
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFeS–biochar and Zn(0)–biochar for remediation of redox-reactive contaminants†
Yong-Deuk Seoa,
Seok-Young Oh *a,
Rajesh Rajagopalb and
Kwang-Sun Ryub
*a,
Rajesh Rajagopalb and
Kwang-Sun Ryub
aDepartment of Civil and Environmental Engineering, University of Ulsan, 93 Daehak-ro, Nam-gu, Ulsan 44610, South Korea. E-mail: quartzoh@ulsan.ac.kr; Fax: +82-52-259-2629; Tel: +82-52-259-2752
bDepartment of Chemistry, University of Ulsan, Ulsan 44610, South Korea
First published on 17th August 2020
Abstract
To enhance the removal of redox-reactive contaminants, biochars including FeS and Zn(0) were developed via pyrolysis. These biochars significantly promoted the removal of 2,4-dichlorophenol (DCP) by means of sorption and reduction. Compared to direct reduction with FeS and Zn(0), the formation of reduction intermediates and product was enhanced from 21% and 22% of initial DCP concentration to 41% and 52%, respectively. 2,4-Dinitrotoluene (DNT), chromate (CrO42−) and selenate (SeO42−) were also reductively transformed to reduction products (e.g., 2,4-diaminotoluene [DAT], Cr3+, and selenite [SeO32−]) after they sorbed onto the biochars including FeS and Zn(0). Mass recovery as DAT, Cr3+ and selenite was 4–20%, 1–3%, and 10–30% under the given conditions. Electrochemical and X-ray analyses confirmed the reduction capability of the biochars including FeS and Zn(0). Fe and S in the FeS–biochar did not effectively promote the reductive transformation of the contaminants. Contrastingly, the stronger reducer Zn(0) yielded faster reductive transformation of contaminants over the Zn(0)-containing biochar, while not releasing high concentrations of Zn2+ into the aqueous phase. Our results suggest that biochars including Zn(0) may be suitable as dual sorbents/reductants to remediate redox-reactive contaminants in natural environments.
1. Introduction
One option to remediate contaminants in natural environments is to immobilize contaminants via sorption onto a carbonaceous material such as activated carbon.1 Owing to its porous structure and high surface area, activated carbon has a strong affinity for contaminants by means of physical and chemical sorption processes such as intraparticular absorption, hydrophobic sorption, surface complexation, ligand exchange, and hydrogen bonding.2 Immobilization with activated carbon appears to be a useful option to decrease the mobility of contaminants in soils and sediments. However, immobilization of organic contaminants via sorption is not an ultimate destruction technology. Contaminants initially immobilized with activated carbon can be subsequently remobilized and released into the natural environment when disturbed by changing physical and chemical conditions such as excavation or introduction of acidic water. Therefore, it is necessary for immobilization to be combined with additional transformation processes that ultimately destroy the contaminants.Among destruction technologies, redox reactions of contaminants have been widely examined in recent decades for environmental remediation of water, soils, and sediments.3 Chemical oxidation with oxidants, including hydrogen peroxide, permanganate, ozone, and persulfate,4 and reductive transformation by non-toxic reductants, such as zero-valent iron [Fe(0)], have been intensively investigated as remediation processes for recalcitrant and redox-reactive contaminants in natural environments.5–7 Moreover, a sequential combination of reduction and oxidation processes has been shown to effectively transform refractory organic contaminants.8 To enhance chemical oxidation processes, the combination of chemical oxidants with other activation methods to generate strongly reactive radicals has also been intensively studied, in a strategy termed advanced oxidation processes (AOPs).9 AOPs can effectively degrade and mineralize recalcitrant organic contaminants in natural and engineered systems.10
Fe(0) reduction processes have become competitive options for treating oxidized organic contaminants owing to their low cost, high reduction potential, and nontoxic corrosion products.11 For some contaminants, however, the Fe(0) reduction process may be too kinetically slow. Some efforts have been made to promote reduction reactions by Fe(0), including bimetallic combinations with catalytic metals (Pd and Ni), and the use of nanoscale metals.12,13 Although these upgrading efforts can accelerate reduction rates, the syntheses of bimetals and nanoscale metals are too expensive, and in many cases nanometals agglomerate, which significantly worsens reaction rates. To prevent agglomeration of nanomaterials such as Fe(0) in the remediation of groundwater and soil, the use of carbonaceous materials such as activated carbon, carbon nanotubes, or biochar as embedding matrixes to separate the nanoparticles has been proposed.2,14–17
Previously, we proposed the inclusion of Fe(0) in biochar (in a material hereinafter referred to as Fe(0)–biochar) as a remediation material, composed of Fe(0) surrounded by biochar.18 Contaminants sorb onto the exterior biochar surface of this material and are subsequently reduced by electrons produced by the corrosion of the interior Fe(0) and transported via electron-conducting iron oxides (e.g., magnetite on Fe(0) surface) to the surface; this electron transfer is facilitated by infiltrated water molecules. Biochar plays the dual roles of electron transfer conduit and catalyst to promote electron transfer to sorbed organic molecules, proving that Fe(0)–biochar may be a competitive material to meet long-term remediation goals regarding redox-reactive organic molecules. It is possible that the electron transfer may not be fully responsible for the reductive transformation of sorbed contaminants. In addition to direct electron transfer, the sorbed contaminants can also be reductively transformed by hydrogen.5 Though the combination of biochar and Fe(0) was suitable for sorption and reduction of contaminants, one problem to overcome with the Fe(0)–biochar material was its somewhat slow reductive transformation of sorbed molecules due to the alkaline pH conditions arising from the basicity of the biochar and the corrosion of Fe. Alternatives to enhance the reduction reactions could be to change the core reductants or to improve the biochar properties, such as by adding chemical groups to improve electrical conductivity or add surface functionality (e.g., surface treatment with acids or oxidants).
Iron sulfide (FeS) and zero-valent zinc [Zn(0)] have been used as reductants to transform oxidized organic contaminants owing to their low redox potentials, and direct reduction by FeS and Zn(0) has been shown to transform redox-reactive contaminants in water.19,20 Formation of Fe2+ (E0 = 0.77 V for oxidation to Fe3+) and S2− (E0 = 0.24 V for oxidation of HS− to SO42−)21 from dissolution of FeS (FeS + H2O → Fe2+ + HS− + OH−) may be favorable for increasing reduction rates because both Fe2+ and HS− could work as reducing agents. Although Zn(0) has a lower reduction potential than Fe(0) (−0.76 vs. −0.44 V), the application of Zn(0) as a reductant has been limited due to the dissolution of Zn2+ produced by oxidation of Zn(0). However, in materials combining Zn(0) and biochar, the release of Zn2+ may be prevented by the surrounding biochar, which would reduce the exposure of the Zn2+ to aqueous environments.
Accordingly, in the present study we synthesized biochars including FeS and Zn(0) (hereinafter FeS–biochar and Zn(0)–biochar, respectively) as metal–biochar composites for environmental remediation. We hypothesized that FeS–biochar and Zn(0)–biochar may be good sorbents and reductants to accelerate the reductive transformation of redox-reactive contaminants. We characterized the physical and chemical properties of FeS–biochar and Zn(0)–biochar, and compared these with those of the previously synthesized Fe(0)–biochar.18 We determined the redox and catalytic properties of FeS–biochar and Zn(0)–biochar by means of cyclic voltammetry (CV) and X-ray absorption near edge structure (XANES) analyses. We tested 2,4-dinitrotoluene (DNT), 2,4-dichlorophenol (DCP), chromate (CrO42−), and selenate (SeO42−) as contaminants; these four contaminants are listed as priority pollutants by the United States Environmental Protection Agency and reductive transformation of these contaminants by reductants has been studied previously.14,15,22,23 We examined the kinetics and pathways of reductive removal of these contaminants in the presence of FeS–biochar and Zn(0)–biochar. Factors affecting the removal of the contaminants in the presence of FeS–biochar or Zn(0)–biochar were investigated and possible reduction mechanisms are discussed herein.
2. Materials and methods
2.1. Chemicals
FeS (−100 mesh, 99.9%) and elemental Zn [Zn(0), −100 mesh, >90%] were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and Kanto Chemicals (Tokyo, Japan), respectively. DNT (97%), 2,4-diaminotoluene (DAT, 98%), DCP (99%), 2-chlorophenol (2CP, 99%), 4-chlorophenol (4CP, 99%), phenol (98%), sodium chromate (Na2Cr2O4, 98%), sodium selenate (Na2SeO4, >95%), sodium selenite (Na2SeO3, 99%), HEPES (N-[2-hydroxyethyl]piperazine-N′-[ethanesulfonic acid]), and N-methyl-2-pyrrolidone (NMP, 99.5%) were purchased from Sigma-Aldrich. Methanol (high performance liquid chromatography [HPLC]-grade) was purchased from Burdick & Jackson (SK Chemicals, Ulsan, Korea). CaCl2·2H2O (>74%), Na2CO3 (>99%), NaHCO3 (>99.5%), and KOH (>85%) were provided by OCI (Seoul, South Korea), Daejung Chemical (Gyeonggi, South Korea), Junsei Chemical (Tokyo, Japan), and DC Chemical (Seoul, South Korea), respectively. All chemicals were used as received.2.2. Synthesis of FeS–biochar and Zn(0)–biochar
Both FeS–biochar and Zn(0)–biochar were synthesized by following a previously reported procedure.18 First, the purchased FeS or Zn(0) was dried in an oven at 105 ± 5 °C for 2 h and stored in a desiccator overnight. At the same time, rice straw collected from a rice farm in the city of Ulsan was dried in the same manner and pulverized to less than 2 mm length using a commercial electric grinder. The dried FeS or Zn(0) and ground rice straw were then mixed completely in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 volumetric ratio using a measuring cylinder. The resulting mixture was then pyrolyzed at 550 °C for 4 h using a tube-type electrical furnace under a 1000 cc min−1 flow of N2. After cooling to <50 °C, the pyrolyzed FeS–biochar or Zn(0)–biochar was removed and dried in a desiccator for at least 12 h. Properties of the synthesized FeS–biochar and Zn(0)–biochar were characterized by means of the following analyses: CHON composition, cation exchange capacity (CEC), pH, Brunauer–Emmett–Teller (BET) surface area, and point of zero charge (PZC); Table 1 lists the results. Scanning electron microscopy (SEM, JSM 600F, JEOL, Japan) images were acquired to determine surface morphology. X-ray diffraction (XRD, Ultima 4, Rigaku, Tokyo, Japan) and Fourier-transform infrared spectroscopy (FT-IR, Nicolet iS5 ThermoFisher Sci., Waltham, MA, USA) analyses were conducted to characterize the metal–biochar composites.
:95 volumetric ratio using a measuring cylinder. The resulting mixture was then pyrolyzed at 550 °C for 4 h using a tube-type electrical furnace under a 1000 cc min−1 flow of N2. After cooling to <50 °C, the pyrolyzed FeS–biochar or Zn(0)–biochar was removed and dried in a desiccator for at least 12 h. Properties of the synthesized FeS–biochar and Zn(0)–biochar were characterized by means of the following analyses: CHON composition, cation exchange capacity (CEC), pH, Brunauer–Emmett–Teller (BET) surface area, and point of zero charge (PZC); Table 1 lists the results. Scanning electron microscopy (SEM, JSM 600F, JEOL, Japan) images were acquired to determine surface morphology. X-ray diffraction (XRD, Ultima 4, Rigaku, Tokyo, Japan) and Fourier-transform infrared spectroscopy (FT-IR, Nicolet iS5 ThermoFisher Sci., Waltham, MA, USA) analyses were conducted to characterize the metal–biochar composites.
| pH | BET SAa (m2 g−1) | CECb (cmol kg−1) | PZCc | Elemental contentsd (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| C | H | O | N | S | |||||
| a BET surface area was analyzed under N2 using a nanoPOROSITY-XQ instrument (Mirae Scientific Instruments, Korea).b Determined by the method of Hesse.27c Determined by the method of Faria et al.28d Determined using the Vario EL Elemental Analyzer (Elementar, GmbH, Germany). | |||||||||
| FeS | 5.72 | 14.6 | 30.2 | 6.26 | 0.07 | 0.01 | 0.00 | 0.06 | 29.0 |
| FeS–biochar | 10.9 | 13.6 | 31.0 | 6.78 | 7.51 | 0.05 | 0.24 | 0.05 | 9.68 |
| Zn(0) | 9.48 | 10.6 | 14.9 | 10.6 | 0.16 | 0.01 | 1.99 | 0.05 | 0.21 |
| Zn(0)–biochar | 10.8 | 12.0 | 20.9 | 12.0 | 12.0 | 0.46 | 1.36 | 0.04 | 0.00 |
| Rice straw biochar | 9.10 | 16.7 | 3.10 | 8.20 | 56.1 | 2.80 | 12.7 | 1.90 | 0.00 |
2.3. Batch experiments
To determine the removal of contaminants by FeS–biochar or Zn(0)–biochar via sorption and reduction, batch experiments were conducted using 40 mL amber vials including 4 g of FeS–biochar or Zn(0)–biochar and 20 mL of contaminant solution. Using volumetric glass flasks (1 L) and high purity deionized water (<3 μS cm−1, Nexpower 1000 deionized system, Human Co., Seoul, South Korea), DNT, DCP, sodium chromate, and sodium selenate solutions were prepared with a magnetic stirrer in the laboratory. Before introduced into the amber vials, each solution was deoxygenated via purging with N2 for 30 min and its pH was controlled at 7.4 using 0.1 M HEPES buffer. The initial concentrations of DNT, DCP, chromate, and selenate were 49.5, 46.7, 36.1, and 61.2 mg L−1, respectively. At predetermined sampling times, duplicate vials were sacrificed and filtered through a 0.22 μm cellulose membrane filter (Millipore, MA, USA) for analytical determination of the contaminants and their reduction products in the aqueous phase by means of HPLC or ion chromatography (IC). For DNT and DCP, FeS–biochar or Zn(0)–biochar remaining on the filter was extracted twice with methanol (8 mL) to determine the sorbed concentrations of the contaminants and their reduction products.24,25 After the second extraction, an additional extraction with methanol did not meaningfully extract sorbed molecules. Similarly, for chromate and selenate determination, FeS–biochar or Zn(0)–biochar remaining on the filter was extracted twice with 1 M CaCl2. Reduction control experiments were conducted using samples of only FeS or Zn(0) under identical conditions. Considering the carbon contents of FeS–biochar and Zn(0)–biochar, sorption control experiments were also conducted.2.4. Chemical analysis
DNT, DCP, and their reduction products were analyzed using a Dionex UltiMate-3000 HPLC (Sunnyvale, CA, USA) equipped with a Dionex Acclaim 120 guard column (4.3 × 10 mm) and an Acclaim 120 C-18 column (4.6 × 250 mm, 5 μm). The analytical methods and conditions used for the quantification of DNT and DAT are described in detail elsewhere.26 For analysis of DCP and its reduction products, a methanol–water mixture (60/40, v/v) was used as the mobile phase and applied at the flow rate of 1.0 mL min−1. The wavelength of the UV detector was set to 224 nm and the injection volume used was 100 μL. The retention times of DCP, 4CP, 2CP, and phenol were 18.22, 8.89, 7.25, and 4.83 min, respectively. Chromate and selenate were analyzed using a Dionex ICS-100 ion chromatograph; the eluent used was a mixture of Na2CO3 (4.8 mM) and NaHCO3 (1.0 mM), the injection volume and flow rate were 10 μL and 1.5 mL min−1, respectively, and the suppressor current was set to 40 mA. Concentration of total Cr was determined using a UV-vis spectrophotometer (DR-2800, Hach Company, Loveland, CO, USA) according to the alkaline hypobromite oxidation method. Cr3+ concentration was determined by subtracting chromate concentration from total Cr concentration. To guarantee precision and accuracy of the obtained data, analytical duplicates, standards, and blank samples were used. Energy dispersive X-ray fluorescence (ED-XRF) spectrometry (SEA 1200 VX, Seiko, Chiba, Japan) was used to determine the inorganic contents of biochars.2.5. Electrochemical and spectroscopic analyses
CV and XANES analyses were conducted to determine the redox properties of FeS–biochar and Zn(0)–biochar. CV analysis was carried out using an IVIUMSTAT instrument with a three-electrode configuration. The working electrode was prepared under ambient conditions as follows. Active material, activated carbon, and polytetrafluoroethylene (PTFE) were mixed in the weight ratio of 80:1:1. A few drops of NMP solution were added to produce a homogeneous slurry. The slurry was drop-coated over acid-treated nickel foil at 100 °C. The resulting electrode was air dried overnight at 80 °C and then used as the working electrode. Thus, electrodes coated with active material were used as the working electrodes, a pure Pt strip was used as the counter electrode, and a saturated KCl-filled Ag/AgCl electrode was used as the reference electrode. Cyclic voltammograms of the electrodes were recorded between the potentials of −0.2 and −1.2 V at the scan rate of 10 mV s−1 in 6 M KOH aqueous electrolyte solution. X-ray absorption spectra for XANES analysis were determined at the Fe K-edge in transmission mode using a Si (1 1 1) double-crystal monochromator at the 10 C beam line at the Pohang Accelerator Laboratory in South Korea.
3. Results and discussion
3.1. Properties of FeS–biochar and Zn(0)–biochar
FeS showed acidic pH (5.72) and a high sulfide content (29%), indicating that reductive transformation could be favorable (Table 1). The synthesized FeS–biochar also had high sulfur content (9.68%), showing promise for reduction reactions. However, its pH was significantly increased to 10.9 due to the surrounding biochar; the FeS was carbonized and attached to the biochar after its co-pyrolysis with excess rice straw. Alkaline pH (9.10) of biochar strongly supported the increase of pH (Table 1). Compared with FeS, the BET surface area (13.6 m2 g−1), CEC (31.0 cmol kg−1), and PZC (6.78) did not change significantly. Similar to FeS–biochar (7.51% C), Zn(0)–biochar showed increased carbon content (12.0%) relative to the base Zn(0) material. Compared with Zn(0), the BET surface area (12.0 m2 g−1), CEC (20.9 cmol kg−1), and PZC (12.0) were slightly increased after co-pyrolysis with rice straw. Considering the carbon content in biochar (56.1%), the portion of biochar for FeS–biochar and Zn(0)–biochar was estimated to be 13.0% and 21.4%, respectively. ED-XRF analysis also showed high contents of Fe (73.5%), S (12.1%) in FeS–biochar and high Zn content (96%) in Zn(0)–biochar (Table S1 in ESI†). Interestingly, the Si and K contents were relatively high in FeS–biochar; these elements probably originated from the rice straw. Minor elements present were Ca, Cl, P, Mn, etc. SEM images of FeS–biochar and Zn(0)–biochar showed that the outside surfaces of FeS and Zn(0) were covered with biochar (Fig. S1†), which was confirmed by energy dispersive X-ray spectroscopy (EDX) analysis and elemental mapping (Fig. S2–S4†). However, some biochars having the form of rice straw chunks were also present because an excess amount of rice straw was added for co-pyrolysis with FeS and Zn(0). EDX analysis indicated that small particles of FeS or Zn(0) were possibly attached to these types of biochar (Fig. S2 and S3†). These results suggest that part of the FeS and Zn(0) in the FeS–biochar and Zn(0)–biochar systems may be directly exposed to the outside. X-ray powder diffraction analysis showed that FeS in FeS–biochar was mostly troilite and that Zn in Zn(0)–biochar were mostly ZnO (Fig. S5†). FT-IR spectra did not show clear stretch of carbon-containing functional groups due to low carbon contents in FeS–biochar and Zn(0)–biochar (Fig. S6†). Instead, FT-IR spectrum of rice straw biochar clearly indicated the existence of C–H bonds (at 2750–2950 cm−1), aromatic bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) (at 1600 cm−1), carboxylic bonds (COOH) (at 1700 cm−1), carbonyl bonds (CO) (at 1720–1730 cm−1), and phenolic bonds (C–O) (at 1200 cm−1).
C) (at 1600 cm−1), carboxylic bonds (COOH) (at 1700 cm−1), carbonyl bonds (CO) (at 1720–1730 cm−1), and phenolic bonds (C–O) (at 1200 cm−1).
CV was carried out to study the redox characteristics of pure FeS and FeS–biochar; the corresponding voltammogram is shown in Fig. 1a. FeS showed clear oxidation peaks at −800 and −0.48 V that respectively confirmed the reductions of Fe to Fe2+ and of Fe2+ to Fe3+. Similarly, we observed small reduction peaks at −1.00 and −0.47 V respectively corresponding to the conversions of Fe2+ to Fe3+ and Fe2+ to Fe. A strong reduction peak at −1.10 V arose due to the Fe to Fe2+ and Fe2+ to Fe3+ processes.18,29,30 Particularly, the oxidation peak at −0.48 V and reduction peak at −1.10 V indicate that the Fe2+ to Fe3+ reduction process occurred at the surface of iron sulfide.30,31 FeS–biochar showed a similar cyclic voltammogram to that of pure FeS. As shown in Fig. 1a, the addition of biochar to FeS shifted the oxidation negatively and the reduction positively; further, the oxidation and reduction peaks were much less intense than those of pure FeS. These results confirmed that the outside biochar fully covered the surface of FeS. The cyclic voltammograms of Zn(0) and Zn(0)–biochar shown in Fig. 1b showed a similar trend. In pure zinc, we observed oxidation peaks at −0.76 and −0.50 V, and a reduction peak at −0.47 V. These oxidation and reduction peaks confirmed the reduction of Zn in to Zn2+; specifically, the oxidation/reduction peak around −0.50 V was attributed to the Zn ↔ Zn2+ reaction.32,33 The outside biochar reduced the intensity of oxidation and reduction peak current for Zn(0)–biochar. Surprisingly, Zn(0)–biochar exhibited a strong reduction peak at −1.12 V due to the reduction of Zn2+ to Zn. This additional peak may have arisen due to the heat treatment of Zn(0) during co-pyrolysis of Zn(0) and rice straw.34
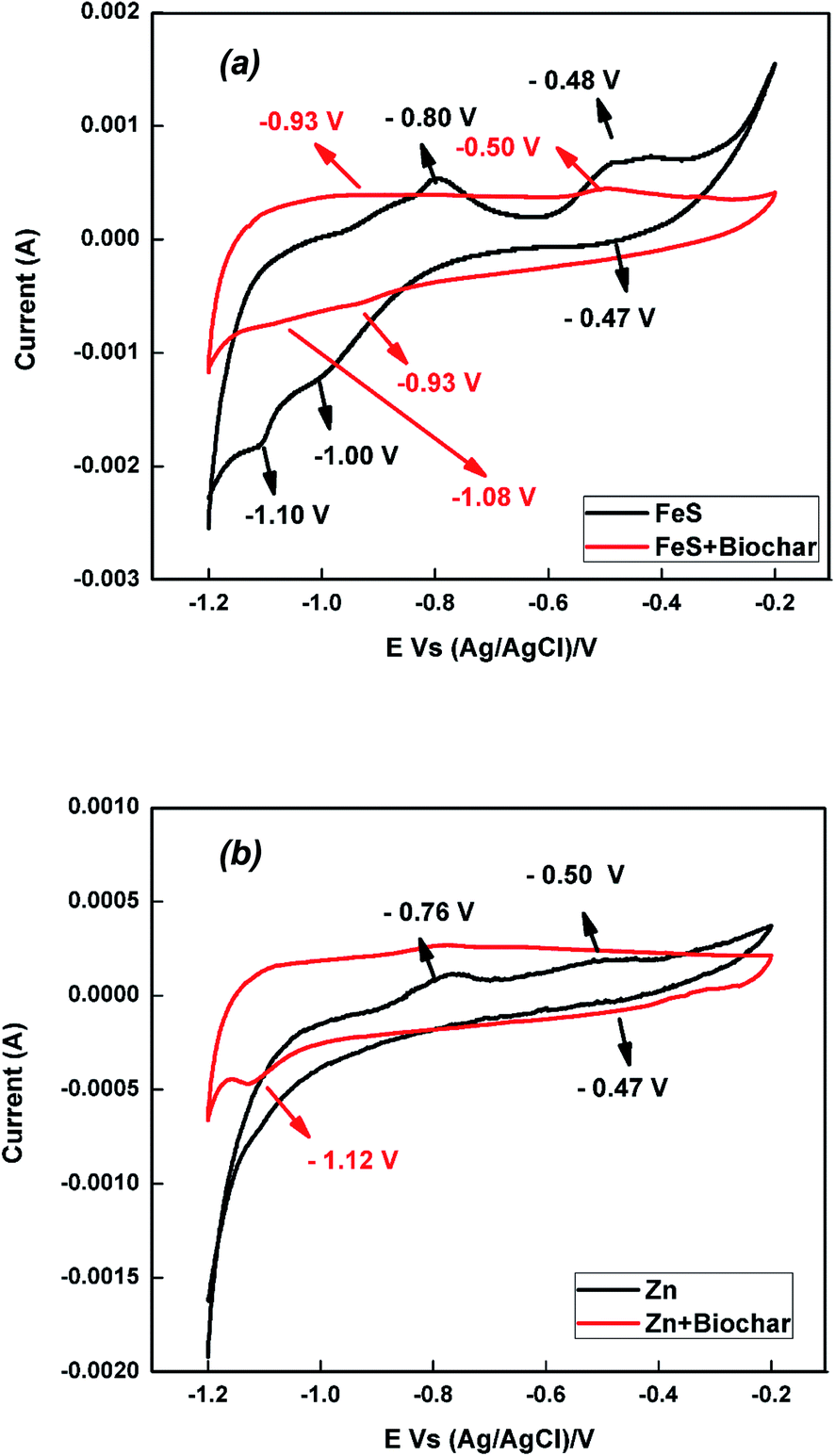 | ||
| Fig. 1 Cyclic voltammograms of (a) FeS–biochar and (b) Zn(0)–biochar. | ||
3.2. Removal of DNT by FeS–biochar and Zn(0)–biochar
In reduction control batches with FeS in systems buffered to pH 7.4, DNT was rapidly and completely removed in 120 min, showing mass recovery of approximately 70% as DAT (Fig. 2a). The incomplete mass recovery was probably due to strong sorption of DAT to the FeS surface or due to the presence of unanalyzed reduction intermediates such as 2-amino-4-nitrotoluene or 4-amino-2-nitrotoluene. Assuming that sufficient amount of FeS was existed, the pseudo-first-order rate was estimated using SimgaPlot (Systat Software Inc., San Jose, CA, USA). The pseudo-first-order rate was 0.096 ± 0.025 min−1 (R2 = 0.938), corresponding to a BET surface area–normalized rate of (3.29 ± 0.87) × 10−5 min−1·L m−2. The reduction rates were similar to previously reported reduction rates for Fe(0).35 In sorption control experiments with biochar under identical conditions, more than 95% of DNT was removed in 2 h from solution (data not shown). However, the reduction intermediates and product were not observed, suggesting that biochar may play a role of only sorbent. FeS–biochar also reductively transformed DNT to DAT. However, the removal was much slower than that for FeS, showing less than 35% of removal in 120 min (Fig. 2a). Considering the rapid removal of DNT by FeS, direct reduction by exposed FeS may not be dominant. However, possible direct reduction by exposed FeS could not be completely ruled out. Mass recovery as DAT slowly increased, and was about 4% at 120 min. Compared with DNT removal with Fe(0)–biochar under identical conditions,18 the reductive removal of DNT with FeS–biochar was somewhat slower, showing a removal rate two orders of magnitude slower than that of the Fe(0)–biochar system (0.003 ± 0.001 vs. 0.28 ± 0.01 min−1).18 This slow removal was probably due to the low carbon content in the metal–biochar composites (7.51% vs. 15.6%), resulting in less sorptive removal of DNT. Another possible explanation of the slow removal observed is that dissolution of FeS in FeS–biochar was limited due to the elevated pH arising from the outside biochar. | ||
| Fig. 2 Removal of DNT by (a) FeS–biochar and (b) Zn(0)–biochar. | ||
Zn(0) could also rapidly transform DNT to DAT (Fig. 2b). Within 10 min, DNT was completely removed and DAT rapidly formed as a reduction product, showing almost complete mass recovery. The reductive removal rate was 0.91 ± 0.05 min−1 (R2 = 0.998), one order of magnitude higher than that of FeS. DNT was also rapidly removed by Zn(0)–biochar; more than 95% of DNT was removed in 120 min, showing the removal rate of 0.175 ± 0.022 min−1 (R2 = 0.982). The removal rate was two orders of magnitude higher than that of FeS–biochar and similar to that of Fe(0)–biochar.18 The high carbon content (12.0%) and strong reduction potential (E0 = −0.76 V) may be attributed to the rapid removal of DNT by Zn(0)–biochar. The formation of DAT was also faster and greater in extent, accounting for about 20% of the initial mass of DNT at 120 min. Overall, FeS–biochar and Zn(0)–biochar were able to remove DNT via sorption and reduction to produce the reduction product DAT, though the extent and kinetics of DNT removal were to some extent different from those of Fe(0)–biochar. DNT was removed by Zn(0)–biochar as rapidly as by Fe(0)–biochar.
3.3. XANES analysis of FeS–biochar and Zn(0)–biochar
To determine whether FeS and Zn(0) in metal–biochar composites were oxidized with DNT, the status of metals in the metal–biochar composites before and after reaction were examined via XANES analysis (Fig. 3). FeS did not show any significant difference in this analysis before and after reaction with DNT, indicating no clear change in the status of Fe. This suggests that rather than the Fe, the S may have accounted for the reductive transformation of DNT in direct reduction by FeS. Contrastingly, spectra of FeS–biochar showed a slight change in the status of Fe. A Fe2+ peak at 7125–7127 eV was slightly shifted to a Fe3+ peak at 7130 eV, indicating that the Fe2+ in FeS was oxidized, and thus responsible for the reduction of DNT.36,37 Spectra of Zn(0) showed the sharp development of Zn2+ (ZnO) at 9661 eV after reaction with DNT, indicating the oxidation of elemental Zn (9659 eV).38–40 Unlike FeS–biochar, Zn(0)–biochar did not show any significant difference after reaction with DNT, implying that the exteriors of the Zn(0) particles were already oxidized before being surrounded by biochar during pyrolysis; this would explain why no additional Zn(0) oxidation in Zn(0)–biochar was clearly observed. It is likely that the removal of DNT with Zn(0)–biochar was mostly via sorption to outside biochar and that the sorbed DNT was further reduced to DAT via reduction by electrons produced from the oxidation of interior Zn(0).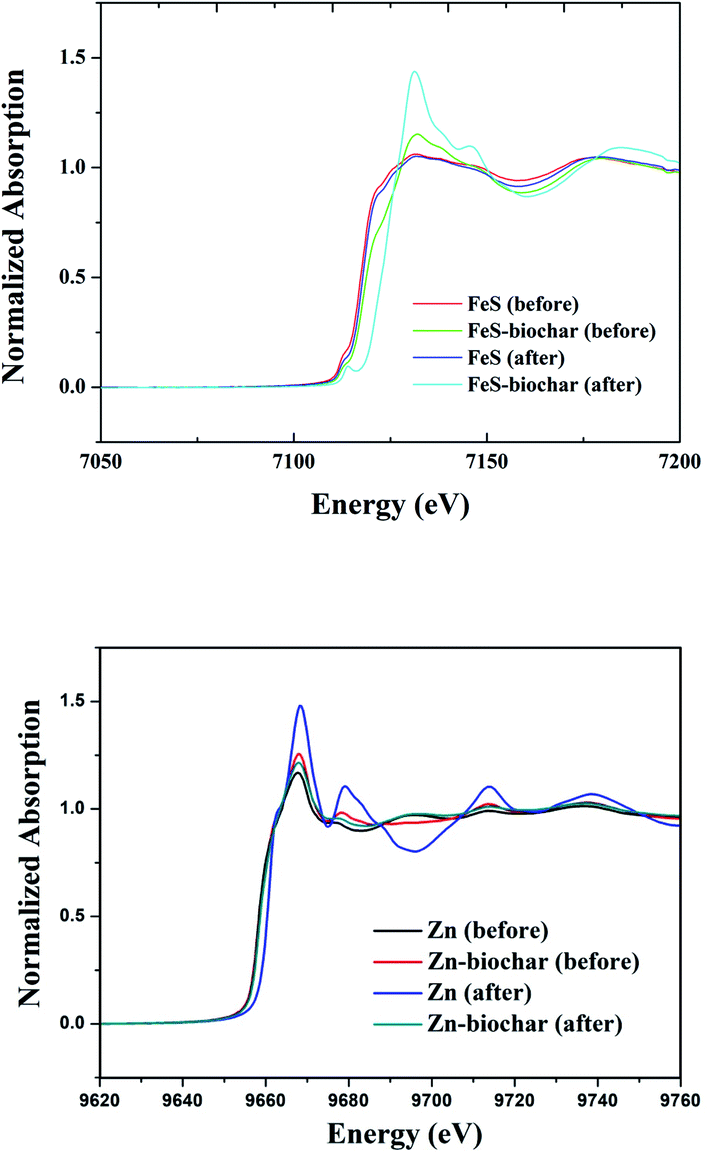 | ||
| Fig. 3 XANES spectra of FeS–biochar and Zn(0)–biochar. | ||
3.4. Removal of DCP by FeS–biochar and Zn(0)–biochar
Experiments on the removal of DCP by FeS–biochar and Zn(0)–biochar more clearly showed the role of biochar because the reduction rates of DCP with FeS and Zn(0) were much slower than those of DNT. In control with biochar under identical conditions, more than 99% of DCP was removed in 24 h. However, reduction daughter products were not formed (data not shown). In direct reduction of DCP with FeS, approximately 80% of the DCP was removed in 1 h with the production of two intermediates, 4CP and 2CP. A small amount of phenol (less than 3% of the initial DCP concentration) was also formed (Fig. 4a). It appears that DCP reduction was somewhat retarded after 5 h. It is likely that DCP was rapidly removed from solution mostly due to initial sorption to the FeS surface the early stage and that further transformation of the sorbed DCP was slow. The molar ratio of the two intermediates was approximately 1:1 in a control with FeS, different from the case for DCP reduction with Fe(0), which predominantly showed 4CP due to dechlorination of a chloro functional group in an ortho position.18 Similarly, FeS–biochar also showed ∼80% removal of DCP in 5 h (Fig. 4b). Two intermediates were formed, 4CP and 2CP, at 21% and 17% of the initial DCP concentration, respectively (Fig. 4b). The concentrations of the two intermediates were about 2 times higher than those in the FeS case, suggesting that the biochar form may somewhat accelerate the reductive transformation of DCP. A small amount of phenol (less than 3% of initial the DCP concentration) was also formed. Unlike the case for Fe(0)–biochar, for FeS–biochar the biochar promoted both ortho and para dechlorination pathways.
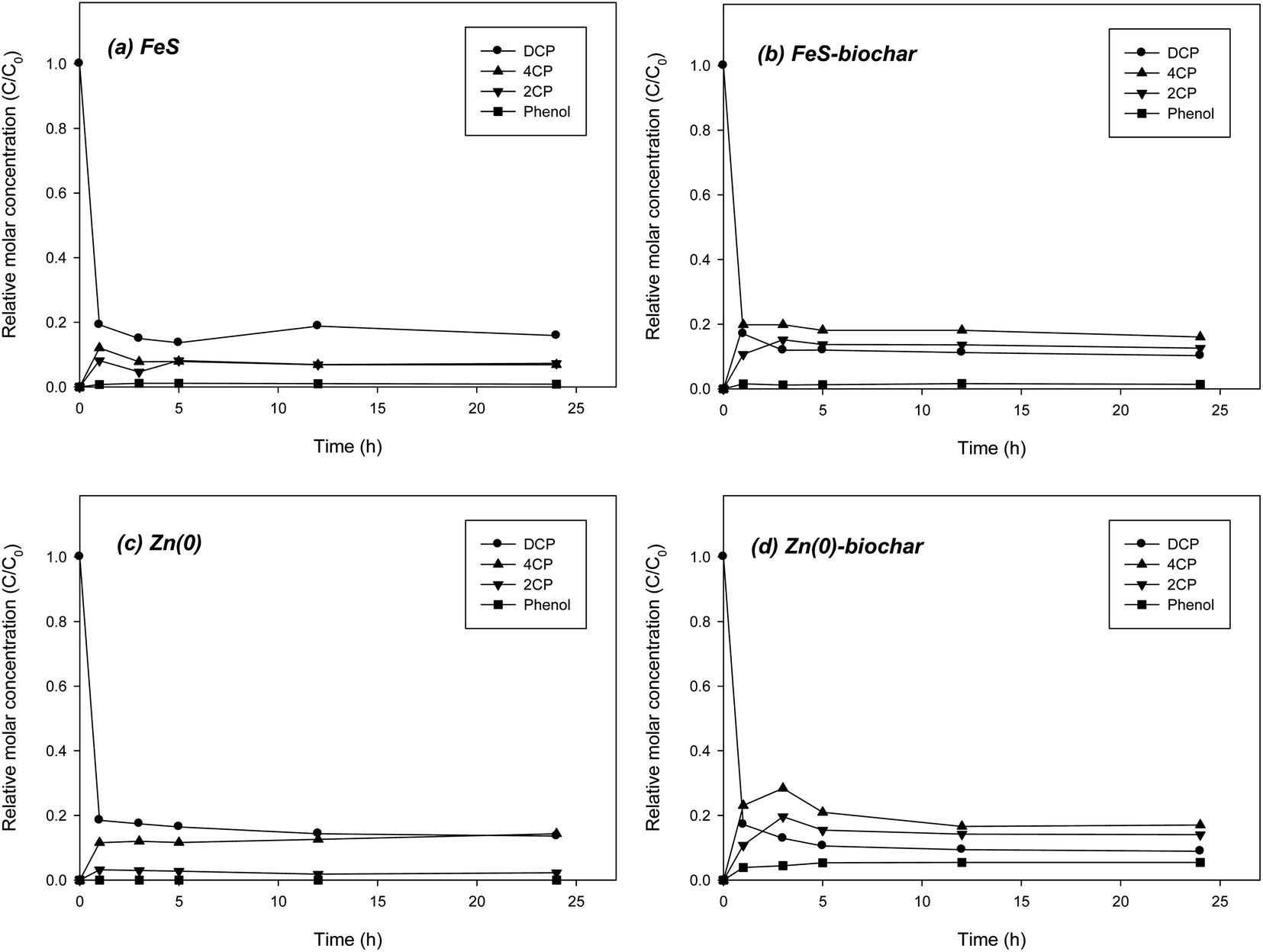 | ||
| Fig. 4 Removal of DCP by (a) FeS, (b) FeS–biochar, (c) Zn(0), and (d) Zn(0)–biochar. | ||
The reduction of DCP with Zn(0) was not rapid either. DCP rapidly moved from solution to the Zn(0) surface, but the reductive transformation of the sorbed molecules proceeded slowly, even after 3 h of reaction time (Fig. 4c). 4CP and phenol were formed as the dominant intermediate and product, respectively, similar to the pathways of DCP reduction with Fe(0).18 Like FeS–biochar, Zn(0)–biochar also enhanced the reductive transformation of DCP. The formation of 4CP and 2CP was significantly increased, showing 28% and 19% of initial DCP, respectively (Fig. 4d). The concentration of phenol was also slightly increased to 5% of initial DCP. These results supported the hypothesis that the addition of biochar to Zn(0) could accelerate the reduction of DCP. Similar to the case for FeS–biochar, dechlorination of the chloro functional groups in both ortho and para positions may have been promoted in the presence of biochar. Overall, the results from batch experiments showed that the addition of biochar to FeS and Zn(0) in the form of metal–biochar composites enhanced the reductive transformations of DNP and DCP, and that the biochar in the metal–biochar composites acted as a catalyst by promoting the transfer of electrons produced by corrosion of core metals to molecules sorbed on the biochar. Previous studies suggest that the catalytic role of biochar may be attributed to the electrical conductivity of its graphitic regions and oxygen-containing surface functional groups.18,41,42 Based on the reductive transformation of DNT and DCP with FeS–biochar and Zn(0)–biochar, the removal of redox-reactive organic contaminants by FeS–biochar and Zn(0)–biochar can be summarized by following steps. First, the contaminant is sorbed to the outside biochar surface of the metal–biochar complexes via possible sorption mechanisms (e.g., π–π electron donor acceptor interaction, hydrophobic sorption, electrostatic sorption, hydrogen bonding, etc.).43,44 Second, at the same time, core metals are corroded by permeated water molecules to generate electrons according to geochemical conditions such as pH and types of metal. Third, the formed electrons are transferred from the core metals to the contaminant sorbed to the outside biochar via the graphitic regions or oxygen-containing surface functional groups (e.g., phenol or carbonyl).41,42 Finally, reduction products are re-partitioned between the sorbed and aqueous phases. Following the suggested removal process, the redox-reactive organic contaminants can be removed from water and soil as well as further transformed to less toxic and more biodegradable compounds in natural environments. Due to many affecting factors, the kinetics of overall reaction cannot be easily predicted in field application of metal–biochar complexes, which still remains to be further explored.
3.5. Removal of chromate and selenate by FeS–biochar and Zn(0)-biochar
The catalytic role of biochar was also examined in the removal of chromate and selenate. Direct reduction of chromate by FeS was extremely rapid, showing complete removal in 5 h (Fig. 5a). Accordingly, Cr3+ was gradually formed as a reduction product. In 24 h, mass recovery as Cr3+ was 71%. Unlike the case for DCP, removal of chromate with FeS–biochar was slower than that with FeS (Fig. 5a). In 24 h, only 20% of the initial chromate was removed by FeS–biochar. It appears that sorptive removal of chromate onto FeS–biochar was not dominant despite the addition of biochar. It is likely that under buffered conditions at pH 7.4, electrostatic sorption between chromate and the biochar surface (PZC = 6.78; Table 1) was not favorable. Rather than electrostatic sorption, other sorption mechanisms may be involved, such as ligand exchange or surface complexation. As chromate sorbed to FeS–biochar, the sorbed chromate was gradually transformed to Cr3+, accounting for 6% of the initial chromate. Similar to the case for FeS and its biochar, the reduction of chromate by Zn(0)–biochar was also slower than that by pure Zn(0) (Fig. 5b). Direct reduction with Zn(0) yielded chromate removal of about 80% and complete mass recovery as Cr3+ at 24 h. Judging by the PZC of Zn(0)–biochar (12.0), electrostatic sorption of chromate onto the positively charged Zn(0)–biochar surface should be dominant. However, in Zn(0)–biochar systems, the reduction of chromate was significantly slowed and the formation of Cr3+ was extremely low (less than 1% of initial chromate). These results suggest that the initial sorption of chromate to Zn(0)–biochar (mostly exterior biochar) may not be electrostatic sorption. It should be noted that direct reduction by exposed FeS or Zn(0) cannot be completely ruled out as a means of reduction of chromate with FeS–biochar or Zn–biochar. Differently from a previous report,45 control experiments with biochar under identical conditions (data not shown) did not produce Cr3+, suggesting that the reduction capability of the biochar in the present study may not be strong enough for this reaction.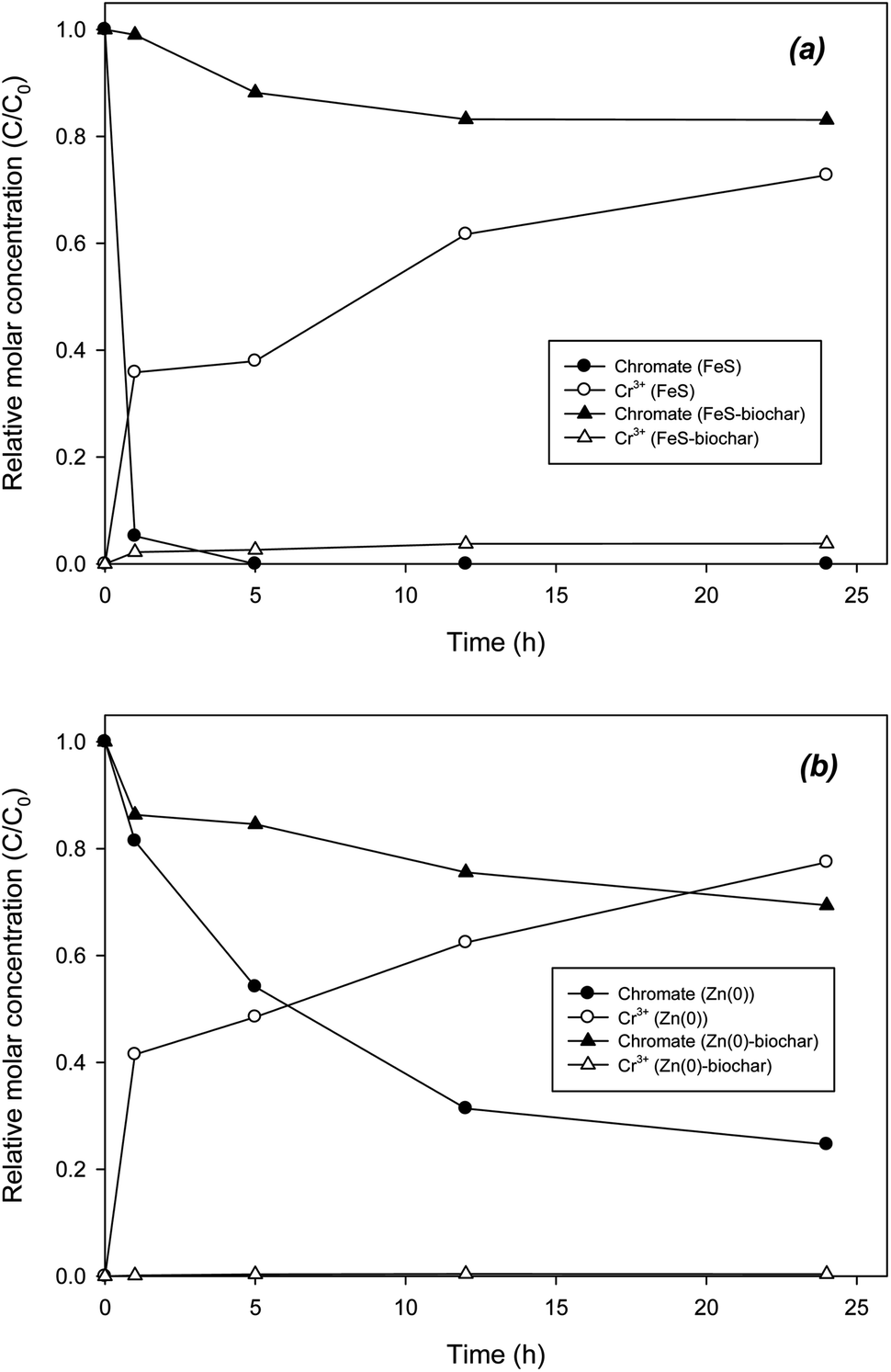 | ||
| Fig. 5 Removal of chromate by (a) FeS–biochar and (b) Zn(0)–biochar. | ||
Similar to chromate, selenate was reduced by FeS–biochar. In direct reduction with FeS, about 50% of the selenate was reductively removed to form selenite in 24 h (Fig. 6a). The sum of selenate and selenite concentrations was ∼90% of the initial selenate concentration at 24 h. The reductive transformation of selenate was also markedly delayed in the presence of FeS–biochar, showing 19% removal in 24 h. Selenite was accordingly formed, representing 10% of the initial selenate concentration. In control with biochar, selenite was not observed under the given conditions (data not shown). The reduction of selenate in the Zn(0) systems was somewhat different. Selenate was completely removed by Zn(0) within 12 h. Judging by the high PZC of Zn(0) (10.6; Table 1), the electrostatic sorption of selenate to Zn(0) could be favorable at pH 7.4. Another possible explanation for the rapid removal of selenate with Zn(0) may be its strong redox potential (E0 = −0.76 V) compared with FeS. The sorbed selenate was rapidly transformed to selenite. However, the selenite formed only accounted for ∼30% of the initial selenate at 1 h. Thereafter, the selenite concentration gradually decreased, indicating that unidentified products may have formed. According to previous studies, selenate could have transformed further in the presence of nanoscale iron and iron sulfide from selenite to elemental selenium (Se0) and selenide (Se2−).22,23,46,47 In control experiments with selenite and Zn(0), selenite was completely removed in 12 h, indicating that selenite was further reductively transformed by Zn(0) (Fig. S7†). Zn(0)–biochar did not show rapid removal of selenate, yielding only 20% removal in 24 h. Similar to the Zn(0) system, selenite was formed and gradually decreased until 24 h, suggesting that selenite was further reduced. Control experiments with selenite and Zn(0)–biochar confirmed the further transformation of selenite (Fig. S7†). Due to the low initial concentration of selenate, XPS analysis of Zn(0)–biochar did not identify other types of selenium reduction products (data not shown), which needs to be further examined in the future.
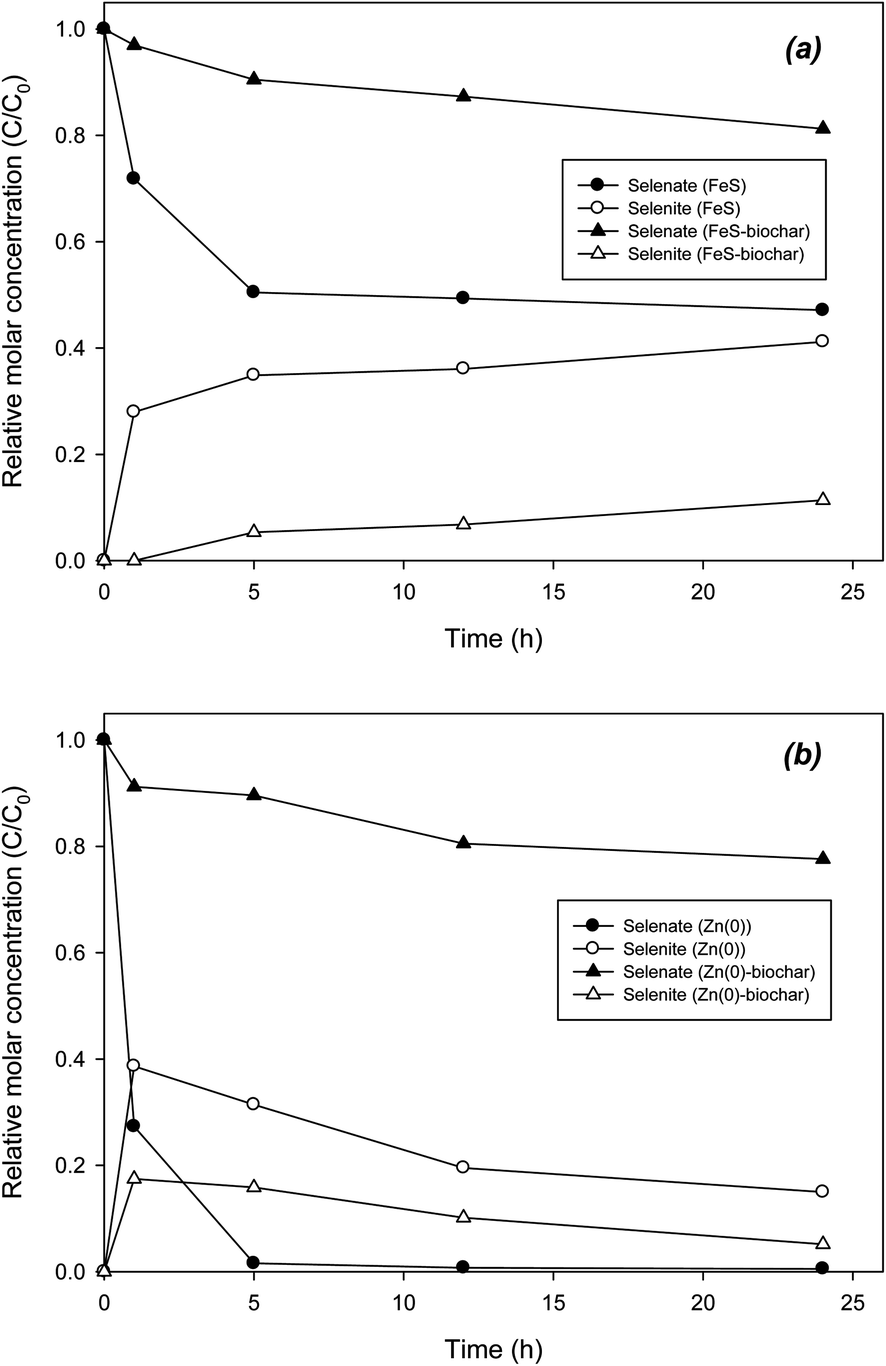 | ||
| Fig. 6 Removal of selenate by (a) FeS–biochar and (b) Zn(0)–biochar. | ||
Overall, the reduction of chromate and selenate with FeS–biochar and Zn(0)–biochar appears to be possible. However the transformation was very slow due to limited sorption onto the exterior biochar surface. Unlike the case for DCP, no catalytic role of biochar in promoting the reduction of chromate or selenate was clearly observed. Instead, similar to the case for DNT, the biochar in FeS–biochar or Zn(0)–biochar could have served as a conduit for electron transfer from corroding interior core metals to exterior sorbed chromates or selenates. We also conducted chromate and selenate reduction experiments with FeS–biochar and Zn(0)–biochar in the presence of DNT or DCP under identical conditions. The results did not show any significant effect of DNT or DCP upon the reductive removal of chromate and selenate (data not shown), suggesting that sorption mechanisms for DNT/DCP and chromate/selenate may differ. Thus, the addition of FeS–biochar or Zn(0)–biochar to contaminated water with co-dissolved DNT/DCP and toxic anions like chromate and selenate could be a possible long-term co-treatment remediation action in natural environmental systems.
It should be noted that the application of Zn(0) to in situ remediation may be of concern due to the possibility of release of Zn2+ to the natural environment. Accordingly, during batch experiments on DNT and DCP removal with Zn(0)–biochar, the Zn2+ concentration was monitored. In a control with Zn(0), the concentration of Zn2+ increased up to 12 and 3 mg L−1 respectively during DNT and DCP reductions (Fig. 7). However, Zn(0)–biochar showed significantly less release of Zn2+ to the aqueous phase, only 0.9–1.3 mg L−1 (Fig. 7), which meets drinking water standards (3 and 5 mg L−1 for South Korea and U.S., respectively). These results confirmed that outside biochar could immobilize the Zn2+ formed by oxidation of Zn(0). It remains to be examined how long the sorption of Zn2+ to biochar in Zn(0)–biochar will last.
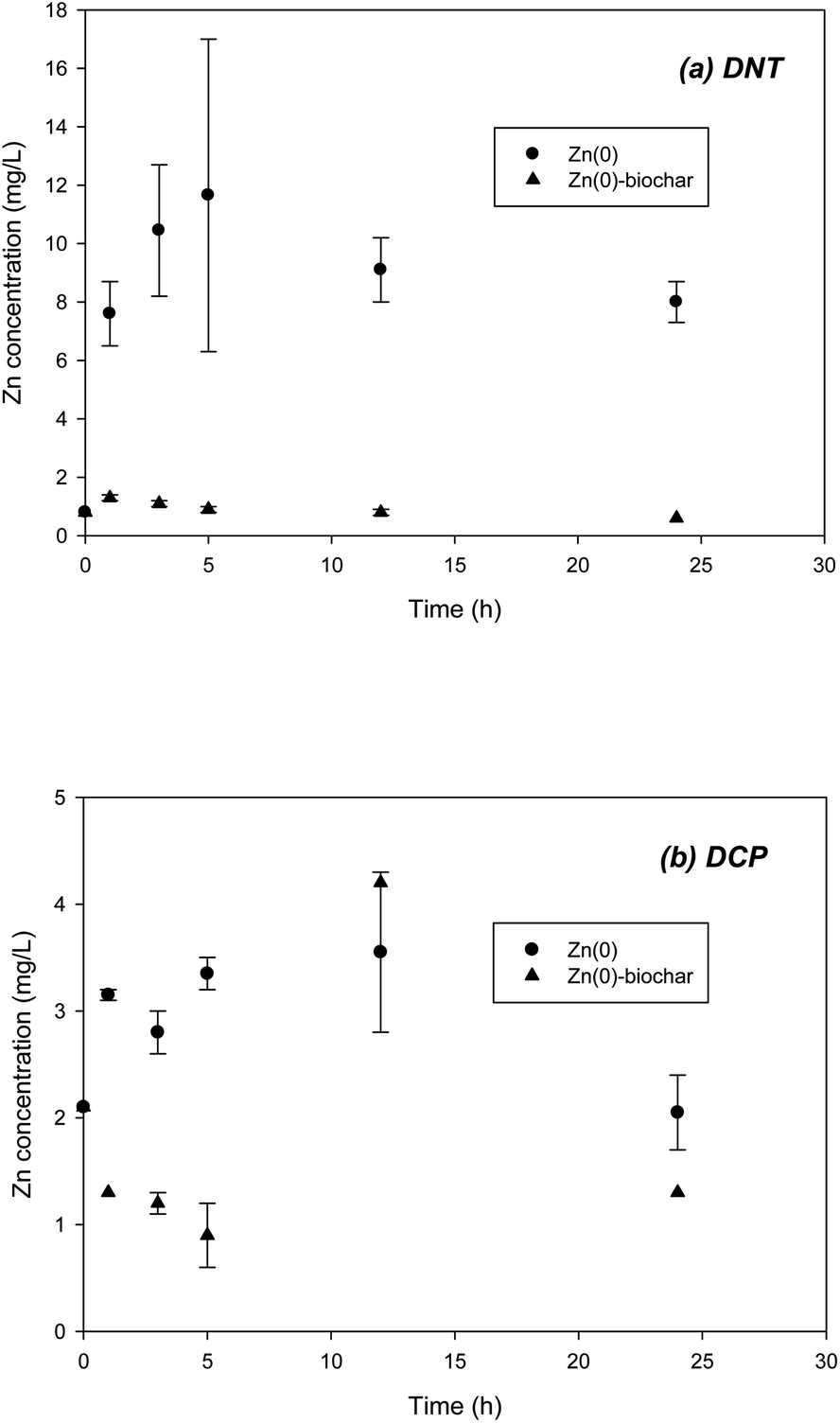 | ||
| Fig. 7 Release of Zn2+ from Zn(0)–biochar systems in the presence of (a) DNT and (b) DCP. | ||
4. Conclusions
Our results showed that novel FeS–biochar and Zn(0)–biochar can be used as combined sorbent/reductant materials to remove redox-reactive contaminants. Due to the rapid removal rates of DNT and the lower sorption capabilities of FeS–biochar and Zn(0)–biochar for DNT, the biochar materials did not clearly show an enhancement in DNT removal. However, a catalytic role of biochar in the metal–biochar composite materials was clearly observed in the removal of DCP. For the toxic anions, chromate and selenate, reductive removal by FeS–biochar or Zn(0)–biochar was limited and the sorbed chromate and selenate reduced to their reduction products slowly. Biochar in FeS–biochar or Zn(0)–biochar could be sorption and reduction sites for electron transfer from core metal corrosion to outside sorbed molecules. Regarding concerns about the environmental release of Zn2+, most Zn2+ remained sorbed to the exterior of the biochar in the short term. Our results suggest that Zn(0)–biochar could be a possible long-term in situ remediation option for natural and engineered systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea grant funded by the Korean government (MOE and MSIT) (2016R1D1A1B03931048, 2020R1A2C1010855). This work was also in part supported by the Korea Institute for Advanced of Technology (KIAT) grant funded by the Korea Government (MOTIE) (P0008421).References
- D. Lin, Y. M. Cho, J. P. Tommerdahl, D. Werner and R. G. Luthy, Bioturbation facilitates DDT sequestration by activated carbon against recontamination by sediment deposition, Environ. Toxicol. Chem., 2018, 37, 2013–2021 CrossRef CAS PubMed.
- S. Wong, N. Ngadi, I. M. Inuwa and O. Hassan, Recent advances in applications of activated carbon from biowaste for wastewater treatment: a short review, J. Cleaner Prod., 2018, 175, 361–375 CrossRef CAS.
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugan and S. Ramakrishna, A review on nanomaterials for environmental remediation, Energy Environ. Sci., 2012, 5, 8075–8109 RSC.
- A. Tsitonaki, B. Petri, C. Crimi, H. Mosbaek, R. L. Siegrist and P. L. Bjerg, In situ chemical oxidation of contaminated soil and groundwater using persulfate: a review, Critical Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- S. Y. Oh, D. K. Cha, B. J. Kim and P. C. Chiu, Effect of adsorption to elemental iron on the transformation of 2,4,6-trinitrotoluene and hexahydro-1,3,5-trinitro-1,3,5-triazine in solution, Environ. Toxicol. Chem., 2002, 21, 1384–1389 CrossRef CAS PubMed.
- R. Mukherjee, R. Kumar, A. Sinha, Y. Lama and A. K. Saha, A review on synthesis, characterization, and applications of nano zero valent iron (nZVI) for environmental remediation, Critical Rev. Environ. Sci. Technol., 2016, 46, 443–466 CrossRef CAS.
- P. K. Tandon and S. B. Singh, Redox processes in water remediation, Environ. Chem. Lett., 2016, 14, 15–25 CrossRef CAS.
- F. Chen, S. Zeng, J. Ma, Q. Zhu and S. Zhang, Degradation of para-nitrochlorobenzene by the combination of zero-valent Iron reduction and persulfate oxidation in soil, Water, Air, Soil Pollut., 2018, 229, 333 CrossRef.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Evaluation of advanced oxidation processes for water and wastewater treatment - A critical review, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- I. A. Ike, K. G. Linden, J. D. Orbella and M. Duke, Critical review of the science and sustainability of persulphate advanced oxidation processes, Chem. Eng. J., 2018, 338, 651–669 CrossRef CAS.
- J. Li, X. Dou, H. Qin, Y. Sun, D. Yin and X. Guan, Characterization methods of zerovalent iron for water treatment and remediation, Water Res., 2019, 148, 70–85 CrossRef CAS PubMed.
- F. He, Z. Li, S. Shi, W. Xu, H. Sheng, Y. Gu, Y. Jiang and B. Xi, Dechlorination of excess trichloroethene by bimetallic and sulfidated nanoscale zero-valent iron, Environ. Sci. Technol., 2018, 52, 8627–8637 CrossRef CAS PubMed.
- D. Jiang, G. Zeng, D. Huang, M. Chen, C. Zhang, C. Huang and J. Wan, Remediation of contaminated soils by enhanced nanoscale zero valent iron, Environ. Res., 2018, 163, 217–227 CrossRef CAS PubMed.
- H. Su, Z. Fang, P. E. Tsang, L. Zheng, W. Cheng, J. Fang and D. Zhao, Remediation of hexavalent chromium contaminated soil by biochar-supported zero-valent iron nanoparticles, J. Hazard. Mater., 2016, 318, 533–540 CrossRef CAS PubMed.
- H. Dong, J. Deng, Y. Xie, C. Zhang, Z. Jiang, Y. Cheng, K. Hou and G. Zeng, Stabilization of nanoscale zero-valent iron (nZVI) with modified biochar for Cr(VI) removal from aqueous solution, J. Hazard. Mater., 2017, 332, 79–86 CrossRef CAS PubMed.
- S. Mortazavian, H. An, D. Chun and J. Moon, Activated carbon impregnated by zero-valent iron nanoparticles (AC/nZVI) optimized for simultaneous adsorption and reduction of aqueous hexavalent chromium: Material characterizations and kinetic studies, Chem. Eng. J., 2018, 353, 781–795 CrossRef CAS.
- X. Liu, D. Lai and Y. Wang, Performance of Pb(II) removal by an activated carbon supported nanoscale zero-valent iron composite at ultralow iron content, J. Hazard. Mater., 2019, 361, 37–48 CrossRef CAS PubMed.
- S. Y. Oh, Y. D. Seo and K. S. Ryu, Reductive removal of 2,4-dinitrotoluene and 2,4-dichlorophenol with zero-valent iron-included biochar, Bioresour. Technol., 2016, 216, 1014–1021 CrossRef CAS PubMed.
- Y. Gong, J. Tang and D. Zhao, Application of iron sulfide particles for groundwater and soil remediation: a review, Water Res., 2016, 89, 309–320 CrossRef CAS PubMed.
- T. Tang, G. Lu, R. Wang, H. Chen, Y. Fang, K. Huang, J. Zheng, M. Zou, X. Tao, H. Yin and Z. Dang, Debromination of polybrominated diphenyl ethers (PBDEs) by zero valent zinc: Mechanisms and predicting descriptors, J. Hazard. Mater., 2018, 352, 165–171 CrossRef CAS PubMed.
- V. L. Snoyenk and D. Jenkins, Water Chemistry, John Wiley & Sons, New York, 1980 Search PubMed.
- M. Kang, F. Chen, S. Wu, Y. Yang, C. Bruggeman and L. Charlet, Effect of pH on aqueous Se(IV) reduction by pyrite, Environ. Sci. Technol., 2011, 45, 2704–2710 CrossRef CAS PubMed.
- I. H. Yoon, K. W. Kim, S. B. Bang and M. G. Kim, Reduction and adsorption mechanisms of selenate by zero-valent iron and related iron corrosion, Appl. Catal., B, 2011, 104, 185–192 CrossRef CAS.
- M. Y. Ahn, J. Dec, J. E. Kim and J. M. Bollag, Treatment of 2,4-dichlorophenol polluted soil with free and immobilized laccase, J. Environ. Qual., 2002, 31, 1509–1515 CrossRef CAS PubMed.
- S. Y. Oh, D. K. Cha and P. C. Chiu, Graphite-mediated reduction of 2,4-dinitrotoluene with elemental iron, Environ. Sci. Technol., 2002, 36, 2178–2184 CrossRef CAS PubMed.
- S. Y. Oh, J. G. Son, O. T. Lim and P. C. Chiu, The role of black carbon as a catalyst for environmental redox transformation, Environ. Geochem. Health, 2012, 34, 105–113 CrossRef CAS PubMed.
- P. R. Hesse, A Textbook of Soil Chemical Analysis, London, John Murry, 1971 Search PubMed.
- P. Faria, J. Órfão and M. Pereira, Adsorption of anionic and cationic dyes on activated carbons with different surface chemistries, Water Res., 2004, 38, 2043–2052 CrossRef CAS PubMed.
- P. Periasamy, B. R. Babu and S. V. Iyer, Cyclic voltammetric studies of porous iron electrodes in alkaline solutions used for alkaline batteries, J. Power Sources, 1996, 58, 35–40 CrossRef CAS.
- E. Bura-Nakić, D. Krznarić, G. R. Helz and I. Ciglenečki, Characterization of iron sulfide species in model solutions by cyclic voltammetry. Revisiting an old Problem, Electroanalysis, 2011, 23, 1376–1382 CrossRef.
- K. Winkler and T. Krogulec, The study of electrode processes of Fe (II)-thiosulphate complexes on mercury electrodes, J. Electroanal. Chem., 1995, 386, 127–134 CrossRef.
- C. Ascensão, L. Ciríaco, M. J. Pacheco and A. Lopes, Metal recovery from aqueous solutions, Port. Electrochim. Acta, 2011, 29, 349–359 CrossRef.
- N. Pootrakulchote, C. Reanprayoon, J. Gasiorowski, N. S. Sariciftci and P. Thamyongkit, A polydiacetylene–nested porphyrin conjugate for dye-sensitized solar cells, New J. Chem., 2015, 39, 9228–9233 RSC.
- H. Shirdel, F. Marandi, A. Jalilzadeh, S. Pourbeyram, S. Huber and A. Pfitzner, Syntheses, structures and properties of a new compound of the type [Zn (4,4′-dmo-2,2′-bpy) 2(CH3COO)] 2[Zn(SCN)4]·H2O with zinc in two cationic and one anionic complexes, Main Group Chem., 2015, 14, 105–114 CAS.
- S. Y. Oh, P. C. Chiu and D. K. Cha, Reductive transformation of 2,4,6-trinitrotoluene, hexahydro-1,3,5-trinitro-1,3,5-triazine, and nitroglycerin by pyrite and magnetite, J. Hazard. Mater., 2008, 158, 652–655 CrossRef CAS PubMed.
- M. Abuín, A. Serrano, J. Chaboy, M. A. García and N. Carmona, XAS study of Mn, Fe and Cu as indicators of historical glass decay, J. Anal. At. Spectrom., 2013, 28, 1118–1124 RSC.
- S. P. Singh, K. Vogel-Mikuš, I. Arcon, P. Vavpetic, L. Jeromel, P. Pelicon, J. Kumar and R. Tuli, Pattern of iron distribution in material and final tissue in wheat grains with contrasting levels of iron, J. Exp. Bot., 2013, 64, 3249–3260 CrossRef CAS PubMed.
- J. P. Veiga and M. O. Figueiredo, A XANES study on the structural role of zinc in ancient tile glazes of Portuguese origin, X-Ray Spectrom., 2008, 37, 458–461 CrossRef CAS.
- M. J. Akhtar, M. Nadeem, S. Javaid and M. Atif, Cation distribution in nanocrystalline ZnFe2O4 investigated using x-ray absorption file structure spectroscopy, J. Phys. Condens. Matter, 2009, 21, 405303 CrossRef CAS PubMed.
- A. K. Adhikari and K. S. Lin, Synthesis, fine structural characterization, and CO2 adsorption capacity of metal organic frameworks-74, J. Nanosci. Nanotechnol., 2013, 13, 1–9 CrossRef PubMed.
- S. Chen, A. E. Rotaru, P. M. Shrestha, N. S. Malvankar, F. Liu, W. Fan, K. P. Nevin and D. R. Lovley, Promoting interspecies electron transfer with biochar, Sci. Rep., 2014, 4, 5019 CrossRef CAS PubMed.
- T. Sun, B. D. A. Levin, J. J. L. Guzman, A. Enders, D. A. Muller, L. T. Angenent and J. Lehmann, Rapid electron transfer by the carbon matrix in natural pyrogenic carbon, Nat. Commun., 2017, 8, 14873 CrossRef CAS PubMed.
- X. Xiao, B. Chen, Z. Chen, L. Zhu and J. L. Schnoor, Insight into multiple and multilevel structures of biochars and their potential environmental applications: a critical review, Environ. Sci. Technol., 2018, 52, 5027–5047 CrossRef CAS PubMed.
- J. J. Pignatello, W. A. Mitch and W. Xu, Activity and reactivity of pyrogenic carbonaceous matter toward organic compounds, Environ. Sci. Technol., 2017, 51, 8893–8908 CrossRef CAS PubMed.
- L. Zhou, Y. Liu, S. Liu, Y. Yin, G. Zeng, X. Tan, X. Hu, X. Hu, L. Jiang, Y. Ding, S. Liu and X. Huang, Investigation of the adsorption-reduction mechanisms of hexavalent chromium by ramie biochars of different pyrolysis temperatures, Bioresour. Technol., 2016, 218, 351–359 CrossRef CAS PubMed.
- L. Liang, W. Yang, X. Guan, J. Li, Z. Xu, J. Wu, Y. Huang and X. Zhang, Kinetics and mechanisms of pH-dependent selenite removal by zero valent iron, Water Res., 2013, 47, 5846–5855 CrossRef CAS PubMed.
- L. Ling, B. Pan and W. X. Zhang, Removal of selenium from water with nanoscale zero-valent iron: mechanisms of intraparticle reduction of Se(IV), Water Res., 2015, 71, 274–281 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Information on XRF, SEM, EDX, XRD, and FT-IR analyses of FeS–biochar and Zn(0)–biochar, and selenite reduction by Zn(0) and Zn(0)-biochar is available. See DOI: 10.1039/d0ra05571a |
| This journal is © The Royal Society of Chemistry 2020 |