DOI:
10.1039/D0RA05343K
(Paper)
RSC Adv., 2020,
10, 28541-28549
Synthesis and properties of a stimulus-responsive block polymer†
Received
18th June 2020
, Accepted 28th July 2020
First published on 4th August 2020
Abstract
In this study, the synthesis of small molecules and use of an improved “one-pot” method to synthesize the reversible addition–fragmentation chain transfer polymerization (RAFT) reagents have been reported. By comparing with the RAFT reagents synthesized by the traditional “step-by-step” method, it was observed that the reagents synthesized by the two methods had the same structure, however, the improved “one-pot” preparation method results in a significantly higher yield. Subsequently, two different macromolecular CTA segments (PVP-CTA-PVP and PDMAEMA-CTA-PDMAEMA) were prepared by RAFT polymerization, followed by the synthesis of the block polymer PDMAEMA-b-PVP-CTA-PVP-b-PDMAEMA. Through FITR, NMR, GPC and DLS analysis of the block polymer, it was observed that the isotacticity gradually became dominant as the degree of polymerization increased. Further, using NMR spectroscopy to study the effect of pH on the block polymer, the ionization degree of the synthesized polymer in the tumor tissue environment was observed to range between 86.32% to 99.50%, which proved that the synthesized polymers exhibit significant prospects in the medical application.
1. Introduction
Living polymerization developed nearly six decades back, has generated significant opportunities for polymer design.1 The original concept and definition of active polymerization was put forward by Szwarc.2–4 It has been found that the group transfer polymerization has the characteristics of living polymerization, thus, further expanding to the concept of living polymerization. Living/controlled polymerization refers to the polymerization reaction which is macroscopically similar to living polymerization, however, cannot actually exclude the chain transfer and chain termination completely.5 In the mid-1980s, Australian Federal Institute of Science and Industry (CSIRO) employed a macromonomer as chain transfer agent in free radical polymerization and named it addition–fragmentation chain transfer (AFCT).6 In 1998, dithioester was used as reagent in living free radical polymerization.7 At the same time, Rhodia also developed a new synthesis method, where xanthate is used as CTA to control the polymerization of various block polymers. This technology was named as macromolecular design by inter-change of xanthates (MADIX).8 In 1999, Chong, Y. K. et al. first proposed reversible addition–fragmentation chain transfer (RAFT) polymerization and its principle.9 Since then, RAFT method has seen a vigorous development.
At present, the methods of free radical living/controlled polymerization mainly include atom transfer radical polymerization (ATRP),10 nitroxide mediated polymerization (NMP)11 and RAFT (including MADIX/RAFT).12–15 Compared with other polymerization methods, RAFT has the advantages of convenient preparation and inexpensive raw materials, along with applicability for polymerization of various monomers, including (meth)acrylate,16,17 styrene,18,19 methacrylamide,20 vinyl acetate,21–23 etc. Moreover, compared with ATRP24 and NMP,25,26 the addition of excessive initiator in RAFT does not affect the polymerization performance. In addition to common bulk and solution polymerization, RAFT can also be applied for polymerization in miniemulsion,18 suspension27 and other special reaction conditions.28 Using RAFT to synthesize the functional materials,7,13,29,30 such as the environmentally sensitive polymers, has become a topic of intense research interest.
Environmentally sensitive polymers, on changing the aqueous solution external environment such as temperature, pressure, pH, CO2, etc.,31–34 usually transform the proportion of their hydrophilic–hydrophobic segments, thus, spontaneously assembling into different aggregated forms. These changes impact the macroscopic properties of the material. This behavior enables the use of the temperature-sensitive polymer aqueous solutions in medical treatment such as drug release, gene loading, etc.,35,36 especially in the treatment of tumors.37 As the microenvironment (pH, temperature, etc.) near the tumor is different from the normal human tissue,38 the unique properties of the temperature-sensitive water-soluble polymers result in an enormous application potential for tumor treatment.39,40
Though RAFT is applicable for polymerization of a wide range of monomers, however, in order to achieve the effectively controlled polymerization, suitable RAFT reagents should be carefully selected for monomers with different activities. PDMAEMA exhibits excellent temperature, pH and ion sensitivity as well as antibacterial properties, however, PDMAEMA is usually used together with other materials. Poly(N-vinyl pyrrolidone) (PNVP) is a polymer with a wide range of applications. Due to its biocompatibility, biodegradability, low toxicity and compounding ability, it can be used in cosmetics, pharmaceuticals and biomedicine.41 However, there are only a few reports on PVP by RAFT, and the research on the synthesis of PVP-b-PDMAEMA block polymer by RAFT is almost inexistent. The complex preparation steps and low yield of low molecular chain transfer agents limit the practical application of RAFT.
In this study, a “one-pot” method was used to synthesize the small molecule chain transfer agents. Compared with the traditional “step-by-step” method, the yield was observed to be significantly improved, and the process required a small extent of post-processing. In addition, the developed transfer agent was used for the first time to synthesize PDMAEMA-b-PVP-CTA-PVP-b-PDMAEMA. The corresponding characterization confirmed that the synthesized block polymer had the dual sensitivity of pH and temperature, thus, exhibiting the potential of application in medical treatment.
2. Results and discussion
2.1. Small molecule CTA
The analysis of small molecules CTA is shown in ESI.†
2.2. Macromolecular CTA-PVP
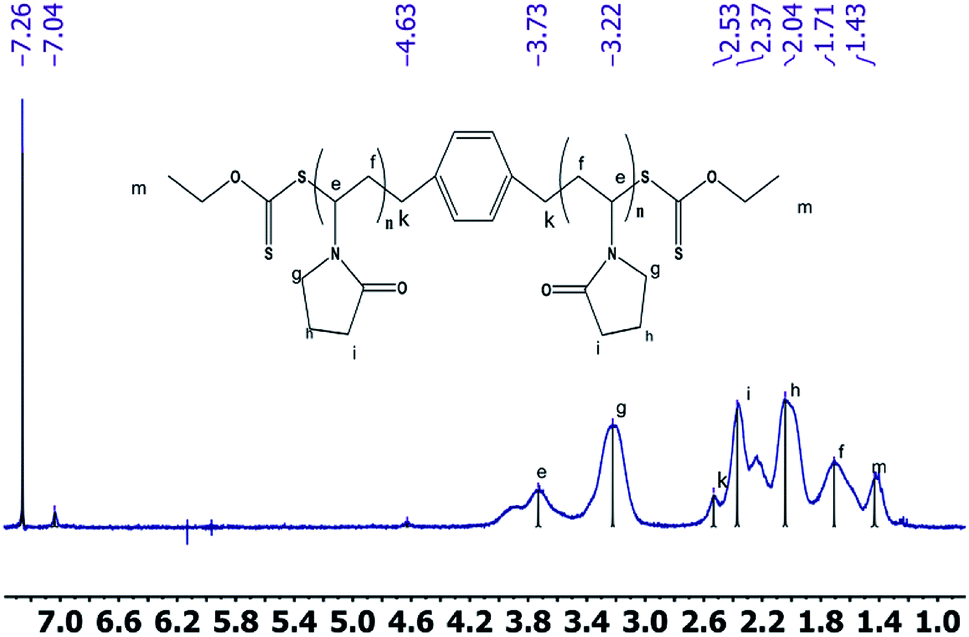
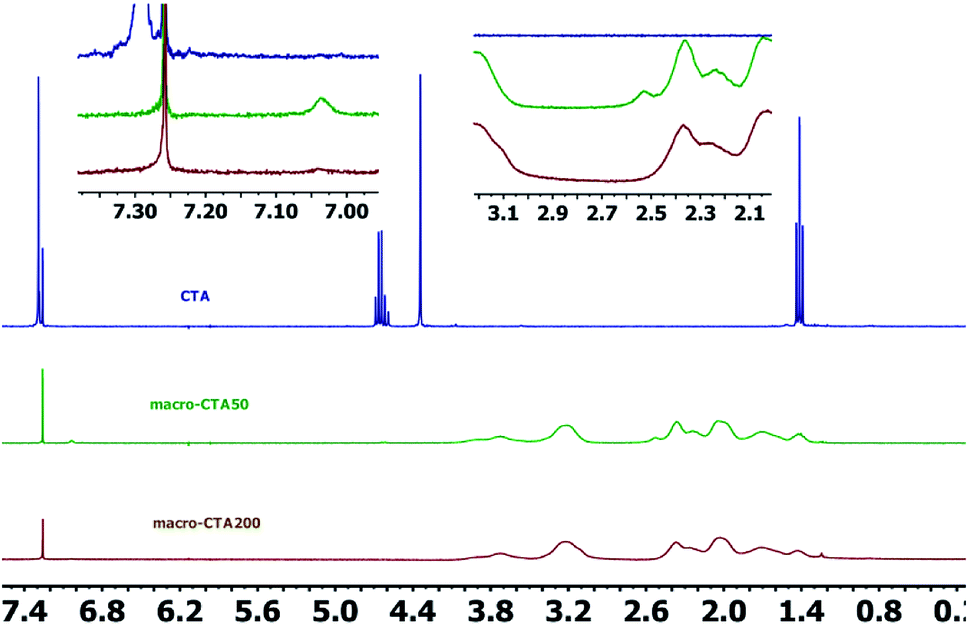
2.2.2 1H NMR spectrum. The 1H NMR spectra of the CTA macro-pvp50 is presented in Fig. 2, with the analysis shown in Table 2. The spectra of the CTA macro-CTA50 and macro-CTA200 are shown in Fig. 3. With the reaction progress after the addition of pyrrolidone, the hydrogen proton absorption peak corresponding to the last methyl group of the phenyl group at 7.29 ppm shifts to a lower value of 7.06 ppm. As the amount of pyrrolidone is increased, the hydrogen proton absorption peak at 7.06 ppm is observed to gradually disappear. At the same time, the hydrogen proton resonance corresponding to the methylene group attached to the methyl group in –OCH2–CH3 at 4.69 ppm, 4.66 ppm, 4.64 ppm and 4.62 ppm are observed to disappear. The peak at 4.34 ppm, corresponding to the proton of the methylene group attached to the phenyl group in 1,4-phenylene(methylene)bis(ethylxanthate), is noted to shift to a lower value, thus, also indicating the intervention of pyrrolidone. These observed findings demonstrated the successful synthesis of the macromolecular CTA.
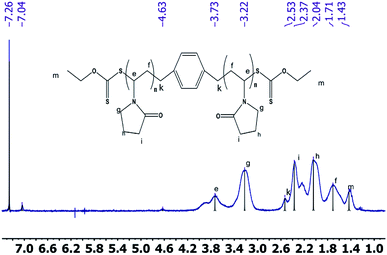 |
| | Fig. 2 1H NMRspectra of the macro-CTA50. | |
Table 2 Analysis of the 1H NMR spectra of the macro-CTA50
| Chemical shift (ppm) |
Type |
| 1.44 |
Hydrogen proton absorption of the terminal methyl group |
| 2.37 |
Hydrogen proton absorption of the methylene group connecting phenyl |
| 4.86 |
Hydrogen proton absorption of the methylene group connecting methyl groups |
| 3.76 |
Hydrogen proton absorption of the last methyl group of the pyrrolidone backbone |
| 2.08 |
Hydrogen proton absorption of the methylene group in pyrrolidone backbone |
| 1.71 2.23 and 3.21 |
Hydrogen proton absorption peak of methylene group in pyrrolidone ring |
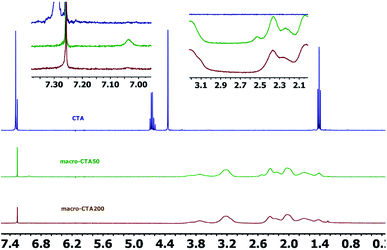 |
| | Fig. 3 1H NMR spectra of the CTA, macro-CTA50 and macro-CTA200. | |
2.3. Block polymer (PDMAEMA-b-PVP-CTA-PVP-b-PDMAEMA)
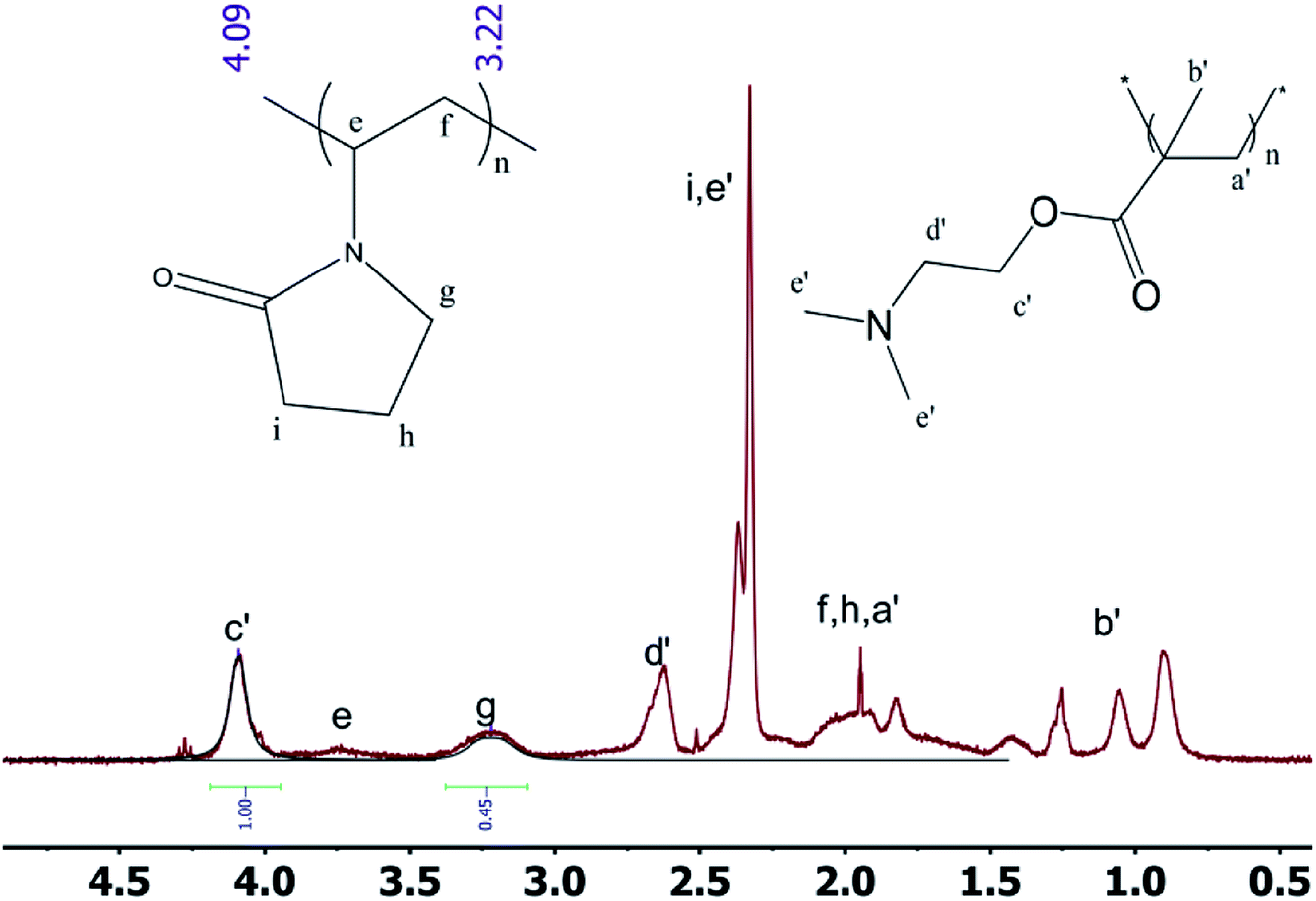
2.3.1 1H NMR spectrum of the block polymer. The 1H NMR spectra of the block polymer are presented in Fig. 4. Based on the computer simulation data and the 1H NMR spectra, it can be observed that the tacticity can be determined by the position of the peak of b′ in PDMAEMA. Moreover, the approximate ratio of the two components can also be calculated. The signal at 4.10 ppm represents the peak of the methylene group adjacent to the carboxylate group of the DMAEMA monomer unit. The peak at 3.20 ppm corresponds to the nitrogen atom in the NVP monomer unit adjacent to the methylene group of lactam. From the 1H NMR spectrum, the length of the repeating unit of the PDMAEMA unit is observed to be ∼194, which is consistent with the expectation from the synthesis method and almost identical to the GPC data.
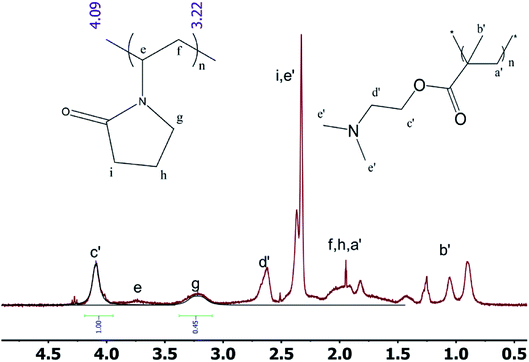 |
| | Fig. 4 1H NMR spectra of the block polymer. | |
1H NMR was used to study the PDMAEMA blocks with different chain lengths. The peaks at 0.9 ppm, 1.1 ppm and 1.3 ppm demonstrate that the isotactic structure gradually acquired the dominant position within the syndiotactic, atactic and isotactic morphologies of the molecule on increasing the degree of polymerization.
2.3.2 Study on sensitivity of pH. The sensitivity of the macromolecular CTA and block polymers to pH was further evaluated. It can be approximately obtained from the equation pH = −lg[H+] and approximate concentration treatment in the low concentration solution as:| |
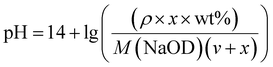 | (1) |
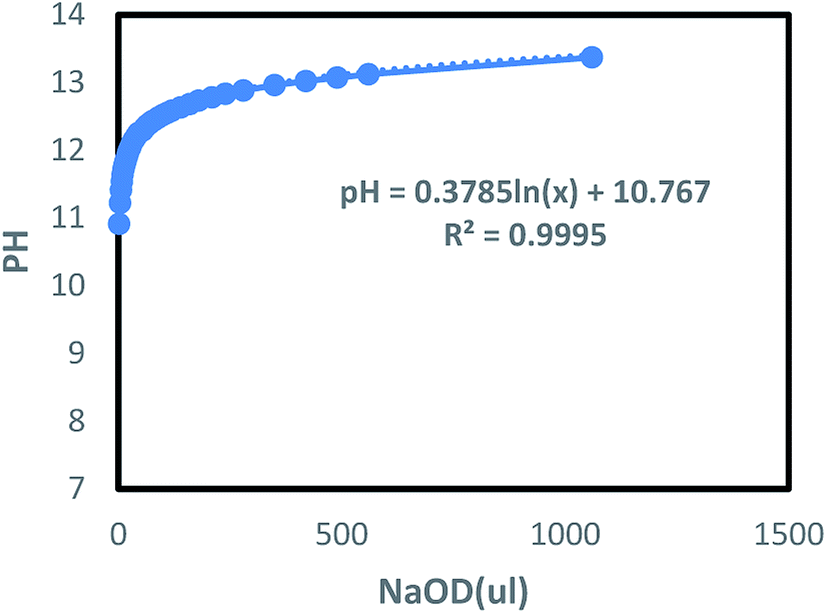
where ρ is the density of NaOD at the current concentration and v is the volume (ml) of the starting liquid. The volume (μl) of x-doped sodium hydroxide (30 wt%) can be derived from the NaOH concentration. For this, a certain volume of deionized water was taken, to which different amounts of NaOD were added, followed by the pH measurement. The results are shown in Fig. 5. It can be observed that the pH of the solution in the low concentration zone is slightly higher than the theoretical value calculated using the relation (1). In the high concentration zone, the measured pH of the solution is noted to be slightly lower than the theoretical value. Based on this theory, the relation (1) is used to fit the change in pH by adding the acidic DCl solution, thus, enabling the study of the effect of pH on the polymer.
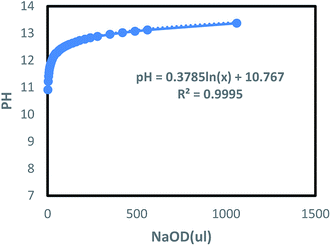 |
| | Fig. 5 The change of pH of aqueous solution with the addition of NaOD. | |
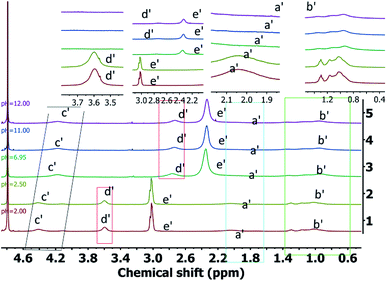
The 1H NMR spectra of PDMAEMA under different pH conditions is shown in Fig. 6 (the peak position is shown on the full spectra, and the peak area is shown on the part spectra). Om increasing the pH value, the acid–base change in the solution is observed to have a significant influence on each peak, with the chemical shifts of the polymer moving to the higher values and the line width becoming wider. The pH of the system can affect the chemical environment around the various groups contained in the polymer. For instance, as the pH increases, the degree of protonation of the tertiary amine groups decreases rapidly, thereby, significantly impairing the ability to attract the electrons. This results in an increase in the shielding effect of the surrounding protons and the movement of the chemical shift to the higher values. At the same time, the interaction between the molecular chain and water molecules is destroyed, thus, the main chain tends to undergo self-assembly. It enhances the mutual shielding effect, and the corresponding attribution peaks almost disappear in comparison. The effect of the protonation process of the tertiary amine group on the line width is observed to include the following aspects:
 |
| | Fig. 6 1H NMR spectra of the PDMAEMA under different pH conditions. | |
(1) The increase in the degree of protonation enhances the interaction between the polymer and D2O solution, thus, improving the mobility of the segment and narrowing the line width;
(2) After protonation, the mutual repulsion between the same charges causes the polymer chains to become dispersed. Thus, the space of mutual movement becomes limited, and the whole system becomes rigid, thereby widening the line width;
(3) In general, although there is a mutual repulsion between the molecular chains after protonation, however, the movement space is not limited (the interaction with the solvent is enhanced, and the tendency of self-assembly is weakened). Thus, the molecular chain mobility is enhanced, and the mutual interference is reduced, thereby narrowing the line width.
Therefore, as the pH decreases, the molecular chain is almost completely protonated instead of the partial protonation originally. Also, the molecular chain structure tends to be single, thus, the molecular chain becomes narrow. It can be seen from Fig. 6 that as the pH decreases, the peaks corresponding to the polymer are significantly narrowed. Thus, as the pH value of the system is reduced, the mutual repulsion between the protonated polymers does not significantly affect the mobility of the polymer segments.
The effect of different pH values on D2O solution of triblock polymer is the same as that of PDMAEMA D2O solution. The introduction of PVP chains leads to stronger interaction between block polymer and D2O solution in acidic conditions, and the molecular chains changes from partially protonated to protonated.
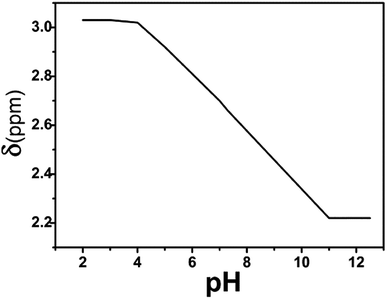
Fig. 7 presents the chemical shift of the long side chain CH3 proton (i.e., e′) of the PDMAEMA segment as a function of the pH value. The triblock polymer, dissolved in water, exhibits a pH of ∼7.4 at a concentration of about 0.25 g L−1, which is consistent with the human body's pH, however, it was slightly higher than value reported in the literature. It may be due to the strong interaction between the PVP segment and water molecules, which destroys the aggregation tendency of PDMAEMA, thus, resulting in a slight increment in ionization and pH.
 |
| | Fig. 7 Chemical shift of the long side chain CH3 proton of the PDMAEMA segment as a function of the pH value. | |
The Henderson–Hasselbalch equation is shown as:
| | |
pKa = pH + lg[α/(1 − α)]
| (2) |
According to eqn (2), as the pH of the system increases, the ionization degree α of the polymer gradually decreases. From the chemical shift of the long side chain CH3 proton of PDMAEMA between pH = 4 and pH = 11, it can be seen that the chemical shifts gradually move to the higher values, which indicates that the relative change in the chemical shift directly reflects the degree of protonation of the polymer. The reason for no change in the chemical shift at pH < 4 and pH > 11 is the pKa value of PDMAEMA of ∼7.3.3 Although the PVP segment is introduced, it has a little impact on the pKa value of the whole segment. Therefore, the equation can be used to estimate the ionization degree α of about 99.96% at pH < 4. As the pH is reduced further, the protonation degree of the segment does not change significantly. Similarly, for pH > 11, the ionization degree reduces to 0.002%, and the deprotonation behavior of the segment does not affect the overall chemical shift, thus, leading to no obvious changes. In addition, as the pH increases, the peak of the short side chain CH3 is noted to be small and insignificant. As the pH gradually decreases, the integrated area of the peak gradually increases. It can be inferred that the PDMAEMA segment aggregates into the nucleus as the pH increases, and the nature and state of the solution changes accordingly. At this stage, the solution apparently changes from clear to turbid, with an obvious phase change behavior. In addition to the change in the chemical shift, the long side chain CH3 proton (i.e., e′) of the PDMAEMA segment exhibits a large decrease in the peak area, leading to eventual disappearance. Two main reasons for this behavior can be the following: firstly, as PVP is highly hydrophilic, its segments are aggregated on the outside after PDMAEME self-assembly of PDMAEME. However, the PVP segments can still bring out a small number of the PDMAEMA segments, and the peak of the short side chain CH3 does not disappear completely. Secondly, the long side chain is hydrophilic, even if the PDMAEMA segment is completely deprotonated into a self-assembled structure. Thus, the inner and outer hydrophilic long side chains of the core shell can still interact with the water molecules.
It can be concluded that the NMR analysis can effectively characterize the pH-sensitive behavior of a substance, along with temperature sensitivity. As the pH of the system decreases, the degree of protonation of the tertiary amine group in the polymer increases. Thus, the mobility of the PDMAEMA segment in the polymer increases significantly. This destabilizes the hydrophilic–hydrophobic balance of the system, and the polymer undergoes significant changes in the hydrophilic–hydrophobic behavior. At the same time, due to the initial tumor stage and differentiation, the pH value is 5.0–6.5, which is lower than the pH value of the normal human tissues.42 With respect to the pH sensitivity of the polymer, comparing the degree of polymer ionization in the normal human tissue environments and tumor, it can be calculated that the degree of ionization of the polymer in the normal human tissues is 55.73% as compared to 86.32–99.50% for the tumor. Thus, it is confirmed that the block polymer has potential applications in the treatment of cancer, along with other medical aspects.43–45
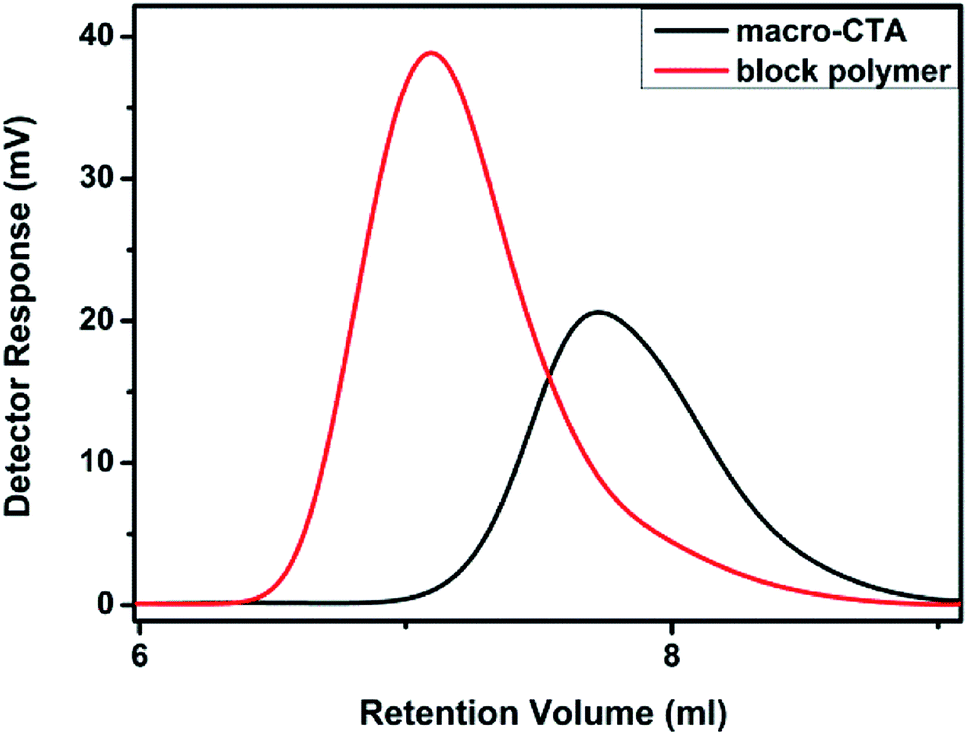
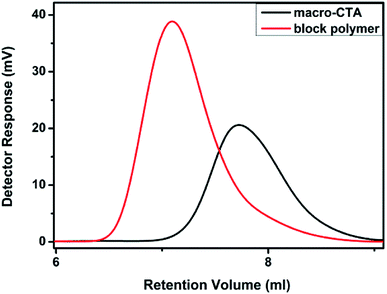
2.3.3 GPC test. GPC tests were carried out on macro-CTA and block polymers, as shown in Fig. 8. It can be observed that the polymers exhibit single peaks and smooth curves with good, thus, proving that the segment lengths in each polymer are close. At the same time, an obvious difference between the two peaks is noted, which proves that the molecular weight of the material increases after the re-reaction. Based on the software analysis, the Mn, Mw and Mz of the macro-CTA are ∼19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1, 22800 g mol−1 and 26100 g mol−1. The polymer dispersity index (PDI, Mw/Mn) is 1.164, based on the calculation results. It confirmed that the polymerization proceeded with the living route, with effective molecular weight control. Similarly, Mn, Mw and Mz of the block polymer obtained in a different reaction are ∼39800 g mol−1, 50000 g mol−1 and 59100 g mol−1, with the calculated PDI of 1.257. As the PDI of the polymers is less than 1.5, it confirms the living polymerization in both cases. The PDI of the block polymer is noted to be slightly larger than the macro-CTA, which may be due to the excessive molecular weight and decreased living polymerization ability during the later stages of polymerization. However, it is still obvious that the chain transfer agent can be used to carry out the living polymerization of PVP and PDMAEMA.
600 g mol−1, 22800 g mol−1 and 26100 g mol−1. The polymer dispersity index (PDI, Mw/Mn) is 1.164, based on the calculation results. It confirmed that the polymerization proceeded with the living route, with effective molecular weight control. Similarly, Mn, Mw and Mz of the block polymer obtained in a different reaction are ∼39800 g mol−1, 50000 g mol−1 and 59100 g mol−1, with the calculated PDI of 1.257. As the PDI of the polymers is less than 1.5, it confirms the living polymerization in both cases. The PDI of the block polymer is noted to be slightly larger than the macro-CTA, which may be due to the excessive molecular weight and decreased living polymerization ability during the later stages of polymerization. However, it is still obvious that the chain transfer agent can be used to carry out the living polymerization of PVP and PDMAEMA.
 |
| | Fig. 8 GPC test of the macro-CTA and block polymer. | |
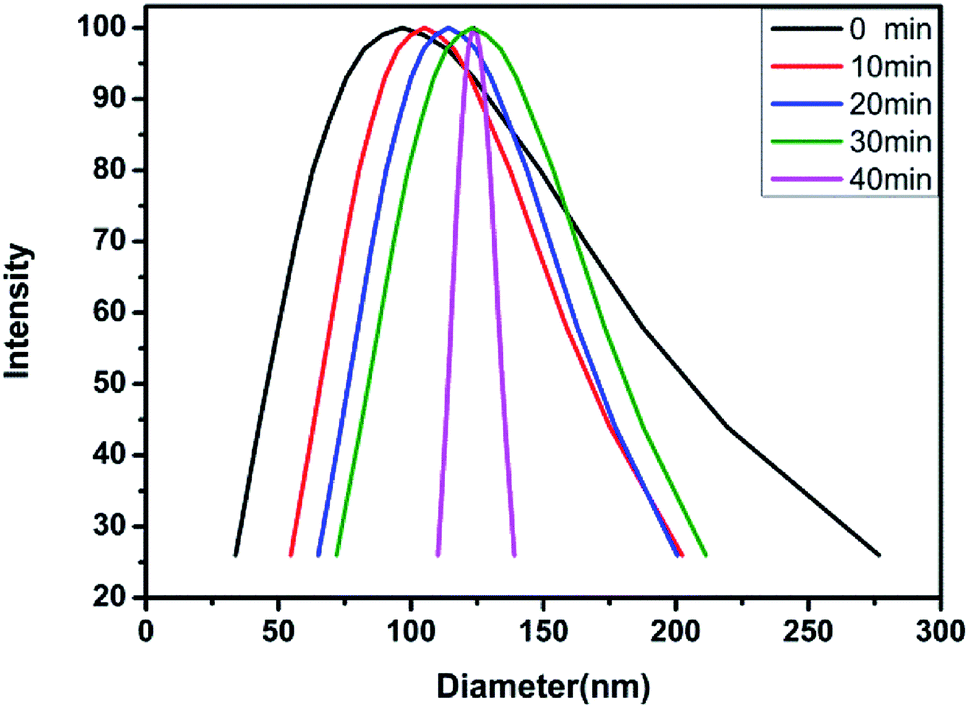
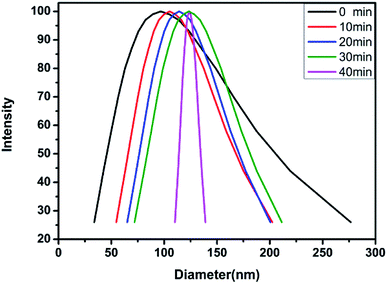
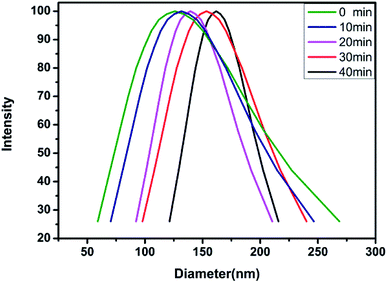
2.3.4 DLS test. The aqueous solution of the block polymer exhibits not only the pH sensitivity, but also the temperature sensitivity. The temperature sensitivity of the polymer solution was analyzed through the macroscopic phenomena, as shown in Fig. 9. The block polymer was dissolved in THF and dialyzed with deionized water (pH = 6.9) for 72 h to prepare a solution with a mass concentration of 0.5 mg ml−1. The solution was filtered through a 1 μm filter membrane and subsequently tested for the particle size at 20 °C, 30 °C, 40 °C and 50 °C. The results indicated no effective hydrodynamic diameter (dH) at 20 °C and 30 °C. As the temperature is increased to 40 °C, the dH of the micelles gradually increases and the polydispersity of the micelles gradually decreases with time (Fig. 10 and Table 3). The reason for the observed behavior is the good solubility of the block polymer with the water phase, thus, forming a homogeneous solution at the low temperature. As the temperature rises to 40 °C, the solubility of the PDMAEMA segments in the polymer deteriorates, thus, forming the hydrophilic–hydrophobic block copolymers. The aggregation tendency of the hydrophobic segments and dissolution tendency of the hydrophilic segments lead to the spontaneous formation of micelles in the aqueous solution. The micelles are not stable at the initial stage of formation, thus, the dispersion coefficient is observed to be large. With time, the micelles with a larger diameter are continuously reduced due to the release of the water phase, while the micelles with a smaller diameter are continuously combined with each other, thus, finally forming the more stable micelles with balanced aggregation and dissolution. Fig. 11 shows the results of test at 50 °C. The same trend as the analysis at 40 °C is observed (Table 4). On increasing the temperature, the dH of the micelles becomes large, which might be due to the effect of temperature on the hydrophilicity of the block polymers.
 |
| | Fig. 9 Photos of PDMAEMA194-PVP87-PDMAEMA194 (0.4 mg ml−1, pH = 7.5) changing at room temperature and 65 °C. | |
 |
| | Fig. 10 Hydrodynamic diameter of block polymer aqueous solution changing with time at 40 °C. | |
Table 3 dH and change in polydispersity with time at 40 °C
| Time (min) |
0 |
10 |
20 |
30 |
40 |
| dH (nm) |
96.7 |
105.1 |
114.1 |
123.8 |
126.7 |
| Polydispersity |
0.504 |
0.172 |
0.125 |
0.113 |
0.075 |
 |
| | Fig. 11 Hydrodynamic diameter of block polymer aqueous solution changing with time at 50 °C. | |
Table 4 dH and change in polydispersity with time at 50 °C
| Time (min) |
0 |
10 |
20 |
30 |
40 |
| dH (nm) |
126.0 |
131.6 |
139.4 |
153.3 |
161.8 |
| Polydispersity |
0.236 |
0.157 |
0.065 |
0.077 |
0.031 |
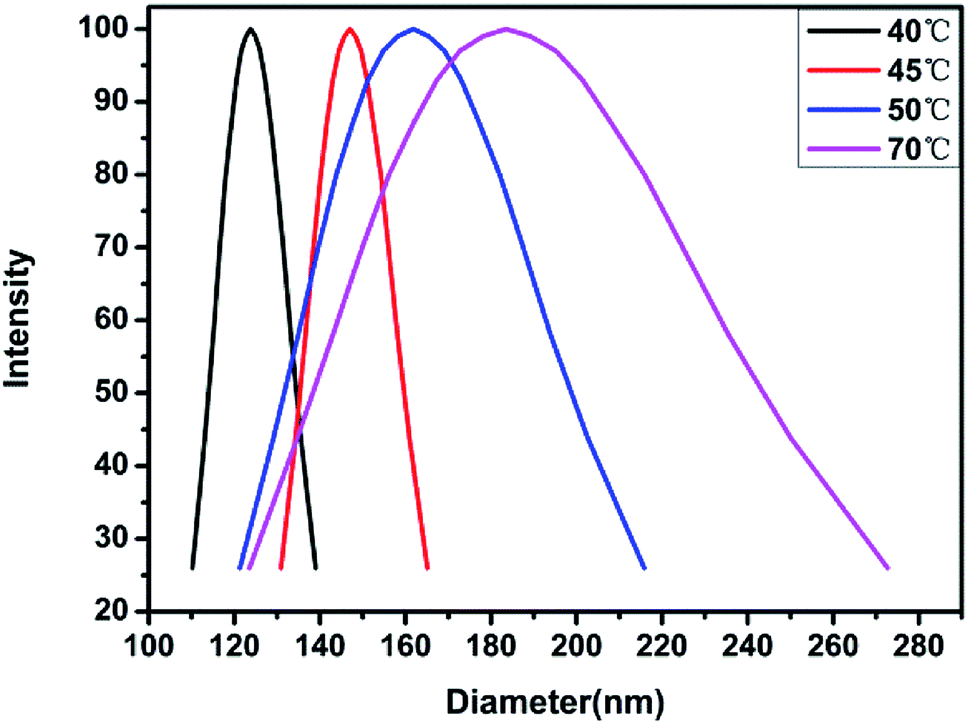
In order to further confirm the observed trend, the analysis was carried out at 45 °C and 70 °C, and the results are shown in Fig. 12. It is observed that the dH of the material gradually increases with temperature, which proves that the temperature has a more obvious effect on the hydrophilicity of the material. Moreover, it is also possible that on increasing the temperature, the pH of the aqueous solution decreases, along with the hydrophobic ability of the PDMAEMA chain segments combined with protons, thereby, resulting in an increase in the micelle diameter. In order to verify this conjecture, a small amount of alkali was added to the sample at 70 °C to observe the micelle change. The hydrodynamic diameter was observed to reach ∼40 nm. This shows that the temperature and pH value exhibit a certain influence on the material, and the influence of the pH value is consistent with the findings from the nuclear magnetic resonance analysis.
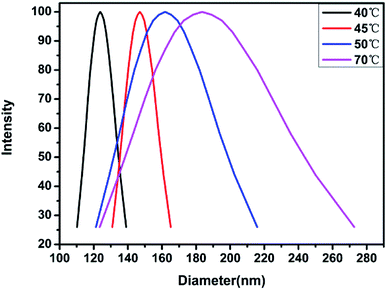 |
| | Fig. 12 Hydrodynamic diameter of block polymer aqueous solution changing with temperature. | |
2.3.5 TEM test. In order to further analyze the hydrophilicity and hydrophobicity of the polymer, the block polymer was dissolved in a small amount of THF. A small amount of water was subsequently added, followed by stirring. The solution was swiftly dropped on a copper mesh (45 °C) for TEM analysis. The TEM results are shown in Fig. 13. It can be seen from Fig. 13(a) that the block polymer is uniformly distributed in the solution, with a uniform micelle size and optimal dispersibility. As seen from Fig. 13(b), the micelles in the solution have a spherical shell structure, thus, the two mutually soluble liquids are separated, with the spheres hollow from inside. It proved that the synthesized block polymer had an excellent hydrophilicity and lipophilicity.
 |
| | Fig. 13 TEM of block polymers: (a) overall effect of 10 microns. (b) Enlarged view of parts. | |
The temperature sensitivity test carried out using laser scattering and transmission electron microscopy revealed that the LCST of the block polymer is not only related to the composition chain length of the block polymer, but also the concentration and pH. The observed multiple sensitivity significantly enhances the potential applications of the developed polymers.
3. Conclusions
PDMAEMA exhibits excellent temperature, pH and ion sensitivity as well as antibacterial characteristics. However, the application of the pure PDMAEMA is rare, and RAFT controlled polymerization of the polymer has rarely been reported. In this study, high-yield RAFT reagents have been synthesized by using a “one-pot” method, with subsequent synthesis of the synthetic block polymers (PDMAEMA-b-PVP-CTA-PVP-b-PDMAEMA). The results of the FITR, NMR and GPC analysis showed that the block polymers with different molecular weights were successfully synthesized. It was observed for the first time that as the degree of polymerization increased, the isotacticity gradually started to dominate. In addition, the further characterization by NMR established the effect of pH on PDMAEMA and block polymers. Further analysis of the developed products using DLS and TEM indicated that the block polymer had dual sensitivity for pH and temperature, with potential applications in medical field, especially cancer treatment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China.
References
- K. Matyjaszewski and A. H. E. Müller, Prog. Polym. Sci., 2006, 31, 1039–1040 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- H. Mark and H. Mark, J. Appl. Polym. Sci., 1970, 14, 859 CrossRef.
- Z. H. Lu, S. Y. Lee and S. H. Goh, Polymer, 1997, 38, 5893–5895 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A: Pure Appl. Chem., 1986, 23, 839–852 CrossRef.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- D. Cummins, P. Wyman, C. J. Duxbury, J. Thies, C. E. Koning and A. Heise, Chem. Mater., 2007, 19, 5285–5292 CrossRef CAS.
- S. Kwan and M. Maric, Polymer, 2016, 86, 69–82 CrossRef CAS.
- C. Almeida, H. Costa, P. Kadhirvel, A. M. Queiroz, R. C. S. Dias and M. Costa, J. Appl. Polym. Sci., 2016, 133, 43993 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- E. Altay and J. Rzayev, Polymer, 2016, 98, 487–494 CrossRef CAS.
- B. B. Yu, Z. Zeng, Q. Y. Ren, Y. Chen, M. Liang and H. W. Zou, J. Mol. Struct., 2016, 1120, 171–179 CrossRef CAS.
- S. Schmidt, M. Koldevitz, J.-M. Noy and P. J. Roth, Polym. Chem., 2015, 6, 44–54 RSC.
- J. Gardiner, C. H. Hornung, J. Tsanaktsidis and D. Guthrie, Eur. Polym. J., 2016, 80, 200–207 CrossRef CAS.
- H. A. Zayas, N. P. Truong, D. Valade, Z. Jia and M. J. Monteiro, Polym. Chem., 2013, 4, 592–599 RSC.
- E. V. Chernikova, S. D. Zaitsev, A. V. Plutalova, K. O. Mineeva, O. S. Zotova and D. V. Vishnevetsky, RSC Adv., 2018, 8, 14300–14310 RSC.
- C. H. Wang, Y. S. Fan, Z. Zhang, Q. B. Chen, T. Y. Zeng, Q. Y. Meng and Y. Z. You, Appl. Surf. Sci., 2019, 475, 639–644 CrossRef CAS.
- C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2015, 36, 2181–2185 CrossRef CAS PubMed.
- D. Hu, Y. Zhang, M. Su, L. Bao, L. Zhao and T. Liu, J. Supercrit. Fluids, 2016, 118, 96–106 CrossRef CAS.
- J. R. Gois, A. V. Popov, T. Guliashvili, A. C. Serra and J. F. J. Coelho, RSC Adv., 2015, 5, 91225–91234 RSC.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- M. A. D. Gonçalves, V. D. Pinto, R. C. S. Dias, J. C. Hernándes-Ortiz and M. R. P. F. N. Costa, Macromol. Symp., 2013, 333, 273–285 CrossRef.
- H. Lee, E. Terry, M. Zong, N. Arrowsmith, S. Perrier, K. J. Thurecht and S. M. Howdle, J. Am. Chem. Soc., 2008, 130, 12242–12243 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- Z. Zhou, F. Guo, N. Wang, M. Meng and G. Li, Int. J. Biol. Macromol., 2018, 116, 911–919 CrossRef CAS PubMed.
- B. Q. Jin, Y. Q. Li, Y. Shi and X. G. Jin, Polymer, 2017, 116, 466–471 CrossRef CAS.
- N. Roka, O. Kokkorogianni and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3776–3787 CrossRef CAS.
- N. Maity, A. Kuila, D. Chatterjee, D. Mandal and A. K. Nandi, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3878–3887 CrossRef CAS.
- X. M. Qian, L. X. Long, Z. D. Shi, C. Y. Liu, M. Z. Qiu, J. Sheng, P. Y. Pu, X. B. Yuan, Y. Ren and C. S. Kang, Biomaterials, 2014, 35, 2322–2335 CrossRef CAS PubMed.
- B. Chen, C. V. Synatschke, V. Jérôme, A. H. E. Müller, R. Freitag and C. Wu, Eur. Polym. J., 2018, 103, 362–369 CrossRef CAS.
- N. Jommanee, C. Chanthad and K. Manokruang, Carbohydr. Polym., 2018, 198, 486–494 CrossRef CAS PubMed.
- V. Varma, H. G. Shivakumar, S. J. Fathima, V. Radha and F. Khanum, RSC Adv., 2016, 6, 105495–105507 RSC.
- Y. Hiruta, Y. Kanda, N. Katsuyama and H. Kanazawa, RSC Adv., 2017, 7, 29540–29549 RSC.
- T. Lei, R. Manchanda, A. Fernandez-Fernandez, Y. C. Huang, D. Wright and A. J. McGoron, RSC Adv., 2014, 4, 17959–17968 RSC.
- R. Nikoletta, K. Olga and P. Marinos, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3776–3787 CrossRef.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 CAS.
- W. Zhang, X. Jin, H. Li, R.-r. Zhang and C.-w. Wu, Carbohydr. Polym., 2018, 186, 82–90 CrossRef CAS PubMed.
- Z. Zhou, G. Li, N. Wang, F. Guo, L. Guo and X. Liu, Colloids Surf., B, 2018, 172, 136–142 CrossRef CAS PubMed.
- A. Singh, K. Vaishagya, R. K. Verma and R. Shukla, AAPS PharmSciTech, 2019, 20, 213 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05343k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
F. Q. Liu
and
F. Q. Liu
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration on pyrrolidone)
O stretching vibration on pyrrolidone)