
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed Pt nanoclusters supported on zeolite-templated carbon for the oxygen reduction reaction†
Raj Kumar Bera *a,
Hongjun Parkab,
Seung Hyeon Koa and
Ryong Ryoo*ab
*a,
Hongjun Parkab,
Seung Hyeon Koa and
Ryong Ryoo*ab
aCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon, 34141, Republic of Korea. E-mail: chemsraj@ibs.re.kr; rryoo@kaist.ac.kr
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea
First published on 1st September 2020
Abstract
The formation of highly dispersed Pt nanoclusters supported on zeolite-templated carbon (PtNC/ZTC) by a facile electrochemical method as an electrocatalyst for the oxygen reduction reaction (ORR) is reported. The uniform micropores of ZTC serve as nanocages to stabilize the PtNCs with a sharp size distribution of 0.8–1.5 nm. The resultant PtNC/ZTC exhibits excellent catalytic activity for the ORR due to the small size of the Pt clusters and high accessibility of the active sites through the abundant micropores in ZTC.
Platinum (Pt) is currently considered one of the best electrocatalysts for the oxygen reduction reaction (ORR), which occurs at the cathode of a fuel cell and is the key process determining the overall performance.1–5 However, the high cost and scarcity of Pt limit its wide commercialization in this field. According to the US Department of Energy, the total Pt loading is required to be below 0.125 mg cm−2, in contrast to a presently used Pt loading of 0.4 mg cm−2 or more for fuel cell application.4 Therefore, reducing the Pt loading without loss or with an improvement of the cathode performance has received significant interest in electrocatalytic research for fuel cell systems.6–10 In this regard, reducing the size of Pt particles to a nanocluster scale (size < 2 nm) and maximizing the Pt dispersion may offer an efficient way to achieve maximum utilization of the Pt electrocatalyst with appropriate consumption.4,11–15
The size of nanomaterials generally plays a critical role in controlling the physical and chemical properties for catalytic applications.16–20 With a decrease in the particle size to the nanoscale, quantum size effects are induced, which alter the surface energy of the material due to unsaturated coordination and change in the energy level of the d orbital of metal atoms, leading to spatial localization of the electrons.17–20 This size-induced effect on the electronic structures at the active sites modifies the capability of binding the reactant molecules in catalytic reactions, thereby altering the activity of the nanocatalyst.20 When the particle contains a few to several dozens of atoms with sizes, ranging from sub-nanometer to 2 nm often termed as nanocluster that bridges nanoparticle and a single atom.21 However, the Pt single atom is not an appropriate electrocatalyst for the ORR in a fuel cell system as the fast four-electron (4e−) pathway for the reduction of O2 to H2O requires at least two neighboring Pt atoms.22,23 Anderson's group demonstrated that the ratio between the production of H2O (product of 4e− process) and H2O2 (2e−) in the ORR strongly depends on the number of atoms in the Pt cluster. Typically, it requires more than 14 atoms in a Pt cluster to produce H2O efficiently through the 4e− pathway of the ORR.24 Therefore, Pt nanoclusters having more than a dozen atoms have proven to be highly efficient ORR electrocatalysts for fuel cell systems.13–15 Upon decreasing the size of the nanoparticles to a nanocluster, the electronic state and structure are known to be changed, leading to an increase of the catalytic activity in the ORR. Therefore, it is highly desirable to synthesize a Pt nanocluster-based material as an ORR electrocatalyst with high catalytic performance. To date, several synthesis strategies, such as wet-chemical, atomic-layer deposition, and photochemical methods, have been applied for the preparation of well-dispersed Pt nanoclusters on different types of support, such as dendrimer, metal oxide, and carbon materials.13–15,25–31
An alternative approach to synthesize Pt nanocluster (PtNC) is the encapsulation of the cluster within nanosized pores, for example, by utilizing microporous (diameters less than 2 nm) carbon materials.32 Among the microporous carbons, zeolite-templated carbon (ZTC) has been attractive for supporting Pt clusters due to its ordered microporous structure.33–37 ZTC is a potentially promising material as catalyst support as it offers the advantages of extremely large surface area and high electrical conductivity of graphene-like carbon frameworks constituting a three-dimensional (3D) interconnected pore structure.36 Moreover, the micropores of ZTC can serve as nanocages for stabilization of the Pt nanoclusters. Coker et al. used Pt2+ ion-exchanged zeolite as a carbon template to synthesize Pt nanoparticles in ZTC with size in a range of 1.3 to 2.0 nm.33 Recently, atomically dispersed Pt ionic species was synthesized via a simple wet-impregnation method on ZTC containing a large amount of sulfur (17 wt%).23 Itoi et al. synthesized PtNC consisting of 4–5 atoms and a single Pt atom in ZTC using the organoplatinum complex.37 Although these methods produced Pt nanoclusters with narrow size distribution and atomic dispersion, they required multi-step processes and/or high-temperature treatment (>300 °C). High-temperature treatment often induces the sintering of nanoclusters to aggregated clusters. Therefore, it is highly desirable to develop a simple and low-cost method for the preparation of PtNC supported on ZTC (PtNC/ZTC) for use as an efficient ORR electrocatalyst. The electrochemical reduction approach offers an alternate and efficient route for the synthesis of PtNC in the micropores of ZTC. The electrochemical method is one of the popular ways to prepare electrocatalysts because it is a simple single-step procedure and ensures electrical contact between the nanoparticles and the support.38,39
Herein, we report a facile electrochemical method for the formation of PtNC with a narrow size range of 0.8–1.5 nm supported on ZTC. The resultant PtNC/ZTC shows higher electrocatalytic activities towards ORR compared to that of commercial Pt/C. Here, ZTC plays two important roles: (i) it provides nanocages to stabilize the PtNC and (ii) it accelerates the ORR activity by enhancing the accessibility of active sites through its abundant micropores.
Fig. 1a shows a schematic representation of the typical electrochemical synthesis of PtNC/ZTC. In the first step, ZTC was impregnated with a Pt-precursor dissolved in a water–ethanol mixture. As ZTC possess ordered micropores (Fig. S1a†) with high Brunauer–Emmett–Teller (BET) surface area of 3400 m2 g−1 (vide infra), the uniform adsorption and anchorage of PtCl62− ions into the micropores of ZTC was favored. After impregnating and drying, the resultant ZTC–PtCl62− was mixed with water–ethanol and Nafion to make the ink for the preparation of the electrode. Using the prepared electrode, a potential of 0.77 V vs. reversible hydrogen electrode (RHE) (Fig. 1b) was applied followed by potential cycling between 1.12 to −0.02 V vs. RHE until the cyclic voltammogram was stabilized. The Pt content of PtNC/ZTC was determined to be ∼10 wt% (Fig. S2†) by thermogravimetric analysis (TGA). The obtained PtNC/ZTC was electrochemically characterized by cyclic voltammetry and electrochemical impedance spectroscopy. The cyclic voltammogram (Fig. 1c) after potential cycling in fresh KOH electrolyte shows the characteristic Pt peaks corresponding to hydrogen adsorption and desorption. The Nyquist plots (Fig. 1d) demonstrate that PtNC/ZTC has lower electrolyte resistance (42 Ω) than that of ZTC (70 Ω), implying an improvement in the conductivity of ZTC by the presence of PtNC. Due to the increase in the conductivity, PtNC/ZTC could facilitate the electron transfer more effectively than ZTC, enhancing its electrocatalytic activity.
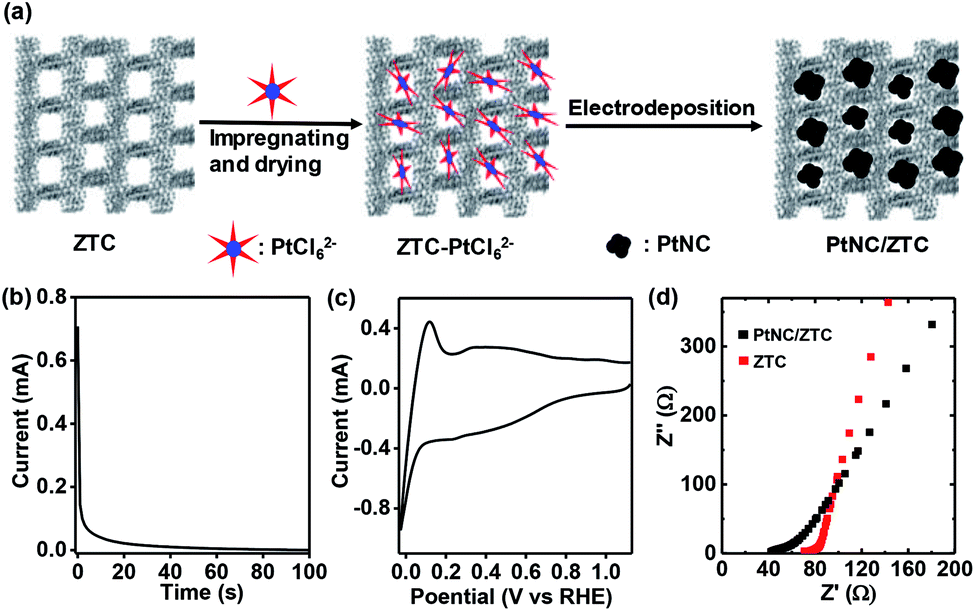 | ||
| Fig. 1 (a) Illustration for the formation of PtNC/ZTC:Pt-precursor was impregnated into ZTC micropores, and then a potential (0.77 V vs. RHE) was exerted on the ZTC–PtCl62− composite in a 0.1 M KOH solution to form PtNC/ZTC (b) Chronoamperometric response of ZTC–PtCl62− at a constant potential of 0.77 V (vs. RHE) in 0.1 M KOH electrolyte. (c) Cyclic voltammogram of PtNC/ZTC in a fresh 0.1 M KOH at a scan rate of 20 mV s−1. (d) Nyquist plots of ZTC and PtNC/ZTC in 0.1 M KOH. | ||
Fig. 2a and b show images from aberration-corrected scanning transmission electron microscope (STEM) with high-angle annular dark-field (HAADF). The HAADF-STEM images exhibit the typical morphology of the final product (PtNC/ZTC) after electrochemical reduction. As shown in Fig. 2a, it is very clear that isolated PtNCs are uniformly dispersed in ZTC. These PtNCs have a homogeneous distribution with a narrow size range (0.8–1.5 nm, Fig. 2b). On further magnification, the STEM image shows a cluster-like structure of Pt (Fig. 2c). The STEM image of selected PtNC (Fig. 2d) reveals that it consists of ∼20 atoms. The number of atom content in PtNC was further determined by matrix-assisted laser-desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry using trans-2-[3-(4-test-butylphenyl)-2-methyl-2-propenylidene]malononitrile as the matrix.40,41 As shown in Fig. S3,† MALDI-TOF measurement produces a mass spectra with a predominant peak centered at ∼3700 Da corresponding to the Pt19 cluster. The TEM image (Fig. S4 a and b†) validates the formation of PtNC with an average size of 0.9 nm. In addition, the energy dispersive X-ray spectrometer (EDS) mapping images clearly shows the uniform dispersion of Pt nanocluster in ZTC (Fig. S4c†). The X-ray powder diffraction (XRD) pattern (Fig. 2e) of PtNC/ZTC showed three broad peaks associated with small size metallic Pt corresponds to (111), (200), and (311) planes (Fig. 2e, inset), along with peaks of ZTC at 2θ = 7.8° and 14.9° corresponding to the ordered microporous structure. Along with the structural analysis, the porous texture of PtNC/ZTC was examined by Ar adsorption (Fig. 2f). PtNC/ZTC had a high BET surface area of 2360 m2 gZTC−1, which is 1.4 times lower than that of pristine ZTC (3400 m2 gZTC−1). The decrease in Ar adsorption capacity after the formation of PtNC in ZTC is interpreted as a result of the filling of ZTC micropores by PtNC. This micropore filling was confirmed in the pore size distributions of the pristine ZTC and the metal-loaded carbon (inset of Fig. 2f). The X-ray photoelectron spectroscopy (XPS) results reveal the signature of Pt in ZTC (Fig. S5†). The elemental survey (Fig. S5a†) shows the signature of C 1s, O 1s, F 1s (Nafion), and Pt 4f. The chemical nature of Pt in PtNC/ZTC was inspected by a detailed Pt 4f XPS analysis. The deconvoluted Pt 4f XPS spectra (Fig. S5b†) reveals the presence of both metallic and ionic Pt species. The peaks observed at 71.0 (4f7/2) and 74.2 (4f5/2) eV correspond to metallic Pt whereas the other peaks positioned at 72.6 (4f7/2) and 76.0 (4f5/2) are attributed to Pt2+ and the peaks at 74.9 (4f7/2) and 77.8 (4f5/2) eV are attributed to Pt4+ originating from the surface oxidation of metallic Pt.42
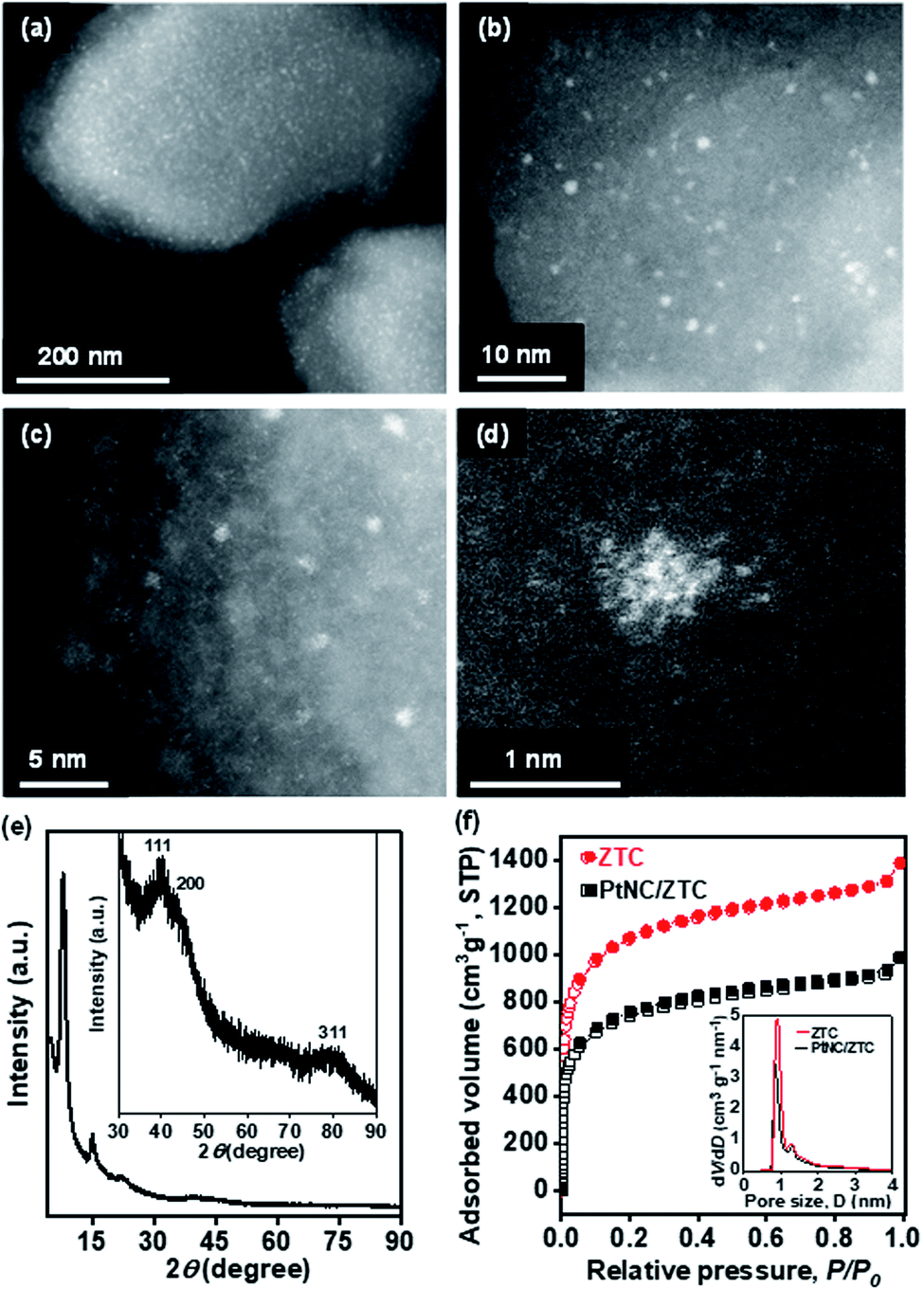 | ||
| Fig. 2 (a–d) Representative spherical aberration-corrected HAADF-STEM images of PtNC/ZTC at various magnifications. (e) XRD pattern of PtNC/ZTC and (f) Ar adsorption–desorption isotherms of ZTC and PtNC/ZTC. Inset in (e) shows a 30 times magnified high-angle region of XRD of PtNC/ZTC. Inset in (f) shows the pore size distributions of the ZTC and PtNC/ZTC. | ||
The formation of narrow sized PtNC by the electrochemical method can be ascribed to the stabilization of PtNC in the ZTC micropores, which serve as cages to impose a spatial limitation on the size of the Pt clusters. For comparison, Pt supported on ZTC was also prepared by the conventional incipient wetness impregnation and subsequent H2-reduction at high temperature (300 °C). The Pt obtained by this incipient wetness impregnation method shows the formation of Pt nanoparticles on the exterior surface of ZTC (PtNP/ZTC) (Fig. S6†). The formation of larger Pt nanoparticles is due to the sintering at high temperature, showing that even ZTC micropores could not prevent the aggregation of PtNCs at high temperatures.
Fig. 3 shows the electrochemical ORR activity of PtNC/ZTC using linear sweep voltammetry (LSV) technique on a rotating disc electrode (RDE) in a 0.1 M KOH solution saturated with O2 at a scan rate of 5 mV s−1. The ORR activity of ZTC (without PtNC) was measured for comparison as well. As shown in Fig. 3a, PtNC/ZTC exhibited higher diffusion limiting current density and higher positive onset and half-wave potential compared to ZTC alone, indicating that PtNC is the active center for the ORR. To investigate the effect of the Pt loading amount on the ORR activity, PtNC/ZTC with various Pt loadings, 2–20 wt%, was used for the measurement of LSV at 1600 rpm. With an increase in Pt content, both the onset and half-wave potential shifted towards more positive potential up to 10 wt% loading of Pt (Fig. 3a and S7†). Upon further increase of loading of Pt on ZTC to 20 wt%, both the onset and half-wave potential of PtNC/ZTC shifted towards less positive potential along with a slight decrease in the diffusion limiting current density (Fig. 3a). The decrease in the ORR activity of PtNC/ZTC at high loading of Pt (20 wt%) was attributed to the decrease in the electrochemically active surface area (Fig. S8†) and decrease in the specific surface area (Fig. S9†). The STEM image clearly shows that the aggregated Pt clusters were formed on the exterior surface of ZTC at 20 wt% loading of Pt (Fig. S10c†), blocking the accessibility of active sites. Therefore, PtNC/ZTC with the optimum loading of 10 wt% of Pt leads to superior ORR activity with a high positive onset potential of 0.99 V, which is similar to commercial Tanaka Pt/C (Pt/C-TKK) (Fig. 3b), and a half-wave potential of 0.87 V, which is ∼10 mV more positive than that of commercial Pt/C-TKK (0.86 V) (Fig. 3b). Compared to the case of PtNC/ZTC, both the onset and half-wave potential of PtNP/ZTC prepared by the conventional incipient wetness impregnation and subsequent H2-reduction with the same loading of Pt exhibited a less positive value (Fig. S11†). The poorer activity of PtNP/ZTC is due to the blockage of active sites by larger PtNPs formed on the exterior surface of ZTC (Fig. S6†).
 | ||
| Fig. 3 (a) RDE ORR polarization curves of PtNC/ZTC with different mass loading of Pt. (b) Comparison of PtNC/ZTC (PtNC10%/ZTC) with commercial Pt/C-TKK at the same loading of 40 μgPt cm−2. (c) RDE ORR polarization curves of PtNC/ZTC at different rotation speeds. Inset in (c) shows the corresponding K–L plots at different potentials. (d) Represents the kinetic current density values of Pt/C-TKK and PtNC/ZTC at the potential of 0.8 V vs. RHE. | ||
To investigate the kinetics of the ORR activity of PtNC/ZTC, LSV measurements were performed with RDE at different rotating rates (Fig. 3c), and the kinetics was analyzed using a Koutecký–Levich (K–L) plot (Fig. 3c, inset). From Fig. 3c, it was observed that the current density increases with the increasing speed of rotation of the electrode, which is characteristic of a diffusion-controlled reaction. The corresponding linear K–L plots (Fig. 3c, inset) with a similar slope at different potentials reveal that the number of transferred electrons was ∼4, indicating that O2 is directly reduced to OH− and the ORR is dominated by the H2O2-free 4e− pathway. To estimate the amount of produced peroxide ion, rotating ring-disc electrode (RRDE) measurement was performed and the produce peroxide ion calculated from RRDE curve was < 4% (Fig. S12†). The kinetic current density (Jk) obtained from K–L plot at the potential of 0.8 V (Fig. 3d) for PtNC/ZTC (Jk = 50 mA cm−2) is 2.2 times higher than that of commercial Pt/C-TKK (Jk = 22 mA cm−2).
As Pt-based electrocatalysts are known to be highly active in an acidic medium, the ORR activity of PtNC/ZTC in O2-saturated 0.1 M HClO4 was also evaluated by comparing it with that of commercial Pt/C-TKK with the same loading of Pt on the electrode surface using RDE at a scan rate of 5 mV s−1. The PtNC/ZTC-based electrode exhibited ORR activity with an onset potential of 0.96 V (Fig. 4), which is close to that of Pt/C-TKK (0.98 V), and half-wave potentials of 0.84 V, which is 20 mV more positive than that of Pt/C-TKK (0.82 V). PtNC/ZTC showed a slightly higher diffusion-limiting current density of ∼5.9 mA cm−2 (0.4–0.7 V) compared with that of the Pt/C-TKK catalyst (∼5.6 mA cm−2). The kinetics of the ORR in an acidic medium was further analyzed using RDE at different rotation rates (Fig. S13†) and it was observed that the current density increases with the increasing speed of rotation of the electrode, as in the case of the alkaline medium. The number of electron involved and the amount of produced H2O2 estimated by RRDE measurement were ∼4 and < 5%, respectively (Fig. S14†). The mass activity of PtNC/ZTC obtained using the mass transport corrected kinetic current at 0.8 V is 0.15 A mg−1, which is 3.2 times higher than that of Pt/C-TKK (0.046 A mg−1).
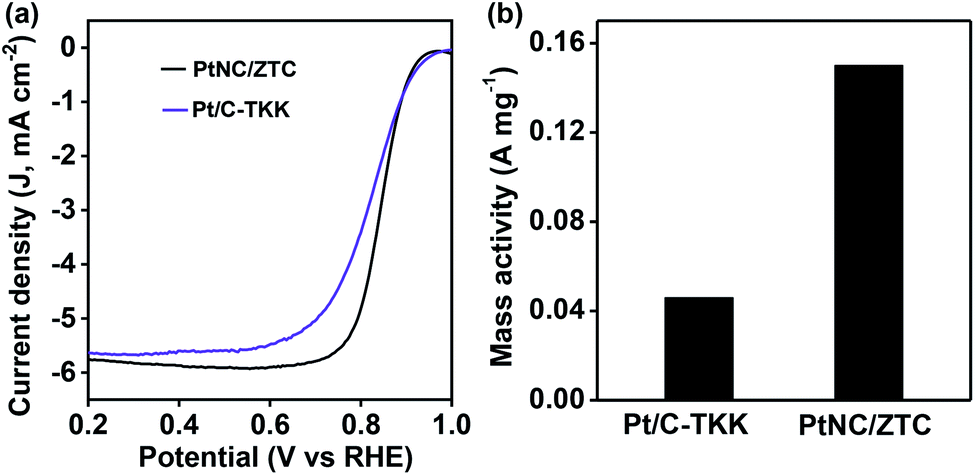 | ||
| Fig. 4 (a) RDE ORR polarization curves at 1600 rpm and (b) mass activity at 0.8 V of PtNC/ZTC and Pt/C-TKK in 0.1 M HClO4. | ||
Furthermore, the methanol tolerance of PtNC/ZTC was assessed by intentionally adding methanol to the oxygen saturated electrolyte solution (both in alkaline and acidic media). The commercial Pt/C-TKK was used for comparison as well. The peak current densities for methanol oxidation with PtNC/ZTC were ∼2.8 and ∼3 times lower than that of Pt/C-TKK in alkaline (Fig. 5a) and acidic (Fig. 5b) media, respectively. These results indicate that PtNC/ZTC has much higher tolerance towards methanol than Pt/C-TKK does. This higher methanol tolerance of PtNC/ZTC can be attributed to the small size of the Pt cluster, which may not be sufficient to catalyze the oxidation of methanol efficiently, as the oxidation of methanol requires Pt ensemble sites.43
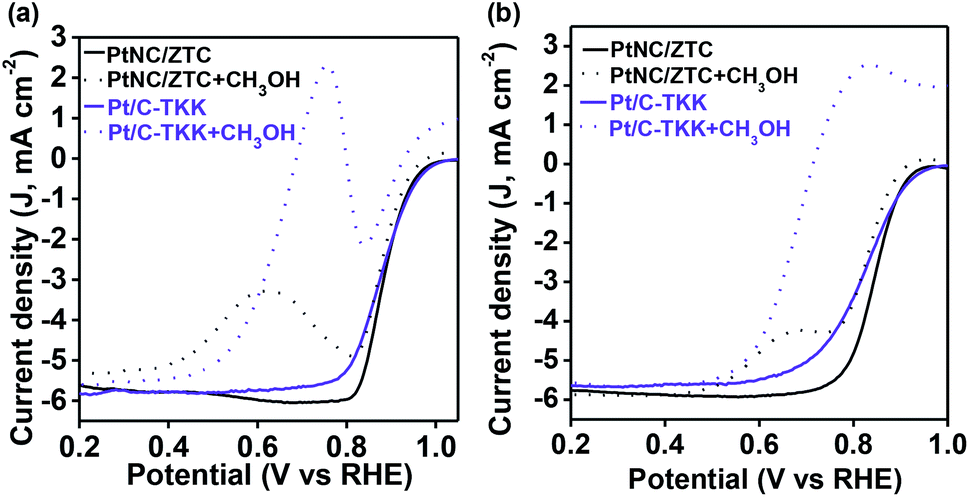 | ||
| Fig. 5 ORR polarization curves of PtNC/ZTC and Pt/C-TKK in the absence (solid line) and presence (dotted line) of 0.1 M of CH3OH at a rotation rate of 1600 rpm in (a) alkaline and (b) acid media. | ||
The durability of PtNC/ZTC was also investigated by the amperometric technique. The test was performed at a constant voltage of the half-wave potential in an O2-saturated alkaline medium and at 0.7 V in an O2-saturated acidic medium at a rotation rate of 1600 rpm (Fig. S15a and b†). The durability of the PtNC/ZTC catalyst in the alkaline medium was higher than that of Pt/C-TKK, exhibiting a 30% decrease compared to a 40% decrease of Pt/C-TKK in 5.5 h of ORR operation (Fig. S15a†). The higher durability of PtNC/ZTC compared to Pt/C-TKK in the alkaline medium may be due to the stabilization of PtNC by pore entrapment. In the acidic medium, however, PtNC/ZTC exhibited a 54% decrease in the initial current after 5.5 h of operation while a 33% decrease was observed in the case of Pt/C-TKK (Fig. S15b†). The decrease in ORR activity in the acidic medium may be due to the leaching out of tiny Pt nanoclusters in acid electrolyte from the ZTC micropores. To understand the decrease in the ORR durability with time, STEM measurements of PtNC/ZTC after 5.5 h of ORR operation were performed. In the alkaline medium, the STEM image of post-ORR PtNC/ZTC shows a slight change in the size of PtNC (Fig. S15c†) while the STEM image of PtNC/ZTC after ORR in the acidic medium exhibited sintering of PtNC into large particles with an average size of 30 nm (Fig. S15d†), resulting in a decrease of the ORR activity. In the alkaline medium, the decrease in ORR activity with time may be due to the oxidation of the ZTC support in KOH.44
We attributed the excellent ORR activity of PtNC/ZTC to the interplay between the following: (1) the structure of the Pt cluster possessing a high ratio of surface atoms that benefits the surface reactions,45–47 (2) the microporous 3D graphene-like structure of the ZTC support that enables easy access of O2 and electrolyte molecules to the active sites,48 and (3) the high conductivity and large accessible surface area of ZTC that facilitates the electron transfer.49–51
Conclusions
In conclusion, we have demonstrated a simple electrochemical approach to synthesize highly dispersed PtNC supported on ZTC as an excellent electrocatalyst for the ORR. The micropores of ZTC served as cages to impose a spatial limitation on the size of the Pt clusters. The formed Pt nanoclusters exhibited enhanced ORR activity by improving the accessibility of active sites through the micropores of ZTC. The resultant PtNC/ZTC showed high ORR activity both in alkaline and acid media. The high ratio of surface atoms resulting from the cluster-like structure and high-surface-area ZTC with uniform microporosity is mainly responsible for the high electrocatalytic activity. The small size of the Pt cluster led to higher methanol tolerance, as compared to that of commercial Pt/C-TKK. Due to the entrapment of PtNC in the ZTC micropores, PtNC/ZTC exhibited high durability both in alkaline and acid media. The developed PtNC/ZTC catalyst will be useful for manufacturing high-performance alkaline anion-exchange membrane (AAEM) and proton-exchange membrane (PEM) fuel cell systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by IBS-R004-D1.Notes and references
- J. Hou, M. Yang, C. Ke, G. Wei and J. Zhang, Nanoscale, 2020, 12, 13858 RSC.
- N. K. Chaudhari, J. Joo, B. Kim, B. Ruqia, S.-I. Choi and K. Lee, Nanoscale, 2018, 10, 20073 RSC.
- C. H. Choi, K. Chung, T.-T. H. Nguyen and D. H. Kim, ACS Energy Lett., 2018, 3, 1415 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C.-j. Liu, J. Mater. Chem. A, 2017, 5, 1808 RSC.
- T. Zhang, S.-C. Li, W. Zhu, Z.-P. Zhang, J. Gu and Y.-W. Zhang, Nanoscale, 2017, 9, 1154 RSC.
- L.-L. Ling, W.-J. Liu, S.-Q. Chen, X. Hu and H. Jiang, ACS Appl. Nano Mater., 2018, 1, 3331 CrossRef CAS.
- S. Fu, C. Zhu, J. Song, P. Zhang, M. H. Engelhard, H. Xia, D. Du and Y. Lin, Nanoscale, 2017, 9, 1279 RSC.
- Y. Wang, K. Yin, J. Zhang, C. Si, X. Chen, L. Lv, W. Ma, H. Gao and Z. Zhang, J. Mater. Chem. A, 2016, 4, 14657 RSC.
- K. Singh, E. B. Tetteh, H.-Y. Lee, T.-H. Kang and J.-S. Yu, ACS Catal., 2019, 9, 8622 CrossRef CAS.
- X. Tian, J. Luo, H. Nan, H. Zou, R. Chen, T. Shu, X. Li, Y. Li, H. Song, S. Liao and R. R. Adzic, J. Am. Chem. Soc., 2016, 138, 1575 CrossRef CAS PubMed.
- K. Kamiya, R. Kamai, K. Hashimoto and S. Nakanishi, Nat. Commun., 2014, 5, 5040 CrossRef CAS PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-j. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397 CrossRef CAS PubMed.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089 CrossRef CAS PubMed.
- T. Imaoka, H. Kitazawa, W.-J. Chun and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9810 CrossRef CAS PubMed.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162 RSC.
- S. E. F. Kleijn, S. C. S. Lai, M. T. M. Koper and P. R. Unwin, Angew. Chem., Int. Ed., 2014, 53, 3558 CrossRef CAS PubMed.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583 RSC.
- J. Zhao, L. Ge, H. Yuan, Y. Liu, Y. Gui, B. Zhang, L. Zhou and S. Fang, Nanoscale, 2019, 11, 11429 RSC.
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058 CrossRef CAS PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- A. von Weber, E. T. Baxter, H. S. White and S. L. Anderson, J. Phys. Chem. C, 2015, 119, 11160 CrossRef CAS.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and A. Katz, Catal. Sci. Technol., 2017, 7, 4259 RSC.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li, M. Lei, C. Zhang, J. Irawan, L.-M. Liu and H. Wu, Nat. Commun., 2017, 8, 1490 CrossRef PubMed.
- S. Srivastava, J. P. Thomas, N. Heinig, M. Abd-Ellah, M. A. Rahman and K. T. Leung, Nanoscale, 2017, 9, 14395 RSC.
- A. Yadav, R. Pandey, T.-W. Liao, V. S. Zharinov, K.-J. Hu, J. Vernieres, R. E. Palmer, P. Lievens, D. Grandjean and Y. Shacham-Diamand, Nanoscale, 2020, 12, 6047 RSC.
- X. Zhao, Y. Hu, H. Jiang, J. Yu, R. Jiang and C. Li, Nanoscale, 2018, 10, 13384 RSC.
- L. L. Zhang, Y. Gu and X. S. Zhao, J. Mater. Chem. A, 2013, 1, 9395 RSC.
- E. N. Coker, W. A. Steen, J. T. Miller, A. J. Kropf and J. E. Miller, J. Mater. Chem., 2007, 17, 3330 RSC.
- H. Itoi, H. Nishihara, S. Kobayashi, S. Ittisanronnachai, T. Ishii, R. Berenguer, M. Ito, D. Matsumura and T. Kyotani, J. Phys. Chem. C, 2017, 121, 7892 CrossRef.
- Y. Kim, D. Lee, Y. Kwon, T.-W. Kim, K. Kim and H. J. Kim, J. Electroanal. Chem., 2019, 838, 89 CrossRef CAS.
- C. Jiang, K. Hara, K. Namba, H. Kobayashi, S. Ittisanronnachai, H. Nishihara, T. Kyotani and A. Fukuoka, Chem. Lett., 2014, 43, 1794 CrossRef.
- H. Itoi, H. Nishihara, S. Kobayashi, S. Ittisanronnachai, T. Ishii, R. Berenguer, M. Ito, D. Matsumura and T. Kyotani, J. Phys. Chem. C, 2017, 121, 7892 CrossRef.
- B. Geboes, J. Ustarroz, K. Sentosun, H. Vanrompay, A. Hubin, S. Bals and T. Breugelmans, ACS Catal., 2016, 6, 5856 CrossRef CAS.
- H. Lim, T. Nagaura, M. Kim, K. Kani, J. Kim, Y. Bando, S. M. Alshehri, T. Ahamad, J. You, J. Na and Y. Yamauchi, RSC Adv., 2020, 10, 8309 RSC.
- T. Imaoka, Y. Akanuma, N. Haruta, S. Tsuchiya, K. Ishihara, T. Okayasu, W.-J. Chun, M. Takahashi and K. Yamamoto, Nat. Commun., 2017, 8, 688 CrossRef PubMed.
- R. Kazan, U. Müller and T. Bürgi, Nanoscale, 2019, 11, 2938 RSC.
- S. Chauhan, G. J. Richards, T. Mori, P. Yan, J. P. Hill, K. Ariga, J. Zou and J. Drennan, J. Mater. Chem. A, 2013, 1, 6262 RSC.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058 CrossRef CAS PubMed.
- H. Park, S. K. Terhorst, R. K. Bera and R. Ryoo, Carbon, 2019, 155, 570 CrossRef CAS.
- G. Schmid, Endeavour, 1990, 14, 172 CrossRef CAS.
- J. D. Aiken and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef CAS.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162 RSC.
- W. Ding, H. Yu, Yu Tang, Z. Li, B. Liu, D. Liu, Y. Wang, S. h. Liu, H. Zhao and D. Mandler, Chem. Commun., 2020, 56, 6277 RSC.
- H. Lee, K. Kim, S.-H. Kang, Y. Kwon, J. H. Kim, Y.-K. Kwon, R. Ryoo and J. Y. Park, Sci. Rep., 2017, 7, 11460 CrossRef PubMed.
- C. O. Ania, V. Khomenko, E. Raymundo-Piñero, J. B. Parra and F. Béguin, Adv. Funct. Mater., 2007, 17, 1828 CrossRef CAS.
- K. Kim, T. Lee, Y. Kwon, Y. Seo, J. Song, J. K. Park, H. Lee, J. Y. Park, H. Ihee, S. J. Cho and R. Ryoo, Nature, 2016, 535, 131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and so on. See DOI: 10.1039/d0ra05654e |
| This journal is © The Royal Society of Chemistry 2020 |