acid and was used to prepare CNCs; however, the CNCs were characterized with low ζ potential and crystallinity (−19.1 mV and 54%, respectively).33 Additionally, neat imidazole or water-imidazole mixtures (25% w/w) were used to prepare CNFs or CNCs, respectively, after heating at 120 °C for 24 h.34
In this work and inspired by previously reported results, a strategy employing the IL [BMIm]Cl, coupled with three different inorganic acids under dilute conditions was investigated for the hydrolysis of cellulose to produce CNCs. As a benchmark comparison, CNCs were prepared by mineral acid hydrolysis under conventional conditions and the physicochemical properties compared to those obtained using dilute acids and ILs. The results are presented and discussed herein.
Experimental
Materials
:20 (w/v). The recovered cellulose fibres were washed thoroughly with water until a colourless eluant was obtained and then oven-dried (70 °C) to a constant mass.Nanocrystal preparation
:20 (w/v). The resulting suspension was quenched by dilution four-fold with ice water and then washed until pH ≥ 5 by successive centrifugation cycles at 16000 × g for 15 min each cycle. The crystals (sCNC) were then dispersed using a 750 W ultrasonic processor (Vibra-Cell probe sonicator, VCX-750, Sonics & Materials) with a power amplitude of 60% (20 kHz frequency) for 5 min. The obtained suspension was centrifuged at 3500 × g for 5 min (to sediment larger particulates and contaminates), filtered under vacuum with a Whatman glass microfiber filter (grade GF/F, 0.7 μm) and dialyzed using a regenerated cellulose dialysis tubing (MW cut-off 10000) for several days until the solution conductivity stabilized for two successive bath changes (measured value < 2 μS cm−1). Hydrolysis with HCl (hCNC) and H3PO4 (pCNC) were similarly conducted using 12% and 74% (w/w) HCl or H3PO4 at 100 °C for 120 and 90 min, respectively, using 75 or 125 mL of the acid solution per gram of cellulose. Work-up and processing conditions were as described for H2SO4 hydrolysis. The suspensions were subsequently stored in a sealed plastic container and refrigerated (4–8 °C) between uses. The yield and concentration of the nanocrystals was determined gravimetrically.:33. The mixture (13.3 g) was added to the extracted cellulose (0.75 g) and stirred for 10 min. After the cellulose fibres were fully suspended the inorganic acid was added for a molar ratio of 1:1:33. (IL:acid:H2O) and the suspension heated to 75 °C for 20 h (or 90 °C, 44 h in the case of H3PO4). Each hydrolysis was then quenched by dilution to a total volume of ∼40 mL with iced water and then centrifuged 12000 × g for 15 min. The IL-containing supernatant was decanted, and the CNC-containing precipitate was repeatedly washed until a turbid supernatant was obtained. Between each wash, ultrasonication was used to re-suspend the precipitate. The obtained suspensions of CNCs from IL-mediated hydrolysis using HCl (hCNCi), H2SO4 (sCNCi), and H3PO4 (pCNCi), collectively denoted IL-CNCs, were dialyzed and stored at 4 °C. The yield and concentration of the nanocrystals was determined gravimetrically.Characterization
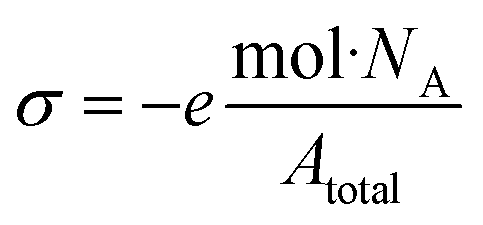 | (1) |
 | (2) |
 | (3) |
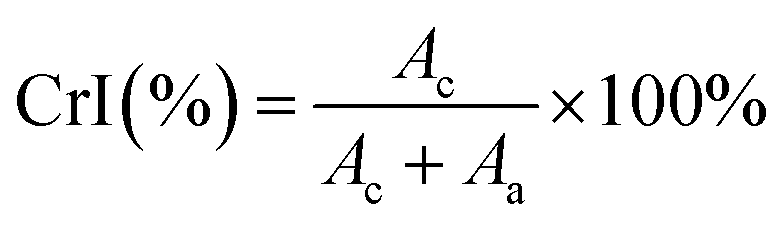 | (4) |
The Scherrer equation (eqn (5)) was used to calculate (a measure) of the crystallite size, L (nm), perpendicular to the (hkl) plane:48
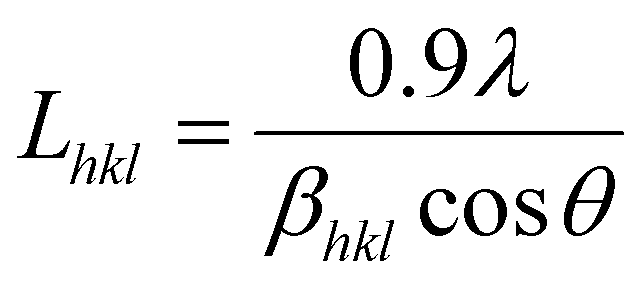 | (5) |
Results and discussion
Extraction of cellulose
Celluloses were extracted using subsequent alkali and bleaching treatments from cotton gin motes (CGM). The starting cotton gin motes were previously shown to contain roughly 67% celluloses and the remaining amount of the material is comprised of lignin (13.5%), and extractables (9.6%), as well as hemicelluloses and other sugar polymers (7.5%).13 Following chemical processing the extracted cellulose powder contained >90% cellulose with small amounts of hemicellulose and lignin (Fig. 1). This represents roughly a 50% increase in the amount of cellulose available for conversion to various CNCs, complete removal of the extractables, and a substantial reduction in the amount of hemicellulose (∼43%) and lignin (∼75%) present in the source material. | ||
| Fig. 1 Composition of extracted celluloses used to prepare CNCs. | ||
Preparation of cellulose nanocrystals
The results of cellulose hydrolysis indicate that roughly one-third of the cellulose was hydrolysed into its constituent sugars during the IL-dilute acid treatment. Additionally, the combination of IL and the inorganic acids generated nanocellulose. Under dilute conditions the combination rendered both fully hydrolysed monosaccharides and incomplete hydrolysis, leading to microcrystalline cellulose. However, these components are readily separated by centrifugation. This is comparable to the results obtained using strong mineral acids. Subsequent work-up to remove the non-hydrolysed cellulosic solid residues (CSR) from the CNC fractions gave slightly lower yields of the obtained CNC suspension in all cases (Table 1). However, these yields are on par with those reported in the literature,10–12 and CSR may be subsequently converted to CNFs.4,49,50 It is apposite to note that many reported conventional work-ups indicated a total yield of isolated non-hydrolysed cellulose without adequately separating CSR from the CNCs, resulting in CNC suspensions that still contain contaminates of MCC and thus leading to substantial opacity at relatively lower concentrations (<1 wt%).| Entry | Sample | Time (h) | Temp (°C) | Acid ratio (equiv. per unitb) | Sol. sugarsc (%) | Yieldd (%) | CSRe (%) |
|---|---|---|---|---|---|---|---|
| a CNCs were prepared with either HCl (hCNC), H2SO4 (sCNC), or H3PO4 (pCNC) as the acid source, while the “i” suffix (e.g. hCNCi, sCNCi, or pCNCi) indicates use of the ionic liquid (1-butyl-3-methylimidizolium chloride) during the preparation.b Per anhydroglucose unit of cellulose.c Soluble sugars total weight were determined from the difference in the mass of total cellulosic residues before and after hydrolysis prior to separation of the CNCs and non-hydrolysable cellulosic solid residues (CSR).d CNC yields were determined gravimetrically after separation of the CSR from the suspension.e CSR yields were determined from the total isolated mass of residual cellulosic solids after exhaustive centrifugation and sonication. | |||||||
| 1 | hCNC | 1.5 | 100 | 48 | 10 | 63 | 27 |
| 2 | sCNC | 1 | 60 | 29 | 44 | 55 | <1 |
| 3 | pCNC | 2 | 100 | 230 | 22 | 67 | 11 |
| 4 | hCNCi | 20 | 75 | 3.5 | 38 | 53 | 9 |
| 5 | sCNCi | 20 | 75 | 3.5 | 34 | 40 | 26 |
| 6 | pCNCi | 44 | 90 | 3.5 | 36 | 43 | 21 |
Hydrolysis with the IL and HCl for 20 h at 75 °C gave CNCs in modest yield (53%) and low residual CSR (9%). Identical conditions were less effective for H3PO4 hydrolysis giving a low 12% yield and substantial amount of CSR (57%). This was not surprising given that typically hydrolysis using concentrated (74%) H3PO4 requires significantly harsher reaction conditions than those that were employed using IL-mediated hydrolysis. The reaction time for H3PO4 hydrolysis was thus increased in the presence of the IL to 44 h and the reaction temperature to 90 °C, which greatly improved the yield (43%) and reduced the CSR to 21% during hydrolysis, with a concomitant increase in the surface functionalisation. For comparison, traditional hydrolysis conditions using 4 M HCl (∼12% w/w) or 11.55 M H3PO4 (74% w/w) were conducted using approximately 48 or 230 equiv. of the acid per anhydroglucose unit of cellulose, whilst hydrolysis using the more dilute acids employed with the IL used only 3.5 equiv. of the acid. This corresponds to a reduction of 93% and 98% in the amount of acid used on a stoichiometric basis. Additionally, milder temperatures (75–90 °C) were employed compared to 100 °C for traditional hydrolysis conditions.
As a possible explanation, Xia et al.7 showed that suspension pretreatment of cellulose with 1.0 M BMIm was more effective when “cellulose dissolving” counterions such as acetate (AcO−), and chloride (Cl−) were used. These anions are strongly solvated (ΔGhdr = −373 and −347 kJ mol−1, respectively)51 suggesting solvation of the anion enhances the efficacy of the IL counterion. For the example containing H2SO4, when H2SO4 is added, the first ionisation (pKa = −3) gives HSO4− (ΔGhdr = −335 kJ mol−1). Collins law of matching water affinities52,53 suggests a greater propensity for HSO4− to form a contact or solvent separated ion pair with BMIm, thereby reducing its effectiveness compared to HCl and Cl−. In the case of H2PO4− (ΔGhdr = −473 kJ mol−1), incomplete ionisation (first pKa = 2.16) suggests despite the greater solvation of H2PO4− there is less available acid to initiate glycosidic bond cleavage, and hence harsher conditions were required.
Overall conditions were thus less improved for H2SO4 hydrolysis of cellulose. Literature yields for H2SO4 hydrolysis typically vary between 20–70% for hydrolysis of soft or hardwood pulps and cellulose from cotton depending on the method of isolation. Under the employed conditions, the IL-mediated hydrolysis gave a slightly lower yield and a significant amount of CSR compared to concentrated acid hydrolysis, providing comparable amounts of nanocellulose, monosaccharides and MCC.16 It is worth noting, the amount of acid was reduced eight-fold, albeit at the expense of longer reaction time and elevated temperature. Thus, while each treatment successfully obtained CNCs, the use of IL-mediated hydrolysis with HCl and H3PO4 proved more economical (in terms of acid consumption and waste products) than isolation of CNCs using concentrated acids. The use of H2SO4 under similar conditions proved a less effective alternative, in agreement with similar work.16
Atomic force microscopy
Nanoparticle dimensions and aspect ratio affect macroscopic properties in end-use applications such as composite fibers,32 reinforcements,28,54 fillers,55 stabilisers,56 and film assembly.57,58 To characterize the nanocrystal dimensions, AFM (Fig. 2) was used to analyse the nanoparticle length, width, and aspect ratio (Table 2). The nanoparticle width was determined with the aid of the section analysis toolkit provided with the AFM software, and ImageJ was used for analysis of the nanoparticle length (ESI: Fig. S2 and S3‡).36 The standard conditions using strong mineral acids promoted a narrower size-distribution and smaller observed crystalline sizes with the majority having lengths of 100–400 nm and widths of 5–15 nm. Specifically, sCNC were obtained with the shortest length, width, and smallest aspect ratio, while differences in the dimensions of hCNC and pCNC were not considered significant. | ||
| Fig. 2 Representative 4 × 4 AFM images from: (a) hCNC; (b) sCNC; (c) pCNC; (d) hCNCi; (e) sCNCi, and; (f) pCNCi. | ||
| Entry | Sample | Lengtha (nm) | Heightb (nm) | Aspect ratioc |
|---|---|---|---|---|
| a Determined using section analysis software on the AFM instrument.b Determined using ImageJ software.c Determined from the ratio of the mean sample length to the mean sample height. | ||||
| 1 | hCNC | 218 ± 69 | 10.4 ± 3.8 | 21.0 |
| 2 | sCNC | 102 ± 51 | 7.2 ± 2.2 | 14.2 |
| 3 | pCNC | 216 ± 77 | 7.5 ± 3.5 | 28.8 |
| 4 | hCNCi | 223 ± 60 | 10.3 ± 3.7 | 21.7 |
| 5 | sCNCi | 198 ± 85 | 9.7 ± 3.7 | 20.4 |
| 6 | pCNCi | 322 ± 149 | 11.4 ± 3.0 | 28.2 |
In contrast, the IL-hydrolysis conditions gave slightly longer nanocrystals in all cases, typically 200–300 nm with some individual crystals up to 800 nm. There was no significant difference observed among IL-CNCs, although the dimensions of sCNCi were comparably longer than sCNC. Generally, the AFM height, or width of the IL-CNCs was marginally larger (approx. 10 nm). In the case of both pCNC and pCNCi a significantly greater aspect ratio was observed (∼28). However, aspect ratios were comparable to literature precedent for those produced by conventional methods. The aspect ratio of CNCs is correlated with an increased elastic modulus and tensile strength, and affects the reinforcement capabilities of CNC-polymer nanocomposites.32,59–61 In film applications, longer CNCs with a greater aspect ratio give more porous surfaces than shorter CNCs with a lower aspect ratio, which provide for more complete surface coverage.62 This directly affects their application in gas adsorption and separations.63 Thus, proper selection of processing conditions is crucial with end-use applications of CNCs in mind.
Conductometry
Hydrolysis with H3PO4 and H2SO4 gives CNCs with phosphate and sulfate half-esters, –OPO32− and –OSO3− respectively, while hydrolysis with HCl gives CNCs with the hydroxyl (–OH) moiety. Typical phosphate content is <40 mmol kg−1, while sulfate content varies from 80–300 mmol kg−1.12,62 Given the high sulfate content that can be expected from traditional hydrolysis conditions, it can be more convenient to achieve colloidally stable CNCs with low surface functionality during synthesis rather than by incorporating a post-synthetic strategy to hydrolyse the surface sulfate groups.40,64 It can be expected, given the significantly reduced concentration of acid employed, that the obtained CNCs from IL-mediated hydrolysis would have low surface functionality, which is beneficial when considering post-functionalization or specific applications. For example, the concentration of –OSO3− is correlated with CNC self-assembly65 and inversely related to CNC thermal stability.10 This limits their use in, for example, melt extrusions;66 moreover, –OPO32− or –OSO3− functional groups impede post-synthetic modification of CNCs.67CNC samples were analysed by conductometry to compare the CNCs prepared under conventional conditions (hCNC, sCNC, and pCNC) and those prepared by IL-mediated hydrolysis (hCNCi, sCNCi, and pCNCi). It should be noted that hCNC and hCNCi bear terminal hydroxyls and are only weakly acidic, thus conductometry was not useful in their characterization. Conductometric titrations (Fig. S1‡) were performed to measure the number of strong acid groups and hence the amount of –OPO32− or –OSO3− present, using procedures reported previously.37–40 The number of –OPO32− or –OSO3− groups detected by the consumption of available protons can then be readily converted to the P or S content (mmol P or S per kg of CNC) at the CNC surface (Table 3). This method does not generally detect P or S that are not acidic or the second proton of the acidic –OPO32− moiety.12 (see ESI‡ for details).
| Entry | Sample | Conductometric titrations | Light scattering | X-ray photoelectron spectroscopy | ||||
|---|---|---|---|---|---|---|---|---|
| S or P content (mmol kg−1) | Sa or Pa (%) | σc (e nm−2) | rHd (nm) | ζ-Potential (mV) | Sf or Pf (%) | Acidic groups per 100 glucose units (n)g | ||
| a Determined from S or P content from conductometric titrations.b The hydrolysis conditions do not impart acidic sulfate or phosphate groups onto the CNC surface.c Calculated from moles of S or P in 1 gram of CNCs assuming a cylindrical shape.d The rH is the apparent hydrodynamic radius obtained from DLS.e Samples flocculated and reliable ζ-potential could not be obtained.f Elemental mass% calculated from atom% from XPS.g Determined from elemental mass% calculated from XPS. | ||||||||
| 1 | hCNC | —b | —b | —b | 82 ± 24 | −15.7 ± 0.9 e | —b | —b |
| 2 | sCNC | 260 ± 6 | 0.83 | −0.436 ± 0.010 | 21 ± 8 | −42.8 ± 1.3 | 0.84 | 4.3 |
| 3 | pCNC | 36 ± 1 | 0.11 | −0.064 ± 0.002 | 63 ± 16 | −20.2 ± 1.4 | 0.25 | 1.3 |
| 4 | hCNCi | —b | —b | —b | 75 ± 20 | −10.2 ± 4.1e | —b | —b |
| 5 | sCNCi | 20 ± 7 | 0.06 | −0.046 ± 0.016 | 75 ± 12 | −24.2 ± 1.7 | 0.20 | 1.0 |
| 6 | pCNCi | 34 ± 4 | 0.11 | −0.092 ± 0.011 | 64 ± 10 | −25.0 ± 2.2 | 0.22 | 1.1 |
Hydrolysis with 62% H2SO4 gave sCNC with 260 ± 6 mmol kg−1 –OSO3−, which is on par with literature precedent.62 IL-mediated hydrolysis with H2SO4, gave sCNCi that contained an order of magnitude less –OSO3− (20 ± 7 mmol kg−1). In contrast, hydrolysis with H3PO4 or IL-mediated hydrolysis with H3PO4 gave a roughly comparable typical concentration of –OPO32− (34 ± 1, and 36 ± 4, respectively).10,12
The values for S or P content were used to calculate the surface charge density of the CNCs using eqn (1), and were used to converted to %S or %P for comparison to the values determined by XPS (vide infra). While the ratio of S found in sCNC compared to sCNCi is 13:1, due to the larger size of the sCNCi and hence smaller surface area per unit volume, the ratio of the surface charge density is reduced (∼9.5:1). The situation is reversed for pCNC and pCNCi, where the ratio of the content of P is approximately 1:1, the surface charge density is 1:1.4 with pCNCi having the larger value of surface charge density.
Light-scattering
DLS is often used as a rapid technique to assess the quality and polydispersity of nanoparticle suspensions, and comparisons of light scattering data have been explicitly applied to CNC suspensions.13,68 In the case of CNCs, the results are expressed as the apparent hydrodynamic radius (rH). This is because DLS relies on Brownian motion to monitor scattering intensity. The particle size is determined based on diffusivity via the Stokes–Einstein equation;69,70 however, this relies on an a constant diffusion coefficient and an assumed spherical hydrodynamic radius. Since the diffusion coefficient perpendicular and parallel to the transverse axis are different, this is not the case for rod-shaped nanoparticles.71,72
Examination of the obtained apparent rH from the different CNC suspensions (Table 3) indicate no statistical differences between hCNC, pCNC, hCNCi, sCNCi, and pCNCi; however, CNCs obtained from H2SO4 hydrolysis (sCNC) where smaller and more uniform in size with a lower apparent rH (21 ± 8) in agreement with the obtained AFM data. These CNCs also exhibited the largest absolute ζ-potential (−42.8 ± 1.3), which is a measure of their colloidal stability determined by electrophoretic light scattering and relates to the surface charge density of the CNCs.
It was previously shown that the amount of surface charge groups (mmol kg−1) is strongly correlated with the ζ-potential of the CNCs (r2 ≈ 0.81).40 However, the association was not perfect because the surface charge density σ, which correlated more strongly with ζ-potential (r2 ≈ 0.87),40 is highly dependent on the size of the CNCs as well as differences arising from transverse location and rotation about the axis of particles during measurement. Thus, although a direct standardization of S or P content from ζ potential cannot be made, the ζ-potential is a useful rapid measure to determine the degree of surface charge and stability of nanoparticle systems.
Generally, CNCs are considered colloidally stable, when the measured absolute zeta(ζ)-potential value is >25 mV,58 while absolute values between 10–25 mV are considered intermittently stable and may flocculate given enough time or added solute (salt),75,76 although neutral solutions may be sterically stabilized.29 All CNC samples analysed exhibited a negative ζ-potential (Table 3). In the case of CNCs hydrolysed with H2SO4 or H3PO4, deprotonation to give either –OPO32− or –OSO3− resulted in highly charged CNCs with a rather large negative ζ-potential (−20.2 and −42.8, respectively). Despite having a weakly ionisable hydroxyl group (pKa of C6 hydroxyl of anhydroglucose ∼12.6), hCNC and hCNCi displayed slightly negative ζ-potentials. This is because the concentration of the CNCs tested, given the pKa of the hydroxyls at neutral pH, resulted in a slight ionisation of the hydroxyls to give ∼13 μmol kg−1 of the resulting alkoxide which were at best intermittently stable. This also accounts for the larger apparent hydrodynamic radius, despite the similarity in absolute size measured by AFM. Samples of sCNCi and pCNCi mediated hydrolysis were at the cusp of colloidal stability with ζ-potentials of −24.2 ± 1.7 and −25.0 ± 2.2, respectively.
X-ray photoemission spectroscopy
X-ray photoemission spectroscopy (XPS) was used for analysis of the nanocrystal elemental composition. In each instance, strong signals were observed at 285 eV and 531 eV, corresponding to C 1s and O 1s, respectively (see ESI, Fig. S4‡). In addition, trace signals were observed at 133 eV or 167 eV, corresponding to the P 2p and S 2p ionisation binding energies in samples containing either phosphate or sulfate half-esters. The summary of results is displayed in Table 3 and compared to the results obtained by conductometric titration. It is shown that for the CNCs with greater functionalisation rates, smaller size, and greater surface area to volume ratio, the value obtained for sCNC by conductometry and XPS are quite similar, 0.84% and 0.83%, respectively. However, for lower functionalisation (sCNCi, pCNC, and pCNCi), a larger amount of S or P was detected in the samples compared to conductometry. There are several contributing factors. First, XPS is a surface sensitive technique that assumes the bulk material is the same as the surface, and second, the lower level of functionalization was near the limit of detection and thus resulted in a lower signal to noise ratio. Finally, the XPS data did not distinguish between residual S or P found in the biomass samples from those introduced as surface acid groups during hydrolysis to produce CNCs.The XPS results confirm low-level functionalization of the CNCs when treated with either H2SO4 or H3PO4. As observed by the conductivity titrations, the surface charge correlates to the number of acidic phosphate esters or sulfate half-esters on the CNC surface. Overall, under the IL-hydrolysis conditions employed, about one-fourth as many anhydroglucose units are functionalised with an –OSO3− group compared to the controls utilising standard H2SO4 hydrolysis conditions. Hydrolysis under harsher conditions with H3PO4 gave similar values for H3PO4 IL-hydrolysis.
Thermogravimetric analysis
Analysis of the TGA and first-derivative (DTG) thermograms for native cellulose, compared to the H2SO4-hydrolysed CNC product (sCNC) and H2SO4/IL-mediated hydrolysis (sCNCi) is shown in Fig. 3. Samples for hydrolysis using HCl, H3PO4, and H2SO4 with and without the IL are shown in the ESI (Fig. S6–S8‡). The TGA thermogram for H2SO4-hydrolysed CNCs displayed an initial degradation temperature of ca. 200 °C corresponding to decomposition of the surface sulfate half-esters, followed by a second DTG local maxima (350 °C) corresponding to the temperature for the decomposition of the cellulose chains.10,13,77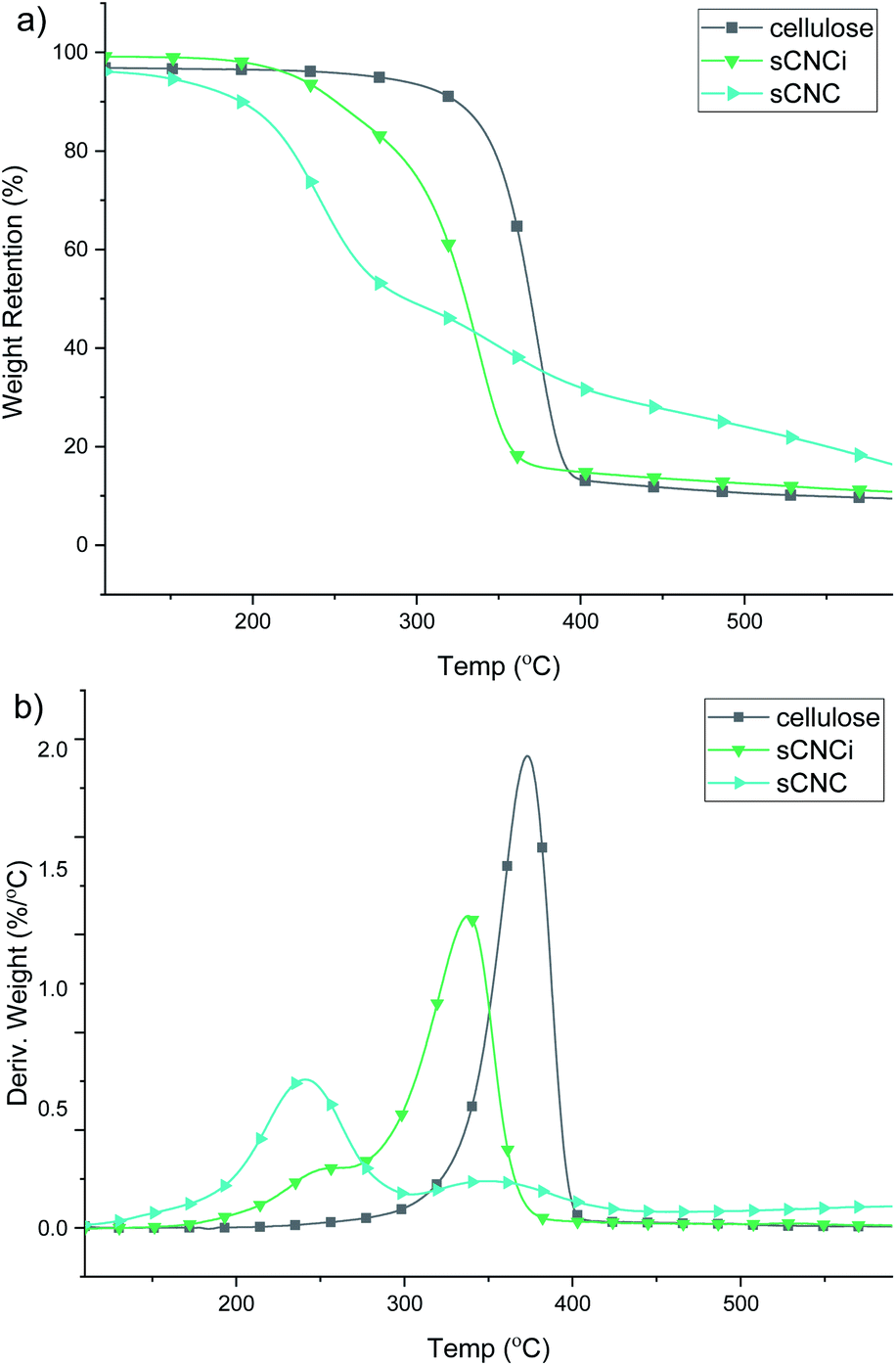 | ||
| Fig. 3 Representative TGA (a) and DTG (b) thermograms for cellulose and sulfate CNCs from traditional (sCNC) and ionic liquid-mediated (sCNCi) hydrolysis of cellulose. Error between individual measurements for each sample was ≤1%. | ||
Very similar thermal stabilities for hCNC and pCNC are indicated based on the TGA thermograms. The initial onset temperature of thermal degradation (Tonset) for native cellulose was 349 °C and the temperature of the peak maxima (Tmax) of the corresponding DTG trace occurred at 374 °C. It has been previously shown that H3PO4-hydrolysed CNCs are more resistant to thermal decomposition, compared to sCNCs.10 One reason for this is that the sulfate half-esters on the CNC surface catalyse the autodegradation of cellulose, especially at higher temperatures,40,77 while in contrast phosphorylated CNCs exhibit a much higher thermal stability.10 A similar effect was observed in this instance, where the Tmax was 349 °C and the Tonset was 323 °C for pCNC. HCl-hydrolysed CNCs displayed intermediate behaviour with a single peak Tmax at 342 °C and initial onset temperatures of 292 °C.
By contrast all IL-hydrolysed CNCs display similar thermal decomposition pathways which are likely the result of partial swelling and dissolution of the cellulose chains during the concurrent IL treatment. This has been linked to a reduction in crystallinity and hence thermal stability of the CNCs. Additionally, IL-hydrolysed CNCs possess a similar abundance of surface charged groups, which do not significantly affect their thermal properties at low functionalization rates. As such, only modest differences were observed; pCNCi from IL-mediated hydrolysis were slightly more stable than sCNCi or hCNCi. The Tonset for IL-hydrolysed sCNCi, hCNCi, and pCNCi was 297, 298, and 308 °C, respectively, while the Tmax was 342, 342, and 356 °C.
Crystallinity
The crystallinity of CNCs has been linked to a toughening effect in polymer composites32 and some pre-treatments, including swelling and hydrolysis methods (e.g. mercerization), are known to affect cellulose crystallinity and/or convert cellulose I to cellulose II,15,42,46,78 which can ultimately affect their solubility, thermal stability, and other physical properties.62,78,79 Therefore, it is important to ascertain the crystalline state of CNCs and the crystallinity index (CrI) when considering potential applications. Samples were analysed for CrI using powder X-ray diffraction (XRD). The MAUD Rietveld refinement program was used for analysis of the resulting diffraction patterns (see ESI Fig. S9‡).46,80 The diffraction patterns for native cellulose, the prepared CNCs and IL-CNCs are shown in Fig. 4a. The predominant reflections observed for the various CNC derivatives and those from IL hydrolysis were of the (1−10), (110), (200) and (004) peaks corresponding to the cellulose 1β diffraction pattern located at approximately 14.8°, 16.5°, 22.8°, and 34.7° 2θ, respectively.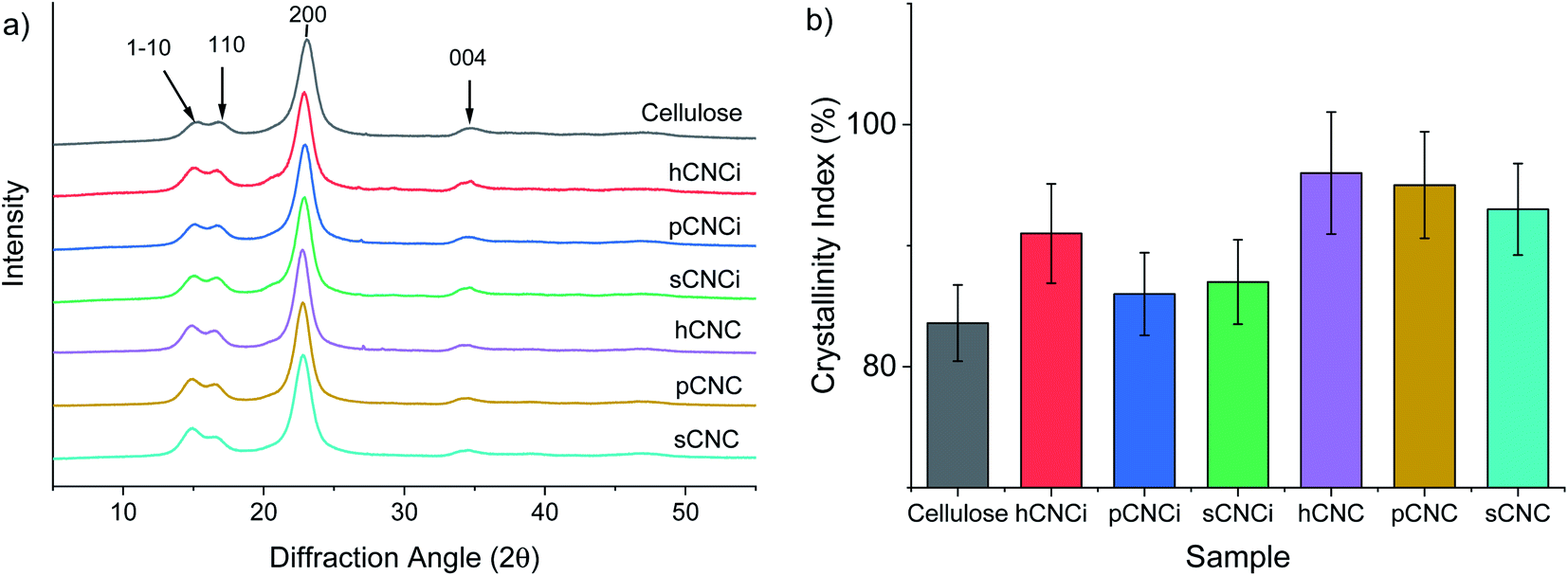 | ||
| Fig. 4 XRD spectra for cellulose and nanocellulose from inorganic acid and IL-mediated hydrolysis of cotton gin motes (a); and, calculated crystallinity index using the MAUD Rietveld refinement program (b). The error bars represent estimated uncertainty in the fit of the calculated diffraction pattern. | ||
The crystallinity (Fig. 4b) and crystallite size were determined using eqn (4) and (5), respectively, while the d-spacings were calculated from the refined unit cell dimensions. CNCs prepared using IL-mediated hydrolysis were highly crystalline with an average crystallinity index of 88%; this is only slightly reduced compared to CNCs produced from mineral acid hydrolysis (95%) and considerably higher than CNCs produced using neat IL from some wood sources26,27 and on par with those reported from cotton linters or cotton pulp.16 This is likely due to the relatively high crystallinity of the native cellulose used for the preparation of CNCs and mild conditions employed during IL-hydrolysis. Harsher conditions, or prolonged swelling prior to acid hydrolysis have previously been shown to severely alter cellulose crystallinity.17,81 Specifically, swelling and dissolution in ILs has been shown to disrupt the crystalline regions of the cellulose chains increasing the amorphous content,82,83 this partially explains the changes in the TGA and DTG thermograms (Fig. S6 and S8‡) as well as the slight reduction in crystallinity observed (Fig. 4b) compared to CNCs from traditional methods.
To further ascertain variations in the crystalline structure as the result of the IL treatment, the crystallite sizes and d-spacings were plotted for the main lattice plains of cotton cellulose (Fig. 5). Compared to native cellulose mineral acid hydrolysis results in increased d-spacing along the (1−10) lattice plane and accordingly an increase in crystallite size along that dimension. This correlated with a decrease in crystallite size along the (110) lattice plane. Under the milder conditions employing the IL, the calculated d-spacings and crystallite sizes were more akin to native MCC than to CNCs produced from mineral acid hydrolysis. Thus, the dilute IL and dilute acid conditions did less to perturb the native cellulose structure than concentrated acid hydrolysis.
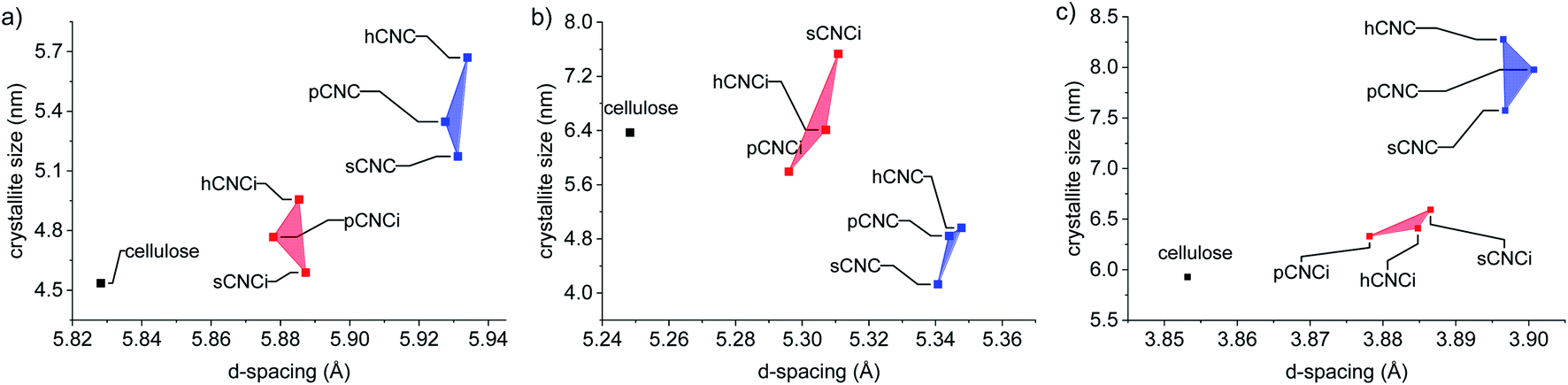 | ||
| Fig. 5 Relationship between d-spacing and crystallite size of the lattice planes: (a) (1−10); (b) (110), and; (c) (200). | ||
Conclusions
This work represents a facile strategy to obtain cellulose nanocrystals using dilute acids in combination with ionic liquids under mild conditions. A single ionic liquid ([BMIm]Cl) was used for the extraction of nanocellulose using three inorganic acids, showing the generality of these conditions. CNCs obtained via this method are larger than CNCs obtained by conventional methods, but with similar aspect ratio, thus, maintaining favourable properties such as high crystallinity and similar crystallite sizes. Moreover, IL-mediated hydrolysis improves the thermal properties of sulfate CNCs, potentially expanding their applications in melt extrusion and polymer composites. Additionally, IL-hydrolysed CNCs with longer aspect ratios may prove beneficial for preparation of gas adsorbents and for emulsion stabilisation. Future work will focus on optimizing the reaction conditions, the selection of the specific IL in combination with different acids to improve yield and CNC recovery and employing IL-hydrolysed CNCs in suitable material applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Xiaodong Zhang and Tulane University, Department of Chemistry, for acquisition of the XPS data, Dr Dongmei Cao at the Shared Instrumentation Facility, Louisiana State University for performing the XRD measurements, Stephanie Thompson, National Center for Agricultural Utilization Research, USDA, for characterization of the extracted cellulose and Dr Matthew B. Hillyer, Southern Regional Research Center, USDA, for preparation of the TOC graphic. Additionally, the authors acknowledge the National Program Staff, the Mid-South Area Director, and the Center Director of the Agricultural Research Service of the U.S. Department of Agriculture for providing the necessary support for the study presented here.Notes and references
- M. J. Earle and K. R. Seddon, in Clean Solvents, American Chemical Society, 2002, vol. 819, ch. 2, pp. 10–25 Search PubMed.
- N. L. Mai, K. Ahn and Y.-M. Koo, Process Biochem., 2014, 49, 872–881 CrossRef CAS.
- A. H. Bhat, I. Khan, M. A. Usmani, R. Umapathi and S. M. Z. Al-Kindy, Int. J. Biol. Macromol., 2019, 129, 750–777 CrossRef CAS.
- H. Xie, H. Du, X. Yang and C. Si, Int. J. Polym. Sci., 2018, 2018, 1–25 Search PubMed.
- Y. Meng, Z. Pang and C. Dong, Carbohydr. Polym., 2017, 163, 317–323 CrossRef CAS.
- A. S. Amarasekara, Isr. J. Chem., 2018, 59, 789–802 Search PubMed.
- S. Xia, G. A. Baker, H. Li, S. Ravula and H. Zhao, RSC Adv., 2014, 4, 10586–10596 CAS.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef CAS.
- L. Brinchi, F. Cotana, E. Fortunati and J. M. Kenny, Carbohydr. Polym., 2013, 94, 154–169 CrossRef CAS.
- S. Camarero Espinosa, T. Kuhnt, E. J. Foster and C. Weder, Biomacromolecules, 2013, 14, 1223–1230 CrossRef CAS.
- O. M. Vanderfleet, M. S. Reid, J. Bras, L. Heux, J. Godoy-Vargas, M. K. R. Panga and E. D. Cranston, Cellulose, 2018, 26, 507–528 CrossRef.
- O. M. Vanderfleet, E. D. Cranston and D. A. Osorio, Philos. Trans. R. Soc., A, 2018, 376, 2112 CrossRef.
- J. H. Jordan, M. W. Easson, B. Dien, S. Thompson and B. D. Condon, Cellulose, 2019, 26, 5959–5979 CrossRef CAS.
- J. Mao, A. Osorio-Madrazo and M.-P. Laborie, Cellulose, 2013, 20, 1829–1840 CrossRef CAS.
- D. Yang, X. W. Peng, L. X. Zhong, X. F. Cao, W. Chen and R. C. Sun, Cellulose, 2013, 20, 2427–2437 CrossRef CAS.
- J. Lazko, T. Sénéchal, N. Landercy, L. Dangreau, J. M. Raquez and P. Dubois, Cellulose, 2014, 21, 4195–4207 CrossRef CAS.
- J. Lazko, T. Senechal, A. Bouchut, Y. Paint, L. Dangreau, A. Fradet, M. Tessier, J. M. Raquez and P. Dubois, Nanocomposites, 2016, 2, 65–75 CrossRef CAS.
- X. Fu, H. Ji, B. Wang, W. Zhu, Z. Pang and C. Dong, Cellulose, 2019, 27, 1289–1299 CrossRef.
- J. Mao, B. Heck, G. Reiter and M. P. Laborie, Carbohydr. Polym., 2015, 117, 443–451 CrossRef CAS.
- X. Y. Tan, S. B. Abd Hamid and C. W. Lai, Biomass Bioenergy, 2015, 81, 584–591 CrossRef CAS.
- N. A. M. Iskak, N. M. Julkapli and S. B. A. Hamid, Cellulose, 2017, 24, 2469–2481 CrossRef CAS.
- Z. Pang, P. Wang and C. Dong, Cellulose, 2018, 25, 7053–7064 CrossRef CAS.
- X. Song, L. Zhou, B. Ding, X. Cui, Y. Duan and J. Zhang, Carbohydr. Polym., 2018, 186, 252–259 CrossRef CAS.
- G. Zhao, F. Wang, X. Lang, B. He, J. Li and X. Li, RSC Adv., 2017, 7, 27017–27023 RSC.
- J. Miao, H. Sun, Y. Yu, X. Song and L. Zhang, RSC Adv., 2014, 4, 36721–36724 RSC.
- J. Miao, Y. Yu, Z. Jiang and L. Zhang, Cellulose, 2016, 23, 1209–1219 CrossRef CAS.
- H. Abushammala, I. Krossing and M. P. Laborie, Carbohydr. Polym., 2015, 134, 609–616 CrossRef CAS.
- S. Pruksawan, S. Samitsu, Y. Fujii, N. Torikai and M. Naito, ACS Appl. Polym. Mater., 2020, 2, 1234–1243 CrossRef CAS.
- J. Leguy, A. Diallo, J.-L. Putaux, Y. Nishiyama, L. Heux and B. Jean, Langmuir, 2018, 34, 11066–11075 CrossRef CAS.
- S. Varanasi, L. Henzel, L. Mendoza, W. Batchelor, G. Garnier, S. Varanasi, R. Prathapan and R. Tabor, Front. Chem., 2018, 6, 409 CrossRef.
- C. Tang, S. Spinney, Z. Shi, J. Tang, B. Peng, J. Luo and K. C. Tam, Langmuir, 2018, 34, 12897–12905 CrossRef CAS.
- S. Shrestha, F. Montes, G. T. Schueneman, J. F. Snyder and J. P. Youngblood, Compos. Sci. Technol., 2018, 167, 482–488 CrossRef CAS.
- A. P. Gonçalves, E. Oliveira, S. Mattedi and N. M. José, Sep. Purif. Technol., 2018, 196, 200–207 CrossRef.
- J. Mao, H. Abushammala, H. Hettegger, T. Rosenau and M.-P. Laborie, Polymers, 2017, 9, 473 CrossRef.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, Denver, CO, 2008 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- S. Beck, M. Méthot and J. Bouchard, Cellulose, 2014, 22, 101–116 CrossRef.
- T. Abitbol, E. Kloser and D. G. Gray, Cellulose, 2013, 20, 785–794 CrossRef CAS.
- L. J. Johnston, Z. J. Jakubek, S. Beck, J. Araki, E. D. Cranston, C. Danumah, D. Fox, H. Li, J. Wang, Z. Mester, A. Moores, K. Murphy, S. A. Rabb, A. Rudie and C. Stephan, Metrologia, 2018, 55, 872–882 CrossRef CAS.
- J. H. Jordan, M. W. Easson and B. D. Condon, Nanomaterials, 2019, 9, 1232 CrossRef CAS.
- F. Jiang, A. R. Esker and M. Roman, Langmuir, 2010, 26, 17919–17925 CrossRef CAS.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS.
- A. Wishard and B. C. Gibb, Supramol. Chem., 2019, 31, 608–615 CrossRef CAS.
- W. Y. Hamad and T. Q. Hu, Can. J. Chem. Eng., 2010, 88, 392–402 CAS.
- A. D. French, Cellulose, 2014, 21, 885–896 CrossRef CAS.
- S. Nam, A. D. French, B. D. Condon and M. Concha, Carbohydr. Polym., 2016, 135, 1–9 CrossRef CAS.
- Z. Ling, T. Wang, M. Makarem, M. Santiago Cintrón, H. N. Cheng, X. Kang, M. Bacher, A. Potthast, T. Rosenau, H. King, C. D. Delhom, S. Nam, J. Vincent Edwards, S. H. Kim, F. Xu and A. D. French, Cellulose, 2019, 26, 305–328 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
- Q. Wang, X. Zhao and J. Y. Zhu, Ind. Eng. Chem. Res., 2014, 53, 11007–11014 CrossRef CAS.
- Q. Wang, J. Y. Zhu and J. M. Considine, ACS Appl. Mater. Interfaces, 2013, 5, 2527–2534 CrossRef CAS.
- Y. Marcus, Ion properties, Marcel Dekker, New York, 1997 Search PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CrossRef CAS.
- V. Mazzini and V. S. J. Craig, ACS Cent. Sci., 2018, 4, 1056–1064 CrossRef CAS.
- M. S. Reid, T. C. Stimpson, E. Niinivaara, M. Villalobos and E. D. Cranston, Ind. Eng. Chem. Res., 2018, 57, 220–230 CrossRef CAS.
- Z. Dastjerdi, E. D. Cranston and M. A. Dube, Int. J. Adhes. Adhes., 2018, 81, 36–42 CrossRef CAS.
- I. Kalashnikova, H. Bizot, P. Bertoncini, B. Cathala and I. Capron, Soft Matter, 2013, 9, 952–959 RSC.
- M. Parit, P. Saha, V. A. Davis and Z. Jiang, ACS Omega, 2018, 3, 10679–10691 CrossRef CAS.
- M. El Achaby, Z. Kassab, A. Barakat and A. Aboulkas, Ind. Crops Prod., 2018, 112, 499–510 CrossRef CAS.
- J. Sapkota, A. Shirole, E. J. Foster, J. C. Martinez Garcia, M. Lattuada and C. Weder, Polymer, 2017, 110, 284–291 CrossRef CAS.
- J. Sapkota, J. C. Natterodt, A. Shirole, E. J. Foster and C. Weder, Macromol. Mater. Eng., 2017, 302, 1600300 CrossRef.
- D. Trache, M. H. Hussin, M. K. M. Haafiz and V. K. Thakur, Nanoscale, 2017, 9, 1763–1786 RSC.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- Q. Cheng, D. Ye, C. Chang and L. Zhang, J. Membr. Sci., 2017, 525, 1–8 CrossRef CAS.
- N. Lin and A. Dufresne, Nanoscale, 2014, 6, 5384–5393 RSC.
- T. Abitbol, D. Kam, Y. Levi-Kalisman, D. G. Gray and O. Shoseyov, Langmuir, 2018, 34, 3925–3933 CrossRef CAS.
- H. Kargarzadeh, M. Mariano, D. Gopakumar, I. Ahmad, S. Thomas, A. Dufresne, J. Huang and N. Lin, Cellulose, 2018, 25, 2151–2189 CrossRef CAS.
- L. Hou, H. Bian, Q. Wang, N. Zhang, Y. Liang and D. Dong, RSC Adv., 2016, 6, 53062–53068 RSC.
- Y. Boluk and C. Danumah, J. Nanopart. Res., 2013, 16, 2174 CrossRef.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS.
- M. Rösslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732–745 CrossRef.
- A. Ortega, J. G. a. d. l. Torre and J. G. a. d. l. Torre, J. Chem. Phys., 2003, 119, 9914–9919 CrossRef CAS.
- J. Wang, Y. Yang, M. Yu, G. Hu, Y. Gan, H. Gao and X. Shi, J. Mech. Phys. Solids, 2018, 112, 431–457 CrossRef CAS.
- S. Skoglund, J. Hedberg, E. Yunda, A. Godymchuk, E. Blomberg and I. Odnevall Wallinder, PLoS One, 2017, 12, e0181735 CrossRef.
- M. R. Gittings and D. A. Saville, Colloids Surf., A, 1998, 141, 111–117 CrossRef CAS.
- R. Prathapan, R. Thapa, G. Garnier and R. F. Tabor, Colloids Surf., A, 2016, 509, 11–18 CrossRef CAS.
- L. Liu, Z. Hu, X. Sui, J. Guo, E. D. Cranston and Z. Mao, Ind. Eng. Chem. Res., 2018, 57, 7169–7180 CrossRef CAS.
- N. Wang, E. Ding and R. Cheng, Polymer, 2007, 48, 3486–3493 CrossRef CAS.
- Y. Yue, C. Zhou, A. D. French, G. Xia, G. Han, Q. Wang and Q. Wu, Cellulose, 2012, 19, 1173–1187 CrossRef CAS.
- Y. Yue, J. Han, G. Han, Q. Zhang, A. D. French and Q. Wu, Carbohydr. Polym., 2015, 133, 438–447 CrossRef CAS.
- Z. Ling, J. V. Edwards, Z. Guo, N. T. Prevost, S. Nam, Q. Wu, A. D. French and F. Xu, Cellulose, 2018, 26, 861–876 CrossRef.
- J. Zhang, N. Luo, X. Zhang, L. Xu, J. Wu, J. Yu, J. He and J. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 4417–4423 CrossRef CAS.
- M. Ghasemi, P. Alexandridis and M. Tsianou, Cellulose, 2017, 24, 571–590 CrossRef CAS.
- M. Ghasemi, P. Alexandridis and M. Tsianou, Biomacromolecules, 2018, 19, 640–651 CrossRef CAS.
Footnotes |
| † This research was supported by the U.S. Department of Agriculture, Agricultural Research Service. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. |
| ‡ Electronic supplementary information (ESI) available: XRD, XPS, TGA, AFM measurements, conductivity titrations and additional experimental details. See DOI: 10.1039/d0ra05976e |
| This journal is © The Royal Society of Chemistry 2020 |