
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDensity functional study on the CO oxidation reaction mechanism on MnN2-doped graphene†
Mingming Luo ,
Zhao Liang,
Chao Liu*,
Xiaopeng Qi,
Mingwei Chen,
Hui Yang and
Tongxiang Liang*
,
Zhao Liang,
Chao Liu*,
Xiaopeng Qi,
Mingwei Chen,
Hui Yang and
Tongxiang Liang*
Faculty of Materials Metallurgy and Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, China. E-mail: liuchao198967@126.com; liang_tx@126.com
First published on 27th July 2020
Abstract
The CO oxidation mechanisms over three different MnN2-doped graphene (MnN2C2: MnN2C2-hex, MnN2C2-opp, MnN2C2-pen) structures were investigated through first-principles calculations. The vacancy in graphene can strongly stabilize Mn atoms and make them positively charged, which promotes O2 activation and weakens CO adsorption. Hence, CO oxidation activity is enhanced and the catalyst is prevented from being poisoned. CO oxidation reaction (COOR) on MnN2C2 along the Eley–Rideal (ER) mechanism and the Langmuir–Hinshelwood (LH) mechanism will leave one O atom on the Mn atom, which is difficult to react with isolated CO. COOR on MnN2C2-opp along the ER mechanism and termolecular Eley–Rideal (TER) mechanism need overcome low energy barriers in the rate limiting step (RLS), which are 0.544 and 0.342 eV, respectively. The oxidation of CO along TER mechanism on MnN2C2-opp is the best reaction pathway with smallest energy barrier. Therefore, the MnN2C2-opp is an efficient catalysis and this study has a guiding role in designing effective catalyst for CO oxidation.
1. Introduction
CO is a common toxic gas that is water-insoluble, which comes from automobile exhaust and insufficient combustion of fossil fuels. CO oxidation at low-temperature has attracted much attention because of its importance in solving environmental pollution and protecting human health.1,2 How to quickly detect and effectively eliminate CO has become one of the most challenging topics today. Converting CO into non-toxic substances has received considerable attention and catalytic oxidation of CO is an effective method. CO oxidation was suitable to reveal the inherent heterogeneous catalytic reaction mechanism.3,4 In previous studies, noble metals such as Pd,5,6 Ag,7 Pt8 and Au9,10 were often used as catalysts for CO oxidation due to chemical stability and excellent catalytic activity. However, their scarcity, high prices and high reaction temperatures have limited application. Therefore, it is valuable to develop low-cost and high-efficiency catalysts to promote CO oxidation at low temperatures.Single-atom catalysts (SACs) with a high level of dispersed atoms and graphene films with high thermal stability have received widespread attention on catalysts.11,12 When size of the transition metal from nanometer to the atomic level, the amount of active site will be dramatically increased and the catalytic ability of transition metal maximize. In 2011, the first single-atom catalyst Pt1/FeOx was prepared for CO oxidation.13 Since then, there have been endless researches and experiments on SACs, which offer an opportunity to efficiently catalyze oxidation of CO. Graphene is a favorable support for SACs due to its high surface area, high stability, low manufacturing cost and excellent electronic properties. The carbon atoms of the original graphene are combined with strong sp2, so the original graphene is chemically inert and has weak catalytic activity. Besides, single-layer graphene was prone to structural defects during the manufacturing process,14 which is an ideal site for anchoring single atoms.15 The catalyst for the experimental synthesis of graphene supported SAC has excellent catalytic performance and does not cause the agglomeration of doped atoms.16,17 A large number of studies have shown that doping transition metal atoms on the original graphene has efficient catalytic performance for CO oxidation at low temperature. Fe,18 Co,19 Ni-doped20 graphene with single vacancy defective (Gra-SV) catalyzed CO showed high activity. The previous research mainly focused on Gra-SV and the double vacancy theory has received less attention. The pentagon in graphene defects can effectively reduce the total energy, and the graphene with double vacancy defect (Gra-DV) is more stable than the single vacancy defect graphene.21 Therefore, the probability of doped atoms being fixed in double vacancy defects is greater in practice. Mn is multi-electron structure and large atomic radius, and Mn-doped Gra-SV will cause significant changes in the structure and make the structure more unstable. In summary, Mn-doped Gra-DV is more advantageous than Gra-SV.
Previous experiments have shown that the FeN2 moieties shows better catalytic activity than the FeN4 moieties due to its better electron transport ability.22,23 Mn and Fe atoms have similar electronic structures and MnN2-doped graphene (MnN2C2) may have high catalytic activity. Orellana24 find that the catalytic activity of the phthalocyanine metal center following the order: Mn > Fe > Co, which indicates that MnN2 moieties doped graphene may show higher catalytic activity for CO oxidation than FeN2 moieties. Previously few studies about CO catalytic on Mn-doped substrate with double vacancy defects,25 CO oxidation along TER mechanism on MnN4 porphyrin-like carbon nanotubes was the most ideal reaction pathway and the energy barrier of RLS was only 0.69 eV.26 MnC4 catalyzes CO along the LH mechanism was the best reaction path and the energy barrier of RLS was 0.41 eV,27 Mn-doped substrate shows good catalytic effect on CO oxidation.28 The experimental results confirm that Mn impurities in graphene have been successfully doped into a single atom form, Mn and N co-doping graphene can effectively improve the catalytic performance of ORR.29 Understanding the intrinsic mechanism of COOR activity of Mn and N co-doped graphene can guide experimenters to design the most effective catalyst.
Inspired by the above research, we carried out DFT calculations to explore the catalytic performance of MnN2C2 for CO oxidation. To the best of our knowledge, there is no research on the mechanism of CO catalyzed by MnN2C2. In this work, the various oxidation mechanisms of CO catalyzed by MnN2C2 were revealed and the optimal reaction pathway of CO oxidation was determined by discussing the reaction energy barrier.
2. Computational methodologies
All calculation results are executed using DMol3 code.30 The exchange correlation functional use the PBE (Perdew–Burke–Ernzerhof) function of GGA (Generalized Gradient Approximation),31,32 The Grimme's dispersion correction has higher accuracy for van der Waals corrections.33 We selected DFT semi-core pseudopotential and a double numerical plus polarization basis set to ensure highly accurate results.34 Synchronization method using conjugate gradient refinement to determine the minimum energy path and transition state,35 all transition states are determined to have only one virtual frequency. Sampling k-point grid using Monkhorst–Pack method36 and Mulliken charge population method to express charge transfer.37We delete atoms in a pure graphene model with periodic boundary conditions to create vacancies, where two undercoordinated carbons are replaced by N atoms (N2C2-vac), the Mn atom bound by the undercoordinated atoms protrudes from the surface after structural optimization. Finally, the three target configurations of MnN2C2 (Fig. 1) were considered: (1) two N atoms on a pentatomic ring (MnN2C2-pen); (2) two N atoms at the opposite sides of Mn atom (MnN2C2-opp); (3) two N atoms on a hexatomic ring (MnN2C2-hex). Previous studies using similar models have shown that it has high catalytic activity for oxygen reduction.38,39
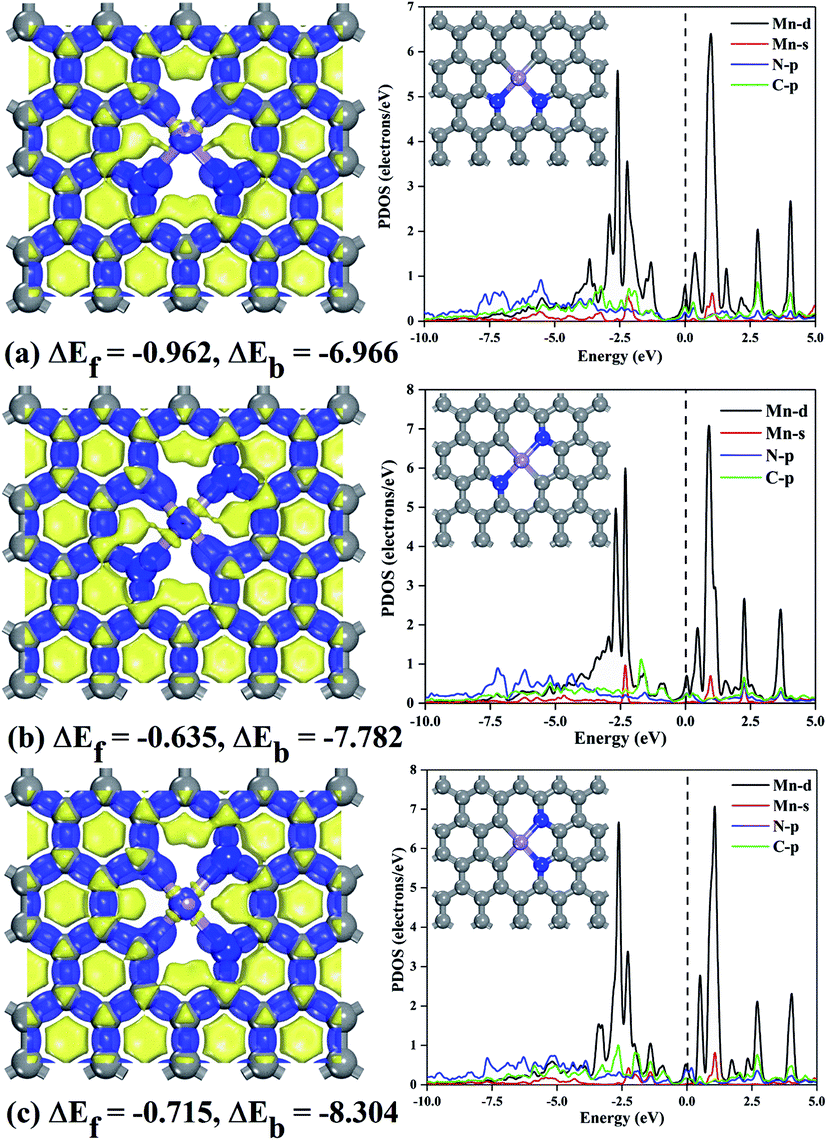 | ||
| Fig. 1 Views of the optimized three configurations and deformation density: (a) MnN2C2-hex, (b) MnN2C2-opp, (c) MnN2C2-pen. ΔEf and ΔEb represent formation energy and binding energy. Where gray, blue and purple represent C, N and Mn atom, respectively. PDOS of MnN2C2, Fermi levels are indicated by dotted lines. | ||
The formation energy (ΔEf) can be used to evaluate the stability of the substrate structure by the following equation
| ΔEf = EMnN2C2 + 4μC − (EGra + 2μN + μMn) | (1) |
The binding energy (ΔEb) can be used to characterize the bonding strength of doped atoms and atoms around vacancies, ΔEb is defined as follows:
| ΔEb = EMnN2C2 − EN2C2-vac − EMn | (2) |
The adsorption energy can evaluate the interaction between gas molecules and MnN2C2 and be calculated by the following method:
| ΔEads = Eadsorbate/MnN2C2 − (EMnN2C2 + Eadsorbate) | (3) |
3. Results and discussion
3.1 Stability of the MnN2C2
Transition metal atoms easily self-aggregate to clusters due to its high cohesive energy, which decreased the catalytic performance and atom utilization rate. Therefore, the bonding strength of the doping atom Mn and surrounding atoms should be analyzed first. The formation energy and binding energy of MnN2C2 are calculated according to formula (1) and (2) as shown in Fig. 1, and negative energy values illustrate the stability of MnN2C2. The binding energy is much stronger (more negative) than the cohesive energy of Mn atom.40 The strong combination of Mn atoms and coordination atoms can effectively prevent the agglomeration and diffusion of Mn atoms. The partial state density (PDOS) can clearly see the orbital overlap between Mn, N and C atoms to further analyze the internal mechanism of interatomic bonding (Fig. 1). The hybridization of d orbits of Mn atoms and p orbits of C and N atoms makes Mn atoms to bind firmly in the substrate, especially there are obvious peaks near the Fermi level, which has an important influence on the catalytic activity and to a certain extent shows that MnN2C2 has high activity. The deformation density is used to evaluate charge transfer within MnN2C2, the consumption and accumulation of electron density are represented by yellow and blue (Fig. 1). Demonstrated that there was a significant electronic overlap between Mn atom and its adjacent four atoms, which further illustrated that there was a strong interaction between Mn atom and adjacent atom. The doping of atoms breaks the local electrical neutrality of the original graphene, and the apparent charge transfer brings favorable conditions for the high activity of the substrate. From the electron display of Fukui function (Fig. S1†), it can be seen more directly that the electrophilic and nucleophilic regions are mainly Mn atoms and N atoms, which is the main reaction region and MnN2C2 may have high activity for free gas molecules. Molecular dynamics simulation from the perspective of thermodynamics to analyze the substrate stability over a period of 2 ps at 1000 K (Fig. 2). The energy of MnN2C2 fluctuates around a fixed value and the energy fluctuation is less than 0.1 eV. There is no chemical bond breakage and formation during the simulation. The slight deformation of MnN2C2 makes the Mn, N and C atoms are almost on the same plane. Above results showed that MnN2C2 were structurally stable and had high activity.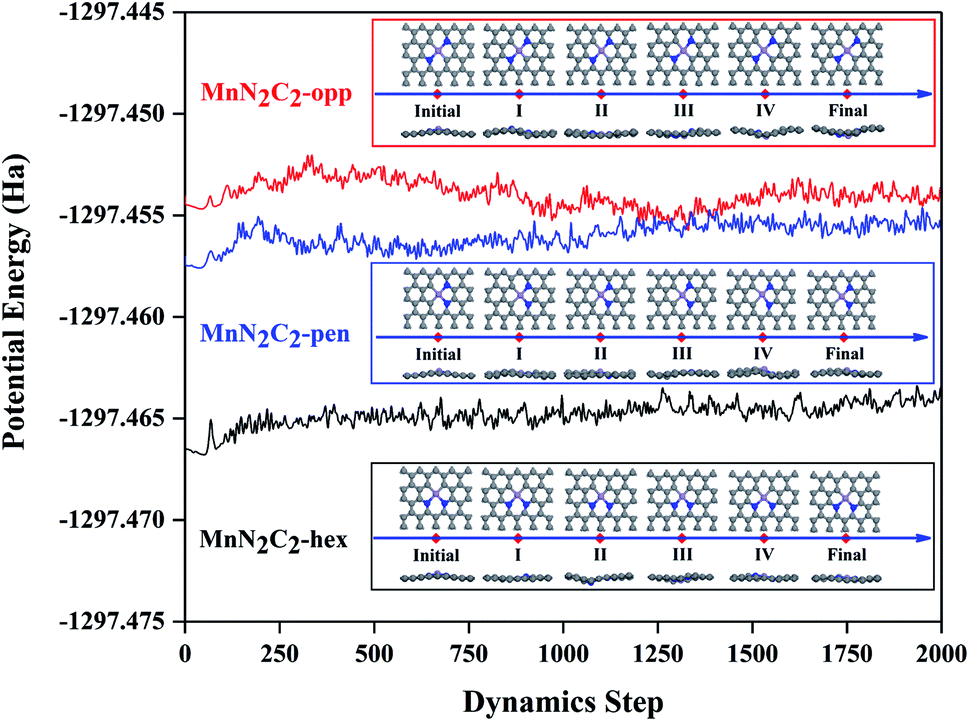 | ||
| Fig. 2 Molecular dynamics trajectory of MnN2C2 at 1000 K with snapshots of intermediates at different times. | ||
3.2 The adsorption properties of MnN2C2
The catalytic mechanism of COOR was revealed based on gas adsorption properties of Mn2N2C2, which include single molecule adsorption (O2, CO, CO2) and bimolecular co-adsorption (CO + O2, 2CO). The most stable adsorption configuration and bond length are shown in Fig. 3, the detailed adsorption energy data are shown in Fig. 4 and Table S1.† Three similar structures have little difference in gas adsorption performance. O2 adsorption is critical for subsequent COOR,41,42 the most stable adsorption configuration of O2 is parallel to the substrate, and its adsorption energy is stronger than that of CO to ensure that the active site is not poisoned by CO when CO and O2 are mixed in equal proportions. After adsorption, activated O2 gains electrons and causes the bond length extension, which is beneficial to CO oxidation. It was found that the adsorbed CO and N atoms would repel each other, and the angle of CO adsorption on MnN2C2-hex and MnN2C2-pen would be inclined. The adsorption energy of CO + O2 is stronger than the adsorption energy of single molecule, but greater than the sum of the two adsorption energies, indicated that when the substrate adsorbs one gas molecule, the activity will decrease, which will lead to weaker adsorption of the second gas. The distance between Mn and CO2 is much larger than the sum of the covalent radii of O and Mn atoms, CO2 is physically adsorbed on MnN2C2 and the catalyst can be recycled. Preliminarily judge that MnN2C2 can be used as catalysts for CO oxidation based on gas adsorption energy.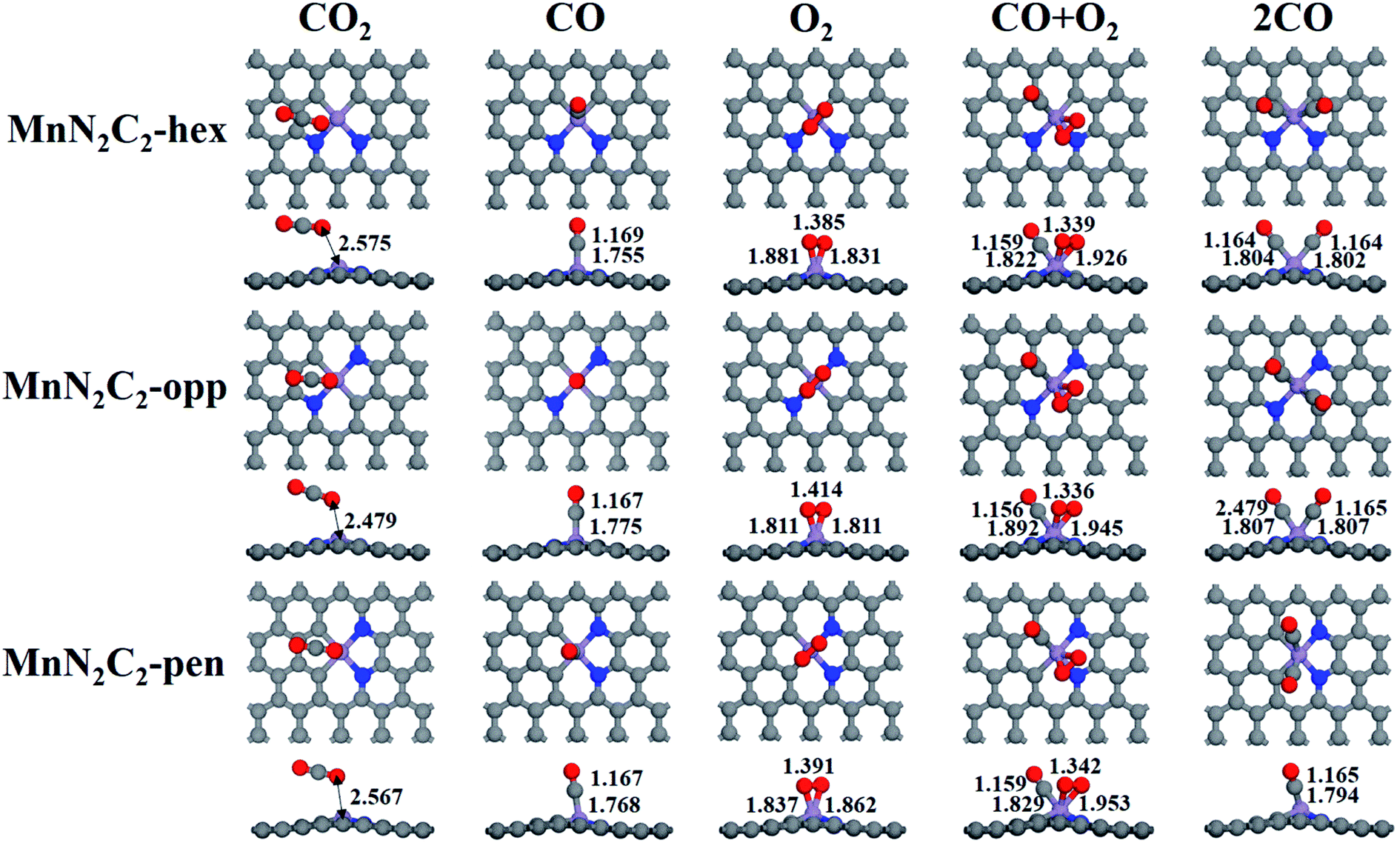 | ||
| Fig. 3 Structure of single molecule adsorption (O2, CO, CO2) and bimolecular co-adsorption (CO + O2, 2CO) on MnN2C2. | ||
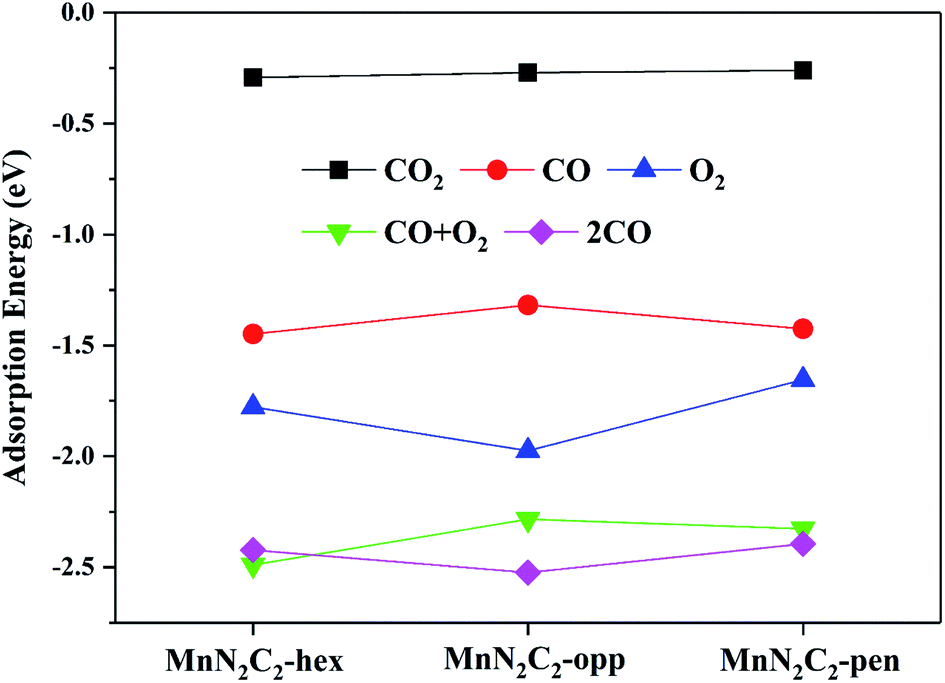 | ||
| Fig. 4 The adsorption energy of gas on MnN2C2. | ||
The pre-adsorption gas determines the type of reaction mechanism for subsequent COOR. When O2 and CO are mixed in equal proportion, O2 can be adsorbed on the substrate in advance, indicating that COOR can proceed along the ER mechanism. O2 and CO co-adsorption reaction mechanism corresponding to COOR is LH mechanism. The adsorption energy of 2CO is stronger than that of O2, the corresponding reaction mechanism (TER) needs to be considered under high CO concentration. The following content will analyze the reaction mechanism of COOR on MnN2C2.
3.3 Reaction mechanism of CO oxidation on MnN2C2
Pre-adsorbed O2 obtains electrons from the substrate to occupy the antibonding π* orbit and subsequently lead to the elongation of the bond length, which may cause O2 to be easily decomposed directly (the relative energy in Fig. 5a, the atomic structure of MnN2C2-opp in Fig. 1c, the atomic structure of MnN2C2-hex and MnN2C2-pen in Fig. S2†). The lowest activation energy of O2 decomposition is 0.854 eV (MnN2C2-opp) among the three structures, the CO oxidation process is difficult to perform at room temperature when the energy barrier is greater than 0.8 eV,43 this means that the kinetics are not conducive to O2 dissociation and O2 adsorbed on MnN2C2 can be effectively activated without decomposition at room temperature.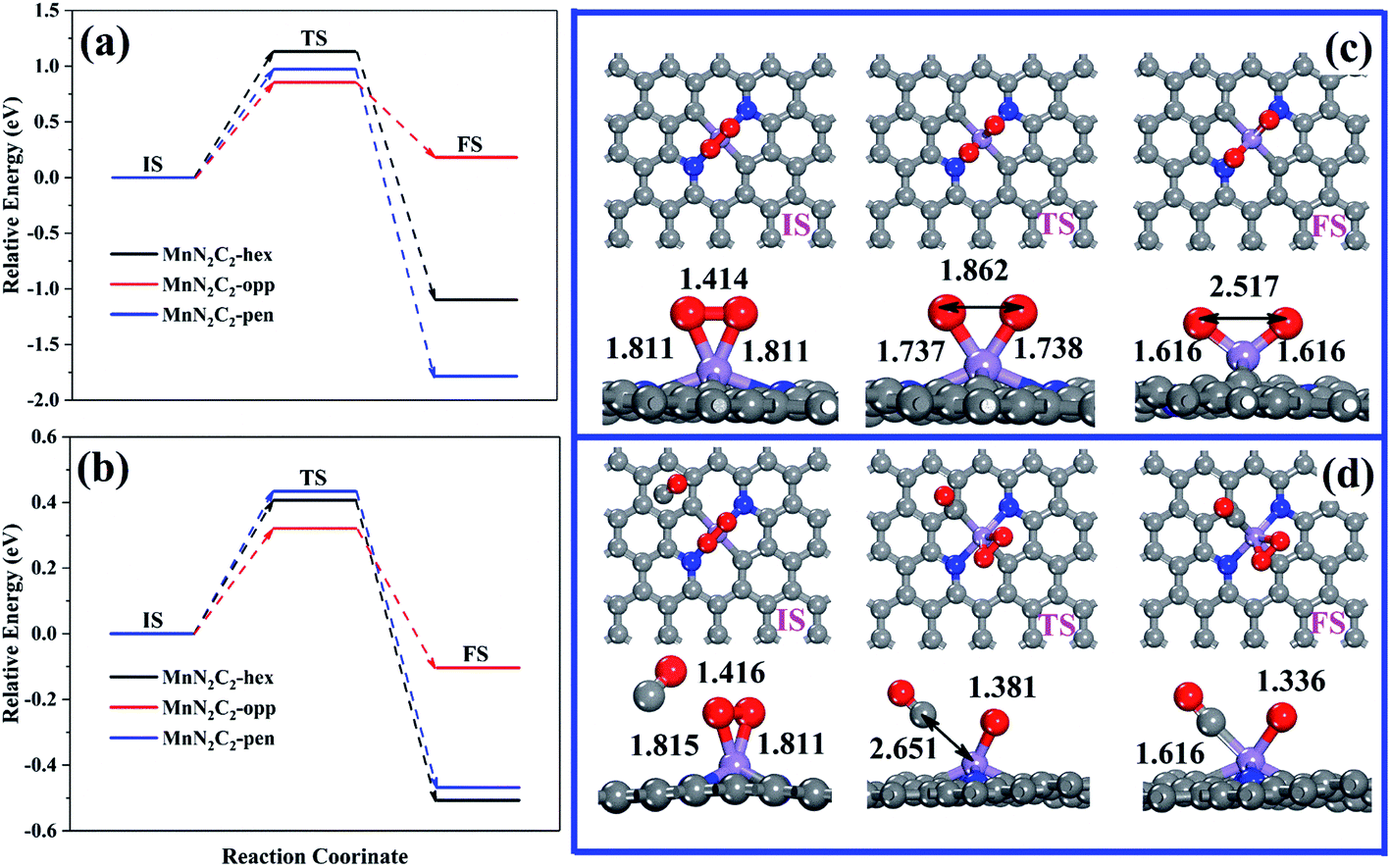 | ||
| Fig. 5 (a) Relative energy of O2 molecule dissociation and (b) the CO + *O2 → *CO + *O2 reaction on MnN2C2. (c) Structure of O2 molecule dissociation and (d) CO + *O2 → *CO + *O2 reaction on MnN2C2-opp. | ||
After adsorbing O2, there will be three cases of CO adsorption. (1) CO and two O atoms are bonded to form silicate-like structure (CO3), the TS search cannot get the CO3 with only a virtual frequency and positive energy barrier on MnN2C2-hex and MnN2C2-pen, which has also appeared in Mn-doped single vacancy graphene.44 (2) CO and an O atom directly generate CO2 and the remaining O atom occupies the Mn site. In both cases, COOR follows the ER mechanism, and its energy barrier and reaction energy are shown in Table S2.† CO reacts with O2 in MnN2C2-opp to form CO2 has a low energy barrier (0.544 eV), but other energy barrier data are not ideal, indicating that the CO oxidation in MnN2C2-hex and MnN2C2-pen is difficult to proceed along the ER mechanism. (3) Isolated CO and O2 co-adsorbed on Mn atom, the energy barrier of the CO adsorption process is shown in Fig. 5b and the atomic structure is shown in Fig. 5d and S3.† Compared with the reaction between isolated CO and pre-adsorbed O2, CO is more likely to form a co-adsorption structure with O2. CO only needs to overcome the low activation energy to adsorb on Mn atoms and the largest energy barrier was as low as 0.434 eV (MnN2C2-pen). In summary, COOR is more powerful along the LH mechanism than the ER mechanism. We also consider that when the active site is occupied by CO, the energy barrier of the process of forming two CO co-adsorption structures (Fig. S4†). The co-adsorption of 2CO on MnN2C2 is accompanied by large exothermic reaction energy and the maximum energy barrier is as low as 0.238 eV (MnN2C2-pen), which is smaller than the energy barrier for the formation of O2 + CO co-adsorption on MnN2C2. The above shows that it is very easy and beneficial to form 2CO co-adsorption configuration on MnN2C2.
Two CO molecules co-adsorbed as initial state of COOR along TER reaction mechanism, which was first study COOR on Au-doped h-BN monolayers by Keke Mao.45 The relative energy of COOR on MnN2C2 along the LH and TER mechanisms are shown in Fig. 6a and b, the corresponding structure are shown in Fig. 6c, d, S5 and S6.† The formed intermediate product OCOO spontaneously decomposes into O and CO2 on MnN2C2-hex at low temperature (350 K), and CO2 can diffuse spontaneously, the energy and structure changes during spontaneous decomposition are shown in the Fig. S7.† COOR on MnN2C2 has similar structural changes, the CO oxidation on MnN2C2-opp along the LH and TER mechanisms will be taken as an example to analyze the reaction mechanism in detail.
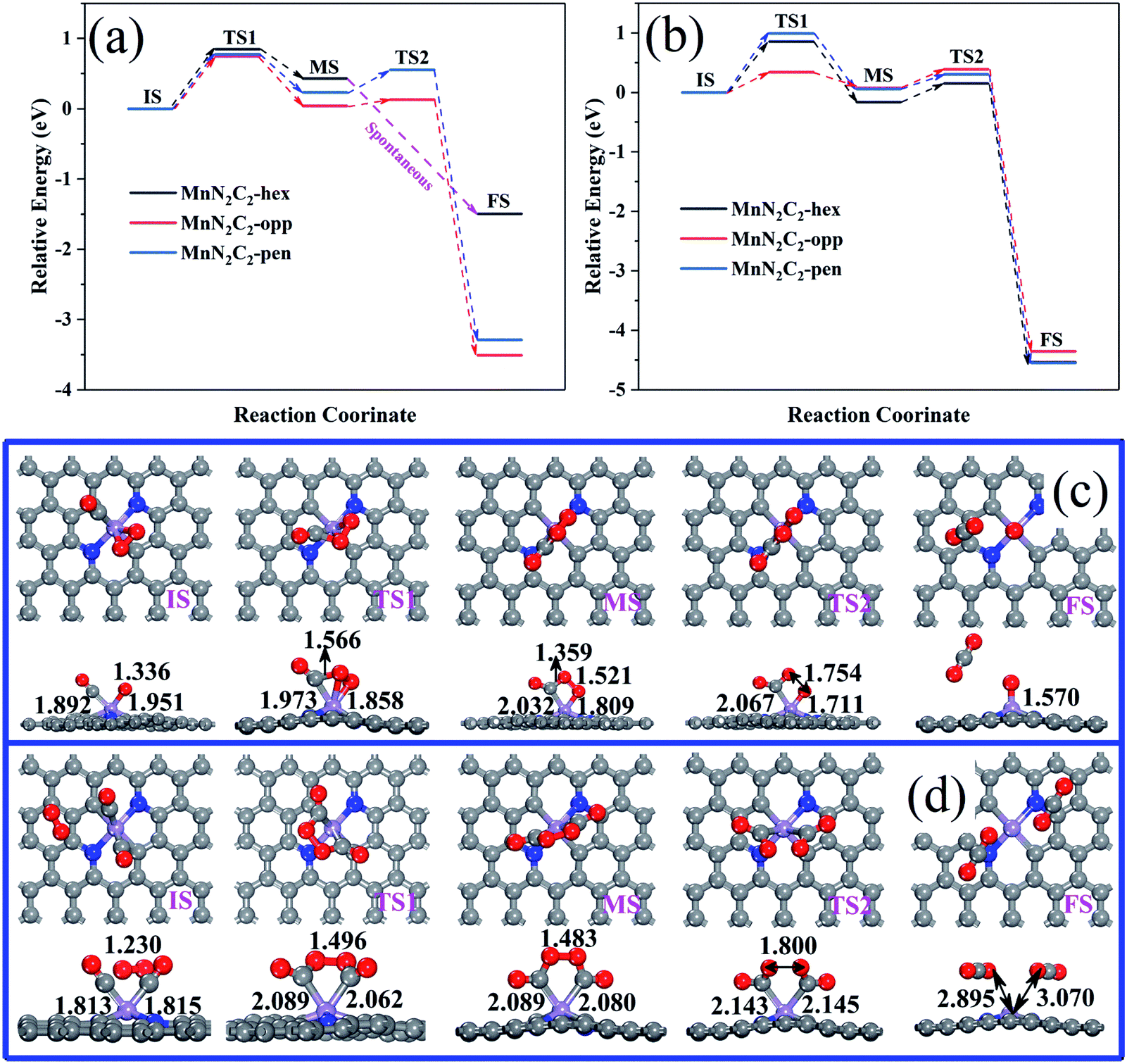 | ||
| Fig. 6 (a) COOR via the LH mechanisms on MnN2C2, (b) COOR via the TER mechanisms on MnN2C2. (c) Structure of the COOR on MnN2C2-opp via the LH and (d) TER mechanisms. | ||
CO and O2 co-adsorption as the initial state of the LH mechanism (Fig. 6c), close to each other can form an experimentally proven intermediate product OCOO.46 The elongation of the O2 bond from 1.336 to 1.458 Å and the Mn–C bond from 1.892 to 1.973 Å, which is conducive to the formation and desorption of CO2. The formation of OCOO needs to overcome the activation energy of 0.747 eV and endothermic reaction energy of 0.038 eV, OCOO only needs to via the activation energy of 0.092 eV to generate CO2 and CO2 can spontaneously release at room temperature.
In the TER mechanism (Fig. 6d), the pre-adsorbed 2CO and free O2 bond to form a five-membered ring intermediate (OCOOCO), the bond length of O2 is stretched to 1.496 Å, which needs to overcome the activation energy of 0.342 eV. The O–O bond of 1.483 Å and the Mn–C bond of 2.089 Å are easily broken to form two physically adsorbed CO2, the OCOOCO decomposition process only requires 0.315 eV of activation energy to prove the above speculation. The energy barriers of the two step reactions of the whole process are small, and emit 4.353 eV heat, which indicates that the oxidation of CO along TER mechanism on MnN2C2-opp is favorable. Table 1 summarizes the energy barriers of RLS for CO oxidation in various precious metal systems. The energy barrier of CO oxidation catalyzing by MnN2C2-opp along the TER mechanism is lower or comparable to that catalyzing by noble metal systems. Moreover, MnN2C2-opp catalyzed CO has lower activation energy than other Mn atoms and N co-doped graphene (MnC3, MnC4 and MnN4–CNT). Therefore, MnN2C2-opp can be used to replace the precious metal as a highly efficient catalyst for CO oxidation.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
The CO oxidation on MnN2C2 via the ER and LH mechanisms to form the first CO2, and the remaining O atoms have strong interactions with Mn atoms. For the strong bond formed by Mn and O atom, CO and O may need to overcome higher energy barrier to complete the reaction. The relative energy of the CO and O reaction and the corresponding structure are shown in Fig. 7. The formation of the second CO2 requires a large activation energy, and the minimum energy barrier on MnN2C2-hen is 0.900 eV, indicating that the last remaining O may remain on the active site, which hinders the next cycle of COOR.
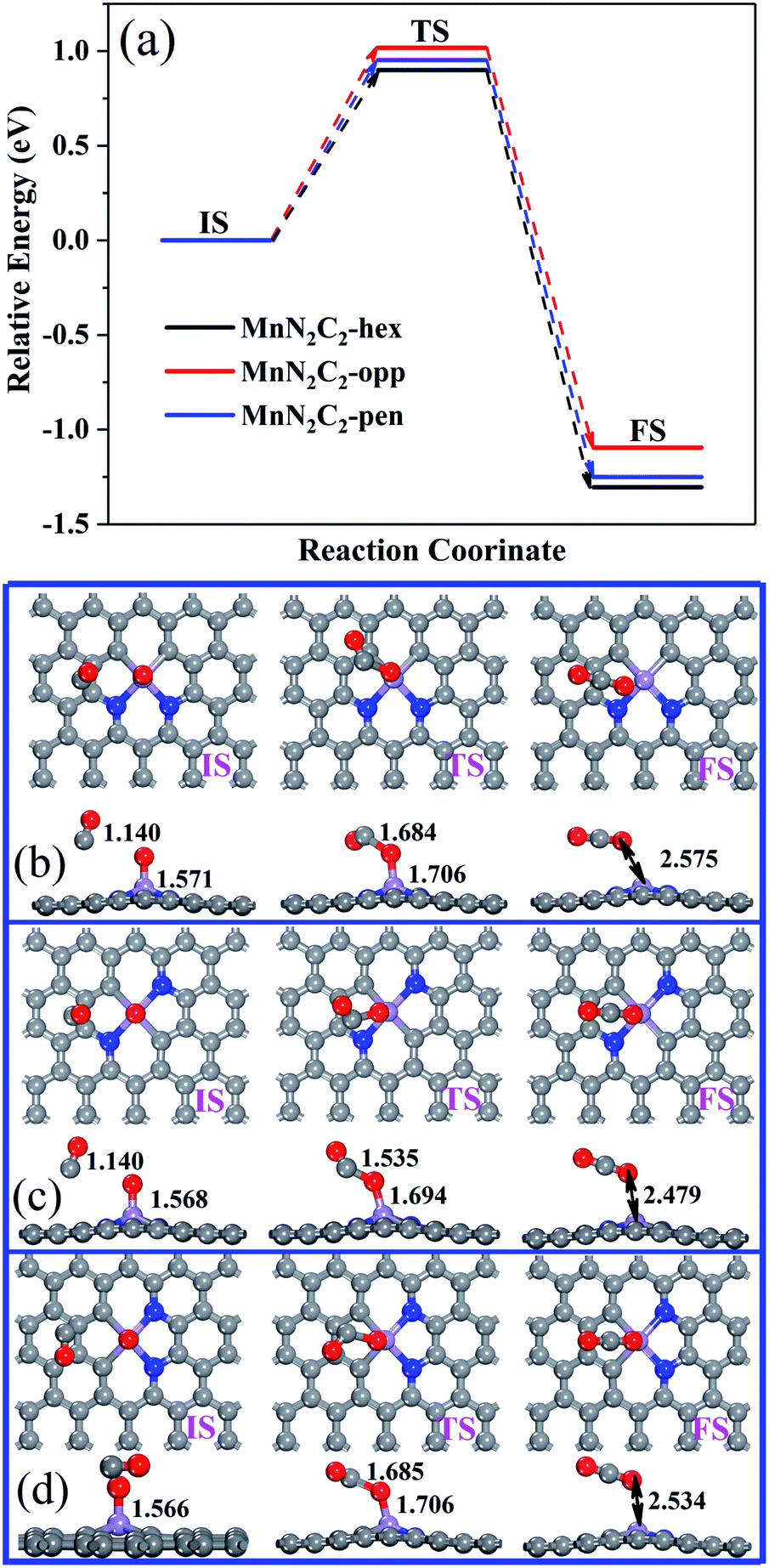 | ||
| Fig. 7 Relative energy (a) and structure of CO and O react to form CO2 on (b) MnN2C2-hex, (c) MnN2C2-opp and (d) MnN2C2-pen. | ||
4. Conclusions
Comprehensive and specific research on the oxidation mechanism of CO catalyzed by MnN2C2 and the oxidation mechanism mainly considers three common mechanisms: ER, LH, TER. The CO oxidation on MnN2C2 prefers to occur via the LH mechanism compared to the ER mechanism. For MnN2C2-hex, the energy barriers of RLS for CO oxidation along the LH and TER mechanisms are 0.850 eV and 0.856 eV, respectively. For MnN2C2-pen, the energy barriers of RLS for CO oxidation along the LH and TER mechanisms are 0.773 eV and 0.900 eV, respectively. MnN2C2-opp has better catalytic performance for CO oxidation, the energy barriers of RLS corresponding to the ER, LH and TER mechanisms are 0.544 eV, 0.747 eV and 0.342 eV, respectively. When CO oxidized on MnN2C2 along the ER and LH mechanism, the remaining O atom is difficult to form a second CO2. In summary, CO oxidation on MnN2C2-opp along the TER mechanism will be the best catalytic pathway. Increasing the proportion of MnN2C2-opp by controlling specific reaction conditions in the catalyst design process may help improve the catalyst's activity in CO oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 51871114), the Research Foundation of the Education Department of Jiangxi Province of China (grant no. GJJ180433 and GJJ180477), the Open Funds of the State Key Laboratory of Metastable Materials Science and Technology, Yanshan University (grant no. 201906), Ganzhou Science and Technology Project (grant no. 201960) and Jiangxi University of Science and Technology Scientific Research Starting Foundation (grant no. jxxjbs17053). High-performance computational resources provided by the National Supercomputer Center on TianHe-2 in LvLiang Cloud Computing Center are also gratefully acknowledged.References
- J. D. Zibrak and A. Ernst, N. Engl. J. Med., 1998, 339, 1603–1608 CrossRef PubMed.
- C. L. Townsend and R. L. Maynard, Occup. Environ. Med., 2002, 59, 708–711 CrossRef CAS.
- H. J. Freund, G. Meijer, M. Scheffler, R. Schlçgl and M. Wolf, Angew. Chem., 2011, 50, 10064–10094 CrossRef CAS.
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650–3653 CrossRef CAS.
- B. Kalita and R. C. Deka, J. Am. Chem. Soc., 2009, 131, 13252–13254 CrossRef CAS.
- H. L. Chen, C. H. Su and H. T. Chen, Chem. Phys. Lett., 2012, 536, 100–103 CrossRef CAS.
- Z. W. Chen, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2017, 121, 3463–3468 CrossRef CAS.
- S. Rashkeev, A. Lupini, S. Overbury, S. Pennycook and S. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 035438 CrossRef.
- C. M. Chang, C. Cheng and C. M. Wei, J. Chem. Phys., 2008, 128, 124710 CrossRef CAS.
- H. T. Chen, J. G. Chang, S. P. Ju and H. L. Chen, J. Comput. Chem., 2010, 31, 256–265 Search PubMed.
- Y. Guo, R. Lang and B. Qiao, Catalysts, 2019, 9, 135 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Crigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- N. Peres, F. Guinea and A. Castro Neto, Ann. Phys., 2006, 321, 1559–1567 CAS.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS.
- H. Wang, Q. Wang, Y. Cheng, K. Li, Y. Yao, Q. Zhang, C. Dong, P. Wang, U. Schwingenschlögl, W. Yang and X. X. Zhang, Nano Lett., 2012, 12, 141–144 CrossRef CAS.
- X. Cui, J. Xiao, Y. Wu, P. Du, R. Si, H. Yang, T. Huanfang, J. Li, W.-H. Zhang, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2016, 55, 6708–6712 CrossRef CAS.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CrossRef CAS.
- Y. Tang, D. Ma, W. Chen and X. Dai, Sens. Actuators, B, 2015, 211, 227–234 CrossRef CAS.
- X. Y. Xu, J. Li, H. Xu, X. Xu and C. Zhao, New J. Chem., 2016, 40, 9361–9369 RSC.
- M. Saito, K. Yamashita and T. Oda, Jpn. J. Appl. Phys., 2007, 46, L1185–L1187 CrossRef CAS.
- F. Jaouen, S. Marcotte, J.-P. Dodelet and G. Lindbergh, J. Phys. Chem. B, 2003, 107, 1376–1386 CrossRef CAS.
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- W. Orellana, Chem. Phys. Lett., 2012, 541, 81–84 CrossRef CAS.
- M. Luo, Z. Liang, M. Chen, S. G. Peera, C. Liu, H. Yang, X. Qi, J. Liu and T. Liang, New J. Chem., 2020, 9402–9410 RSC.
- Z. Lu, M. Yang, D. Ma, P. Lv, S. Li and Z. Yang, Appl. Surf. Sci., 2017, 426, 1232–1240 CrossRef CAS.
- Q. Jiang, J. Zhang, Z. Ao, H. Huang, H. He and Y. Wu, Front. Chem., 2018, 6, 187 CrossRef.
- M. Luo, Z. Liang, S. Gouse Peera, M. Chen, C. Liu, H. Yang, J. Liu, U. Pramod Kumar and T. Liang, Appl. Surf. Sci., 2020, 525, 146480 CrossRef CAS.
- J. Kang, H. Wang, S. Ji, J. Key and R. Wang, J. Power Sources, 2014, 251, 363–369 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 77, 3865–3868 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- W. C. Setyawan and S. Curtarolo, Comput. Mater. Sci., 2010, 49, 299–312 CrossRef.
- F. G. Célia, H. Jan-Willem, B. Evert Jan and B. F. Matthias, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- G. Zhu, F. Liu, Y. Wang, Z. Wei and W. Wang, Phys. Chem. Chem. Phys., 2019, 21, 12826–12836 RSC.
- Y. Yang, K. Li, Y. Meng, Y. Wang and Z. Wu, New J. Chem., 2018, 42, 6873–6879 RSC.
- L. Gao, F. Wang, M. Yu, F. Wei, J. Qi, S. Lin and D. Xie, J. Mater. Chem. A, 2019, 7, 19838–19845 RSC.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- F. Li, X. Liu and Z. Chen, Small Methods, 2019, 1800480 CrossRef.
- L. Xu, L. M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed.
- K. Mao, L. Li, W. Zhang, Y. Pei, X. C. Zeng, X. Wu and J. Yang, Sci. Rep., 2014, 4, 5441 Search PubMed.
- H. Huber, D. McIntosh and G. A. Ozin, Inorg. Chem., 1977, 16, 975–979 CrossRef CAS.
- A. Bongiorno and U. Landman, Phys. Rev. Lett., 2005, 95, 106102 CrossRef.
- Y. Lu, M. Zhou, C. Zhang and Y. Feng, J. Phys. Chem. C, 2009, 113, 20156–20160 CrossRef CAS.
- J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398–10406 CrossRef CAS PubMed.
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566–16572 RSC.
- G. Xu, W. Ran, Y. Feng, D. Ma, Z. Yang and Z. Lu, Carbon, 2017, 118, 35–42 CrossRef CAS.
- G. Xu, R. Wang, Y. Ding, Z. Lu, D. Ma and Z. Yang, J. Phys. Chem. C, 2018, 122, 23481–23492 CrossRef CAS.
- Q. G. Jiang, Z. M. Ao, S. Li and Z. Wen, RSC Adv., 2014, 4, 20290–20296 RSC.
- Q. G. Jiang, J. Zhang, H. Huang, Y. Wu and Z. M. Ao, J. Mater. Chem. A, 2020, 8, 287–295 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05287f |
| This journal is © The Royal Society of Chemistry 2020 |