
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-assisted synthesis of mesoporous calcium carbonate with a controlled specific surface area and its potential to ferulic acid release
Kazunori Kadota a,
Toi Ibeb,
Yuto Sugawarab,
Hitomi Takanob,
Yus Aniza Yusofcd,
Hiromasa Uchiyamaa,
Yuichi Tozukaa and
Shinya Yamanaka*b
a,
Toi Ibeb,
Yuto Sugawarab,
Hitomi Takanob,
Yus Aniza Yusofcd,
Hiromasa Uchiyamaa,
Yuichi Tozukaa and
Shinya Yamanaka*b
aDepartment of Formulation Design and Pharmaceutical Technology, Osaka University of Pharmaceutical Sciences, 4-20-1 Nasahara, Takatsuki, Osaka 569-1094, Japan
bDivision of Applied Sciences, Muroran Institute of Technology, Mizumoto-cho 27-1, Muroran 050-8585, Japan. E-mail: syama@mmm.muroran-it.ac.jp
cDepartment of Process and Food Engineering, Faculty of Engineering, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
dLaboratory of Halal Services, Halal Products Research Institute, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
First published on 28th July 2020
Abstract
A carbonation process to control the specific surface area of mesoporous calcium carbonate and the dissolution profile of ferulic acid on mesoporous carbonate particles are presented. The effects of water content on the physicochemical properties, specific surface area, pore size, crystallinity, and morphology are evaluated. Mesoporous calcium carbonate particles are synthesised with well-controlled specific surface areas of 38.8 to 234 m2 g−1. Each of the submicron-size secondary particles consists of a primary particle of nano-size. During secondary particle formation, primary particle growth is curbed in the case with less water content. By contrast, growth is promoted via dissolution and recrystallisation in the presence of water. The release rates of ferulic acid are gradually enhanced with increasing specific surface area of the mesoporous calcium carbonate, that reflects crystallinity of ferulic acid.
Introduction
Porous inorganic materials have received considerable attention due to the fascinating characteristics of biological stability and controlled release properties of macromolecules. Several porous substances such as silica,1 synthetic zeolite,2 hydroxyapatite,3 and calcium carbonate4 are used as host porous particles for controlled drug (guest) release. Several attempts to control the dissolution profiles of bioactive compounds such as polyphenols and flavonoids have been made using porous substances.5 One method is to improve the dissolution rate of a bioactive compound with poor water-soluble properties by maintaining it in the amorphous state. This strategy enlarges the interfacial surface area between bioactive compounds and the release medium since the dissolution rate has a linear relationship with the specific surface area and the concentration difference between the surface of the dissolving bioactive compound and the medium follows the Noyes–Whitney equation.6 By contrast, incorporation or encapsulation of a bioactive compound into a porous substance may realise its sustained release.7 Particularly, calcium carbonate has been employed as a pharmaceutical excipient and a food additive. It is used in solid dosage forms as a diluent, a buffering agent, and a dissolution aid.8 In recent years, porous calcium carbonate has been studied for encapsulation purposes because calcium carbonate may control the different crystal forms and several morphologies such as needle-like aragonite, rhombic calcite morphologies, and the unstable vaterite.9Ferulic acid (FA) is a family of phenolic acids that are abundant in rice, wheat, and vegetables.10 FA has recently received much attention due to a wide range of health-promoting properties, including antioxidant and anti-inflammatory activities. In addition, it has potential to prevent Alzheimer's disease.11 Despite the attractive pharmacological effects of FA, its bioavailability is not optimal due to the limited aqueous solubility.12 However, its solubility could be improved by inclusion in macromolecules with a huge specific surface area (e.g., nanopores).
Among various porous materials, calcium carbonate has promise as a host particle due to its biodegradability and biocompatibility.13 Although porous calcium carbonate has been reported as a host particle for controlled release of macromolecules, it has a relatively low specific surface area. The specific surface area of porous calcium carbonate host particles is about 10 m2 g−1.14 This value is one or two orders of magnitude smaller than that of other porous inorganic materials (several hundred to thousand m2 g−1).
Recently, R. Sun et al., synthesized amorphous calcium carbonate (ACC) with a specific surface area of ca. 360 m2 g−1.15 The porous ACC was evaluated as a potential drug host particle. The drug release rates were significantly enhanced for itraconazole and celecoxib. They clearly demonstrated the possibility of calcium carbonate for various applications where the surface area is important.15 To clarify the effect of the host surface area on the release rate of a loaded substance with a poor solubility, the production of a well-controlled and wide-ranged specific surface area of calcium carbonate remains a challenge.
To date, there are two main processes to produce porous calcium carbonate. One is surface crystallisation on templates.16 The other is non-classical crystallisation via colloidal intermediates.17 We have presented a scalable production strategy for mesoporous calcium carbonate by means of a modified industrial carbonation process.18,19 ACC is initially formed and then stepwise transformations or the ageing stage (ACC → vaterite → calcite) takes place. The ageing stage is critical for mesopore formation where self-assembled nanostructure formation occurs via Brownian aggregation of nanoscale building units in a colloidal intermediate dispersion of ACC. Based on this mechanism, it can be concluded that the water content in the ACC dispersion plays a crucial role in the transformation from vaterite to calcite.19 Hence, the water content has potential to control the specific surface area of mesoporous calcium carbonate.
Herein we report the effect of the water content on the specific surface area of mesoporous calcium carbonate during the ageing process. In addition, mesoporous calcium particles are applied to FA release. The physicochemical properties of calcium carbonate are evaluated, including pore size, specific surface area, crystallinity, and morphology. Then the dissolution profile of FA from mesoporous calcium carbonate is evaluated when applying the porous calcium carbonate to control the release behaviour of bioactive compounds such as polyphenols.
Results and discussion
Table 1 shows the particle properties. The specific surface area and pore structure of the resultant calcium carbonate particles were highly dependent on the water content. The specific surface area of a prepared particle without water addition was 234 m2 g−1, which was consistent with that of our previous study (207 ± 10 m2 g−1).18 As the added amount of water increased from 0.0 to 1.0, 5.0, 6.0, and 7.0 wt%, the specific surface area decreased to 150, 81.5, 55.4, and 38.8 m2 g−1, respectively. Fig. 1 shows the N2 adsorption/desorption isotherm, and pore size distribution of calcite particles. The average pore size increased to 8.63 nm (0 wt%), 14.8 nm (1.0 wt%), 20.7 nm (5.0 wt%), 28.1 nm (6.0 wt%), and 39.7 nm (7.0 wt%).| Sample | Watera [wt%] | SSAb [m2 g−1] | Pore volume [cm3 g−1] | Average pore size [nm] | Equivalent diameterc [nm] | Crystallite sized [nm] | Secondary sizee [nm] |
|---|---|---|---|---|---|---|---|
| a This ratio is the added weight of water to the total weight of the dispersion. Note that the expected stoichiometric amount of water is in accordance with the following reaction: Ca(OH)2 + CO2 → CaCO3 + H2O. Reaction mass of water is 1.2 wt%, stoichiometrically.b Specific surface area.c Equivalent spherical diameter is equal to 6/(ρp × SSA), where ρp = 2.7 g cm−3 is the density of calcite.d Calculated from the full width at half maximum (FWHM) of the corrected diffraction profile. A pseudo-Voigt fitting is conducted to obtain the FWHM for the (104) calcite diffraction.e Number mean diameter of 100 particles and its population standard deviation is from each SEM image. | |||||||
| Calcite 1 | 0 | 234 | 0.470 | 8.63 | 9.5 | 15.5 | 430 ± 60 |
| Calcite 2 | 1.0 | 150 | 0.555 | 14.8 | 15 | 18.5 | 430 ± 70 |
| Calcite 3 | 5.0 | 81.5 | 0.422 | 20.7 | 27 | 22.5 | 430 ± 60 |
| Calcite 4 | 6.0 | 55.4 | 0.389 | 28.1 | 40 | 22.5 | 410 ± 60 |
| Calcite 5 | 7.0 | 38.8 | 0.385 | 39.7 | 57 | 23.3 | 420 ± 70 |
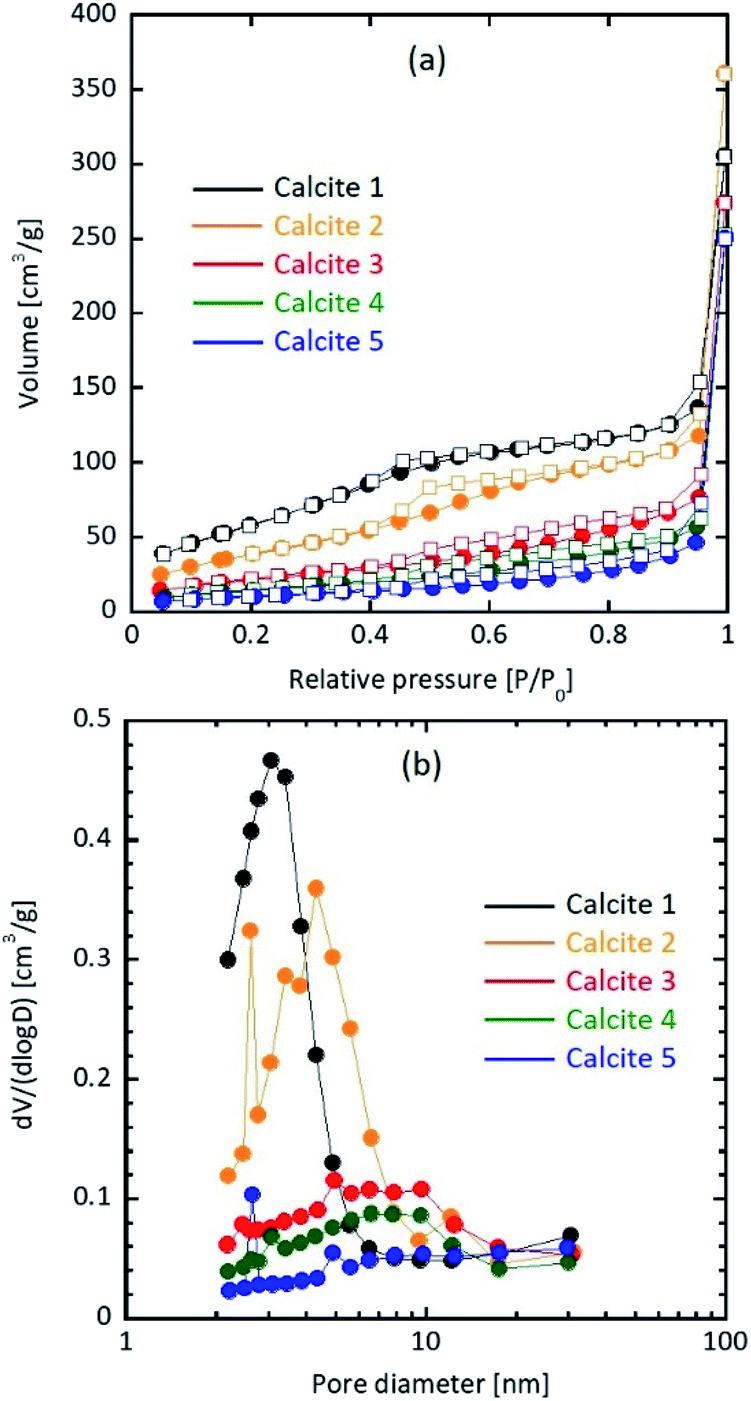 | ||
| Fig. 1 (a) N2 adsorption (circle)/desorption (square) isotherm and (b) pore size distribution of calcite particles. | ||
Because crystalline calcium carbonate polymorphs such as calcite, aragonite, and vaterite likely crystallise via unstable amorphous calcium carbonate (ACC),17,20 the polymorphs of the mesoporous calcium carbonate particles were determined. The peak positions of the XRD profiles agreed with that of the calcite (ICDD#05-0586) crystal for all samples (Fig. 2). The intensities of the calcite diffraction peaks increased as the water content increased, indicating that water promoted crystal growth. We then calculated the crystallite size based on Scherrer's equation. The calculated crystallite sizes of calcite (104) were 15.5 nm (0 wt%), 18.5 nm (1.0 wt%), 22.5 nm (5.0 wt%), 22.5 nm (6.0 wt%), and 23.3 nm (7.0 wt%) (Table 1). That is, the size gradually increased with increasing water content. These sizes were roughly consistent with each equivalent spherical diameter, as shown in Table 1. The equivalent diameter was equal to 6/(ρp × SSA), where ρp = 2.7 g cm−3 is the density of calcite and SSA is each specific surface area from the Brunauer–Emmett–Teller (BET) measurement.
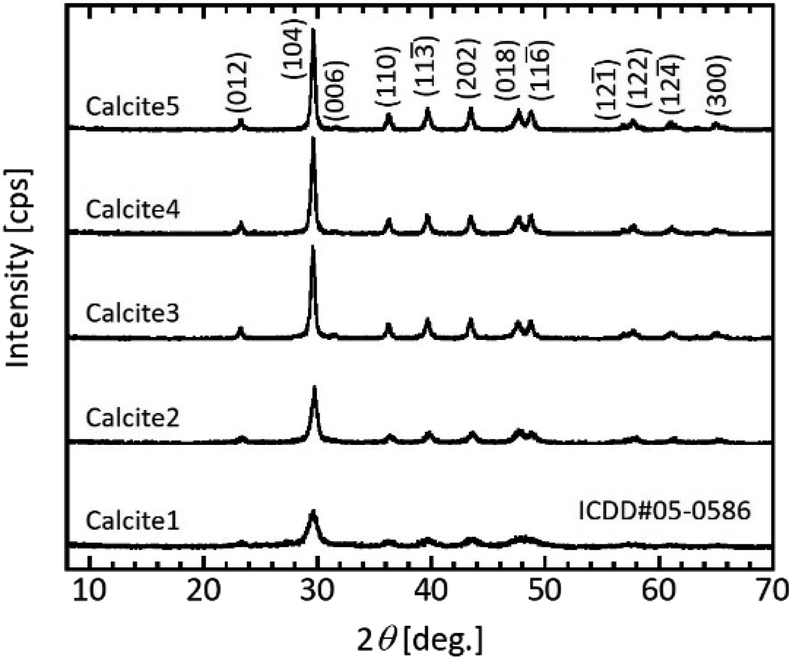 | ||
| Fig. 2 Typical XRD patterns of the prepared particles after the ageing treatment. Sample obtained with and without water addition. Labelling refers to Table 1. | ||
Generally, the initially formed ACC was immediately transformed into calcite via vaterite.21 In addition, the crystal was grown via dissolution and re-crystallisation of calcium carbonate.16 In the water additive-free condition, water was produced by the following reaction: Ca(OH)2 + CO2 → CaCO3 + H2O. Because the carbonation process in this study contained a small amount of water, the dissolution and re-crystallisation of calcite crystal (i.e., crystal growth) hardly occurred. On the other hand, water addition should promote crystal growth.
Fig. 3 shows typical SEM images of the mesoporous calcium carbonate particles with water contents of 0–7.0 wt%. All samples formed an ellipsoidal shape with a similar size of 430 ± 60 nm (0 wt%), 430 ± 70 nm (1.0 wt%), 430 ± 60 nm (5.0 wt%), 410 ± 60 nm (6.0 wt%), and 420 ± 70 nm (7.0 wt%) (Table 1).
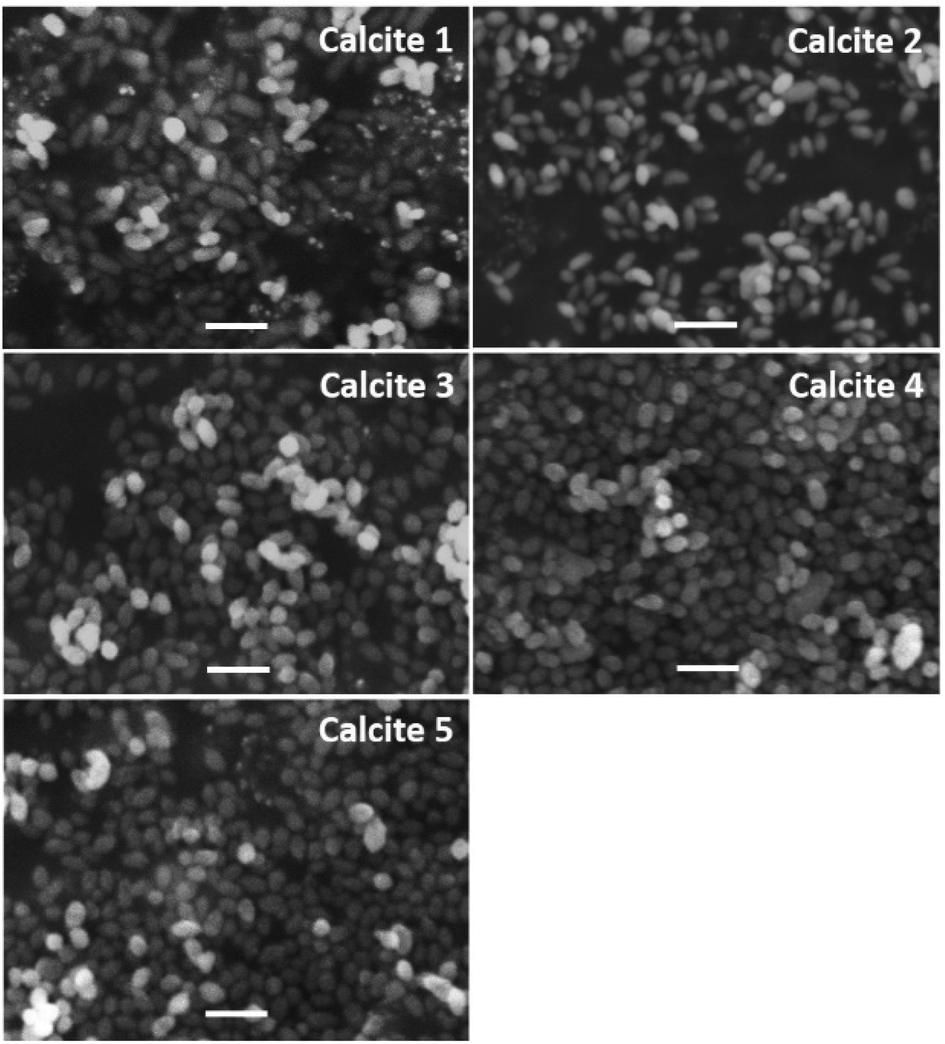 | ||
| Fig. 3 Typical SEM images of porous calcium carbonate particles obtained with various amounts of additional water. Scale bar is 1 μm. | ||
Since the particle size from the SEM images in Fig. 3 differed from its crystallite size and calculated equivalent size, the pores in mesoporous calcium carbonate were formed by the void spaces between the primary particles. The typical TEM images suggest the primary particle size depended on the water addition (Fig. 4). The primary size of calcite 1 (∼10 nm), calcite 3 (20–30 nm) and calcite 5 (∼50 nm) is consistent with each equivalent diameter listed in Table 1. During secondary particle formation, primary particle growth was curbed in the case with less water content. By contrast, growth was promoted via dissolution and recrystallisation in the presence of water.
 | ||
| Fig. 4 Typical TEM images of the prepared mesoporous calcium carbonate. (a–c) Calcite 1, (d–f) calcite 3 and (g–i) calcite 5. Labelling refers to Table 1. | ||
Fig. 5 schematically illustrates the effect of water addition on pore formation of porous calcium carbonate particles. The ACC in the colloidal dispersion aggregated was associated with the van der Waals attractive force (Fig. 5a) because the electrostatic interactions were negligible in such a relatively low dielectric constant medium.22 Simultaneously, the phase transition from unstable ACC to stable calcite occurred (Fig. 5b). In the last stage of particle formation, primary calcite crystals were growing to a larger size via dissolution and the re-crystallisation process. When the system included less water, the small primary calcite particles formed as secondary particles (Fig. 5c). As the water content increased, secondary particles consisted of the larger primary particles (Fig. 5d). As mentioned above, the pores in secondary particles were the void spaces between the primary particles. Therefore, the pore size depended on the primary particle size, suggesting that the larger the primary particles, the larger the pore size (magnified images of Fig. 5c and d).
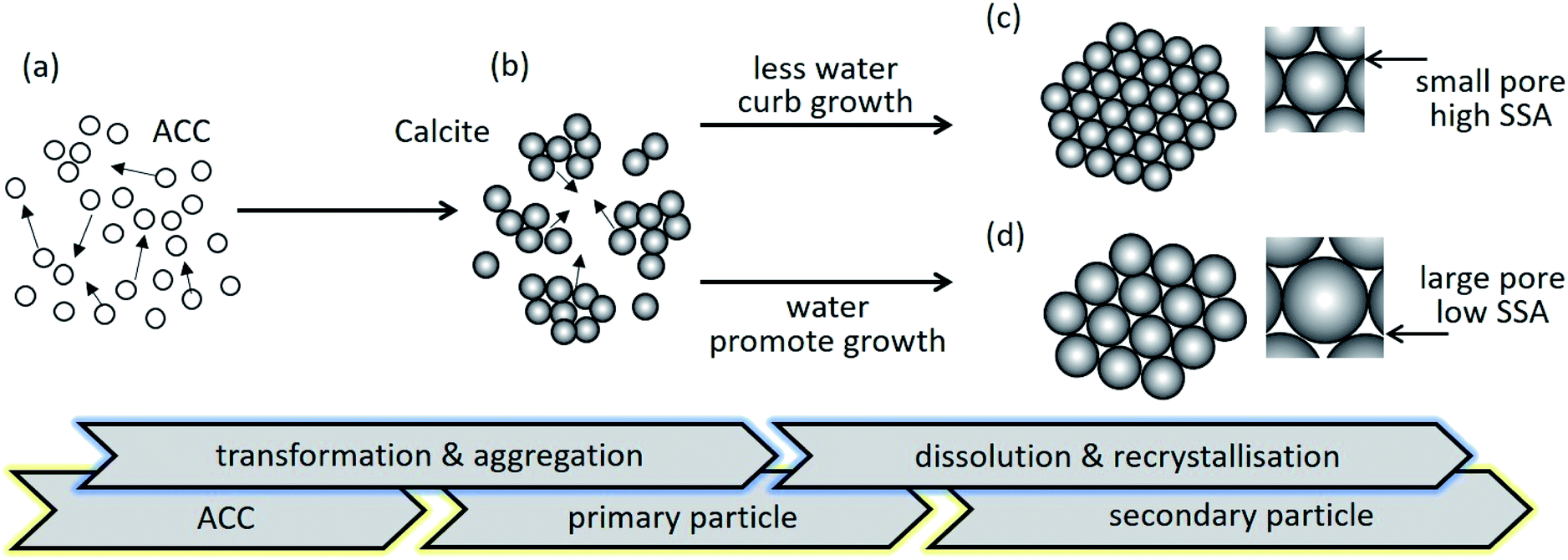 | ||
| Fig. 5 Schematic illustration for mesoporous particle formation. (a) Precursor ACC aggregates due to the attractive force. (b) After ageing the ACC dispersion for a predetermined amount of time, the primary ACC particles transform to calcite crystal and aggregate. (c) In the case of less water content, the slightly grown particles transform into a more preferable self-assembly to form ellipsoidal structures. (d) In the presence of water, secondary particles with a larger primary size of particles are formed via dissolution and recrystallisation. | ||
Because the state of compounds in porous substances affects the compound's solubility and the dissolution rate, the dissolution profiles of bioactive compounds must be controlled. The states of loaded compounds in mesoporous calcium carbonate were examined using 1-NPA as a fluorescence probe.
1-NPA, which is a representative compound, exhibited different fluorescence spectra due to varying crystalline and molecular states.23 Fig. 6 shows the fluorescence emission spectra of the 1-NPA crystal and 1-NPA-loaded calcium carbonate with five different properties. The emission peak of 1-NPA was around 430 nm, while samples with 1-NPA–loaded calcium carbonate showed a blue-shifted emission spectrum. As the specific surface area of calcite decreased, the redshift increased gradually. This may be due to the slow crystallisation of 1-NPA on calcite. In particular, most of 1-NPA-loaded calcite 1 and 2 existed in the molecular state in the porous calcium carbonate, while several portions of 1-NPA loaded in other calcite samples existed in the crystal state on the porous calcium carbonate.
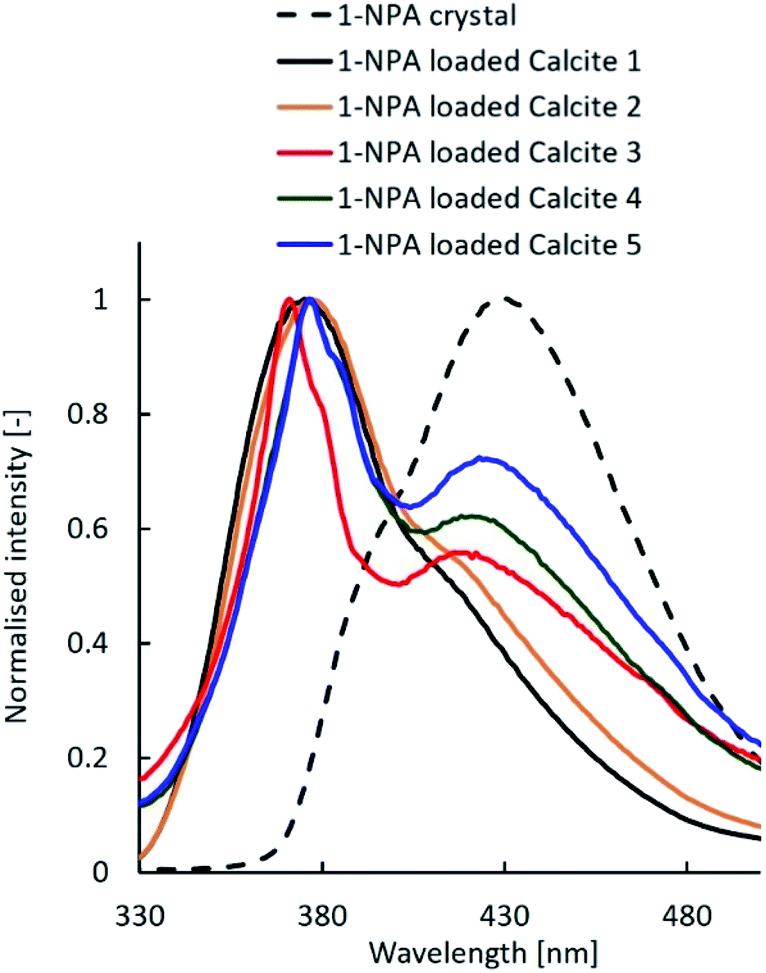 | ||
| Fig. 6 Fluorescence emission spectra of the 1-NPA crystal and 1-NPA–loaded calcium carbonate with five different properties. | ||
Previous studies reported that the dissolution rate of poorly water-soluble compounds from porous substances could be improved by decreasing the crystallinity, including the amorphous state, increasing the contact area between compounds and the dissolution media, and enhancing the wettability of the formulations.24 Fig. 7 shows the dissolution profiles of untreated FA alone and the FA-loaded porous calcium carbonates. Although both samples had the same amount of FA, their properties differed. The dissolution rate of the FA crystal was about 60% after 120 min. On the other hand, FA was immediately and completely released within 5 min for FA-loaded calcite 1, while FA was gradually and completely released over 120 min for FA-loaded calcite 2. There were several differences between calcite 1 and calcite 2 (Table 1). In particular, the specific surface area and the average pore size may influence the release behaviour of FA from porous calcium carbonates.25 The three other formulations, which were similar, did not achieve a 100% release rate of FA. Several portions of FA loaded in other calcites might have existed in the crystal state on the porous calcium carbonate as explained in Fig. 6.
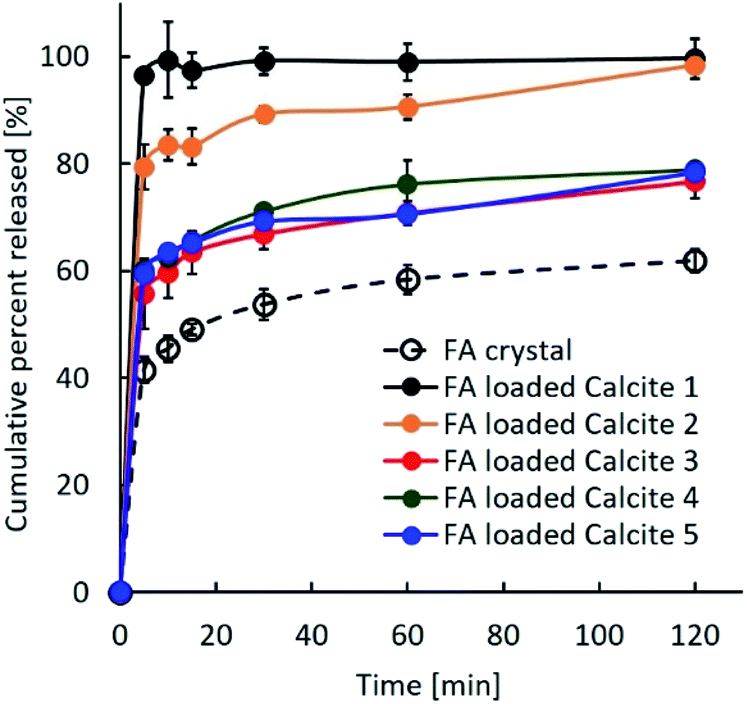 | ||
| Fig. 7 Dissolution profiles of untreated crystal FA and the FA-loaded porous calcium carbonates with different properties. | ||
We believe that the porous calcium carbonate presented here may be a promising carrier material of drugs and bioactive compounds due to its characteristic superstructure and very high specific surface area.
Experimental
Materials
Calcium hydroxide (Nacalai Tesque, Kyoto, Japan), ethylene glycol (Kanto Chemical, Tokyo, Japan), ethanol (Kanto Chemical, Tokyo, Japan), ferulic acid (FA; Tokyo Chemical Industry, Tokyo, Japan), and 1-naphthoic acid (1-NPA; Wako Pure Chemical Industries, Osaka, Japan) were used as received. All other chemicals and solvents were reagent or high-performance liquid chromatography grade.Synthesis of porous calcium carbonate
Mesoporous calcium carbonate was prepared using our original carbonation method.18,19,26 This method consists of two stages: the preparation stage of colloidal dispersion and the ageing stage.In the preparation stage of the colloidal dispersion, calcium hydroxide was carbonated in an ethanol–ethylene glycol organic solvent mixture with 70 wt% ethanol. After 25.0 g of calcium hydroxide was mixed with 475.0 g of the mixture in a reactor, mixed gas (CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 = 30:70 vol%) flowed into the mixture at a rate of 1.0 L min−1, while stirring at a rate of 400 rpm. A water bath controlled the carbonation reaction temperature at 20 °C. Afterwards, the suspension was centrifuged at 3540g for 20 min to remove the unreacted calcium hydroxide. The resulting supernatant was transparent in which colloidal amorphous calcium carbonate was dispersed.18 We used the colloidal dispersion for the subsequent ageing stage.
:N2 = 30:70 vol%) flowed into the mixture at a rate of 1.0 L min−1, while stirring at a rate of 400 rpm. A water bath controlled the carbonation reaction temperature at 20 °C. Afterwards, the suspension was centrifuged at 3540g for 20 min to remove the unreacted calcium hydroxide. The resulting supernatant was transparent in which colloidal amorphous calcium carbonate was dispersed.18 We used the colloidal dispersion for the subsequent ageing stage.
To demonstrate the effect of moisture on the particle properties, especially the specific surface area of porous calcium carbonate, the amount of water content prior to the ageing stage was regulated as 0, 1.0, 5.0, 6.0, or 7.0 wt% distilled water against the total amount of the colloidal dispersion. After the washing and drying processes, the resultant dry powder was used in the characterisation and FA release tests.
Loading of FA and 1-NPA into porous calcium carbonate
Herein 1-NPA was used as a model fluorescence compound to study the molecular state of 1-NPA on porous calcium carbonate.27,28 Evaporation was used to load 1-NPA into 200 mg of porous calcium carbonate. 1-NPA crystals (50 mg) were dissolved in 300 mL of ethanol in a round bottom flask. Then the ethanol and water were removed by rotary evaporation (Rotavapor R-3; Nihon Büchi) to obtain evaporated 1-NPA particles under reduced pressure at 50 °C.For the solvent-evaporated solid dispersions, 50 mg of FA was dissolved in 1 mL of ethanol. To this solution, 200 mg of porous calcium carbonate at a specified drug to calcium carbonate ratio (1:4) was added. Afterwards, the ethanol and water were removed by rotary evaporation (Rotavapor R-3; Nihon Büchi) to obtain evaporated particles of FA on calcium carbonate under reduced pressure at 50 °C.
Characterisation of porous calcium carbonate
The specific surface area and pore size distribution of the resultant calcium carbonate samples were determined by nitrogen gas adsorption based on the multi-point BET method and the BJH method, respectively. Both analyses were conducted on Autosorb-1-c/MK2 (Quantachrome, USA). Prior to the measurements, the samples were degassed for 2 h at 200 °C under a vacuum to remove adsorbed solvent molecules. X-ray diffraction (XRD, MultiFlex, Rigaku, Japan) powder patterns of the samples were obtained with CuKα radiation (40 kV, 40 mA) to determine the polymorphs of calcium carbonate and the crystallite size. SEM and TEM studies were performed using a JEOL JSM-6380A and JEOL JFM-2100F, respectively.Fluorescence spectroscopy was performed to investigate the incorporation of organic compounds into the porous calcium carbonate. An RF-5300 PC fluorescent spectrometer (Shimadzu, Kyoto, Japan) was used to measure the fluorescence spectra of the powder samples, which were placed in a solid sample holder attached to the instrument. The excitation wavelength was fixed at 330 nm. The widths of the excitation and emission slits were fixed at 5 and 10 nm, respectively.
FA release tests
Release studies of untreated FA and evaporated samples of FA with five different calcium carbonates were conducted in 900 mL with pH 6.8 phosphate buffer saline. All studies were performed in triplicate using the paddle method. Nearly 350 mg of FA was used for each test and dispersed in pH 6.8 phosphate buffer saline, which was stirred constantly at 50 rpm and maintained at a temperature of 37 ± 0.5 °C. Next, 2 mL samples were collected at 5, 10, 15, 30, 45, 60, 90, and 120 min. Each sample was immediately passed through 0.2 μm Millipore filter.The UV absorbance was based on the results measured at 318 nm for FA using an ultraviolet spectrometer (UV-2900, Hitachi, Tokyo, Japan). The FA present in the supernatant was quantified. Compared to the background buffer solution, the CaCO3 particles did not contribute to the measured absorbance at this wavelength. Prior to a measurement, the baseline was established in the buffer solution (background solution) for approximately 5 min. As a minimum, all measurements were performed in triplicate.
| FA released (%) = (Absorbance of sample/Absorbance of standard) × 100 |
Conclusions
We proposed a carbonation process to synthesise the calcite phase of mesoporous calcium carbonate with a well-controlled specific surface area (38.8–234 m2 g−1). The water content in the precursor of ACC dispersion played a crucial role on pore formation. Then we demonstrated the release tests of FA on mesoporous calcium carbonate. The mesoporous calcium carbonate with the highest specific surface area of 234 m2 g−1 had the highest release of FA. The fluorescence spectroscopy study showed the model fluorescence compound was dispersed in the molecular state when using a higher specific surface area of mesoporous calcium carbonate. This result reflected the release behaviour of FA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grant number JP19K05117). We wish to thank Dr Miki Fujimori and Ayano Nagayama, Osaka University of Pharmaceutical Sciences, for the technical support on the measurements of release tests.Notes and references
- Z. Z. Li, L. X. Wen, L. Shao and J. F. Chen, J. Controlled Release, 2004, 98, 245 CrossRef CAS PubMed; K. Suzuki, T. Yumura, Y. Tanaka and M. Akashi, J. Controlled Release, 2001, 75, 183 CrossRef PubMed; M. Otsuka, K. Tokumitsu and Y. Matsuda, J. Controlled Release, 2000, 67, 369 CrossRef PubMed; P. Kortesuo, M. Ahola, M. Kangas, T. Lenio, S. Laakso, L. Vuorilehto, A. Yli-Urpo, J. Kiesvaara and M. Marvola, J. Controlled Release, 2001, 76, 227 CrossRef PubMed.
- K. A. Fisher, K. D. Huddersman and M. J. Taylor, Chem.–Eur. J., 2003, 9, 5873 CrossRef CAS PubMed; L. Bacakova, M. Vandrovcova, I. Kopova and I. Jirka, Biomater. Sci., 2018, 6, 974 RSC.
- H. W. Kim, J. C. Knowles and H. E. Kim, Biomaterials, 2004, 25, 1279 CrossRef CAS PubMed; A. Haider, S. Haider, S. S. Han and I. K. Kang, RSC Adv., 2017, 7, 7442 RSC.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Günther, A. I. Petrov, D. B. Shenoy and H. Mohwald, J. Mater. Chem., 2004, 14, 2073 RSC; W. Wei, G. H. Ma, G. Hu, D. Yu, T. Mcleish, Z. G. Su and Z. Y. Shen, J. Am. Chem. Soc., 2008, 130, 15808 CrossRef CAS PubMed; M. Fujiwara, K. Shiokawa, M. Araki, N. Ashitaka, K. Morigaki, T. Kubota and Y. Nakahara, Cryst. Growth Des., 2010, 10, 4030 CrossRef.
- W. Wichaita, D. Polpanich and P. Tangboriboonrat, Ind. Eng. Chem. Res., 2019, 58, 20880 CrossRef CAS.
- M. L. Johnson, D. Noreland, P. Gane, J. Schoelkopf, C. Ridgway and A. M. Fureby, Food Funct., 2017, 8, 1627 RSC.
- A. Maleki, H. Kettiger, A. Shoubben, J. M. Rosenholm, V. Ambrogi and M. Hamidi, J. Controlled Release, 2017, 262, 329 CrossRef CAS PubMed.
- D. Evans, P. B. Webb, K. Penkman, R. Kroger and N. Allison, Cryst. Growth Des., 2019, 19, 4300 CrossRef CAS.
- C. J. Ridgway, P. A. C. Gane and J. Schoelkopf, Colloids Surf., A, 2004, 236, 91 CrossRef CAS.
- C. Mancuso and R. Santamgelo, Food Chem. Toxicol., 2014, 65, 185 CrossRef CAS PubMed.
- N. Kumar and V. Pruthi, Biotechnol. Rep., 2014, 4, 86 CrossRef PubMed; E. Graf, Biol. Med., 1992, 3, 435 Search PubMed.
- C. M. Hsu, S. C. Yu, F. J. Tsai and Y. Tsai, Colloids Surf., B, 2019, 180, 68 CrossRef CAS PubMed.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306 CrossRef CAS PubMed.
- D. V. Volodkin, A. I. Petrov, M. Prevt and G. B. Sukhorukov, Langmuir, 2004, 20, 3398 CrossRef CAS PubMed; M. Fujiwara, K. Shiokawa, K. Morigaki, Y. Zhu and Y. Nakahara, Chem. Eng. J., 2008, 137, 14 CrossRef.
- R. Sun, P. Zhang, É. G. Bajnóczi, A. Neagu, C.-W. Tai, I. Persson, M. Strømme and O. Cheung, ACS Appl. Mater. Interfaces, 2018, 10, 21556 CrossRef CAS PubMed.
- D. Walsh and S. Mann, Nature, 1995, 377, 320 CrossRef CAS; T. Tomioka, M. Fuji, M. Takahashi, C. Takai and M. Utsuno, Cryst. Growth Des., 2011, 12, 771 CrossRef; H. D. Yu, S. Y. Tee and M. Y. Han, Chem. Commun., 2013, 49, 4229 RSC.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef PubMed.
- S. Yamanaka, T. Oiso, Y. Kurahashi, H. Abe, K. Hara, T. Fujimoto and Y. Kuga, J. Nanopart. Res., 2014, 16, 2266 CrossRef.
- S. Yamanaka, Y. Sugawara, T. Oiso, T. Fujimoto, Y. Ohira and Y. Kuga, CrystEngComm, 2015, 17, 1773 RSC.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959 CrossRef CAS; S. F. Chen, H. Cölfen, M. Antonietti and S. H. Yu, Chem. Commun., 2003, 49, 9564 RSC; M. Faatz, F. Grohn and G. Wegner, Adv. Mater., 2004, 16, 996 CrossRef; J. Ihli, W. C. Wong, E. H. Noel, Y. Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 CrossRef PubMed; D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergströmc and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348 RSC.
- H. Wei, Q. Shen, Y. Zhao, D. J. Wang and D. F. Xu, J. Cryst. Growth, 2003, 250, 516 CrossRef CAS; Y. S. Han, G. Hadiko, M. Fuji and M. Takahashi, J. Cryst. Growth, 2005, 276, 541 CrossRef; J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265 RSC.
- J. N. Israelachvili, Intermolecular and surfaces forces, Academic Press, London, UK, 2nd edn, 1992 Search PubMed.
- Y. Tozuka, C. Yokohama, K. Higashi, K. Moribe and K. Yamamoto, J. Drug Delivery Sci. Technol., 2009, 19, 401 CrossRef CAS.
- Q. Wei, C. M. Keck and R. H. Müller, Int. J. Pharm., 2017, 518, 253 CrossRef CAS PubMed.
- M. Manzano, M. Colilla and M. Vallet-Regi, Expert Opin. Drug Delivery, 2009, 117, 1383 CrossRef PubMed.
- T. Oiso and S. Yamanaka, Adv. Powder Technol., 2018, 29, 606 CrossRef CAS.
- K. Y. Lai, T. M. Chu, F. C. Hong, A. Elangovan, K. M. Kao, S. W. Yang and T. I. Ho, Surf. Coat. Technol., 2006, 200, 3283 CrossRef CAS.
- K. Kadota, A. Senda, T. Ito and Y. Tozuka, Eur. J. Pharm. Sci., 2015, 79, 79 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |