
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of the surface polarity of nanomaterials on their interaction with complement protein gC1q†
Shuai Wangab,
Xinwen Oub,
Yanee Wutthinitikornkitb,
Ming Yi*ac and
Jingyuan Li *b
*b
aCollege of Informatics, Huazhong Agricultural University, Wuhan 430070, China
bDepartment of Physics, Zhejiang University, Hangzhou 310027, China. E-mail: jingyuanli@zju.edu.cn
cSchool of Mathematics and Physics, China University of Geosciences, Wuhan 430074, China. E-mail: mingyi@cug.edu.cn
First published on 18th November 2020
Abstract
There are increasing studies about the biocompatibility of nanomaterials (NMs) as their applications in biomedicine become more widespread. The biocompatibility of NMs involves the recognition by the immune system including complement protein gC1q. In this work, the interaction of graphene oxide (GO) and self-assembled monolayer (SAM)-coated gold (111) surface with gC1q was studied by molecular dynamics (MD) simulations. The impacts of surface polarity of GO on its interaction with gC1q and the possible immune response were discussed by comparing the binding behavior of gC1q to the GO sheets with different oxidation degrees (i.e., C5O1(OH)1 and C20O1(OH)1). We find the ghB module of gC1q tends to bind to GO sheet (C5O1(OH)1) with strong surface polarity, as the ghB module forms more hydrogen bonds with this GO sheet. On the other hand, the ghC module of gC1q tends to bind to GO (C20O1(OH)1) with weak surface polarity, as the ghC module tends to form pi–pi stacking and stronger hydrophobic interaction with this GO sheet. Similar phenomena are also found in the adsorption of gC1q with SAM: ghC prefers to bind to hydrophobic CH3-SAM, and ghB prefers to bind to charged COO-SAM. The different binding modules of gC1q may result in different activation levels of complement system. Our findings suggest that the surface polarity of NMs regulates the interaction of NMs with gC1q and the subsequent immune response. In other words, the biocompatibility of NMs may be regulated by adjusting their surface polarity.
Introduction
Because of their applications in biomedicine and biotechnology,1–8 the studies on the biocompatibility of nanomaterials (NMs), especially graphene oxide (GO)9–12 and self-assembled monolayer (SAM) modified surfaces,13–15 have received intensive attention. One important factor affecting the biocompatibility of NMs is their interaction with the immune system and the resulting immunological effects.16–21 There is growing evidence that the immunological effects of NMs are influenced by their surface polarity.17,22,23 For example, the polyvinyl alcohol-coated poly(lactic-co-glycolic acid) (PLGA) nanoparticles (PLGA-NPs) with neutral surface show higher phagocytosis than charged PLGA-NPs coated with chitosan or poloxamer 188.17 And there have been a variety of experimental works showing the impacts of oxidation degree of GO on the interaction with proteins and the resulting inflammation response and cytotoxicity in macrophages.24–28The complement system consisting of three complement pathways (i.e., classical, alternative and lectin pathway) is an important part of human immune system. The activation of immune response through the classical complement pathway is initiated by complement protein gC1q when it senses foreign intruders.29 gC1q is composed of three modules, i.e., ghA, ghB, and ghC. Both ghB and ghC modules can recognize foreign intruders, though the immune response initiated by ghB is stronger than that by ghC.30 There are two distinct recognition modes of gC1q with foreign intruders: ghC module is likely to have a central role in the binding of gC1q with hydrophobic targets,31,32 and gC1q bind polyanions is largely dependent on the ghB module.31,33 Therefore, the surface properties of foreign intruders, especially the surface polarity, should affect the recognition mode of gC1q and subsequent activation level of complement system. Thielens et al.34 found that only anionic polydiacetylene micelles can bind complement proteins and activate the complement system. Wibroe et al.35 found that the activation level of complement system is positively correlated with the oxidation degree of GO.
Molecular dynamics (MD) simulations have been widely used to study the interaction between NMs and immune-related proteins and to discuss the impacts of surface properties of NMs. For example, Yu et al.36 investigated the interaction of both pristine and modified fullerenes with leukocyte common antigen (CD45) and found their distinct binding modes. Xie et al.37 studied the adsorption behavior of lysozyme which plays an important immunomodulatory role in human innate immune system,38 and they explored the effects of surface polarities of self-assembled monolayer (SAM) on Au (111) on their interaction with lysozyme. In addition, previous computational studies on the interaction of NMs with the complement protein gC1q investigated the effects of the size of NMs on the binding stability of gC1q.39 To our knowledge, there are no computational researches on the interaction mode between gC1q and NMs, especially the comparison of two recognition modules, i.e., ghB and ghC modules. The impacts of the surface polarities of NMs on the recognition behavior of gC1q and the resulting immune response remain largely elusive.
In this study, MD simulations were performed to investigate the interaction of complement protein gC1q with the GO sheet and self-assembled monolayer (SAM)-coated gold (111) surface. We focused on the impacts of the surface polarity of the GO and SAM on the binding mode of gC1q as well as the corresponding immune response. The ghB module of gC1q recognizes GO with high oxidation degree (C5O1(OH)1), whereas the ghC module of gC1q recognizes GO with low oxidation degree (C20O1(OH)1). There are up to 22 charged residues distributed on the surface of ghB module engendering the preference for GO with higher polarity. Such recognition mode may trigger strong complement activation level and immune response. On the other hand, the surface of ghC module shows a predominance of non-polar residues (∼40%) including several aromatic residues. Thus, ghC can form pi–pi stacking and hydrophobic interaction with the GO with lower polarity and induce a weak immune response. Similar phenomena are also found for gC1q adsorption on self-assembled monolayer (SAM)-protected gold nanoparticle with distinct surface polarities (i.e. hydrophobic CH3-SAM and charged COO-SAM). The ghB prefers to bind to COO-SAM, and the ghC prefers to bind to the CH3-SAM. Our results suggest that the recognition mode of gC1q and the activation of corresponding immune response can be modulated by turning the surface polarity of GO and SAM.
Methods
A. Protein and surface models
The crystal structure of gC1q protein was obtained from the Protein Data Bank (PDB ID: 1PK6 (ref. 40)). As shown in Fig. 1A, gC1q protein is a compact, almost spherical heterotrimeric assembly consisting of 394 residues. As the O/C ratio of graphene oxide (GO) can be effectively adjusted in a range of ∼11–∼51% in experiments,41–44 two kinds of GO sheets with distinct O/C ratios, i.e., C5O1(OH)1 (Fig. 2A), and C20O1(OH)1 (Fig. 2B) were constructed based on the Lerf–Klinowski model.45 The oxygen-containing groups (epoxy and hydroxyl functional groups) were randomly distributed on both sides of the GO sheet. Both kinds of GO sheets have the same size of 7.1 × 7.5 nm2. In addition, self-assembled monolayer (SAM)-protected gold nanoparticles with distinct surface polarities were constructed using 10-amino-1-decanethiol (–S(CH2)10-R), where R stands for a functional group, i.e. –CH3, –COO. All alkanethiol chains of SAM were packed on the gold (111) surface with a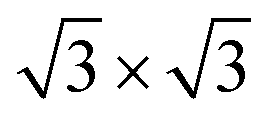 lattice structure.46,47 And all the SAMs have dimensions of 7.2 nm × 7.4 nm.
lattice structure.46,47 And all the SAMs have dimensions of 7.2 nm × 7.4 nm.
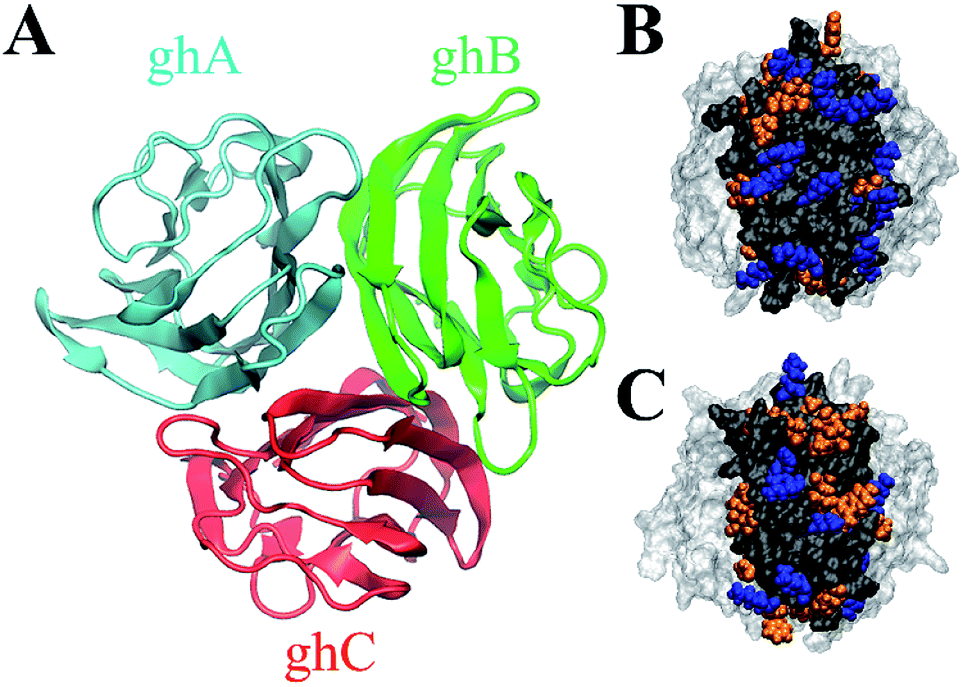 | ||
| Fig. 1 Top view of the structure of gC1q, comprising three modules: ghA (cyan), ghB (green) and ghC (red) (A). Side view of the hydrophobic (orange) and positively charged (blue) residues on the surface of two binding modules (highlighted in black) of gC1q: ghB (B) and ghC (C). | ||
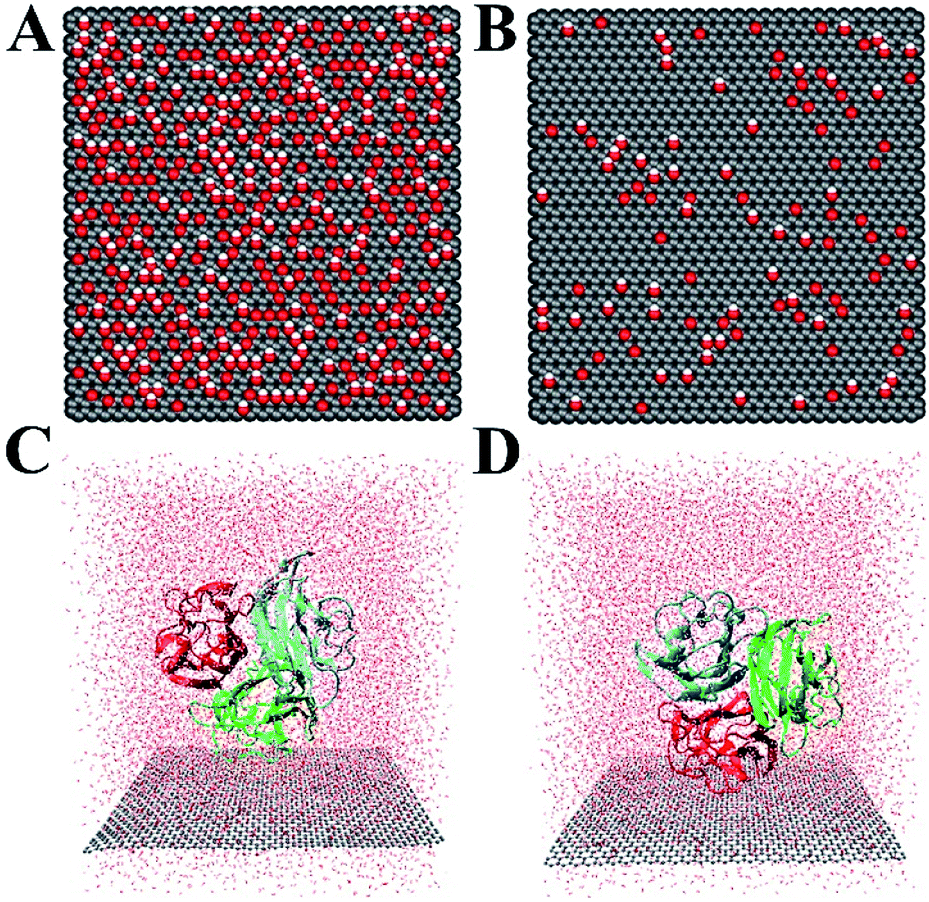 | ||
| Fig. 2 Top view of the graphene oxide: C5O1(OH)1 (A) and C20O1(OH)1 (B). Atoms in graphene oxide sheet are shown as vdW spheres (C: gray, O: red, H: white). The initial configurations of protein gC1q with ghB (C) and ghC (D) facing the GO sheet. Water molecules are shown as red dots. | ||
B. MD simulations
In our simulations, OPLS-AA force field48 was applied for the protein and SAM. We adopted the force field for graphene nanosheets and oxygen-containing groups from previous studies,49,50 wherein the sp2 carbon atoms were modelled as uncharged Lennard-Jones particles with σ = 0.340 nm and a depth of potential well ε = 0.233 kJ mol−1. The GO is electrically neutral. Detailed force field parameters of sp3-hybridized carbon, hydroxyl and epoxy groups are described in the Table S1.†The ghB and ghC modules of gC1q were facing the two GO surfaces (Fig. 2C and D) separately to obtain four different initial configurations of gC1q on the GO sheet (i.e., ghB/C5O1(OH)1, ghB/C20O1(OH)1, ghC/C5O1(OH)1, and ghC/C20O1(OH)1). The initial closest distance of heavy atoms between the protein and GO is 0.5 nm. All gC1q/GO systems were embedded in a box of 7.4 × 7.7 × 8.5 nm3 and solvated with TIP3P51 water molecules. The distance between the protein or GO sheet and the boundary of the box was at least 1.0 nm. Afterwards, nine chloride ions were added to neutralize the system, and then appropriate numbers of chloride and sodium ions were added to reach a physiological salt concentration of 0.15 M at neutral pH. Finally, all the simulations were performed using GROMACS 2018.3 package.52
For each run, the simulation was first energy-minimized by using the steepest descent method, and then the system was pre-equilibrated with a 1 ns equilibration in the NVT ensemble to relax the solvent molecules with position restraints imposed on the GO sheet and the backbone of protein. After that, the system was pre-equilibrated with 1 ns constant pressure equilibration in the NPT ensemble. Finally, production run was performed in the NPT ensemble for 50 ns. The temperature of all systems was maintained at 310 K using a v-rescale thermostat53 with a 0.1 ps coupling constant, and the pressure was kept at 1 bar with semi-isotropic coupling directions (X + Y, Z) using the Berendsen's algorithm.54 The standard periodic boundary conditions (PBC) were applied in all directions (X, Y and Z). Solute bonds involving hydrogen atoms were constrained by the LINCS algorithm.55 Both the van der Waals (vdW) interaction and real-space Coulomb interaction were calculated using a cutoff distance of 1.0 nm, while the long-range electrostatic interaction was handled by the particle mesh Ewald (PME) method.56 For all simulations, the integration time step was 2.0 fs, and snapshot was recorded every 5 ps. The neighbor list generated with a cutoff distance of 1.0 nm was updated every 20 steps. Visualization and analysis of MD trajectories were implemented by VMD1.9.3.57
Results and discussion
We first investigated the adsorption of gC1q with its ghB module facing the graphene oxide (GO) sheets. The adsorption on C5O1(OH)1 and C20O1(OH)1 was studied separately. And the number of contacts with GO was calculated to illustrate the adsorption process (a contact is counted when a heavy atom in the protein is within 0.5 nm of a carbon/oxygen atom in GO). It should be noted that the contacts of gC1q are totally attributed to ghB.As shown in Fig. 3A, the number of contacts of gC1q increases in a stepwise manner as the adsorption proceeds. For example, during the adsorption process on C5O1(OH)1, the number of contacts progressively increases to ∼100 (t = 2.5 ns), ∼200 (t = 6 ns), and ∼229 (t = 23 ns). Similar adsorption process is also exhibited on C20O1(OH)1. In both systems, the number of contacts reaches a plateau after t = 25 ns. And the average numbers of contacts over the interval of t = 25 ns to 50 ns are 229.9 (C5O1(OH)1) and 123.0 (C20O1(OH)1). This suggests that the adsorption of ghB on C5O1(OH)1 is stronger than on C20O1(OH)1. The distinct adsorption behaviors are also reflected in the representative snapshots of the adsorbed protein. ghB module adopts flat structure and establishes more intimate contact with C5O1(OH)1 than with C20O1(OH)1 (Fig. 3B).
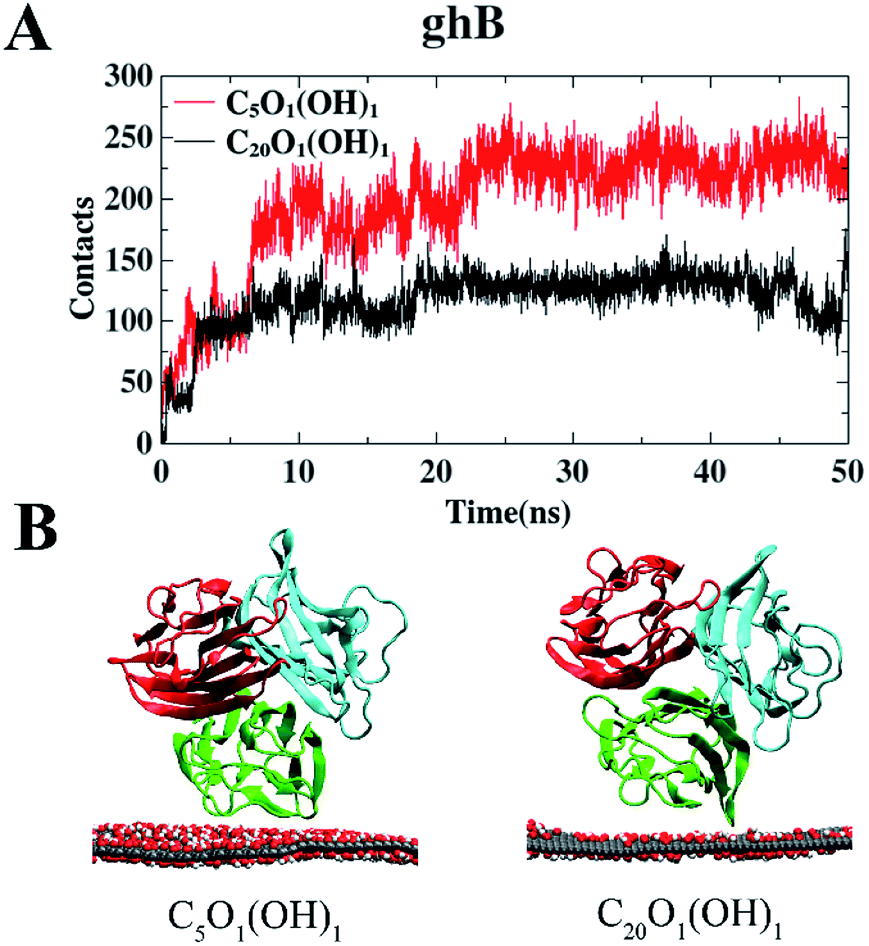 | ||
| Fig. 3 The number of contact heavy atoms of gC1q (ghB) with GO (A). Representative structures of gC1q with ghB adsorbed on GO (B). | ||
The interaction energies between ghB and GO sheets were also calculated. The Coulomb interaction potential of ghB module with C5O1(OH)1 (−192.9 kJ mol−1) is stronger than that with C20O1(OH)1 (−25.4 kJ mol−1). In addition, the average number of hydrogen bonds formed between ghB module and C5O1(OH)1 is 7.5, much higher than that formed with C20O1(OH)1 (0.7). Taken together, the binding preference of the ghB module for C5O1(OH)1 is largely attributed to the favorable electrostatic interaction.
We also studied the adsorption behavior of gC1q with its ghC module initially facing the GO sheets. In these cases, the contacts of gC1q with GO are totally attributed to the ghC module. As shown in Fig. 4A, the numbers of contacts of ghC with both GO sheets reach a plateau before t = 30 ns. Thereafter they oscillate around the mean values of ∼213 (C20O1(OH)1) and ∼141 (C5O1(OH)1). Besides, the interaction energy between ghC and C20O1(OH)1 is stronger than that between ghC and C5O1(OH)1 (−411.1 kJ mol−1 vs. −320.5 kJ mol−1). Such difference is mainly attributed to the stronger van der Waals energy between ghC and C20O1(OH)1 than that between ghC and C5O1(OH)1 (−311.8 kJ mol−1 vs. −217.6 kJ mol−1). In order to comprehensively evaluate the binding stability of protein, we further calculated free energy of protein binding using MM-PBSA method.58 The binding free energy between ghC and C20O1(OH)1 is also stronger than that between ghC and C5O1(OH)1 (−368.9 kJ mol−1 vs. −308.2 kJ mol−1). Consequently, gC1q can adsorb on GO sheets through its ghC module. And the adsorption on C20O1(OH)1 is clearly stronger than on C5O1(OH)1. As can be seen in the representative snapshots of the adsorbed protein, ghC module adopts flat configuration and establishes more intimate contact with C20O1(OH)1 than with C5O1(OH)1 (Fig. 4B).
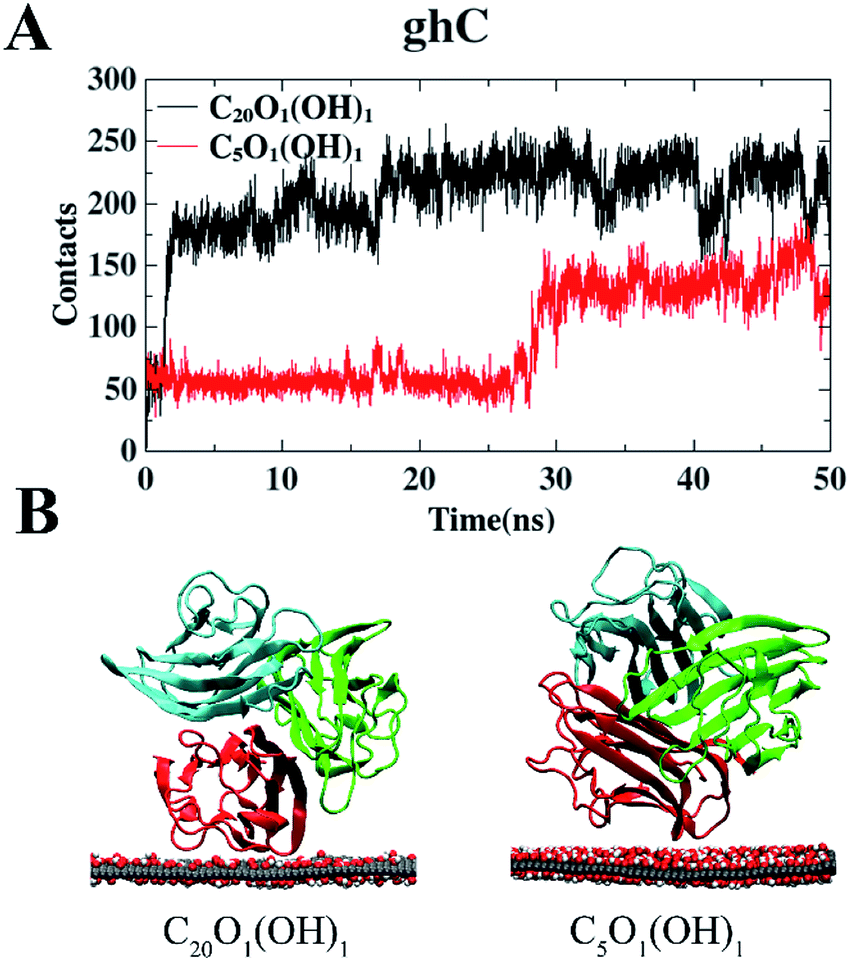 | ||
| Fig. 4 The number of contact heavy atoms of gC1q (ghC) with GO (A). Representative structures of gC1q with ghC adsorbed on GO (B). | ||
While there are more contact atoms of ghC with C20O1(OH)1 than with C5O1(OH)1, the Coulomb interaction potential of ghC with these two GO sheets are very close: −102.9 kJ mol−1 (C5O1(OH)1) vs. −99.3 kJ mol−1 (C20O1(OH)1). In addition, the average numbers of hydrogen bonds formed between ghC and oxygen-containing groups on GO sheets are also modest: 4.5 (C5O1(OH)1) vs. 3.7 (C20O1(OH)1). In short, the interaction between polar groups, e.g. hydrogen bond interaction, may not account for the distinct adsorption behaviors of ghC on these two GO sheets. The binding preference of ghC for C20O1(OH)1 with weak surface polarity may be attributed to the hydrophobic interaction.
Taken together, gC1q exhibits distinct behaviors on GO sheet with various oxidation degrees: the ghB module of gC1q prefers to bind to GO sheet with high oxidation degree (C5O1(OH)1), whereas the ghC module of gC1q tends to bind to GO sheet with low oxidation degree (C20O1(OH)1). In other words, the binding modes of gC1q are markedly affected by surface polarity of GO sheet.
In order to further validate the impacts of the surface polarity of nanomaterials on their interaction with gC1q, we have also studied the binding behavior of gC1q on self-assembled monolayer (SAM)-protected gold nanoparticles with two distinct surface polarities (i.e. hydrophobic CH3-SAM and charged COO-SAM). The number of contacts of gC1q was calculated to compare the binding preference of the protein. It can be seen from Fig. S2† that the average number of contacts of ghC with the CH3-SAM is significantly higher than that with the COO-SAM (17.0 vs. 0). This result suggests that ghC prefers to bind to the hydrophobic SAM, which is consistent with the result that ghC prefers to bind to C20O1(OH)1 with weak surface polarity. As for the case of ghB (Fig. S1†), the average number of contacts with SAMs are 34.9 (COO-SAM) and 9.8 (CH3-SAM), separately. The binding preference of ghB to COO-SAM is considerably higher than that to CH3-SAM, which is also in agreement with the result that ghB prefers to bind to C5O1(OH)1 with more oxidized groups. The results of both SAM and GO suggest that the surface polarity of nanomaterials affect the adsorption mode of gC1q: the ghC prefers to bind to nanomaterials with weak surface polarity, and the ghB prefers to bind nanomaterials with strong surface polarity.
We notice the adsorption behavior of protein in response to the surface polarity of GO and SAM is largely ascribed to the distinct residue composition (i.e., positively charged, negatively charged, polar, and non-polar residues) of the surface of ghB and ghC modules (Table S2†). Charged residues are more abundant on the surface of ghB than on the surface of ghC (22 vs. 16), especially positively charged residues (14 vs. 9). On the other hand, the number of non-polar residues in ghC is much higher than in ghB (27 vs. 18). The distinct residue composition is also illustrated in Fig. 1B and C. In short, ghC exhibits much lower surface polarity than ghB.
To further study the relationship between the surface residue composition of gC1q and its adsorption behavior on the GO sheet with varying surface polarity, the number of contact residues (i.e., the contact residues are the ones that establish at least one contact atom with GO sheet) with GO sheets were assessed. We focused on the two preferential binding modes, i.e., ghB module on C5O1(OH)1 and ghC module on C20O1(OH)1. In the case of ghB on C5O1(OH)1, the average number of positively charged, negatively charged, polar, and non-polar residues in contact with GO are 6.5, 3.0, 3.0, and 1.9, respectively (Fig. 5A). Clearly, there are much more positively charged residues contacting with this GO than the other three types of residues. The fraction of positively charged residues in the contact residues is 45.1% (6.5 out of 14.4), considerably higher than their “intrinsic” proportion (22.9%, 14 out of 61) on the surface of ghB. Hence, the positively charged residues are preferred in this binding mode.
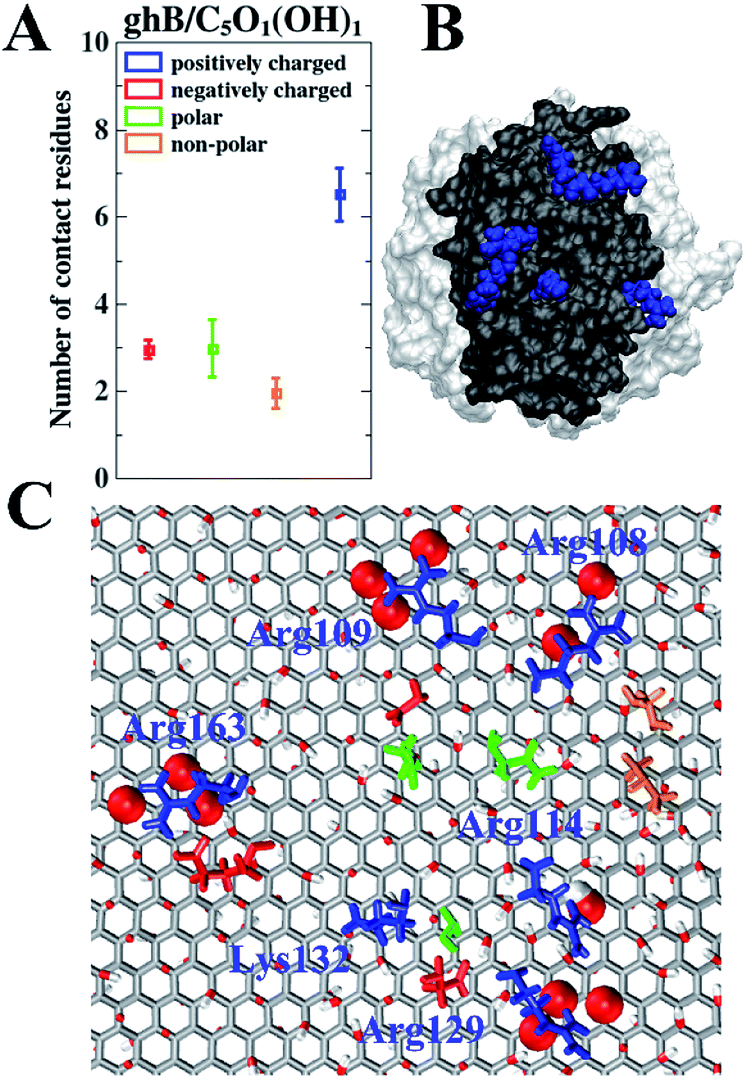 | ||
| Fig. 5 The number of contact residues of ghB module with the C5O1(OH)1 (A). Location of the positively charged residues of ghB module (highlighted as blue region in black surface) adsorbed on C5O1(OH)1 (B). Top view of the highly-involved residues whose contact probability is more than 50% (C). The sidechains of adsorbed residues are shown in licorice representation following the same color scheme in (A). | ||
We further identified the highly-involved residues (i.e., the residues with the contact probability more than 50%). As can be seen in Fig. 5C, the highly-involved residues include six positively charged residues (Arg108, Arg109, Arg114, Arg129, Lys132 and Arg163), three negatively charged residues (Asp110, Glu127 and Glu162) as well as three polar residues (Gln111, Ser130 and Gln198) and two non-polar residues (Val105 and Pro106). As mentioned above, the binding mode of ghB to C5O1(OH)1 is mainly attributed to a favorable electrostatic interaction. The results about the highly-involved residues also agree with previous experimental study, wherein the ghB module mainly recognizes foreign intruders through the charged residues (i.e., Arg114, Arg129, Arg163, and Glu162).31,33 Our simulation further suggests that three positively charged residues (i.e. Arg108, Arg109 and Lys132) may also be involved in the intruder recognition. Therefore, the adsorption of ghB on C5O1(OH)1 is mainly through its charged residues, especially the positively charged ones.
Such binding mode is also consistent with the nature of C5O1(OH)1 with strong surface polarity: there are more hydroxyl groups and epoxy groups on C5O1(OH)1 than on C20O1(OH)1. And there are more charged residues distributed on the surface of ghB module than on ghC (22 vs. 16). These charged residues, especially positively charged ones (Fig. 5B) form favorable electrostatic interaction with the hydroxyl groups and epoxy groups of GO (Fig. 5C, highlighted as vdW spheres). Hence, the Coulomb interaction potential of ghB on C5O1(OH)1 is stronger than on C20O1(OH)1 (−192.9 kJ mol−1 vs. −25.4 kJ mol−1).
Taken together, the binding preference of ghB module for C5O1(OH)1 is mainly owing to a favorable interaction through hydrogen bonds. Similar phenomenon was also found for the adsorption of virus protein R (Vpr13-33) from HIV-1 on GO, whose adsorption affinity is also attributed to its positively charged residues.59
As for ghC on C20O1(OH)1, the average numbers of the positively charged, negatively charged, polar, and non-polar contact residues are 1.9, 1.7, 6.9, and 5.9, respectively (Fig. 6A). Clearly, there are much more non-polar and polar residues contacting with GO than charged residues. In addition, we find the number of non-polar and polar contact residues of ghC on C20O1(OH)1 are 3.1 times (5.9 vs. 1.9) and 2.3 times (6.9 vs. 3.0) more than that of ghB on C5O1(OH)1. On the other hand, the number of charged contact residues of ghC on C20O1(OH)1 is only 37.8% (3.6 vs. 9.5) of ghB on C5O1(OH)1. Apparently, non-polar and polar residues are preferred for this binding mode.
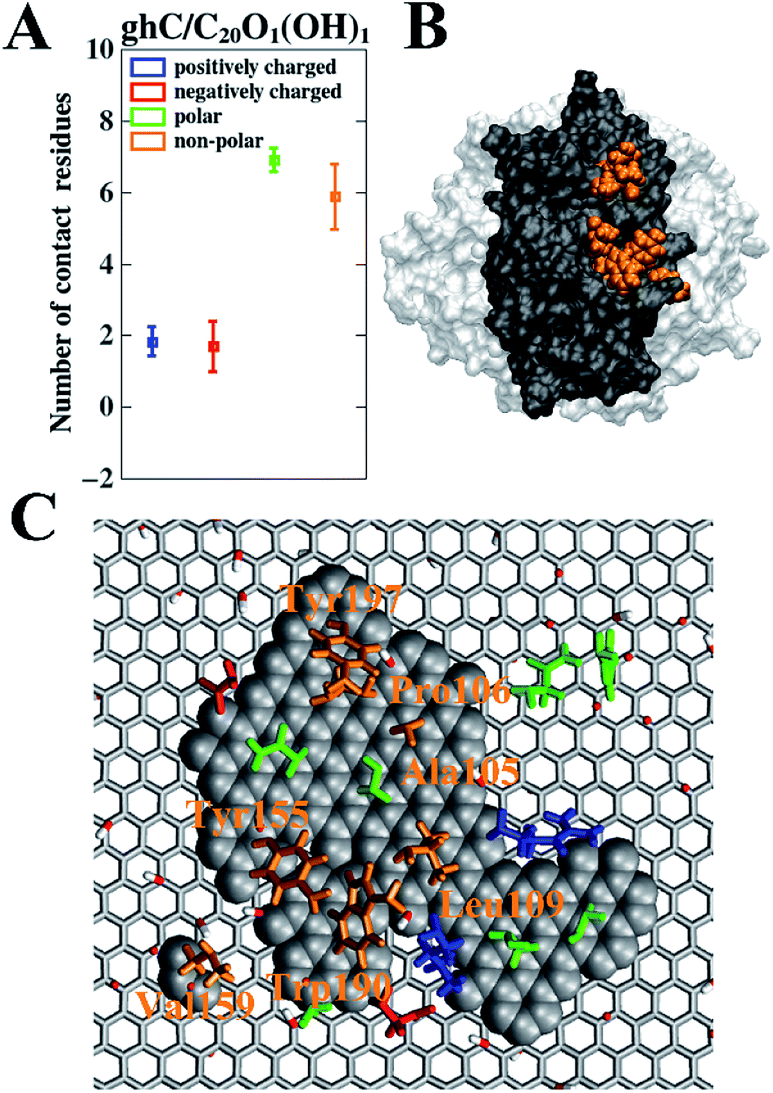 | ||
| Fig. 6 The number of contact residues of ghC module with C20O1(OH)1 (A). Location of the contact non-polar residues of the ghC module (highlighted as yellow region in black surface) with C20O1(OH)1 (B). Top view of the highly-involved residues whose contact probability is more than 50% (C). The adsorbed residues are shown in licorice representation following the same color scheme in (A). | ||
We also analyzed the highly-involved residues responsible for their adsorption following the same criterion described above. As can be seen in Fig. 6C, the highly-involved residues include seven non-polar residues (Ala105, Pro106, Leu109, Tyr155, Val159, Trp190 and Tyr197), seven polar residues (His101, Gln102, Asn107, Ser108, Ser126, Thr127 and Ser157) as well as two positively charged residues (Arg111 and Lys129) and two negatively charged residues (Glu188 and Asp195). The results about the highly-involved residues agree well with previous experimental study, wherein ghC module recognizes hydrophobic targets through its non-polar residues (i.e., Ala105, Pro106, Tyr155 and Trp190).31,32 Our simulation further indicates that three non-polar residues (Tyr197, Leu109, and Val159) may also be involved in the intruder recognition. Hence, the adsorption of ghC on C20O1(OH)1 is mainly through its non-polar residues. Such binding mode is consistent with the nature of C20O1(OH)1, featuring weak surface polarity. And there are more non-polar residues distributed on the surface of ghC than on ghB (27 vs. 18). As a result, of the highly-involved residues of ghC, all seven non-polar (Fig. 6B, highlighted in orange) and 57.1% polar residues (4 out of 7) bind to the non-oxidized region of GO (Fig. 6C, highlighted as vdW grey spheres). And these polar residues mainly bind to the non-oxidized region of GO through their hydrophobic carbonaceous groups (Fig. 7).
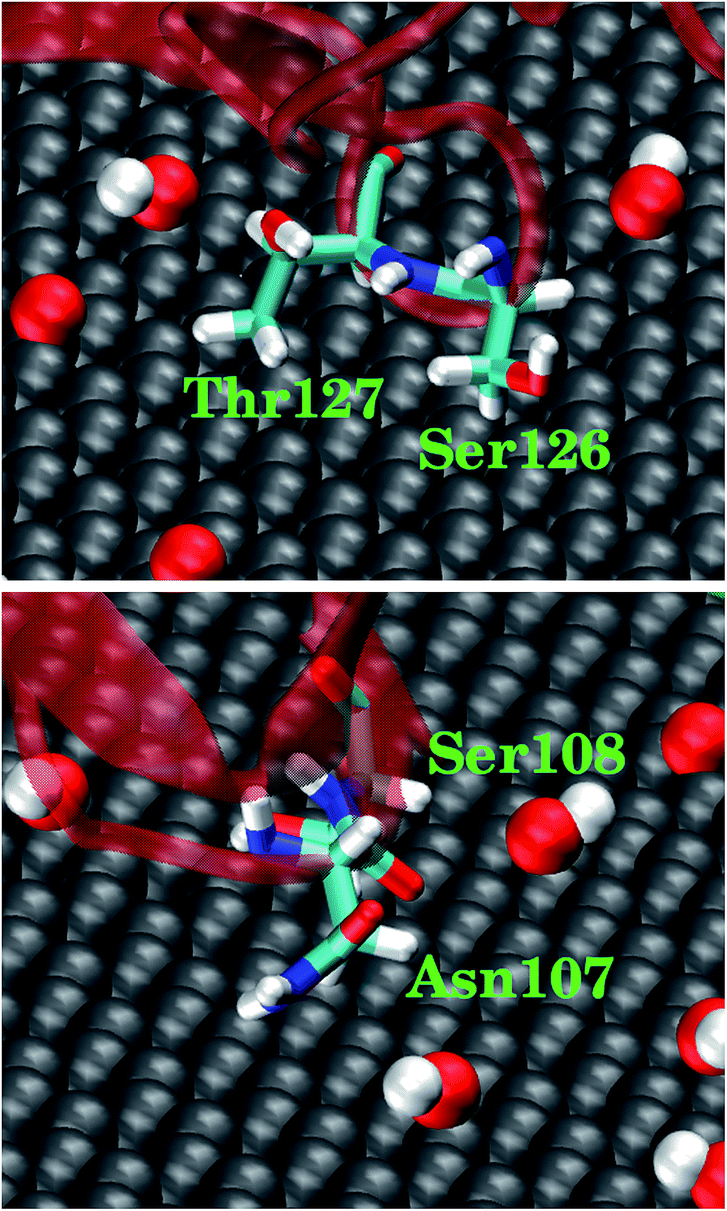 | ||
| Fig. 7 Representative snapshots for four polar residues in ghC adsorbed on C20O1(OH)1. The polar residues contact the non-oxidized region of GO through their hydrophobic carbonaceous groups. | ||
In order to illustrate the interaction of highly-involved residues of ghC module with C20O1(OH)1 in a more quantitative way, we calculated the interaction energies of different types of these residues (i.e., positively charged, negatively charged, polar, and non-polar residues) with C20O1(OH)1. The interaction energy of uncharged residues is stronger than charged residues (−186.4 kJ mol−1 vs. −118.4 kJ mol−1) owing to their stronger van der Waals interaction energy (−180.1 kJ mol−1 vs. −34.6 kJ mol−1), which is mainly attributed to the pi–pi stacking of the aromatic residues with the non-oxidized regions of C20O1(OH)1 (Fig. 6C). In addition, we noticed that multiple non-polar residues of ghC form favorable hydrophobic interaction with extended non-oxidized regions of C20O1(OH)1, which is not reflected in the interaction energy. Taken together, ghC module binds to GO with low oxidation degree primarily through pi–pi stacking and hydrophobic interactions.
Conclusions
In this work, the recognition mode of complement protein gC1q with graphene oxide (GO) and self-assembled monolayer (SAM)-coated gold (111) surface and the impacts of surface polarity of these two typical nanomaterials were investigated using molecular dynamics (MD) simulations. The protein gC1q preferentially bind to GO with strong surface polarity (C5O1(OH)1) through its ghB module. On the other hand, gC1q tends to bind to GO with weak surface polarity (C20O1(OH)1) through its ghC module. Similar results are also observed in the adsorption of gC1q on SAM: ghC prefers to bind to nonpolar CH3-SAM and ghB prefers to bind to charged COO-SAM. The dependence of recognition mode on the surface polarity of GO is mainly attributed to the distinct surface residue composition of ghB and ghC module. There are more charged residues distributed on the surface of ghB module, facilitating formation of hydrogen bonds with GO with higher oxidation degree. By contrast, the surface of ghC module is dominated by non-polar residues, resulting in its tendency to form hydrophobic interactions and pi–pi stacking with GO with large non-oxidized region.As shown in our work, complement protein gC1q can recognize a variety of foreign intruders with different surface polarities. Moreover, its recognition mode and subsequent immune response can be regulated by surface polarities of intruders. Our results reveal the structural basis of gC1q for the dependence of its recognition mode and the resulting immune response on the surface polarities of nanomaterials (NMs). These findings will be helpful to improve the biocompatibility of NMs by regulating its immune response and provide some theoretical guidance for the application of NMs in the field of biomedicine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Ruhong Zhou, Zonglin Gu, Zhi He, Yichong Lao and Rui Ye for providing comments. This work was supported by National Nature Science Foundation of China (11722434 and 11874319 to J. L., 11675056 and 91730301 to M. Y., 22003057 to X. O.).References
- B. Jana, A. Biswas, S. Mohapatra, A. Saha and S. Ghosh, Chem. Commun., 2014, 50, 11595–11598 RSC.
- T. Bonnard, A. Jayapadman, J. A. Putri, J. Cui, Y. Ju, C. Carmichael, T. A. Angelovich, S. H. Cody, S. French, K. Pascaud, H. A. Pearce, S. Jagdale, F. Caruso and C. E. Hagemeyer, ACS Nano, 2018, 12, 6988–6996 CrossRef CAS.
- C. Wang, D. Wen and Z. Gu, Bioconjugate Chem., 2018, 29, 702–708 CrossRef CAS.
- P. Feng, Y. Chen, L. Zhang, C.-G. Qian, X. Xiao, X. Han and Q.-D. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 4359–4368 CrossRef CAS.
- A. Y. Mochizuki, I. M. Frost, M. B. Mastrodimos, A. S. Plant, A. C. Wang, T. B. Moore, R. M. Prins, P. S. Weiss and S. J. Jonas, ACS Chem. Neurosci., 2018, 9, 11–28 CrossRef CAS.
- R. Chen and J. E. Riviere, Wiley Interdiscip. Rev. Nanomed., 2017, 9, e1440 CrossRef.
- K. Pradhan, G. Das, J. Khan, V. Gupta, S. Barman, A. Adak and S. Ghosh, ACS Chem. Neurosci., 2019, 10, 1535–1543 CrossRef CAS.
- B. Jana, G. Mondal, A. Biswas, I. Chakraborty, A. Saha, P. Kurkute and S. Ghosh, Macromol. Biosci., 2013, 13, 1478–1484 CrossRef CAS.
- M. Xu, J. Zhu, F. Wang, Y. Xiong, Y. Wu, Q. Wang, J. Weng, Z. Zhang, W. Chen and S. Liu, ACS Nano, 2016, 10, 3267–3281 CrossRef CAS.
- S. F. Kiew, L. V. Kiew, H. B. Lee, T. Imae and L. Y. Chung, J. Controlled Release, 2016, 226, 217–228 CrossRef CAS.
- J. Chen, X. Shi, L. Ren and Y. Wang, Carbon, 2017, 111, 18–27 CrossRef CAS.
- X. Jing, H.-Y. Mi, X.-F. Peng and L.-S. Turng, Carbon, 2018, 136, 63–72 CrossRef CAS.
- C.-M. Tîlmaciu, M. Mathieu, J.-P. Lavigne, K. Toupet, G. Guerrero, A. Ponche, J. Amalric, D. Noël and P. H. Mutin, Acta Biomater., 2015, 15, 266–277 CrossRef.
- K. Nomura, S. Mikuni, T. Nakaji-Hirabayashi, M. Gemmei-Ide, H. Kitano, H. Noguchi and K. Uosaki, Colloids Surf., B, 2015, 135, 267–273 CrossRef CAS.
- S. Klein, M. Kızaloğlu, L. Portilla, H. Park, T. Rejek, J. Hümmer, K. Meyer, R. Hock, L. V. R. Distel, M. Halik and C. Kryschi, Small, 2018, 14, 1704111 CrossRef.
- M.-A. Shahbazi, M. Hamidi, E. M. Mäkilä, H. Zhang, P. V. Almeida, M. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 7776–7789 CrossRef CAS.
- S. Barillet, E. Fattal, S. Mura, N. Tsapis, M. Pallardy, H. Hillaireau and S. Kerdine-Römer, Nanotoxicology, 2019, 13, 606–622 CrossRef CAS.
- X. Yuan, X. Zhang, L. Sun, Y. Wei and X. Wei, Part. Fibre Toxicol., 2019, 16, 18 CrossRef.
- N. B. Alsaleh and J. M. Brown, Curr. Opin. Toxicol., 2018, 10, 8–14 CrossRef.
- D. Boraschi, P. Italiani, R. Palomba, P. Decuzzi, A. Duschl, B. Fadeel and S. M. Moghimi, Semin. Immunol., 2017, 34, 33–51 CrossRef CAS.
- B. Fadeel, Front. Immunol., 2019, 10, 133 CrossRef CAS.
- R. M. Visalakshan, M. N. MacGregor, S. Sasidharan, A. Ghazaryan, A. M. Mierczynska-Vasilev, S. Morsbach, V. Mailänder, K. Landfester, J. D. Hayball and K. Vasilev, ACS Appl. Mater. Interfaces, 2019, 11, 27615–27623 CrossRef CAS.
- T. Bertok, E. Dosekova, S. Belicky, A. Holazova, L. Lorencova, D. Mislovicova, D. Paprckova, A. Vikartovska, R. Plicka, J. Krejci, M. Ilcikova, P. Kasak and J. Tkac, Langmuir, 2016, 32, 7070–7078 CrossRef CAS.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef CAS.
- R. Li, L. M. Guiney, C. H. Chang, N. D. Mansukhani, Z. Ji, X. Wang, Y.-P. Liao, W. Jiang, B. Sun, M. C. Hersam, A. E. Nel and T. Xia, ACS Nano, 2018, 12, 1390–1402 CrossRef CAS.
- Y. Bai, Z. Ming, Y. Cao, S. Feng, H. Yang, L. Chen and S.-T. Yang, Colloids Surf., B, 2017, 154, 96–103 CrossRef CAS.
- M. Orecchioni, D. Bedognetti, L. Newman, C. Fuoco, F. Spada, W. Hendrickx, F. M. Marincola, F. Sgarrella, A. F. Rodrigues, C. Ménard-Moyon, G. Cesareni, K. Kostarelos, A. Bianco and L. G. Delogu, Nat. Commun., 2017, 8, 1109 CrossRef.
- P. C. Henriques, A. T. Pereira, A. L. Pires, A. M. Pereira, F. D. Magalhães and I. C. Gonçalves, ACS Appl. Mater. Interfaces, 2020, 12, 21020–21035 CrossRef CAS.
- M. V. Carroll and R. B. Sim, Adv. Drug Deliv. Rev., 2011, 63, 965–975 CrossRef CAS.
- C. Gaboriaud, P. Frachet, N. M. Thielens and G. J. Arlaud, Front. Immunol., 2012, 2, 1–8 Search PubMed.
- U. Kishore, R. Ghai, T. J. Greenhough, A. K. Shrive, D. M. Bonifati, M. G. Gadjeva, P. Waters, M. S. Kojouharova, T. Chakraborty and A. Agrawal, Immunol. Lett., 2004, 95, 113–128 CrossRef CAS.
- U. Kishore, S. K. Gupta, M. V. Perdikoulis, M. S. Kojouharova, B. C. Urban and K. B. M. Reid, J. Immunol., 2003, 171, 812–820 CrossRef CAS.
- A. S. Zlatarova, M. Rouseva, L. T. Roumenina, M. Gadjeva, M. Kolev, I. Dobrev, N. Olova, R. Ghai, J. C. Jensenius, K. B. M. Reid, U. Kishore and M. S. Kojouharova, Biochemistry, 2006, 45, 9979–9988 CrossRef CAS.
- N. M. Thielens, A. Belime, E. Gravel, S. Ancelet, C. Caneiro, E. Doris and W. L. Ling, Int. J. Pharm., 2018, 536, 434–439 CrossRef CAS.
- P. P. Wibroe, S. V. Petersen, N. Bovet, B. W. Laursen and S. M. Moghimi, Biomaterials, 2016, 78, 20–26 CrossRef CAS.
- Y. Yu, H. Sun, T. Hou, S. Wang and Y. Li, RSC Adv., 2018, 8, 13997–14008 RSC.
- Y. Xie, W. Gong, J. Jin, Z. Zhao, Z. Li and J. Zhou, Appl. Surf. Sci., 2020, 506, 144962 CrossRef CAS.
- S. A. Ragland and A. K. Criss, PLoS Pathog., 2017, 13, 1–22 Search PubMed.
- M. Saint-Cricq, J. Carrete, C. Gaboriaud, E. Gravel, E. Doris, N. Thielens, N. Mingo and W. L. Ling, Nano Lett., 2017, 17, 3409–3415 CrossRef CAS.
- C. Gaboriaud, J. Juanhuix, A. Gruez, M. Lacroix, C. Darnault, D. Pignol, D. Verger, J. C. Fontecilla-Camps and G. J. Arlaud, J. Biol. Chem., 2003, 278, 46974–46982 CrossRef CAS.
- Y. Yuan, X. Gao, Y. Wei, X. Wang, J. Wang, Y. Zhang and C. Gao, Desalination, 2017, 405, 29–39 CrossRef CAS.
- H. Yang, J.-S. Li and X. Zeng, ACS Appl. Nano Mater., 2018, 1, 2763–2773 CrossRef CAS.
- P. Sun, Y. Wang, H. Liu, K. Wang, D. Wu, Z. Xu and H. Zhu, PloS One, 2014, 9, e111908 CrossRef.
- C. Chen, Y.-C. Chen, Y.-T. Hong, T.-W. Lee and J.-F. Huang, Chem. Eng. J., 2018, 352, 188–197 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- R. Tian, M. Luo and J. Li, Phys. Chem. Chem. Phys., 2018, 20, 68–74 RSC.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- C.-J. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2012, 28, 235–241 CrossRef CAS.
- J. A. L. Willcox and H. J. Kim, ACS Nano, 2017, 11, 2187–2193 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS.
- S. Zeng, G. Zhou, J. Guo, F. Zhou and J. Chen, Sci. Rep., 2016, 6, 24906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05493c |
| This journal is © The Royal Society of Chemistry 2020 |