
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a recombination neutral protease I from Aspergillus oryzae to improve enzyme activity at acidic pH†
Yucheng Hu ab,
Tong Liab,
Zhui Tuabc,
Qinghua Heabc,
Yanping Li*abc and
Jinheng Fuab
ab,
Tong Liab,
Zhui Tuabc,
Qinghua Heabc,
Yanping Li*abc and
Jinheng Fuab
aState Key Laboratory of Food Science and Technology, Nanchang University, No. 235 Nanjing East Road, Nanchang 330047, China. E-mail: liyanping@ncu.edu.cn; Fax: +86-791-8333708
bSino-German Joint Research Institute, Nanchang University, No. 235 Nanjing East Road, Nanchang 330047, China
cJiangxi Province Key Laboratory of Analytical Sciences, Nanchang University, No. 235 Nanjing East Road, Nanchang 330047, China
First published on 19th August 2020
Abstract
Extracellular neutral proteases (NPs) in Aspergillus oryzae (A. oryzae) play a role in hydrolyzing soybean proteins into smaller peptides at pH about 7.5. The optimum pH of moromi fermentation (The second stage of soy sauce fermentation.) is 4.5–5.5. NPI is acid sensitive. To decrease the pH optimum of NPI, we got a mutant NPI-Y122FK246ID382V from the error-prone PCR library that showed optimal activity at pH 5.5. The specific activity at 40 °C of the NPI-Y122FK246ID382V mutant was 1383.50 U mg−1, which was 2.75-fold that of wild-type (503.09 U mg−1). The Michaelis constants of the mutant decreased from 22.13 mM (wild-type) to 19.98 mM (NPI-Y122FK246ID382V). The residues at positions 122 and 246 are important in influencing hydrolytic activity at pH 5.5 through site-directed mutagenesis. And the pH optimum of double amino acid mutants (Y122FK246I) shifted dramatically to an acidic pH compared to those of single amino acid substitution. Molecular models and structural comparisons of native and mutant provided further insight on the basis to improve catalytic efficiency at acidic pH. These results indicated that we modified the neutral protease I of Aspergillus oryzae, which can effectively improve the application of the neutral protease in industrial production, and finally lay the foundation for improving the utilization rate of raw protein.
1. Introduction
The types of soy sauce mainly include high-salt, low-salt, and salt-free soy sauce. Some recent reviews have reported that excessive intake of dietary salt (>10 g per day) can cause hypertension in the population.1,2 Low-salt and salt-free soy sauce are beneficial to patients with heart disease, kidney disease, and hypertension. The whole fermentation of soy sauce involves two steps.3 In the first step (koji fermentation) the mixture of cooked soybean and wheat are inoculated with spores of Aspergillus oryzae and solid state fermentation is performed for 2 days to make koji. During this process the pH drops to about 5.5 because A. oryzae produces small organic acids through the glycolysis pathway and tricarboxylic acid cycles. In the second step (moromi fermentation) the koji mixed with brine ferments lasting for over 6 months. In the second period fermentation, yeasts and lactic acid bacteria produce lactic acid, so the pH drops from an initial value of 6.5–7.0 to 5.5 even 4.7–4.8.4–6 The ethanol or KCl is added to the finished koji instead of brine to reduce the sodium content in the fermentation of low-salt and salt-free soy sauce.7,8 Furthermore, the result of decreasing salt brine contributes in higher acidity and lower pH.9 Low pH value is one of the desired characteristics of soy sauce.10,11A. oryzae extracellular proteases hydrolyze soy protein in whole stages. Furthermore, the salt-free soy sauce is a product fermented by A. oryzae in an ethanol medium, thus the flavor impact is quite similar to that from commercial soy sauce.7 According to the pH optimum, the proteases secreted by A. oryzae are divided into three categories: acid protease, neutral protease and alkaline protease.12 Alkaline protease will be gradually lost activity under the acidic condition.13 Because the optimum pH of acid protease and alkaline protease is 4.0 or 9.0 separately, neutral proteases (NPs) that show the optimal activity at pH 7.0 should play a leading role especially in the second fermentation stage. NPs of A. oryzae is a class of metallic-endoproteinases, containing one zinc atom per molecule.14 The genomic sequencing result of A. oryzae RIB40 showed there are three NPs: NPI, NPII, and NPIII. NPI has been studied more in applied research. The molecular weight of NPII, 19.8 kDa determined by gel filtration, is much smaller than that of NPI.15 Moreover, NPII has low activities on substrates (such as milk casein, hemoglobin, albumin and gelatin) routinely used in the laboratory, while NPI has high activities on them.16 NPIII is also a secreted zinc protease, which is classified in the M36 protein family, and its expression is regulated by the prtT transcription factor. NPI gene has an open reading frame of 1905 bp, encoding 634 amino acid residues including single peptide (1–18 aa), propeptide (19–245 aa), and mature polypeptide (246–634 aa). It was expressed at high levels in Pichia pastoris and glycosylation analysis in P. pastoris has been described.17,18 The optimal pH of NPI is 7.5, but it was inactivated under the acidic condition and its enzyme activity quickly decreased at pH < 6.0.17 So acidic pH will severely restrict the enzymatic activity of NPs and reduces the utilization efficiency of the raw materials in the whole fermentation. Thus it's crucial to improve the activities of Nps at acidic pH for the production of high quality low-salt and salt-free soy sauce using appropriate breeding technology.
Directed evolution has become a major technology in studying the enzyme's structure–function relationships.19–22 In order to obtain more useful properties, this technology has been used to amend the properties or activity of neutral enzymes. Hence, in vivo laboratory evolution, including of rational design and directed evolution, were regarded as the biggest contributor in explorations of non-natural properties and used by many researchers.23,24 By rational design method Candida rugosa lipases (CRL) can have the most thermostable mutant, Asp457Phe, exhibiting a 5.5-fold longer half-life at 50 °C and a 10 °C increase in optimal temperature.25 Zhou et al. performed structure-based rational design of a thermostable phytase (PhyAn), and acquired the mutant Y65H enzyme that shifted the pH optimum from 5.0 to 3.0 and enhanced stability in more acidic environments.26
To adapt the harsh industrial environment, in this paper, a random mutated library of NPI gene with 2 × 104 variants was constructed by error-prone PCR, and 50% of the variants still had neutral protease activity. A mutated strain enzyme which the pH optimum was pH 5.5 was screened out. The mutation of three amino acids was found by DNA sequencing. In order to verify which site had the greatest impact on the optimal reaction pH, site-specific mutation was performed at three sites respectively. Recombinant NPI gene was firstly mutated by error-prone PCR with an expectation of improving catalytic efficiency at acidic pH, which could be applied in fermentation industry better.
2. Materials and methods
2.1 Materials
The Escherichia coli strain, DH5α, was used for DNA manipulation. And the Pichia pastoris GS115 strain (Invitrogen) was used for expression. Plasmid pPIC9K-NPI-6His was previously constructed and used as the template.182.2 Construction of error-prone PCR mutant library
Error-prone PCR was carried out using the plasmid pPIC9K-NPI-6His as a template, the NPI_EcoRI_f and NPI6His_NotI_r as primers. Each 100 μL of reaction mixture contained 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 5 mM MgCl2, 5 ng of plasmid DNA, 5 units of Taq polymerase, and different concentrations of dNTPs (TAKARA, Dalian, China). The four combined concentrations of dNTPs were separately used for error-PCR, then we mixed the four groups of PCR products to get a large enough mutation library. The concentrations of dNTPs were shown in ESI Table 1. PCR was performed at 94 °C for 30 s, and 68 °C for 2 min, with a total 30 cycles. The PCR products were digested by EcoRI and NotI before connect to digest pPIC9K vector.The resulting DNA mixture was introduced into E. coli DH5α using Gene Pluser Xcell (BioRad, USA) and plated on LB plates containing Ampicillin 100 μg mL−1. Transformants were collected after incubating at 37 °C overnight, and then transferred into fresh LB-ampicillin liquid medium to incubate at 37 °C for 8 h. Plasmids were linearized with SalI, then transferred into P. pastoris GS115 by electroporation method. Transformants were cultivated on minimal dextrose (MD) medium agar plates and incubated at 30 °C for 3 days.
2.3 Screening for mutants with acid pH optimum in enzyme activity
Single colonies from the mutant library were grown in 96-well plates with buffered glycerol complex medium (BMGY) at 30 °C, 200 rpm for 24 h and then shifted to buffered minimal methanol medium (BMMY). After 72 h of further incubation (200 rpm, 30 °C, 1% (v/v) methanol as an inducer), the supernatant was collected by centrifuging and transferred into casein identification plates (1% casein, 1% agar) with various pH from 5.0 to 8.0, incubated at 40 °C for 6 h. Strains with the transparent circle not less than the wild type's (pH 7.5) were selected as the result of the first-round screening. The value of H/C can reflect the ability of strains producing protease in milk medium (H: the diameter of the transparent circle; C: the diameter of the colony).At the second-round screening, the positive ones from original plates were cultured in shake flasks. Supernatant of each culture was collected and neutral protease activities were measured at 40 °C with various pH. And those which had higher activity at lower pH than 7.5 were selected.
2.4 Sequence analysis
The genomic DNA was extracted with the yeast genome DNA extraction kit (Tiangen, Tianjin, China). The mutated NPIgene was amplified by PCR with Primer STAR HS DNA Polymerase (TAKARA, Dalian, China), the primers were same as the error-prone PCR. DNA sequence was carried out in Genscript (Nanjing, China).2.5 Enzyme purification
Methanol was added to the final concentration of 0.5% every 24 hours in BMMY medium. After 72 hours of induction expression, the cell culture supernatant was collected by centrifuging at 8000 rpm for 15 min at 4 °C. It was treated with (NH4)2SO4 at 70% saturation and purified by nickel-affinity chromatography column and homogeneity was detected by SDS-PAGE.27 The protein concentration was determined according to Bradford assay, BSA was used as standard.282.6 Measurement of pH optimum
The pH optimum of purified neutral protease was carried out by measuring the activity at different pH from 4.5 to 8.5. The protease activity was measured at 40 °C as the standard method.29 Each sample was repeated three times. One unit (U) of protease activity was defined as the amount of enzyme releasing 1.0 μg of tyrosine equivalent per minute.2.7 Effect of metal ions, EDTA, and SDS on protease activity
The enzyme solution was incubated in 0.1 M phosphate buffer (pH 7.5) with 5 mM different metal ions (Cu2+, Zn2+, Mg2+, Fe3+, Li+, Ca2+), ethylenediaminetetraacetic (EDTA) or sodium dodecyl sulfonate (SDS) at 40 °C for 60 min to probe the impact of metal ions on the enzyme activity. The residual activity was measured using 0.5% casein as a substrate. The activity of enzyme without any treatments was considered as 100% control. Each sample was repeated three times.2.8 Kinetic studies
The protease (0.2 mg mL−1) was incubated with the casein ranging from 0.2% to 1.0%. And the enzyme activity was measured with different substrate concentrations as described previously at 40 °C, 1.01 × 105 Pa. Kinetic parameters of all enzymes were determined at their optimal pH. The kinetic parameters (Km and Vmax) were determined by the method of Lineweaver–Burk. Each sample was repeated three times.2.9 Site-directed mutagenesis (SDM)
Compared to the wild type, three pairs of bases in the mutant are mutated, resulting in three amino acid residue changes (Y122F, K246I, D382V). Overlap extension PCR was used to achieve site-directed mutagenesis.30 The primers used are listed in ESI Table 2. The PCR products were digested by EcoRI and NotI, and attached to plasmid pPIC9K. The recombinant plasmids were transformed into P. pastoris GS115.2.10 Three-dimensional structure analysis of wild type and the mutants
The blastp program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to aligned protein sequence. Homology modeling of recombinant NPI was built by the SWISS-MODEL program (https://swissmodel.expasy.org/interactive) and I-TASSER serve using the extracellular metalloproteinase from Aspergillus fumigatus (Protein Data Bank [PDB] code 4k90.1) as a structure template.31,323. Results
3.1 The isolation of mutants with acidic pH optimum in enzyme activity by error-prone PCR mutagenesis
Error-prone PCR product digested by EcoRI and NotI was ligated with digested pPIC9K vector and the ligation was transformed into E. coli. The plasmids from the transformants were electroporated into P. pastoris GS115 to construct a random mutated error-prone PCR library with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 clones. Sequence analysis showed this library had 1.4 base substitutions per kb and approximately 50% of colonies displayed neutral protease activity. To isolate the mutants with acid pH optimum in enzyme activity, about 6000 clones in the random mutagenesis library were screened. The preliminary selection used the casein identical plates, 256 mutants with the same-sized transparent zones at pH 5.0 and 7.0 were selected. Next, in a second screening, all the 256 mutants were induced to express by methanol in shake flasks and NPI was pelleted at saturation of 70% ammonium sulfate. Five mutants displayed the lower optimum pH compared to the wild type when measuring the enzyme activities at pH 5.0–8.0. Then, the five mutants were purified by nickel-affinity chromatography column. Only one mutant can keep enough enzyme activity under acidic condition. Its pH optimum was 5.5 and two-unit pH reduced against to the wild type. The genomic DNA of the mutant was extracted and the amino acid sequence was analyzed (Fig. 1a). Three bases change (TAC–TTC; AAA–ATA; GAT–GTT) resulted in the replacement of three amino acids residue (Tyr122-Phe; Lys246-Ile; Asp382-Val) was found and chosen for further analysis.
000 clones. Sequence analysis showed this library had 1.4 base substitutions per kb and approximately 50% of colonies displayed neutral protease activity. To isolate the mutants with acid pH optimum in enzyme activity, about 6000 clones in the random mutagenesis library were screened. The preliminary selection used the casein identical plates, 256 mutants with the same-sized transparent zones at pH 5.0 and 7.0 were selected. Next, in a second screening, all the 256 mutants were induced to express by methanol in shake flasks and NPI was pelleted at saturation of 70% ammonium sulfate. Five mutants displayed the lower optimum pH compared to the wild type when measuring the enzyme activities at pH 5.0–8.0. Then, the five mutants were purified by nickel-affinity chromatography column. Only one mutant can keep enough enzyme activity under acidic condition. Its pH optimum was 5.5 and two-unit pH reduced against to the wild type. The genomic DNA of the mutant was extracted and the amino acid sequence was analyzed (Fig. 1a). Three bases change (TAC–TTC; AAA–ATA; GAT–GTT) resulted in the replacement of three amino acids residue (Tyr122-Phe; Lys246-Ile; Asp382-Val) was found and chosen for further analysis.
 | ||
| Fig. 1 (a) Protein sequences of mature NPI and mutant NPI-230. The mutated positions were signed with a single underline, and the mutated amino acids were in the brackets. (b) SDS-PAGE analysis of NPs. Lane M: protein marker, Lane 1: wild-type NPI; Lane 2: mutant NPI-230 (Y122FK246ID382V). Expected size of glycosylated NPI and NPI-230 are 55 kDa. (c) SDS-PAGE analysis of mutants. Lane M: protein marker; Lane 1: Y122F; Lane 2: K246I; Lane 3: Y122FK246I; Lane 4: D382V. Expected size of glycosylated Y122F, K246I, Y122FK246I, and D382V proteins are 55 kDa. | ||
3.2 Purification and characterization of the mutated neutral protease I
The mutated NPI-230 (Y122FK246ID382V) and the wild-type were purified by nickel-affinity chromatography column as described above, and both of the enzymes showed a band of 55 kDa determined by SDS-PAGE analysis (Fig. 1b). At 40 °C, the specific activity was measured at pH 5.5 of NPI-230 mutant (1383.50 U mg−1), which was 2.75-fold of the wild-type (503.09 U mg−1) at pH 7.5. The pH optimum of the mutant enzyme was 5.5, which showed 2 units lower than the wild type (Fig. 2a).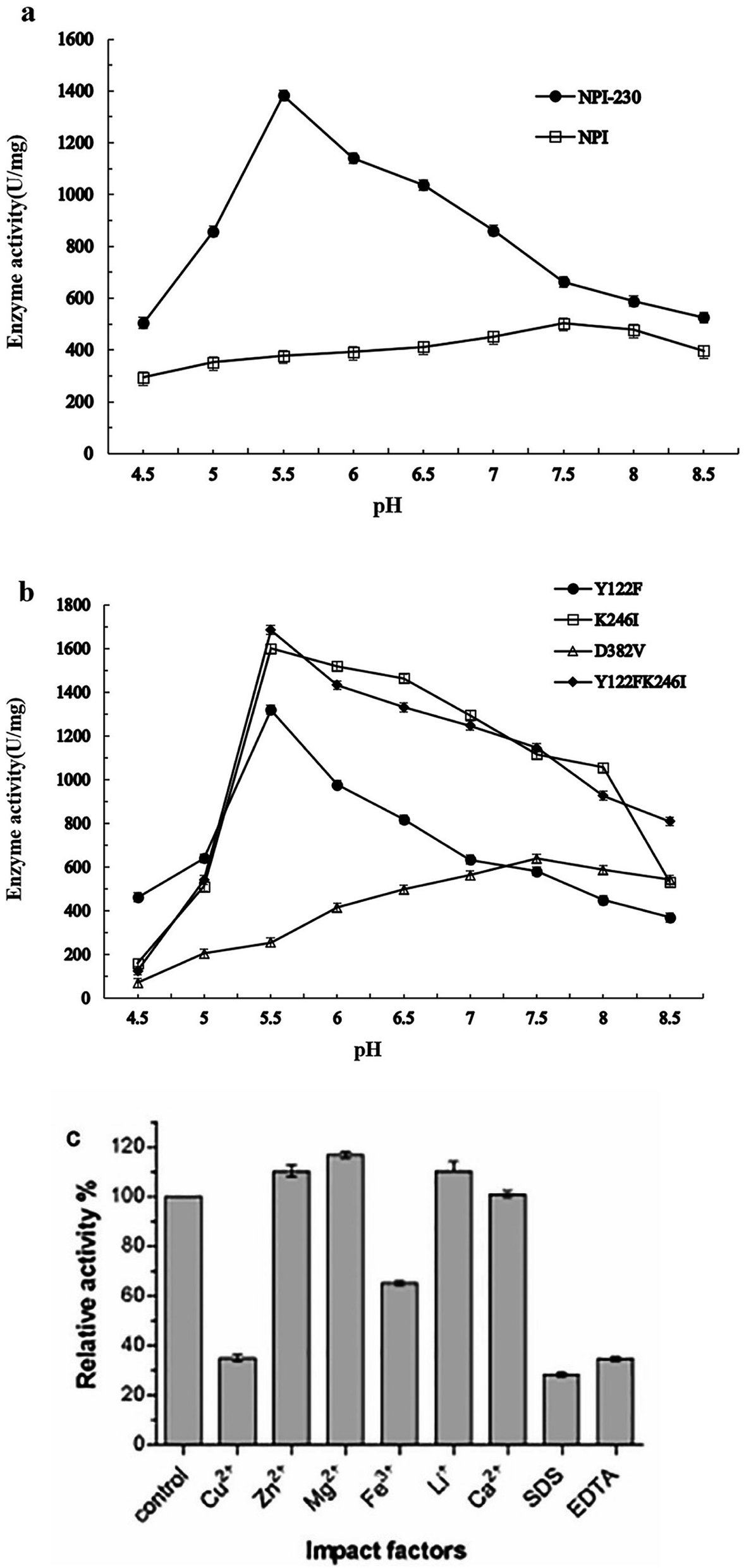 | ||
| Fig. 2 (a) Effects of pH on the activity of the wild type (NPI) and the mutant (NPI-230); (b) effects of pH on the activity of the mutant Y122F, K246I, D382V, and Y122FK246I. Each sample was repeated three times. The error bars indicate standard deviations. (c) Effect of metal ions, protease inhibitors, and surfactants on protease activity of NPI-230. Relative activity was given as a percentage compared with the most active sample. | ||
3.3 Effect of metal ions, EDTA, and SDS on protease activity
The protease separately was treated with different metal ions, protease inhibitors and surfactants. As demonstrated in Fig. 2c, Cu2+ and EDTA were strong inhibitors of NPI-230 of the enzyme.17 We could also find that Fe3+ and SDS inhibited protease activity. Ca2+ had no significant effect on protease activity.33 The presence of Zn2+, Mg2+, and Li+ slightly increased the enzyme activity as compared to the control.3.4 Enzyme kinetics of the protease
The kinetic parameter of NPI-230 was measured at pH 5.5 and the kinetic parameter of NPI was measured at pH 7.5. The Michaelis constant (Km) and maximum reaction velocity (Vmax) were 19.98 ± 0.25 mM and 196.75 ± 12.50 nM mg−1 s−1 for NPI-230 and 22.13 ± 0.33 mM and 90.21 ± 7.08 nM mg−1 s−1 tyrosine per min per mL for NPI, respectively (Table 1). The Vmax of the mutant increased by 118% compared to the wild-type.| Vmax (nM mg−1 s−1) | Km (mM) | |
|---|---|---|
| a Kinetic parameters of all enzymes were determined at their optimal temperatures and are expressed as mean ± standard deviation. Each sample was repeated three times. | ||
| NPI-230 | 196.75 ± 12.50 | 19.98 ± 0.25 |
| NPI | 90.21 ± 7.08 | 22.13 ± 0.33 |
3.5 Site-directed mutagenesis
Three amino acid residue substitutions were found through comparison with the wild type and the mutant (Y122F, K246I, and D382V). Three variants with single-mutations and double-mutation (Y122FK246I) were successfully constructed. The recombinants confirmed by sequencing analysis in Genscript (Nanjing, China). The 55 kDa protein band was observed by SDS-PAGE analysis (Fig. 1c). The recombinant NPI was purified by nickel-affinity chromatography column, then the activity was determined at 40 °C with different pH. The protease activity of the mutants at different reaction pH is shown in Fig. 2b. The specific activity levels of mutants Y122F, K246I and Y122FK246I reached 1320.94, 1600.21 and 1686.25 U mg−1 at pH 5.5 respectively. Otherwise, the pH optimum of D382V mutant was 7.5, and it had lower activity (639.72 U mg−1) at pH 5.5. Site-directed mutagenesis showed that the mutant Y122FK246I had higher activity than those of mutants Y122F and K246I at pH 5.5.3.6 Structural analysis of NPI-230 and NPI
To obtain a deeper insight into the structural basis of thermal stability, the modeled three-dimensional structures of the mutant and wild type were built, using the 4K90_A as a template. As shown in Fig. 3, the amino acids of 122, 246 and 382 are located at the surface of the protein, which was far from the catalytic center of NPI (His184, His188 and Glu185) and the structure surface view of the NPI-230 protein was shown in blue. The active area of the wild type and mutant NPI-230 was shown in Fig. 3a and b. In addition, polarity hydrophilic amino acids Tyr at 122-site was replaced by non-polar hydrophobic amino acid Phe. At the 246-site, positively charged amino acid Lys was instead of Ile which was a non-polar hydrophobic amino acid. The polarity 382-site with negatively charged amino acids Asp was instead of Val which was a non-polar hydrophobic amino acid. Because the above amino acids located at the surface of enzyme with weaker polarity and low charge, the enzyme will show better substrate affinity and the hydrolysis in the acidic conditions.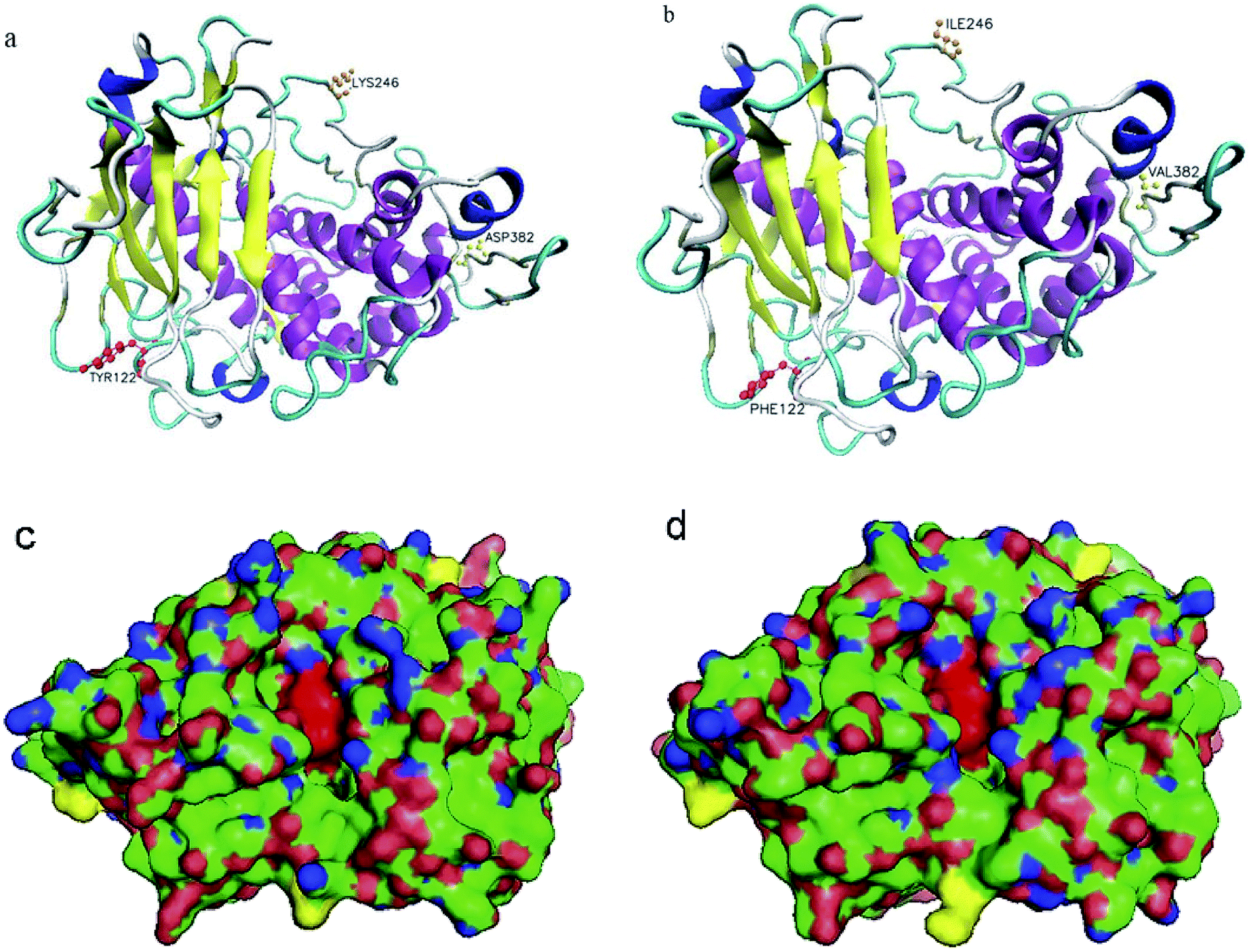 | ||
| Fig. 3 Structural analysis of the wild-type NPI (a) and NPI-230 (b). The mutated residues were shown in red (122), orange (246), and yellow (382). Structural representation of the wild-type NPI (c) and NPI-230 (d). The mutated residue was shown in yellow, the catalytic region was shown in red. | ||
4. Discussion
In our previous works, plasmid pPIC9K-NPI-6His was constructed and transformed into P. pastoris GS115 to express recombination NPI. When induced by 0.5% methanol for 72 hours in BMMY media, the culture supernatant was collected.18 The activity of the neutral protease reached 46 U mL−1. More than 95% of the neutral protease precipitated after final concentration of 70% saturated (NH4)2SO4 treatment. The pH optimum of purified recombinant NPI was 7.5 and it showed the stability at pH 7.0. The optimal temperature of recombinant NPI was 55 °C and there was little influence to the enzyme activity under 50 °C. The recombinant NPI was proved to be N-glycosylated by PAS staining and Endo H digestion.16 And the apparent molecular weight of the deglycosylated protein decreased from 54.5 kDa to 43.3 kDa proved by SDS-PAGE and MALDI-TOF-MS. The hyper glycosylation extent was 20.5%.In the brewing process, with the continuous fermentation of low-salt and salt-free soy sauce, pH value in the system will gradually decline to 5.5 even 4.7. Because of low stability or low specific activity of nature neutral protease at pH 5.5, their enzyme activity reduced rapidly.17 To get a mutant, which has higher enzyme activity at acidic pH, a mutation library of NPI was constructed by error-prone PCR and using plasmid pPIC9K-NPI-6His as template. By the different reaction conditions on a PCR to reduce the specificity of Taq DNA Polymerase and improve the probability of mismatched base pairs in the reaction process, the participation frequencies of base mismatches were increased by changing the concentration of dNTPs in the PCR system.34 The concentration of Mg2+ was increased from 2 mM to 5 mM to stabilize non-complementary base pairs, and Taq DNA Polymerase was used for error-prone PCR.35,36
We successfully constructed a random NPI mutation library in E. coli with a capacity of 4 × 104 colony forming units (cfu). After the extracted plasmid was transferred into P. pastoris, a total of 2 × 104 yeast transformants were obtained. Before the screening of NPI random mutation P. pastoris library, we should evaluate base-pair mutation rate and the library capacity. The average base random mutation frequencies of the library were 1.4‰, which was consistent with the base mutation probability of 1–2‰ in the previous reports.37,38 Because the length of NPI fragment was about 1883 bp and the average base mutation rate was 1.4‰, no more than 1428 yeast transformants were screened theoretically, that is, the whole DNA fragment sequence could be covered. Therefore, a total of 6000 yeast transformants was screened in this study, and 50% of transformants retained neutral protease activity. After first-round screening single colonies from the mutant library grown in 96-well plates, we further precipitated enzyme by (NH4)2SO4 at 70% saturation and purified by nickel-affinity chromatography column to identify mutants' pH optimum. According to above procedure, we finally found a mutant NPI-230 (Y122FK246ID382V) which has higher enzyme activity at pH 5.5 to meet the needs of industry. The pH optimum of the variants (NPI-230) enzyme was 5.5, significantly lower than wild type (the pH optimum 7.5), and the specific activity had significantly improved than the wild-type at 40 °C. Aside from the change in pH optimum and activity, mutant showed a change in Michaelis constants (Km), from 22.13 to 19.98 mM. It shows that the mutant has a higher affinity to the substrate than wild type. The Vmax of the mutant of NPI increased by 118% compared to the wild-type (Table 1). These characteristics make mutant more suitable for industrial production.
To understand amino acid which played a major role in enzyme activity at acidic pH, overlap PCR was used to site-directed mutagenesis to find more information about the influence of Y122, K246 and D382. Compared with the wild-type enzyme, mutants NPI-Y122F and NPI-K246I showed lower optimum pH (5.5) and higher activity than wild type at 40 °C. But the mutant NPI-D382V has no obvious improvement of the wild-type of optimum pH and enzymatic activity. The three substituted residues were far from the locus of substrate-binding (Fig. 3). The pH optimum of the three mutant enzymes (Y122F, K246I and D382V) was 5.5, 5.5 and 7.5 respectively. We speculate that the mutation of 122 and 246-site contributed to the main change on the optimal reaction profile. The recombinant protein NPI-Y122FK246I has the specific neutral protease activity (1686.25 U mg−1) at pH 5.5, 40 °C. We haven't got a mutation whose neutral protease has a lower optimal pH than 5.5 and the pH optimum of the wild-type enzyme at pH 7.5 showed in Fig. 2a. The enzymatic activity of every mutant was higher than the wild type at pH 7.5. And the comparison of enzymatic activity among each mutant and wild type at pH 5.5 and 7.5 was shown in Fig. 2a and b. These data reveal the residues at positions 122 and 246 are more important in influencing hydrolytic activity at pH 5.5 than that at 382. And the pH optimum of double amino acid mutants (Y122FK246I) shifted dramatically to an acidic pH than those of single amino acid substitution.
The simulated three-dimensional structure of mutants evidenced that the substituted sites were located on the surface and far from the catalytic center. These residues far from the catalytic center had some influence on subtilisin's property.39,40 In addition, three displacements are not located in the core of the β-barrel of the enzyme, which are critical for the conformational stability of the enzyme.41 The increase in hydrogen bonds and salt bridges around the catalytic domain can significantly improve α-amylase stability and catalytic efficiency at low pH.42 In our study, the three active sites are at H184, H188, and E263. But the above three amino acids (Y122F, K246I, D382V) were located at the surface of NPI with weaker polarity and were far from the catalytic center. The substitution of surface polarity or charged amino acids is beneficial to the binding of NPI to the substrate.43 This may help the enzyme reduce the impact of the charge in the environment, which is beneficial to the hydrolysis of NPI under acidic conditions.
5. Conclusions
In conclusion, we mutated NPI of A. oryzae by construction randomized library using error-prone PCR method. A mutant NPI-230 (Y122FK246ID382V) from expression library of P. pastoris was successfully selected out and increased catalytic activity at pH 5.5. Examining activity of mutants at reside 122, K246 and D382 by site-directed mutagenesis is helpful to give us new insights into which residue is more important. This is beneficial for producing low-salt, and salt-free soy sauce.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was funded by the National Natural Science Foundation of P. R. China (grant numbers 31260389), the State Key Laboratory of Food Science and Technology, Nanchang University (No. SKLF-ZZB-201925, SKLF-ZZA-201912), and the Science and Technology Innovation Platform Project of Jiangxi Province (No. 20192BCD40001).References
- A. V. Chobanian and M. Hill, Hypertension, 2000, 35, 858–863 CrossRef CAS PubMed.
- L. A. Wyness, J. L. Butriss and S. A. Stanner, Publ. Health Nutr., 2012, 15, 254–261 CrossRef PubMed.
- A. Kaewkrod, N. Niamsiri, T. Likitwattanasade and S. Lertsiri, LWT–Food Sci. Technol., 2018, 89, 735–739 CrossRef CAS.
- T. Hamada, T. Ishiyama and H. Motai, Appl. Microbiol. Biotechnol., 1989, 31, 346–350 CrossRef CAS.
- N. X. Hoang, S. Ferng, C. H. Ting, W. H. Huang, R. Y. Y. Chiou and C. K. Hsu, LWT–Food Sci. Technol., 2016, 74, 242–250 CrossRef.
- J. Sulaiman, H. M. Gan, W.-F. Yin and K.-G. Chan, Front. Microbiol., 2014, 5, 556 Search PubMed.
- X. L. Zhu, K. Watanabe, K. Shiraishi, T. Ueki, Y. Noda, T. Matsui and K. Matsumoto, Peptides, 2008, 29, 338–344 CrossRef CAS PubMed.
- R. Y. Y. Chiou, S. Ferng and L. R. Beuchat, Int. J. Food Microbiol., 1999, 48, 11–20 CrossRef CAS PubMed.
- Y. R. Song, D. Y. Jeong and S. H. Baik, Food Microbiol., 2015, 51, 171–178 CrossRef CAS PubMed.
- K. S. Choi, J. D. Choi, H. C. Chung, K. I. Kwon and W. S. Kim, Korean J. Food Sci. Technol., 2000, 32, 174–180 CrossRef.
- T. Nakadai and S. Nasuno, J. Ferment. Technol., 1988, 66, 525–533 CrossRef CAS.
- C. Sandhya, A. Sumantha, G. Szakacs and A. Pandey, Process Biochem., 2005, 40, 2689–2694 CrossRef CAS.
- C. Ma, X. Ni, Z. Chi, L. Ma and L. Gao, Mar. Biotechnol., 2007, 9, 343–351 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS PubMed.
- H. Sekine, Agric. Biol. Chem., 1972, 36, 198–206 CAS.
- H. Sekine, Agric. Biol. Chem., 1973, 37, 1765–1767 CrossRef CAS.
- Y. Ke, W. Q. Huang, J. Z. Li, M. Q. Xie and X. C. Luo, J. Agric. Food Chem., 2012, 60, 12164–12169 CrossRef CAS PubMed.
- D. Lei, Y. Xu, Q. He, Y. Pan, B. Chen, L. Xiong and Y. Li, Biotechnol. Lett., 2013, 35, 2121–2127 CrossRef CAS PubMed.
- Y. Fang, Y. Lu, F. Lv, X. Bie, H. Zhao, Y. Wang and Z. Lu, Enzyme Microb. Technol., 2009, 44, 84–88 CrossRef CAS.
- B. L. Gong, R. Q. Mao, Y. Xiao, M. L. Jia, X. L. Zhong, Y. Liu, P. L. Xu and G. Li, Enzyme Microb. Technol., 2017, 106, 97–105 CrossRef CAS PubMed.
- W. Zhang, Z. Liu, S. Zhou, H. Mou and R. Zhang, Enzyme Microb. Technol., 2019, 124, 70–78 CrossRef CAS PubMed.
- X. Wu, Z. Tian, X. Jiang, Q. Zhang and L. Wang, Appl. Microbiol. Biotechnol., 2018, 102, 249–260 CrossRef CAS PubMed.
- R. D. Chen, Trends Biotechnol., 2001, 19, 13–14 CrossRef CAS PubMed.
- M. S. Kim and X. G. Lei, Appl. Microbiol. Biotechnol., 2008, 79, 69–75 CrossRef CAS PubMed.
- G. Li, Y. Chen, X. Fang, F. Su, L. Xu and Y. Yan, RSC Adv., 2018, 8, 1948–1957 RSC.
- S. Zhou, Z. Liu, W. Xie, Y. Yu, C. Ning, M. Yuan and H. Mou, Int. J. Biol. Macromol., 2019, 131, 1117–1124 CrossRef CAS PubMed.
- J. Wang, A. Xu, Y. Wan and Q. Li, Appl. Microbiol. Biotechnol., 2013, 170, 2021–2033 CAS.
- T. Zor and Z. Seliger, Anal. Biochem., 1996, 236, 302–308 CrossRef CAS PubMed.
- F. Moradian, K. Khajeh, H. Naderi-Manesh and M. Sadeghizadeh, Appl. Microbiol. Biotechnol., 2009, 159, 33–45 CAS.
- M. Waneskog and P. Bjerling, Anal. Biochem., 2014, 444, 32–37 CrossRef PubMed.
- D. Fernandez, S. Russi, J. Vendrell, M. Monod and I. Pallares, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1946–1957 CrossRef CAS PubMed.
- Y. Zhang, BMC Bioinf., 2008, 9, 40 CrossRef PubMed.
- X. Gao, Y. Yin, J. Yan, J. Zhang, H. Ma and C. Zhou, J. Sci. Food Agric., 2019, 99, 3359–3366 CrossRef CAS PubMed.
- M. Fromant, S. Blanquet and P. Plateau, Anal. Biochem., 1995, 224, 347–353 CrossRef CAS PubMed.
- R. C. Cadwell and G. F. Joyce, PCR Methods Appl., 1992, 2, 28–33 CrossRef CAS PubMed.
- Y. H. Zhou, X. P. Zhang and R. H. Ebright, Nucleic Acids Res., 1991, 19, 6052 CrossRef CAS PubMed.
- Z. G. Zhang, Z. L. Yi, X. Q. Pei and Z. L. Wu, Bioresour. Technol., 2010, 101, 9272–9278 CrossRef CAS PubMed.
- S. B. Zhang, X. Q. Pei and Z. L. Wu, Bioresour. Technol., 2012, 117, 140–147 CrossRef CAS PubMed.
- G. E. Sroga and J. S. Dordick, Protein Eng., 2001, 14, 929–937 CrossRef CAS PubMed.
- L. You and F. H. Arnold, Protein Eng., 1996, 9, 77–83 CrossRef CAS PubMed.
- B. Hocker, C. Jurgens, M. Wilmanns and R. Sterner, Chem. Rev., 2001, 105, 4038–4055 Search PubMed.
- H. Yang, L. Liu, H.-d. Shin, R. R. Chen, J. Li, G. Du and J. Chen, J. Biotechnol., 2013, 164, 59–66 CrossRef CAS PubMed.
- S. M. Asghari, M. Pazhang, S. Ehtesham, H. R. Karbalaei-Heidari, M. Taghdir, M. Sadeghizadeh, H. Naderi-Manesh and K. Khajeh, Protein Eng., Des. Sel., 2010, 23, 599–606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05462c |
| This journal is © The Royal Society of Chemistry 2020 |