
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLessons learned in engineering interrupted adenylation domains when attempting to create trifunctional enzymes from three independent monofunctional ones†
Taylor A.
Lundy‡
,
Shogo
Mori‡
 and
Sylvie
Garneau-Tsodikova
*
and
Sylvie
Garneau-Tsodikova
*
Department of Pharmaceutical Sciences, University of Kentucky, College of Pharmacy, Lexington, KY 40536-0596, USA. E-mail: sylviegtsodikova@uky.edu
First published on 15th September 2020
Abstract
Interrupted adenylation (A) domains are fascinating examples of multifunctional enzymes. They are found in nonribosomal peptide synthetases (NRPSs), which biosynthesize nonribosomal peptides (NRPs), a major class of medically relevant natural products (NPs). Interrupted A domains contain the catalytic portion of another domain within them, typically a methylation (M) domain, thus combining both adenylation and methylation capabilities. In recent years, interrupted A domains have demonstrated tremendous enzyme engineering potential as they are able to be constructed artificially in a laboratory setting by combining the A and M domains of two separate NRPS proteins. A recent discovery and characterization of a naturally occurring interrupted A domain that harbored two M domains back-to-back, a trifunctional protein, showed the ingenuity of Nature to both N- and O-methylate amino acids, the building blocks of NRPs. Since we have shown that a single M domain could be added to an uninterrupted A domain to create an artificial interrupted A domain, we set out to investigate if: (i) an A domain could be engineered to contain two back-to-back M domains and (ii) the added M domains would have to reflect the pattern in Nature, a side chain (O-) methylating M domain (Ms) followed by a backbone (N-) methylating M domain (Mb), or if the order of the M domains could be reversed. To address these questions, we set out to create our own AMsMbA and AMbMsA engineered interrupted A domains. We evaluated these engineered proteins connected (in cis) and/or disconnected (in trans) from the native thiolation (T) domain, through a series of radiometric assays, high performance liquid chromatography (HPLC), and mass spectrometry (MS) for adenylation, loading, and methylation ability. We found that although adenylation activity was preserved in both versions (AMsMbA and AMbMsA), addition of the M domains, in natural and unnatural order, did not result in the desired added methylation capability. This study offers valuable insights into the limits of constructing engineered interrupted A domains as potential tools for modifications of NRPs.
Introduction
Enzyme engineering is emerging as a powerful tool in many areas of research, especially with regard to natural products (NPs). NPs are molecules synthesized by living organisms that serve a variety of functions in Nature and thus display profound structural diversity, chemical novelty, and biological activity. NPs have inspired many medications today, accounting for almost 50% of all new approved drugs.1 A major and diverse class of NPs is nonribosomal peptides (NRPs), which are derived from both proteinogenic and non-proteinogenic amino acids. NRPs are biosynthesized by mega-enzymes termed nonribosomal peptide synthetases (NRPSs), which string together amino acids or amino acid-like building blocks to make a final NRP product. NRPSs act in an assembly-line fashion due to their modular nature. These mega-enzymes can be subdivided into individual domains responsible for a particular function. There are three core domains: adenylation (A), thiolation (T), and condensation (C).2The biosynthesis of NRPs occurs through a well-choreographed sequence of events. The T domain is expressed in its inactive (apo) form and must be converted to its active (holo) form before it can participate in the assembly-line. To prime the T domain, a 4′-phosphopantetheinyl transferase (PPTase) transfers the 4′-phosphopantetheine (Ppant) prosthetic group from coenzyme A (CoA) onto a serine residue of the T domain, providing a long flexible arm to carry amino acids from one active site of the assembly-line to the next.3 In order to attach an amino acid to the Ppant arm of the T domain, the amino acid first must be activated, a process called adenylation, where adenosine monophosphate (AMP) is attached to the carboxyl terminus of an amino acid/amino acid-like substrate. This activated substrate is then transferred to the Ppant arm of the T domain through formation of a thioester bond, thus resulting in a loaded T domain. The C domain is then tasked with forming an amide bond between two amino acid/amino acid-like substrates on two surrounding T domains. This cycle: activation, loading, and condensation, is repeated until the biosynthesis of an NRP is complete and it is cleaved from the NRPS, commonly through the action of a terminal thioesterase (TE) domain. There can be additional auxiliary domains (e.g., methylation (M), epimerization (E), halogenation (Hal), etc.) that adorn the NRP scaffold with additional moieties to create more structural diversity.
Due to the manner by which NRPs are biosynthesized, the organization of the modules of an NRPS dictates the final structure of the NRP product.2 For this reason, many A domains are typically selective for only one amino acid/amino acid-like substrate and thus, are considered to be the gatekeepers of diversity. As such, A domains have attracted a significant amount of attention from researchers to expand or unlock A domains as potential tools to enhance the structural diversity of NRPs, typically by altering their substrate specificity.4,5 Another particularly fascinating aspect of A domains is that they can contain within them a portion of another domain, most commonly an M domain.6 These types of A domains are termed interrupted A domains.6 These M domains, which utilize the methyl donor S-adenosyl-L-methionine (SAM), are N-, O-, or S-methyltransferases, which can methylate either the backbone (N-), Mb (where “b” indicates backbone methylation), or side chain (O- or S-), Ms (where “s” indicates side chain methylation), of amino acids.6–9 Based on the ten conserved sequence motifs of A domains (a1–a10),10 these characterized M domain interruptions are found to be either between a2–a3 or a8–a9.6 By far the most common type of M domain interruption is between the a8–a9 region.6 A possible explanation for this propensity was illuminated by the structure of an a8–a9 interrupted A domain, which showed that the M domain protruded from the side of the A domain within the small subdomain.11 This allows the small subdomain to remain in the same position as it is in uninterrupted A domains, preserving the catalytic ability of the A domain. In fact, it was shown that an uninterrupted A domain could be interrupted with either a backbone or side chain methylating M domain between the a8–a9 region and yield completely functional and specific interrupted A domains.12 Additionally, a di-interrupted A domain was generated that contained an M domain between a2–a3 and one between a8–a9, a scaffold not found in Nature, which could produce N,S-diMe-L-Cys.13 However, Nature has its own version of an interrupted A domain containing two M domains. These two M domains are positioned back-to-back between the a8–a9 region of the A domain. This arrangement is observed in an NRPS of the columbamides biosynthetic pathway,14 ColG(AMsMbA), and was recently biochemically characterized by our laboratory.15 These recent findings evoked two very compelling questions: (i) can an uninterrupted A domain be engineered to have both a non-cognate side chain and a non-cognate backbone methylating M domain (all three from different NP assembly-lines) inserted between a8–a9? And (ii) does the order of the M domains (MsMbversus MbMs) inserted into the uninterrupted A domain matter for activity? More specifically, based on the fact that in naturally occurring back-to-back interrupted A domains (e.g., ColG(AMsMbA)14,15 and DidJ(AMsMbA)16) the order of the M domains is MsMb, we wondered if MbMs would work equally as well or better than MsMb when producing a dimethylating back-to-back interrupted A domain (Fig. 1).
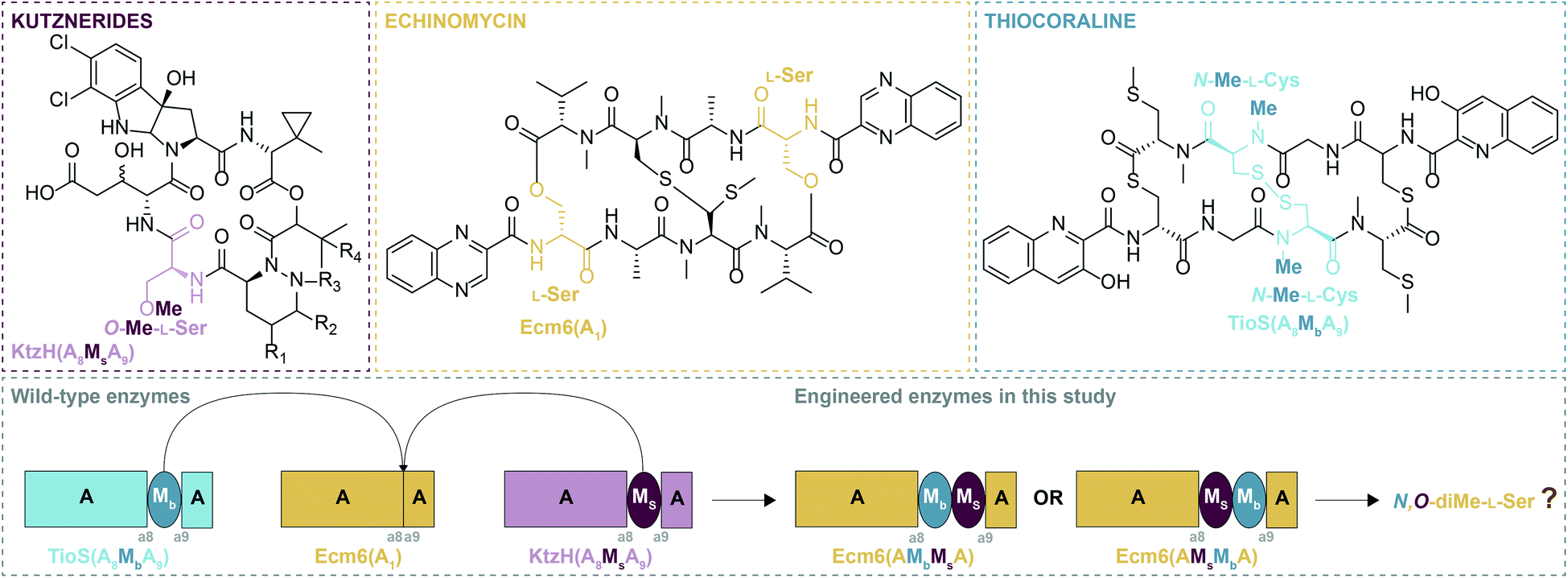 | ||
| Fig. 1 The NPs kutznerides, echinomycin, and thiocoraline with the moieties incorporated by KtzH(A8MsA9), Ecm6(A1), and TioS(A8MbA9) highlighted in pink, dark yellow, and teal, respectively. A cartoon representation of the Mb and Ms added to the uninterrupted A domain and resulting engineered back-to-back interrupted A domains. | ||
To investigate the above questions, we modified our previously successfully engineered interrupted A domain system12 using the interruption point that we demonstrated was capable of withstanding insertions of these single M domains individually. We used the uninterrupted A domain Ecm6(A1) known to activate L-Ser from the echinomycin biosynthetic pathway (Fig. 1) as the M domain acceptor.12 The M domain donors were the side chain O-methylating M domain (Ms) located between the a8–a9 region of the interrupted A domain found in the kutznerides biosynthetic pathway, KtzH(A8MsA9) (previously called KtzH(A4aMHA4b), but simplified for this manuscript),12 and the backbone N-methylating M domain (Mb) located between the a8–a9 region of the interrupted A domain TioS(A8MbA9) (previously called TioS(A3aM3SA3b), but simplified for this manuscript)12 involved in thiocoraline biosynthesis (Fig. 1). These were combined to make Ecm6(A8MsMbA9) and Ecm6(A8MbMsA9) as well as Ecm6(A8MsMbA9T1) and Ecm6(A8MbMsA9T1). For simplification in this manuscript, we will use a shorthand version of the names of these engineered enzymes, Ecm6(A8MsMbA9)/Ecm6(A8MsMbA9T1) will be Ecm6(AMsMbA)/Ecm6(AMsMbAT1). Likewise, Ecm6(A8MbMsA9)/Ecm6(A8MbMsA9T1) will be denoted as Ecm6(AMbMsA)/Ecm6(AMbMsAT1). Both of these engineered enzymes were evaluated through radiometric assays, high performance liquid chromatography (HPLC), and mass spectrometry (MS) assays for their various activities.
Results and discussion
Cloning, heterologous co-overexpression, and co-purification of Ecm6(AMsMbA), Ecm6(AMsMbAT1), Ecm6(AMbMsA), and Ecm6(AMbMsAT1) with the MbtH-like protein (MLP) Ecm8, as well as overexpression and purification of Ecm6(T1)
To establish if an A domain could be engineered to contain a back-to-back side chain and backbone M domain interruption, we cloned Ecm6(AMsMbA). We were also curious if the order of the M domains in the interruption was essential as the back-to-back interrupted A domains found in Nature are characterized or predicted to be side chain first and backbone second.14,16 Thus, we reversed the order of the M domains and constructed Ecm6(AMbMsA). We also generated Ecm6(AMsMbAT1) and Ecm6(AMbMsAT1) to evaluate if the A domain activity was altered by engineering the interrupted A domain with or without the T domain as A/T interactions of naturally occurring interrupted A domains are known to be able to act both in cis and in trans.11 To generate these proteins, we cloned them in a multistep process; we used overlapping PCR (Fig. S1, Tables S1 and S2†) to generate the MsMb or MbMs sections of the constructs. We used the previously reported pecm6(A8MbA9T1)-pET28a (originally called pecm6(A1aM3SA1bT1)-pET28a)12 as a template to first clone pecm6(A8MbA9)-pET28a and pecm6(T1)-pET28a. The Mb section of pecm6(A8MbA9)-pET28a was removed via restriction digest, and the new MsMb or MbMs fragments were ligated into the A/AT domain scaffold. The annotated amino acid sequences for the Ecm6(AMsMbA) and Ecm6(AMbMsA) constructs are presented in Fig. S2.† As previously reported,12 these interrupted A domains were co-overexpressed and co-purified with the native MLP Ecm8 in E. coli BL21 (DE3) ybdz::aac(3)IV.17 In recent years, MLPs have been shown to be an essential component for some A domains18 to yield successful heterologous expression of soluble and active A domains, including Ecm6(A1), from which these interrupted A domains were derived. MLPs bind to the A domains19 and this interaction was shown to remain unperturbed in the structure of an A domain with a single interruption.11 Therefore, we suspect the previous findings regarding MLPs' influence on expression/activity17 and substrate specificity20,21 to hold true with the interrupted A domains generated in this study.Evaluation of the adenylating activity of Ecm6(AMsMbA), Ecm6(AMbMsA), Ecm6(AMsMbAT1), and Ecm6(AMbMsAT1)
The A domain activity of Ecm6(AMsMbA) and Ecm6(AMbMsA) as well as the influence on adenylation activity of MsMbvs. MbMs interruptions were evaluated by using well-established ATP-[32P]PPi exchange assays.22 The substrate profiles of these two enzymes were first determined with the 20 common amino acids as well as N- and O-methylated derivatives of L-Ser (Fig. 2). The natural substrate of the wild-type (wt) Ecm6(A1T1) is known to be L-Ser (presented in Fig. S4†for comparison purposes only).12,23 The results of the substrate profiles showed that both Ecm6(AMsMbA) and Ecm6(AMbMsA) were very specific for L-Ser. This along with other engineering studies12,13 indicate that the natural substrate selectivity is not dramatically altered by insertion of one or two M domains into the a8–a9 region of an uninterrupted A domain. Additionally, the interruption order of the two M domains, MbMs or MsMb, does not influence the substrate specificity of the A domain. Next, Ecm6(AMsMbA) and Ecm6(AMbMsA) were compared to their Ecm6(AMsMbAT1) and Ecm6(AMbMsAT1) counterparts to determine if the addition of the T domain impacted the adenylation ability (Fig. 3). Although it is known that A and T domains can be separated in NRPS systems and investigated independently,24 some constructs are impaired by improper division of the A and T domains,25,26 therefore we sought to verify that this was not the case with our engineered interrupted A domain constructs. We found that there was a negligible difference in the relative percent of the adenylation activity with L-Ser between the two enzyme sets (Fig. 3). Therefore, we carried on with Ecm6(AMsMbA) and Ecm6(AMbMsA) for loading and methylation assays because the protein production yields were higher, and the concentration of the T domain alone was far greater than that of Ecm6(AMsMbAT1) and Ecm6(AMbMsAT1).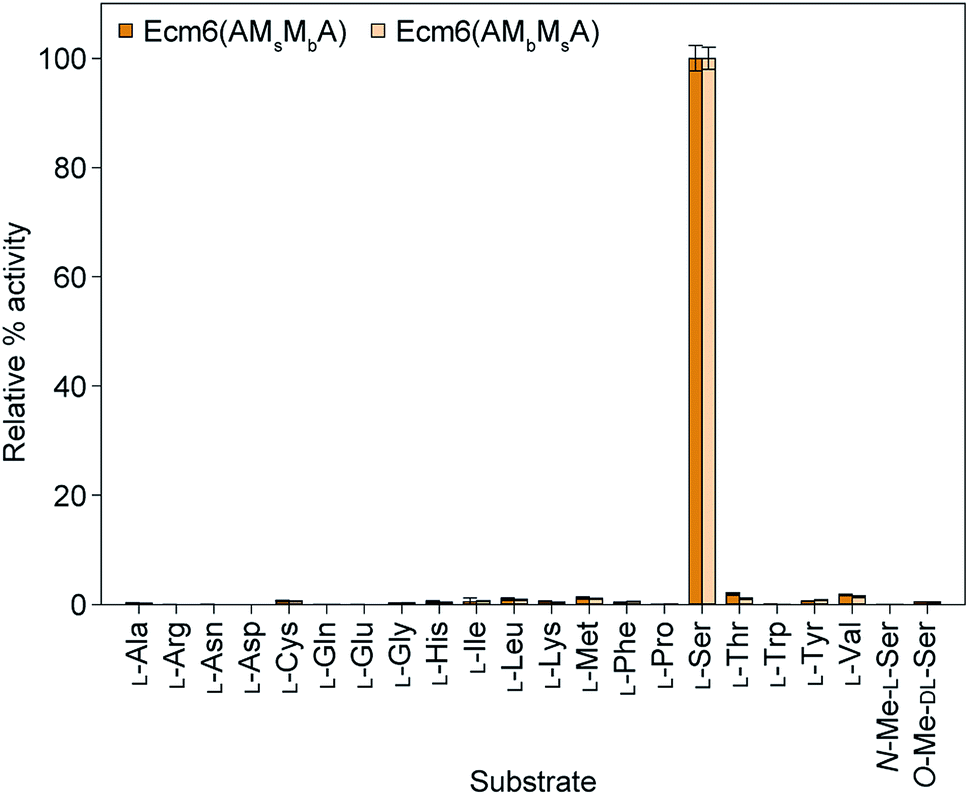 | ||
| Fig. 2 Substrate profiles of Ecm6(AMsMbA) (orange bars) and Ecm6(AMbMsA) (peach bars) determined via ATP-[32P]PPi exchange assays with a 2 h end point with all 20 natural amino acids as well as N-Me-L-Ser and O-Me-D,L-Ser. The substrate profile of the wt uninterrupted Ecm6(A1T1) is presented in Fig. S4†for comparison purposes only and was originally published in ref. 12. | ||
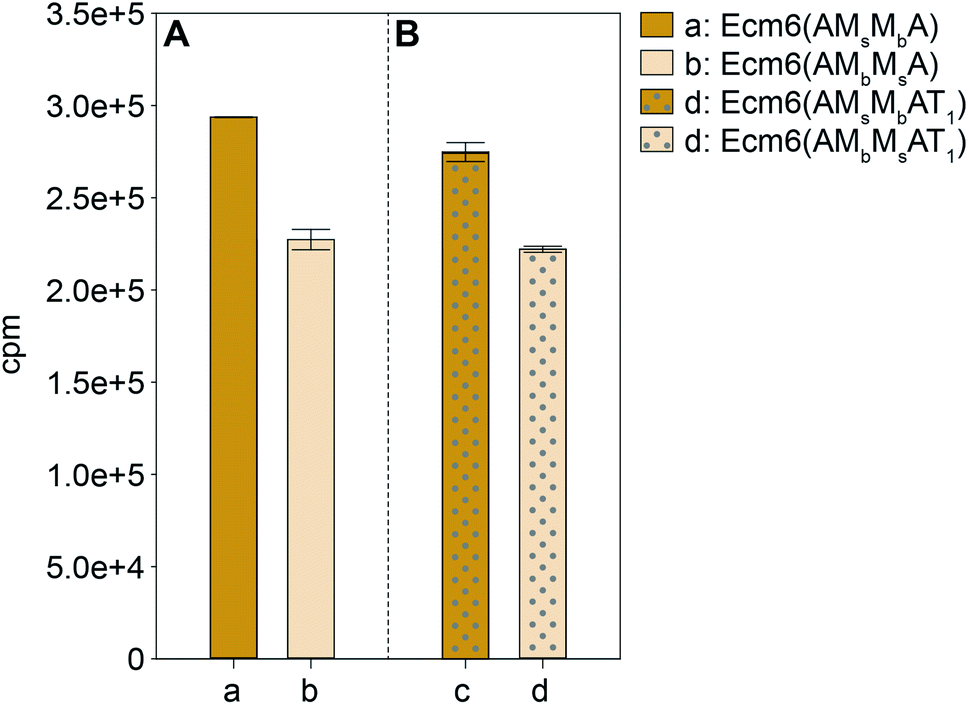 | ||
| Fig. 3 Comparison of adenylation of L-Ser by (A) Ecm6(AMsMbA) (orange bar) and Ecm6(AMbMsA) (peach bar) to that by their (B) Ecm6(AMsMbAT1) (orange bar with gray dots) and Ecm6(AMbMsAT1) (peach bar with gray dots) counterparts. | ||
When compared to the wt Ecm6(A1T1), previously engineered interrupted A domains, Ecm6(A8MsA9T1) and Ecm6(A8MbA9T1), both displayed an increased Km, but the kcat was decreased in Ecm6(A8MsA9T1) and increased in Ecm6(A8MbA9T1). As a result, the kcat/Km of Ecm6(A8MbA9T1) was the same as that of the wt.12 Therefore, the activities of Ecm6(AMsMbA) and Ecm6(AMbMsA) were quantitatively compared to each other by measuring and calculating Michaelis–Menten kinetic parameters (Table 1 and Fig. S5†). Since proper folding and positioning of the core N-terminal domain and a small C-terminal subdomain of the A domain must be maintained for adenylation activity by interrupted A domains,11,27 we hypothesized that the order of M domains could significantly affect the kinetics. This could perhaps be a reason as to why we only see one arrangement in Nature, because it is necessary for proper positioning and activity of the A domain. However, both the substrate affinity (Km = 7.2 ± 0.1 mM(S) and 7.6 ± 0.2 mM(S) for Ecm6(AMsMbA) and Ecm6(AMbMsA), respectively) and maximum velocity (Vmax = 18.8 ± 0.1 μM(E) min−1 and 12.0 ± 0.1 μM(E) min−1 for Ecm6(AMsMbA) and Ecm6(AMbMsA), respectively) were not significantly affected (note: mM(S) and μM(E) represent concentrations of substrate and enzyme, respectively). Although we were unable to calculate the turnover rate (kcat) because of difficulties of removing all impurities or degradation products, the concentrations of these two enzymes in the purified solutions were almost identical considering the degradation pattern of the proteins observed by SDS-PAGE (Fig. S3†). This result strongly suggests that inserting MsMb or MbMs in an uninterrupted A domain does not alter the overall structure of the A domain portion. Therefore, the naturally occurring order of two M domains (Ms followed by Mb) does not appear to be a result of selection pressure for A domain activity.
| Protein | Substrate | K m (mM(S)) | V max (μM(E) min−1) | V max/Km (μM(E) mM(S)−1 min−1) |
|---|---|---|---|---|
| a V max (instead of kcat) was measured because the enzyme concentrations were not accurately obtained. b The kinetic parameters for the wt Ecm6(A1T1) as well as engineered Ecm6(A8MsA9T1) and Ecm6(A8MbA9T1) are presented here for comparison purposes only and these data were originally published in ref. 12. | ||||
| Ecm6(AMsMbA) | L-Ser | 7.2 ± 0.1 | 18.8 ± 0.1 | 2.61 ± 0.04 |
| Ecm6(AMbMsA) | L-Ser | 7.6 ± 0.2 | 12.0 ± 0.1 | 1.57 ± 0.04 |
Attempts at evaluation of the methylation activity of Ecm6(AMsMbA) and Ecm6(AMbMsA)
Despite our best efforts to detect any form of a methylated L-Ser product from the engineered back-to-back interrupted A domains, Ecm6(AMsMbA) and Ecm6(AMbMsA), we were unable to ascertain evidence of methylation activity. Initially, we performed radioactive methylation assays using trichloroacetic acid (TCA) precipitation with [methyl-3H]SAM, a method our laboratory previously successfully used to evaluate interrupted A domain methylation capabilities.12,13,15 We found no discernible difference in M domain activity when looking at the cpm (counts per minute) values of the reactions by the engineered interrupted A domains compared to the no substrate negative control. After verifying that the [methyl-3H]SAM stock had not degraded as a result of long-term storage and/or multiple freeze–thaw cycles, we thought perhaps the issue lied in the ability of the engineered interrupted A domains to activate and load L-Ser onto the T domain, for if the T domain cannot be loaded, methylation cannot take place.9 Therefore, we supplemented the reactions with wt Ecm6(A1), which is able to load the T domain with L-Ser,12 however, we were still unable to detect increasing methylation compared to the no substrate negative controls. To assess if perhaps loading of the T domain was the issue, we also investigated the engineered interrupted A domain's ability to load L-Ser onto Ecm6(T1) by HPLC. Doing so, we were able to detect a change in the retention times of the loaded and unloaded T domain. First, we verified that there was a difference in the retention times for the holo Ecm6(T1) and the L-Cys-S-Ecm6(T1) by utilizing TioN, which is known to activate and load L-Cys onto Ecm6(T1) (Fig. 4A, reactions 2 and 3).12 (Note: we used L-Cys and N-Me-L-Cys loaded onto Ecm6(T1) by TioN as a control to determine if the change in retention time was sensitive enough to detect a single methyl group using HPLC described below.) After confirming that there was a detectable difference in the retention times of loaded and unloaded T domain, we analyzed the ability of Ecm6(AMsMbA) and Ecm6(AMbMsA) to load L-Ser onto Ecm6(T1). There was minimal conversion of Ecm6(T1) from holo to L-Ser-S-Ecm6(T1), indicating that the loading was very poor and certainly incomplete, even after an overnight reaction (Fig. 4B, reactions 6–9). This incomplete loading indicates that perhaps the engineered interrupted A domains are unable to properly adopt the thiolation conformation that requires a 140° domain rotation28–30 and, therefore, cannot load the substrate onto the T domain sufficiently. Additionally, hinderance of this critical and dynamic movement may be the same reason methylation was not observed. Thus, we reached the conclusion that loading of L-Ser was certainly impaired by the addition of two back-to-back M domains into an uninterrupted A domain (Fig. 4B, reactions 6 and 7). Additionally, in an attempt to evaluate methylation using HPLC, we determined if there was a difference in the retention times for L-Cys-S-Ecm6(T1) and N-Me-L-Cys-S-Ecm6(T1), as controls also using TioN to activate and load L-Cys and N-Me-L-Cys onto Ecm6(T1) as it is known to do.12 However, we could not confidently determine which HPLC peak was N-Me-L-Cys-S-Ecm6(T1) (Fig. 4A, reaction 1). There was no shift in peaks observed when SAM was added to the reactions with Ecm6(AMsMbA) and Ecm6(AMbMsA), indicating no methylation was occurring (Fig. 4B, reactions 4 and 5).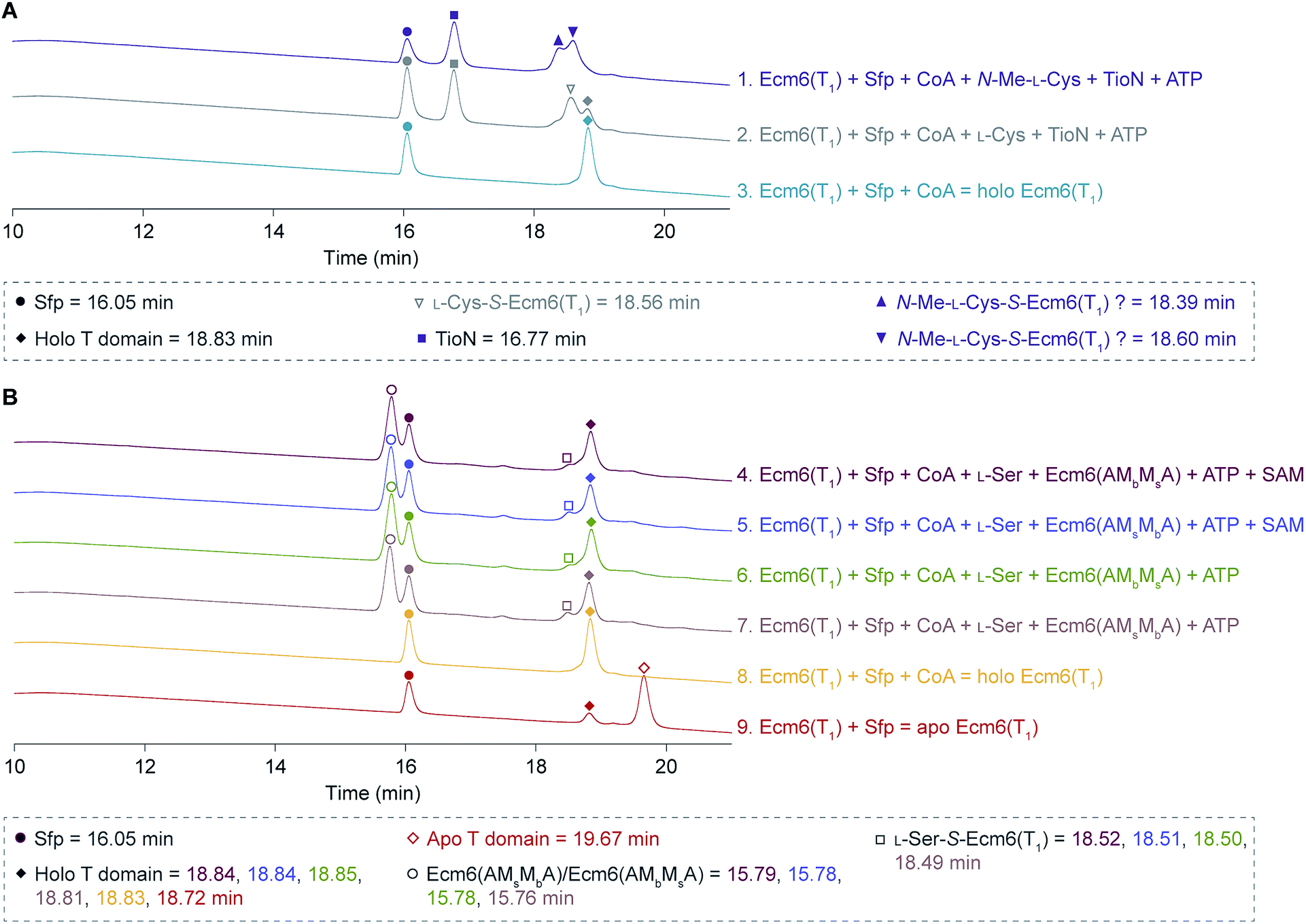 | ||
| Fig. 4 HPLC traces of (A) holo Ecm6(T1), L-Cys-S-Ecm6(T1), and N-Me-L-Cys-S-Ecm6(T1) using TioN to activate and load L-Cys and N-Me-L-Cys, and (B) loading and methylation reactions carried out by Ecm6(AMsMbA) and Ecm6(AMbMsA), compared to apo and holo Ecm6(T1). The peaks for Sfp are indicated by filled circles, Ecm6(AMsMbA) and Ecm6(AMbMsA) by empty circles, and TioN by filled squares. Apo and holo Ecm6(T1) are indicated by empty and filled diamonds, respectively. L-Cys-S-Ecm6(T1) is indicated by an empty triangle, and possible N-Me-L-Cys-S-Ecm6(T1) by filled and filled upside down triangles. The L-Ser-S-Ecm6(T1) is indicated by empty squares. The retention times are listed in the key beneath the traces. These HPLC traces were adjusted to align the Sfp peaks as described in the ESI.† | ||
Finally, to verify the absence of methylation, we analyzed the products of the enzymatic reactions by mass spectrometry (MS) and tandem mass spectrometry (MS2), another method that we have previously demonstrated sensitive enough to detect dimethylated amino acids (Fig. S6†).13,15 In this reaction, the interrupted A domain was allowed to activate the amino acid, load it onto the T domain, and covalently bind it to the Ppant arm where it was then attempted to be methylated. From there, the amino acid was cleaved using KOH, and the resulting product was then extracted with ethyl acetate and analyzed via MS and MS2. However, N,O-diMe-L-Ser was not detected (Fig. S6†) despite several rounds of reaction and extraction optimization. It is important to note that we have a synthetic standard of N,O-diMe-L-Ser that we have demonstrated could be ionized and fragmented in the positive mode (calculated [M + H]+ for N,O-diMe-L-Ser is 134.0812) (Fig. S6A†).15 This mass was not detected by simple visualization via MS. For good measure, we still conducted MS2 on products with [M + H]+ of 134.0812 (Fig. S6D–F†), but we did not obtain the fragmentation pattern for N,O-diMe-L-Ser (Fig. S6A and C†), instead it matched that of the no interrupted A domain controls (Fig. S6B and D†). Therefore, we were forced to conclude that, while two back-to-back M domains can be inserted into an A domain between a8–a9 without demolishing the A domain activity, the intricacies of coordinating two M domains are more challenging than inserting a single M domain in that position. Perhaps instead of combing three different proteins together, this lack of methylation activity could be overcome by using a native set of Mb and Ms from a naturally occurring back-to-back interrupted A domain (e.g., the MsMb of ColG(AMsMbA)) and transplanting the entire back-to-back set into Ecm6(A1). This would, however, address a much simpler hypothesis “can one transfer a naturally occurring back-to-back MsMb into an uninterrupted A domain, effectively combining two enzymes rather than three” than the hypotheses we attempted to address in this manuscript: (i) can one create AMMAs from three independent systems and (ii) is the MsMb order essential for dimethylation activity. As we learned in this study, the A domain portion can accommodate large insertions, making the insertion the limiting factor for engineering. In the future, as generating a functional AMMA from three different systems proved more intricate than originally thought based on the ease of creating other bi- and tri-functional enzymes,12,13,31 we could investigate this by utilizing natural MsMb domain sets. Additionally, this could shed light on the possible linkers between the various domains or other key differences discussed below.
Alternatively, it is possible that although what we originally thought were negligible differences in the two M domains compared to a natural back-to-back interruption like that found in ColG(AMsMbA), the differences were more significant than we first believed. For example, there are several gaps in the M domains of an amino acid sequence alignment of Ecm6(AMsMbA) and ColG(AMsMbA) (Fig. 5). The “missing” residues could provide essential spacing and proper positioning. Additionally, the Mb of ColG(AMsMbA) is longer than that of the Mb inserted into Ecm6, which could perhaps contribute to the overall functionality of the interrupted A domain. These small differences could play roles that we do not yet recognize and therefore were overlooked in our original design. Finally, another possibility is that the active sites of the M domains were positioned too far away from the T domain for the Ppant arm to reach, which could potentially be solved by the addition of a pseudo A subdomain, as recently observed in naturally occurring A/KR domains.32
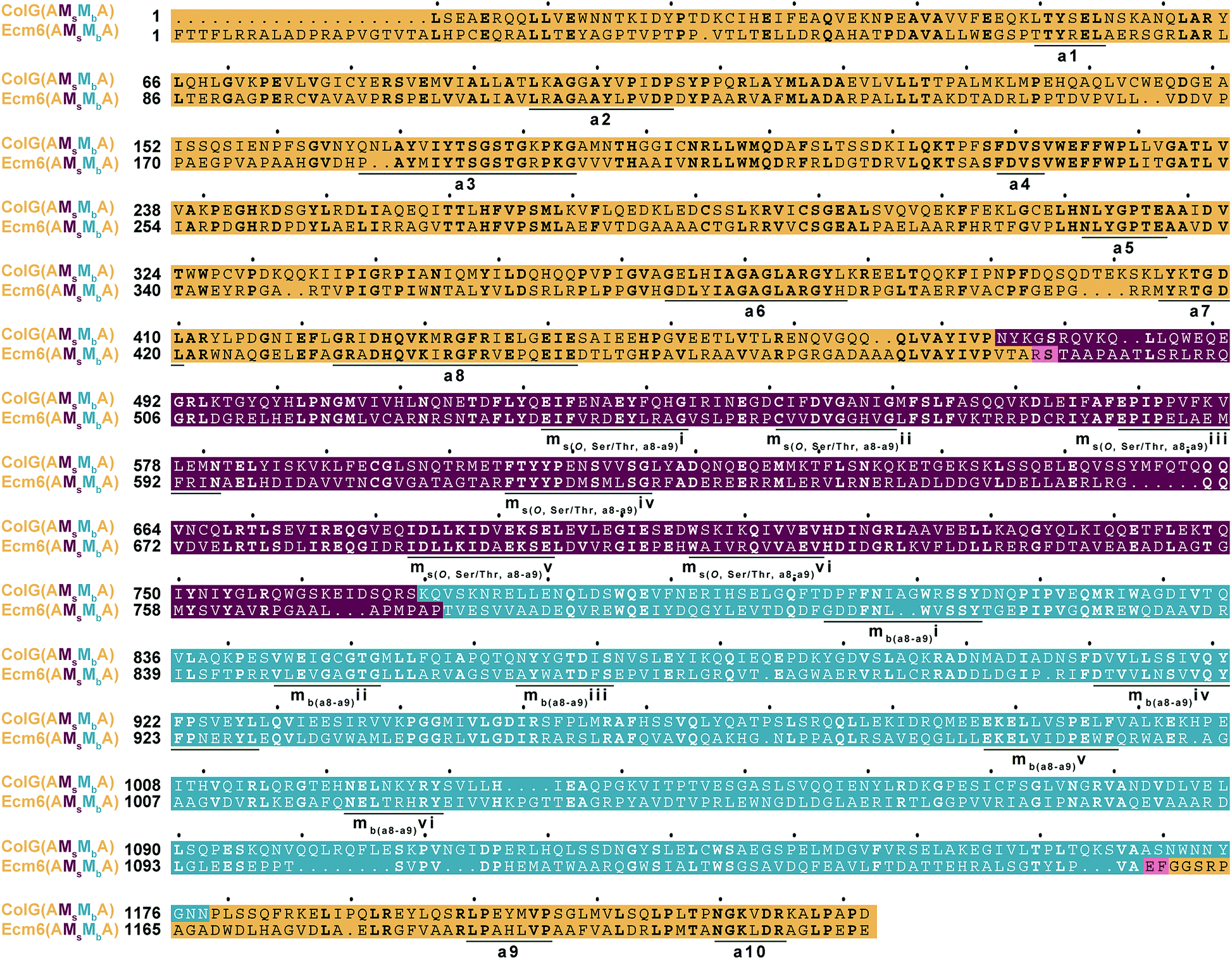 | ||
| Fig. 5 Multiple sequence alignment of ColG(AMsMbA) and Ecm6(AMsMbA). The A domain is highlighted in dark yellow, the Ms in burgundy, and the Mb in teal. The amino acid residues in Ecm6(AMsMbA) added as a result of the addition of the restriction sites used to generate the engineered construct are highlighted in pink. The conserved A domain motifs are underlined and labeled a1–a10. The conserved Ms domain motifs are underlined and labeled ms(O, Ser/Thr, a8–a9)i–vi. The Mb domain conserved motifs are underlined and labeled mb(a8–a9)i–vi. | ||
Conclusion
In summary, we have created two back-to-back interrupted A domains from an uninterrupted A domain. While we were unable to detect methylation activity from the engineered interrupted A domains, we believe this work provides valuable insight into the limitations of engineering interrupted A domains. Perhaps the most impactful and translatable lesson we have learned from this work is that the region between the a8 and a9 conserved motifs is incredibly tolerant of artificial insertions. This location could even be considered a privileged location for interrupting A domains without concern for loss of adenylation activity and specificity, an incredible obstacle in enzyme engineering. This also sheds light as to why this is the most common location for interruptions of not only length of interruption, but type of M domain as well.6,33 The two M domains that were added between the a8–a9 region were a total of 670 amino acids in length, and adenylation activity was still observed, indicating size may not be a serious constraint in engineering interrupted A domains. This study demonstrates the remarkable ability of the A domain to maintain the folding and positioning of the A subdomain, which is known to form the open and closed state of the A domain by moving in and out of the A domain binding pocket. Therefore, the limits of engineering interrupted A domains is not the A domain itself, rather what is inserted, perhaps opening the door for even more unnatural or innovative insertions to be added to A domains. For the engineered A domains generated in this study, while the A domain activity was maintained, the problem of theses engineered interrupted A domains lies with the downstream processes of loading and methylation. As we know, for interrupted A domains to successfully operate, the A domain must first activate the substrate, then load it onto the T domain, and only then is the substrate able to be methylated.9 Therefore, the lack of methylation activity could result from an inability of the loaded T domain to reach the active sites of the M domains due to improper alignment of the two M domains. This is a significant challenge to overcome for engineering back-to-back interrupted A domains, which was not a problem when only one M domain was artificially inserted.12,13 Until there is a crystal structure solved, we will not know for sure how close together the active sites are for back-to-back interrupted A domains. Alternatively, it could be that the two M domains used in this project were not compatible or did not communicate or coordinate well with each other or the A and T domains, halting methylation. This highlights the importance of potential communication or linking regions between these domains. Linking regions are critical to the proper coordination of different domains in NRPS synthesis and successful module exchanges.26,34,35 There could perhaps be a yet to be identified linker domain needed between the two M domains. It is possible that this could be overcome by using a native set of Mb and Ms from a naturally found back-to-back interrupted A domain and transplanting the entire insertion rather than combing three different proteins together. Ultimately, this manuscript demonstrates the immense ability of A domains to withstand insertions of substantial size while retaining the adenylation activity, and by extension, the A domain folding. Our laboratory is pursuing other ways to generate multifunctional interrupted A domains with the ultimate goal of creating targeted modifications to NRPs.Abbreviations
| A | Adenylation |
| AMP | Adenosine monophosphate |
| C | Condensation |
| CoA | Coenzyme A |
| cpm | Counts per minute |
| E | Epimerization |
| Hal | Halogenation |
| HPLC | High performance liquid chromatography |
| M | Methylation |
| MLP | MbtH-like protein |
| MS | Mass spectrometry |
| MS2 | Tandem mass spectrometry |
| NP | Natural product |
| NRP | Nonribosomal peptide |
| NRPS | Nonribosomal peptide synthetase |
| PCR | Polymerase chain reaction |
| Ppant | 4′-Phosphopantetheine |
| PPTase | 4′-Phosphopantetheinyl transferase |
| PPi | Inorganic pyrophosphate |
| SAM | S-Adenosyl-L-methionine |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| T | Thiolation |
| TCA | Trichloroacetic acid |
| TE | Thioesterase |
| wt | Wild-type |
Author contributions
T. A. L. and S. G.-T. designed the study. T. A. L. designed, cloned, expressed, and purified the proteins. S. M. conducted adenylation and kinetics assays. T. A. L. investigated methylation activity. T. A. L., S. M., and S. G.-T. prepared all figures and wrote the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Science Foundation (NSF) CAREER Award MCB-1149427 (to S. G.-T.) and by startup funds from the University of Kentucky College of Pharmacy (to S. G.-T.). T. A. L. was in part supported by a 2018–2019 and a 2019–2020 Pharmaceutical Sciences Excellence in Graduate Achievement Fellowship from the College of Pharmacy at the University of Kentucky as well as a 2019–2020 Pre-doctoral Fellowship in Pharmaceutical Sciences from the American Foundation of Pharmaceutical Education (AFPE). We thank Dr Nishad Thamban Chandrika for synthesizing the N,O-dimethyl-L-Ser15 used as a standard in the MS and MS2 studies presented herein. We thank Dr Atefeh Garzan for synthesizing the N-methyl-L-Cys9 used in the HPLC studies presented in this manuscript.References
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the 30 years from 1981 to 2010, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS.
- R. Finking and M. A. Marahiel, Biosynthesis of nonribosomal peptides, Annu. Rev. Microbiol., 2004, 58, 453–488 CrossRef CAS.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, A new enzyme superfamily - The phosphopantetheinyl transferases, Chem. Biol., 1996, 3, 923–936 CrossRef CAS.
- K. Zhang, K. M. Nelson, K. Bhuripanyo, K. D. Grimes, B. Zhao, C. C. Aldrich and J. Yin, Engineering the substrate specificity of the DhbE adenylation domain by yeast cell surface display, Chem. Biol., 2013, 20, 92–101 CrossRef CAS PubMed.
- J. Thirlway, R. Lewis, L. Nunns, M. Al Nakeeb, M. Styles, A. W. Struck, C. P. Smith and J. Micklefield, Introduction of a non-natural amino acid into a nonribosomal peptide antibiotic by modification of adenylation domain specificity, Angew. Chem., 2012, 51, 7181–7184 CrossRef CAS.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Interrupted adenylation domains: Unique bifunctional enzymes involved in nonribosomal peptide biosynthesis, Nat. Prod. Rep., 2015, 32, 641–653 RSC.
- O. E. Zolova and S. Garneau-Tsodikova, KtzJ-dependent serine activation and O-methylation by KtzH for kutznerides biosynthesis, J. Antibiot., 2014, 67, 59–64 CrossRef CAS.
- A. H. Al-Mestarihi, G. Villamizar, J. Fernandez, O. E. Zolova, F. Lombó and S. Garneau-Tsodikova, Adenylation and S-methylation of cysteine by the bifunctional enzyme TioN in thiocoraline biosynthesis, J. Am. Chem. Soc., 2014, 136, 17350–17354 CrossRef CAS.
- S. Mori, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Deciphering Nature’s intricate way of N,S-dimethylating L-cysteine: Sequential action of two bifunctional adenylation domains, Biochemistry, 2017, 56, 6087–6097 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Structural basis for backbone N-methylation by an interrupted adenylation domain, Nat. Chem. Biol., 2018, 14, 428–430 CrossRef CAS PubMed.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Engineering bifunctional enzymes capable of adenylating and selectively methylating the side chain or core of amino acids, ACS Synth. Biol., 2018, 7, 399–404 CrossRef CAS.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Probing the limits of interrupted adenylation domains by engineering a trifunctional enzyme capable of adenylation, N-, and S-methylation, Org. Biomol. Chem., 2019, 17, 1169–1175 RSC.
- K. Kleigrewe, J. Almaliti, I. Y. Tian, R. B. Kinnel, A. Korobeynikov, E. A. Monroe, B. M. Duggan, V. Di Marzo, D. H. Sherman, P. C. Dorrestein, L. Gerwick and W. H. Gerwick, Combining mass spectrometric metabolic profiling with genomic analysis: A powerful approach for discovering natural products from cyanobacteria, J. Nat. Prod., 2015, 78, 1671–1682 CrossRef CAS PubMed.
- T. A. Lundy, S. Mori, N. Thamban Chandrika and S. Garneau-Tsodikova, Characterization of a unique interrupted adenylation domain that can catalyze three reactions, ACS Chem. Biol., 2020, 15, 282–289 CrossRef CAS PubMed.
- Y. Xu, R. D. Kersten, S. J. Nam, L. Lu, A. M. Al-Suwailem, H. Zheng, W. Fenical, P. C. Dorrestein, B. S. Moore and P. Y. Qian, Bacterial biosynthesis and maturation of the didemnin anti-cancer agents, J. Am. Chem. Soc., 2012, 134, 8625–8632 CrossRef CAS.
- E. A. Felnagle, J. J. Barkei, H. Park, A. M. Podevels, M. D. McMahon, D. W. Drott and M. G. Thomas, MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases, Biochemistry, 2010, 49, 8815–8817 CrossRef CAS PubMed.
- M. Wolpert, B. Gust, B. Kammerer and L. Heide, Effects of deletions of mbtH-like genes on clorobiocin biosynthesis in Streptomyces coelicolor, Microbiology, 2007, 153, 1413–1423 CrossRef CAS.
- D. A. Herbst, B. Boll, G. Zocher, T. Stehle and L. Heide, Structural basis of the interaction of MbtH-like proteins, putative regulators of nonribosomal peptide biosynthesis, with adenylating enzymes, J. Biol. Chem., 2013, 288, 1991–2003 CrossRef CAS PubMed.
- R. A. Schomer and M. G. Thomas, Characterization of the functional variance in MbtH-like protein interactions with a nonribosomal peptide synthetase, Biochemistry, 2017, 56, 5380–5390 CrossRef CAS.
- S. Mori, K. D. Green, R. Choi, G. W. Buchko, M. G. Fried and S. Garneau-Tsodikova, Using MbtH-like proteins to alter the substrate profile of a nonribosomal peptide adenylation enzyme, ChemBioChem, 2018, 19, 2186–2194 CrossRef CAS.
- G. F. Bryce and N. Brot, Studies on the enzymatic synthesis of the cyclic trimer of 2,3-dihydroxy-N-benzoyl-L-serine in Escherichia coli, Biochemistry, 1972, 11, 1708–1715 CrossRef CAS.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. Wang, H. Oguri and H. Oikawa, Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed.
- P. H. Weinreb, L. E. Quadri, C. T. Walsh and P. Zuber, Stoichiometry and specificity of in vitro phosphopantetheinylation and aminoacylation of the valine-activating module of surfactin synthetase, Biochemistry, 1998, 37, 1575–1584 CrossRef CAS PubMed.
- D. E. Ehmann, C. A. Shaw-Reid, H. C. Losey and C. T. Walsh, The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: Sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2509–2514 CrossRef CAS PubMed.
- B. R. Miller, J. A. Sundlov, E. J. Drake, T. A. Makin and A. M. Gulick, Analysis of the linker region joining the adenylation and carrier protein domains of the modular nonribosomal peptide synthetases, Proteins, 2014, 82, 2691–2702 CrossRef CAS PubMed.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S, EMBO J., 1997, 16, 4174–4183 CrossRef CAS.
- A. S. Reger, R. Wu, D. Dunaway-Mariano and A. M. Gulick, Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate CoA ligase, Biochemistry, 2008, 47, 8016–8025 CrossRef CAS PubMed.
- A. M. Gulick, Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase, ACS Chem. Biol., 2009, 4, 811–827 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Structures of two distinct conformations of holo-non-ribosomal peptide synthetases, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- S. K. Shrestha and S. Garneau-Tsodikova, Expanding substrate promiscuity by engineering a novel adenylating-methylating NRPS bifunctional enzyme, ChemBioChem, 2016, 17, 1328–1332 CrossRef CAS.
- D. A. Alonzo, C. Chiche-Lapierre, M. J. Tarry, J. Wang and T. M. Schmeing, Structural basis of keto acid utilization in nonribosomal depsipeptide synthesis, Nat. Chem. Biol., 2020, 16, 493–496 CrossRef CAS.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, A thorough analysis and categorization of bacterial interrupted adenylation domains, including previously unidentified families, RSC Chem. Biol., 2020 10.1039/d0cb00092b.
- S. Farag, R. M. Bleich, E. A. Shank, O. Isayev, A. A. Bowers and A. Tropsha, Inter-modular linkers play a crucial role in governing the biosynthesis of non-ribosomal peptides, Bioinformatics, 2019, 35, 3584–3591 CrossRef CAS.
- M. L. Nielsen, T. Isbrandt, L. M. Petersen, U. H. Mortensen, M. R. Andersen, J. B. Hoof and T. O. Larsen, Linker flexibility facilitates module exchange in fungal hybrid PKS-NRPS engineering, PLoS One, 2016, 11, e0161199 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the molecular cloning, protein expression and purification, substrate profile, kinetics, and methylation assays performed. Table of primers (Table S1), overlapping PCR for MsMb and MbMs fragments (Table S2), cloning strategy (Fig. S1), amino acid sequences of the engineered proteins (Fig. S2), SDS-PAGE gels of purified proteins used in this study (Fig. S3), substrate profile of the wild-type Ecm6(A1T1) for comparison purposes only (Fig. S4), Michaelis–Menten kinetic plots (Fig. S5), and MS2 analysis of enzymatic extracts (Fig. S6). See DOI: 10.1039/d0ra05490a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |