
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn energetic composite formed of wrinkled rGO sheets wrapped around copper azide nanowires with higher electrostatic safety as a green primary explosive†
Xuwen Liu ab,
Yan Hu*ab,
Tingting Liab,
Yinghua Yeab and
Ruiqi Shenab
ab,
Yan Hu*ab,
Tingting Liab,
Yinghua Yeab and
Ruiqi Shenab
aSchool of Chemical Engineering, Nanjing University of Science and Technology, China. E-mail: huyan@njust.edu.cn
bMicro-Nano Energetic Devices Key Laboratory, Ministry of Industry and Information Technology, China
First published on 19th August 2020
Abstract
A green primary explosive with high energy density and electrostatic safety was synthesized in this work. A precursor consisting of wrinkled reduced graphene oxide sheets wrapped around copper nanowires (CuNWs@rGO) was fabricated through a facile one-pot hydrothermal approach. The as-prepared precursor was deposited on a silicon wafer by electrophoretic deposition technology, which significantly reduced the safety risks of directly handling the powder sample in the azide reaction. Wrinkled rGO sheets wrapped around copper azide nanowires (CANWs@rGO) were prepared in situ by reaction of the precursor with HN3 gas. The initiation capability was tested by using it to detonate hexogen (RDX) against a lead plate with a thickness of 5 mm, and its detonation performance was found to be better than that of commercial diazodinitrophenol (DDNP). The electrostatic sensitivity of the CANWs@rGO composite was investigated, and the result shows that the discharge energy at 50% (E50%) of CANWs@rGO was 0.96 mJ, which indicates that it has a much higher electrostatic safety than that of pure copper azide (0.05 mJ).
1. Introduction
A primary explosive is an essential element in explosive systems which is extremely sensitive to stimuli.1–3 Once initiated, the primary explosive can rapidly transition from deflagration to detonation, and consequently initiate a more substantial charge of a less sensitive secondary explosive that is usually safer to handle. Due to their excellent initiation efficiency and high flame sensitivity, lead azide (LA) and LS are the most commonly used primary explosives which have been used for more than one century.4–7 However, the use of these explosives is harmful to the environment and public health because of the toxicity and carcinogenicity of lead. Moreover, the energy density of lead-containing primary explosives can hardly meet the requirements of the micro-initiator for energy due to the miniaturization of the ignition device.8–12 Hence, developing a green substitute for lead-based primary explosives with high safety and energy density is urgently needed.Copper azide (CA) as a replacement of primary explosives containing lead has shown advantages in terms of initiation properties and environmental friendliness.13–16 Its high output energy enables CA to meet the dosage limitation and assembly requirements in miniaturized explosive systems.17–21 Unfortunately, its high electrostatic sensitivity, which is a crucial parameter to assess safety performance, limits the wide range of applications of CA, only 0.05 mJ discharge energy caused by collision and friction will induce an explosion. Previous studies have investigated some strategies for reducing the sensitivity of several existing primary explosives.22–24 For instance, graphene and carbonized metal–organic frameworks (MOFs) have been utilized as a carbon conductive matrix by us and others to prepare energetic composites with low sensitivity.1,25,26 Copper oxide particles were loaded into oriented carbon nanotubes with open ends prepared using a self-supporting alumina film as a template to reduce copper oxide to copper in a hydrogen atmosphere.27 In order to simplify the preparation of CA@CNT composites, copper particles were also deposited in oriented carbon nanotubes directly by electrochemical deposition and then underwent azidation by hydrogen azide gas.28 In spite of these developments, there are still few reports of an insensitive energetic composite based on CA.29,30 Moreover, most of these methods require tedious synthesis processes and involve high costs and safety risks during preparation.
The crucial factor for promoting the properties of CA is the precursor.31–34 CA can be in situ fabricated by a gas–solid phase azide reaction. Nanowires or nanorods provide the essential advantages of a high surface area and features that facilitate self-assembly.35–38 The effectiveness of the nanowire morphology to react with gaseous HN3 appears to be particularly advantageous where particle morphology sometimes fails.39 The application prospects of self-assembled functionalized graphene sheet nanocomposites in energetic materials are extensive.40,41 CuNWs@rGO with its extraordinary electrical properties and small charge transfer resistance value provided by rGO,42 as the precursor, can effectively depress static electricity accumulation on the surface of CA, thereby reducing the electrostatic sensitivity of the energetic composite.
The work presented in this paper used CuNWs@rGO obtained through a simple one-pot hydrothermal reduction as the precursor. The high sensitivity of CA posed significant safety risks. Hence, the CuNWs@rGO precursor was deposited on a silicon substrate by electrophoretic deposition, a binder-free coating method,43 which greatly reduced the uncertainty of directly handling the powder sample. After 48 h of the in situ azide reaction, the CuNWs@rGO precursor was reacted with HN3 gas and completely transformed into copper azide nanowires (CANWs)@rGO. The samples were characterized and tested by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), differential scanning calorimetry (DSC), Raman spectroscopy, and an electrostatic sensitivity tester. The detonation capability was also evaluated by testing its ability to detonate secondary explosives (RDX).
2. Experimental
2.1 Synthesis of CuNWs@rGO
The GO (15 mL, 5 mg mL−1) was dispersed in ultrapure water and ultrasonicated for 20 min. Cu(NO3)2 (30 mL, 0.1 M) was added into the beaker containing GO. The mixture was stirred unceasingly for 30 min under magnetic stirring. Cu2+ ions were attached to the functional groups on the GO surface through electrostatic interactions. Then EDA (15 mL, 0.13 M) was added, leading to a color change to deep violet (for Cu(EDA)22+) with stirring for 15 min (Fig. S1†). The as-prepared mixed solution was transferred to a three-necked flask containing NaOH (300 mL, 15 M) and heated in a water bath at 80 °C for 1 h. Catechin (0.6 g) was added to the solution and used as a reducing agent, then ultrasonicated again in a water bath at 80 °C for 1 h. Upon hydrothermal treatment, rGO was obtained by reducing graphene oxide and simultaneously CuNWs grew readily using EDA as the complexing agent for Cu2+. The solution was cooled to room temperature naturally, then the CuNWs@rGO composite was precipitated through centrifugation. After repeated centrifugation/washing cycles, the unreacted catechin and excess EDA were removed. The samples were re-dispersed and ultrasonicated in ultrapure water for 15 min and then transferred to centrifuge tubes. All samples prepared were kept under N2 in a glovebox.2.2 Electrophoretic deposition
In the general process of depositing the composites by electrophoretic deposition (Fig. 1), a monocrystalline silicon wafer was adopted as the cathode while graphite was used as an anode material. Aluminum nitrate (2.4 mg) was dispersed in a mixture of ethanol (10 mL) and acetone (20 mL), then NaOH (100 mL, 0.005 M) was added into the solution to adjust the PH. The electrophoretic suspension was prepared by adding the as-prepared CuNWs@rGO (80 mg) composites to the solution with stirring for 15 min and ultrasonicating for 20 min. The two electrodes were inserted vertically into the suspension and placed parallel to each other with a constant distance of 20 mm. The CuNWs@rGO films were electrophoretically deposited at 25 V for 20 min on a monocrystalline silicon wafer (the size was 30 mm × 13 mm × 0.2 mm) from the CuNWs@rGO suspension. Through the deposition–drying–deposition treatment, the thickness of the as-deposited CuNWs@rGO film can be controlled.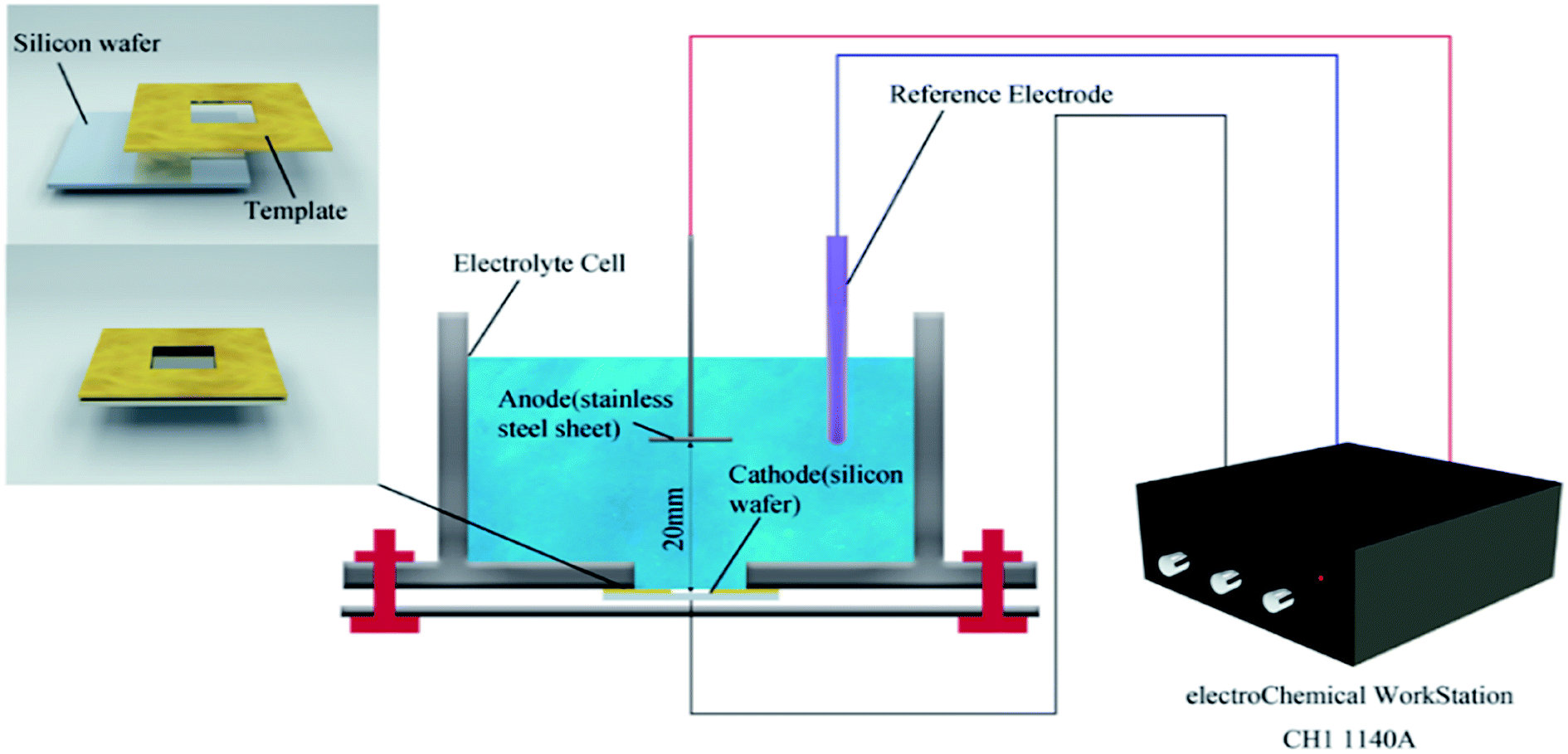 | ||
| Fig. 1 A schematic diagram of the electrophoretic deposition process. | ||
2.3 In situ synthesis of CANWs@rGO
The in situ gas–solid phase azidation reaction was carried out in a custom device for 48 h. Ultrapure sodium azide (0.2 g mL−1) was dispersed in deionized water to a total volume of 10 mL. A glass dish containing the solution was placed at the bottom of the device. The sample was placed on a polyurethane sieve fixed in the middle of the reaction vessel. The azidation reaction was air-sensitive in terms of the energetic products, so all joints must be greased to prevent contamination by O2. The right neck of the device was connected to a glass tube inserted into a beaker containing KOH, which had the effect of filtrating the unreacted HN3. Before the reaction, N2 was introduced from the left neck to ensure an inert atmosphere in the reaction vessel. HNO3 (30 mL, 4 M) was added dropwise (from a dropping funnel) to the glass dish. The reaction vessel was heated by an oil bath at 100 °C for 48 h. Once the azidation reaction has started, the air-tight seal of the device must be ensured. In order to take out the azide product safely after the reaction, a positive flow of inert gas must be introduced again to ultimately ensure there is no more unreacted gaseous HN3 in the device.3. Results and discussion
3.1 Structural characterization of the CuNWs@rGO and CANWs@rGO composite
The morphologies of the fabricated energetic composites depend primarily on the morphologies of the CuNWs@rGO precursors.The presence of EDA is necessary for the anisotropic growth of CuNWs. Fig. S3–S7† display the representative TEM images of CuNWs and urchin-like copper nanoparticles grown using constant concentrations of Cu(NO3)2 (30 mL, 0.1 M) and catechin (6 mM) and increasing concentrations of EDA. In the composite, CuNWs with fewer tiny nano-thorns were prepared in the presence of 1.8 mM EDA. When a higher concentration of EDA (3.6 mM) was applied, the nano-thorns on CuNWs became denser and thicker. Nanowires are approximately 1 μm and 100 nm in length and diameter. As depicted in Fig. S7,† in the presence of a higher concentration of EDA (7.2 mM), copper can be found in the form of urchin-like nanoparticles with diameters about 400 nm. These results suggest that nano-thorns were grown from the exposed side faces of CuNWs under low concentrations of EDA, showing that EDA molecules preferentially cover their side faces. The higher the EDA concentration, the more molecules cap the end of the wire, leading to the formation of urchin-like nanoparticles.
To provide meaningful insights into the growth mechanism of CuNWs, we also studied the effect of reaction time on the morphology of copper. To investigate the anisotropic growth characteristics and propose a possible formation mechanism of CuNWs, we evaluated the effect of reaction time on the morphology change of CuNWs. Under optimal conditions, the mixture was continuously stirred at 80 °C for various reaction time durations (15 min, 30 min, and 60 min), and the products were further characterized by electron microscopy, respectively. A possible formation mechanism of CuNWs was proposed by studying the morphology evolution of copper under different reaction times. TEM images of these precipitates (Fig. 2) revealed the anisotropic growth process of the CuNWs. After reaction for 15 min, urchin-like Cu nanoparticles 1.5 μm in diameter were observed (Fig. 2(a)). Upon heating the solution for 30 min, as shown in Fig. 2(b), the urchin-like seeds underwent a rolling process, conical copper wires that grew out of nanoparticles were formed. After being reacted for 60 min, CuNWs with thick nano-thorns were found to be longer and thinner and no nanoparticles were observed, as depicted in Fig. 2(c).
 | ||
| Fig. 2 TEM images of the copper morphology after (a) 15; (b) 30; and (c, e and f) 60 min of reaction; (d) HRTEM of CuNWs@rGO; (g) TEM-EDS mapping of CANWs@rGO; and SEM images of (h) CuNWs@rGO and (i) CANWs@rGO. | ||
Fig. 2(e and f) displays higher magnification TEM images of the CuNWs@rGO precursor composite, revealing that the CuNWs were wrapped with an ultra-thin layer of wrinkled sheets of rGO, supported by the SEM image depicted in Fig. 2(h). From the high-resolution transmission electron microscopy (HRTEM) image (Fig. 2(d)), the interplanar spacings of the composite are determined to be 0.34 nm and 0.208 nm, which correspond to the crystallization of rGO and the (111) plane of Cu. The bright lattice fringes indicate that the composite material has high-grade crystallinity.
The nitric acid solution reacts with NaN3 to produce gaseous HN3 at normal temperatures (reaction eqn (1)). The gas–solid phase azide reaction of CuNWs with gaseous HN3 can synthesize CA (reaction eqn (2)).
| HNO3 + NaN3 → HN3 + NaNO3 | (1) |
| Cu + 3HN3 → Cu(N3)2 + NH3 + N2 | (2) |
After the azidation reaction, the SEM images reveal that the morphology of the nanowire precursor was still retained (Fig. 2(i)). The diameter of the CuNWs precursor increased and more protrusions were formed on the surface due to grain growth. The microstructure and compositional distribution of the energetic composites were further investigated by TEM-energy dispersive spectroscopy (EDS) mapping. Elemental mapping indicates that the CANWs@rGO composite was mainly composed of C, N and Cu (Fig. 2(g)). Cu and N were homogeneously distributed in the CANWs, and the density was relatively high. Due to the coating of rGO, C was widely spread in the CANWs@rGO composite.
Fig. 3(a) shows the XRD pattern of the azide product of the CuNWs@rGO composite. It can be seen from the figure that strong characteristic diffraction peaks appeared at 11.785°, 16.371°, 27.946°, 31.936°, and 36.727°, which correspond to the (110), (120), (230), (021) and (131) crystal planes of Cu(N3)2, respectively, which indicates that Cu(N3)2 was formed after 48 h of the gas–solid phase azide reaction. The sample was characterized when the reaction progressed to 36 hours, and the results show that the product was cuprous azide (Fig. S13†).
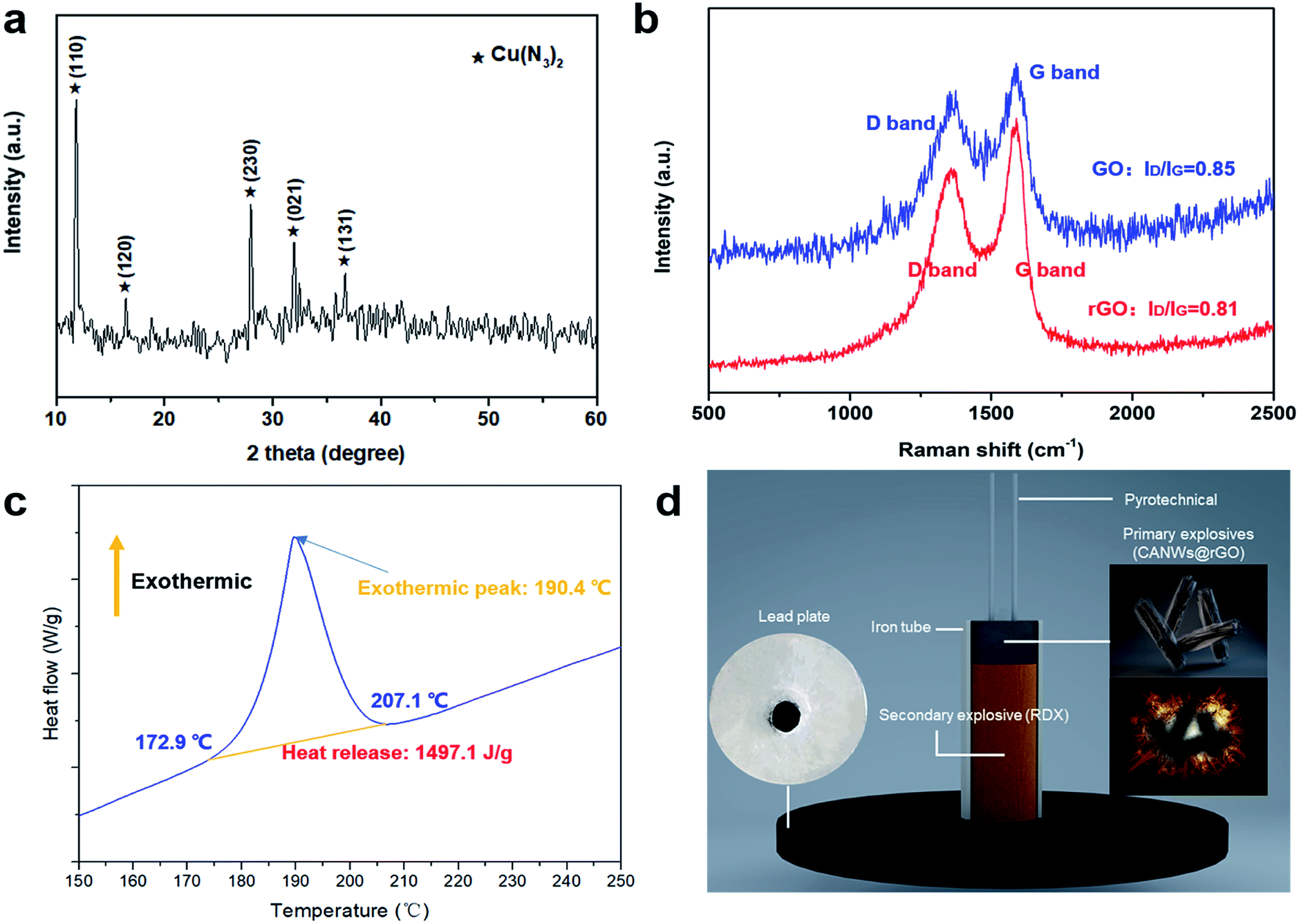 | ||
| Fig. 3 (a) The PXRD pattern of CANWs@rGO (JCPDS card no. 21-0281); (b) the Raman spectrum of GO and CANWS@rGO; (c) the DSC curve of CANWs@rGO; and (d) a schematic diagram of the detonation ability test process of CANWs@rGO. | ||
We acquired the Raman spectrum of GO and CuNWs@rGO, which revealed that GO was reduced into rGO using catechin as the reducing agent. The oxygen anions of catechin readily opened the functional groups on the surfaces of GO through SN2 nucleophilic substitution. As displayed in Fig. 3(b), the Raman spectra reveal two distinct peaks corresponding to the D band (due to the doubly degenerate E2g mode at the Brillouin zone center) and the G band (arising from the defect-mediated zone-edge phonons). The D- and G-bands are at 1347 and 1584 cm−1, respectively, for GO, while they are at 1339 and 1592 cm−1, respectively, for CuNWs@rGO. The D-band and G-band intensity ratios (ID/IG) of the GO and CuNWs@rGO are 0.85 and 0.81, respectively, revealing that some of the oxygen-containing groups can be removed when a prolonged reduction is performed under heating, leading to a decrease in the ID/IG value. The electric conductivity of graphene is strongly dependent on the quality of its sp2 carbon domain, which can be evaluated by ID/IG. The electrical transport property of GO was enhanced by removing the oxygenic groups. The electrical transport property of GO was enhanced by removing the oxygenic groups. This result indicates that the graphitization degree of GO was improved during the reduction process.
3.2 Properties of the CANWs@rGO composite energetic materials
Fig. 3(c) shows the results of the DSC analysis of the CANWs@rGO composite energetic materials and the heating rate was 5 °C min−1. It can be seen from Fig. 3(c) that there is only one exothermic peak in the heating process, the exothermic reaction starts from 172.9 °C, the termination temperature is 207.1 °C, the peak temperature is about 190.4 °C, and the shaded portion of the exothermic peak curve has been integrated and the exotherm is about 1497.1 J g−1. The results show that CANWs@rGO has a sharp heat release process. In this work, 0.14 mg of the sample was tested by DSC. The DSC results for a smaller amount (0.1 and 0.12 mg) of the samples shows that the onset (172.7 and 173.1 °C) and peak temperature (187.3 and 189.2 °C) of the exothermic process are slightly advanced, and the amount of the exotherm remains basically unchanged (1479.8 and 1489.8 J g−1) (Fig. S14†). The DSC result shows that, as a composite energetic material, the energy release of CANWs@rGO is lower than that of the pure CA film previously reported (1650 J g−1),15 but higher than that of the CuN3 film (1200 J g−1).16As a detonator, the two most notable performances are detonation capability and safety. In order to test the detonation ability of CANWs@rGO, it was used as a primary explosive to detonate the secondary explosive (RDX) loaded in the detonator. The detonation effect was evaluated through the experiment of the lead plate. The detonator designed in this work is shown in Fig. 3(d). The diameter of the iron tube was about 6 mm, and the thickness of the lead plate was about 5 mm, pyrotechnics were used to stimulate the CANWs@rGO primary explosive. The experimental results showed that 50 mg of the CANWs@rGO primary explosive could successfully detonate 500 mg of RDX. Compared with commercial DDNP (70 mg),44 CANWs@rGO showed a better detonation performance, which emphatically proves that CANWs@rGO as a primary explosive has an excellent ability to detonate secondary explosives.
In order to test how much the electrostatic safety of the CANWs@rGO composite energetic materials has been improved compared to pure CA, we conducted a test of electrostatic sensitivity (Fig. S9, Tables S1 and S2†). As illustrated in Fig. 4, the 50% electrostatic ignition energy of the composite energetic material was 0.96 mJ, significantly higher than that of pure Cu(N3)2 (0.05 mJ).1 For the CANWs@rGO composite energetic materials, due to the conductivity of rGO, the electrostatic charge did not accumulate on the CA surface, which caused the primary explosive to explode, and the charge was dispersed on the surface of rGO. Therefore, the electrostatic safety of CANWs@rGO can be effectively improved.
 | ||
| Fig. 4 A schematic diagram of the mechanism of electrostatic sensitivity for CA and CANWs@rGO. | ||
4. Conclusions
CuNWs provide the essential advantage of a relatively high surface area. The effectiveness of the nanowire morphology to react with gaseous HN3 and be wrapped by wrinkled rGO sheets appears to be particularly advantageous. The work presented in this paper used CuNWs@rGO obtained through a simple one-pot hydrothermal reduction as the precursor. After 48 h of the in situ gas–solid phase azide reaction, the precursor completely transformed to an energetic composite, rGO with its extraordinary electrical properties can effectively depress static electricity accumulation on the surface of CA, thereby reducing the electrostatic sensitivity of the energetic composite. As a primary explosive, CANWs@rGO also demonstrated a better detonating ability than commercial DDNP. In summary, we have designed and obtained a high-energy and insensitive primary explosive, which shows broad application prospects.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for the financial support from the Postgraduate Research & Practice Innovation Program from Jiangsu Science and Technology Department under Grant number KYCX19_0320. We are grateful to Jiang Lin for his support in visualization. We are grateful for the support of the Instrument and Equipment Fund of Nanjing University of Science and Technology.References
- Q. Wang, X. Feng, S. Wang, N. Song, Y. Chen, W. Tong, Y. Han, L. Yang and B. Wang, Adv. Mater., 2016, 28, 5837–5843 CrossRef CAS PubMed.
- C. He and J. M. Shreeve, Angew. Chem., Int. Ed., 2016, 55, 772–775 CrossRef CAS PubMed.
- Z. Yi, Q. Ang, N. Li, C. Shan, Y. Li, L. Zhang and S. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 8584–8590 CrossRef CAS.
- T. M. Klapotke and N. Mehta, Propellants, Explos., Pyrotech., 2014, 39, 7–8 CrossRef.
- M. Huynh, M. A. Hiskey, T. J. Meyer and M. Wetzler, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 5409–5412 CrossRef CAS PubMed.
- M. Zhou, Z. Li, Z. Zhou, T. Zhang, B. Wu, L. Yang and J. Zhang, Propellants, Explos., Pyrotech., 2013, 38, 569–576 CrossRef CAS.
- Z. Yan, L. Yang, J. Han, N. Song and J. Liu, RSC Adv., 2020, 10, 14347–14352 RSC.
- Z. Zheng, W. Zhang, C. Yu, G. Zheng, K. Ma, Z. Qin, J. Ye and Y. Chao, RSC Adv., 2018, 8, 2552–2560 RSC.
- Q. Yan, Z. Yang, X. Zhang, J. Lyu, W. He, S. Huang, P. Liu, C. Zhang, Q. Zhang, G. He and F. Nie, J. Mater. Chem. A, 2019, 7, 17806–17814 RSC.
- C. Rossi, Propellants, Explos., Pyrotech., 2018, 43, 1–16 CrossRef.
- W. Zhang, B. Yin, R. Shen, J. Ye, J. A. Thomas and Y. Chao, ACS Appl. Mater. Interfaces, 2013, 5, 239–242 CrossRef CAS PubMed.
- A. Nicollet, G. Lahiner, A. Belisario, S. Souleille, M. D. Rouhani, A. Esteve and C. Rossi, J. Appl. Phys., 2017, 121, 034503 CrossRef.
- Q. Yan, M. Gozin, F. Zhao, A. Cohen and S. Pang, Nanoscale, 2016, 8, 4799–4851 RSC.
- C. Rossi, Propellants, Explos., Pyrotech., 2014, 39, 323–327 CrossRef CAS.
- C. Yu, W. Zhang, S. Guo, B. Hu, Z. Zheng, J. Ye, S. Zhang and J. Zhu, Nano Energy, 2019, 66, 104135 CrossRef CAS.
- C. Yu, Z. Zheng, W. Zhang, B. Hu, Y. Chen, J. Chen, K. Ma, J. Ye and J. Zhu, ACS Sustainable Chem. Eng., 2020, 8, 3969–3975 CrossRef CAS.
- Q. Wang, J. Han, Y. Zhang, Z. Yan, E. Velasco, L. Yang, B. Wang and S. Zang, ACS Appl. Mater. Interfaces, 2019, 11, 8081–8088 CrossRef CAS PubMed.
- R. Wu, K. Zhou, C. Y. Yue, J. Wei and Y. Pan, Prog. Mater. Sci., 2015, 72, 1–60 CrossRef CAS.
- M. Cartwright and J. Wilkinson, Propellants, Explos., Pyrotech., 2010, 35, 326–332 CrossRef CAS.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- X. Liu, J. George, S. Maintz and R. Dronskowski, Angew. Chem., Int. Ed., 2015, 54, 1954–1959 CrossRef CAS PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- I. A. Rubtsov, E. R. Pruuel and K. A. Ten, J. Phys. Conf., 2017, 899, 092012 CrossRef.
- H. Li, H. Rui, Q. Jiao, S. Du and L. Yu, Propellants, Explos., Pyrotech., 2016, 4, 126–135 CrossRef.
- X. Ma, Y. Zhu, S. Cheng, H. Zheng, Y. Liu, Z. Qiao, G. Yang and K. Zhang, Chem. Eng. J., 2020, 392, 123719 CrossRef CAS.
- X. Liu, Y. Hu, H. Wei, B. Chen, Y. Ye and R. Shen, Micromachines, 2020, 11, 575 CrossRef PubMed.
- V. Pelletier, S. Bhattacharyya, I. Knoke, F. Forohar, M. Bichay and Y. Gogotsi, Adv. Funct. Mater., 2010, 20, 3168–3174 CrossRef CAS.
- Y. Wamg, F. Zhang, L. Zhang, Z. Zhang, R. Han and X. Sun, Chin. J. Energetic Mater., 2016, 24, 386–392 Search PubMed.
- L. Zhang, F. Zhang, Y. Wang, R. Han, J. Chen, R. Zhang and E. Chu, Mater. Lett., 2019, 238, 130–133 CrossRef CAS.
- B. Li, M. Li, Q. Zeng and X. Wu, J. Energ. Mater., 2016, 34, 123–128 CrossRef CAS.
- Q. Yu, M. Li, Q. Zeng, J. Guo and X. Wu, Mater. Lett., 2018, 224, 18–21 CrossRef CAS.
- G. Zhang, H. Wang and L. Yang, Initiators Pyrotech., 2018, 1, 54–56 Search PubMed.
- Q. Yu, M. Li, Q. Zeng, X. Wu and Z. Zhang, Appl. Surf. Sci., 2018, 442, 38–44 CrossRef CAS.
- Y. Shen, J. Xu, N. Li, J. Dai, C. Ru, Y. Ye and R. Shen, Chem. Eng. J., 2017, 326, 1116–1124 CrossRef CAS.
- A. R. Rathmell, S. M. Bergin, Y. Hua, Z. Li and B. J. Wiley, Adv. Mater., 2010, 22, 3558–3563 CrossRef CAS PubMed.
- R. Shende, S. Subramanian, S. Hasan, S. Apperson, R. Thiruvengadathan, K. Gangopadhyay and S. Gangopadhyay, Propellants, Explos., Pyrotech., 2008, 33, 122–130 CrossRef CAS.
- R. Thiruvengadathan, A. Bezmelnitsyn, S. Apperson, C. Staley, P. Redner, W. Balas, S. Nicolich, D. Kapoor, K. Gangopadhyay and S. Gangopadhyay, Combust. Flame, 2011, 158, 964–978 CrossRef CAS.
- S. Apperson, R. V. Shende, S. Subramanian, D. Tappmeyer, S. Gangopadhyay, Z. Chen, K. Gangopadhyay, P. Redner, S. Nicholich and D. Kapoor, Appl. Phys. Lett., 2007, 91, 243109 CrossRef.
- F. Zhang, Y. Wang, Y. Bai and R. Zhang, Mater. Lett., 2012, 89, 176–179 CrossRef CAS.
- R. Thiruvengadathan, C. Staley, J. M. Geeson, S. Chung, K. E. Raymond, K. Gangopadhyay and S. Gangopadhyay, Propellants, Explos., Pyrotech., 2015, 40, 729–734 CrossRef CAS.
- R. Thiruvengadathan, S. W. Chung, S. Basuray, B. Balasubramanian, C. S. Staley, K. Gangopadhyay and S. Gangopadhyay, Langmuir, 2014, 30, 6556–6564 CrossRef CAS PubMed.
- A. P. Periasamy, J. Liu, H. M. Linc and H. Chang, J. Mater. Chem. A, 2013, 1, 5973–5981 RSC.
- Y. Liang, Z. Sun, J. Chen, X. Liu and Y. Zhou, J. Am. Ceram. Soc., 2010, 93, 1916–1921 CAS.
- W. Huang, Y. Tang, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2020, 142, 3652–3657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05403h |
| This journal is © The Royal Society of Chemistry 2020 |