
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of quinolactacin-H from marine-derived Penicillium sp. ENP701 and biological activities†
Jin-xin Zhu,
Yaojia Lu,
Jun Chen,
Jianwei Chen,
Huawei Zhang ,
Xiaoze Bao,
Xinyi Ye* and
Hong Wang*
,
Xiaoze Bao,
Xinyi Ye* and
Hong Wang*
College of Pharmaceutical Science & Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, 310014, Hangzhou, China. E-mail: hongw@zjut.edu.cn; xinyiye1020@zjut.edu.cn
First published on 25th June 2020
Abstract
The quinolactacins are a family of pyrroloquinoline-type natural products from Penicillium sp. From the organic extract of Penicillium sp. ENP701 fermentation broth, a microorganism from the east China sea, one new quinolactacin was isolated and named quinolactacin-H. The structure of quinolactacin-H was determined by spectroscopic analysis and the absolute configurations by X-ray crystallographic analysis. Enantioselective total synthesis of (R)-(+)-quinolactacin-H and (S)-(−)-quinolactacin-H was achieved. When assayed through crystal violet (CV) microtiter plate biofilm, both (R) and (S)-quinolactacin-H showed a strong inhibition and dispersion of Pseudomonas aeruginosa PAO1 biofilms. Thus, quinolactacins could be proposed and developed as natural anti-bioflm agents in order to solve the problem of microbial resistance in future.
Quinolones have a wide range of biological activities like anticancer, antibacterial,1 antimalarial,2 antifungal3 and anti-HIV activities (Fig. 1a).4 Nalidixic acid is the first generation of 4-quinolone antibiotics, which was discovered as a byproduct during the synthesis of chloroquine.5 Therein, 4-quinolone antibiotics have become the second dominant drugs used in anti-infective chemotherapy (Fig. 1b).6 Natural sources of quinolones have adequate quinolone motifs that act as structural subunits of a large number of complexes in natural products,1b but the only problem is the high cost of obtaining quinolones from nature. Rahme and co-workers reported 3,4-dihydroxy-2-heptylquinoline (PQS) as a signal molecule for Pseudomonas aeruginosa to coordinate their population behaviour (Fig. 1c).7
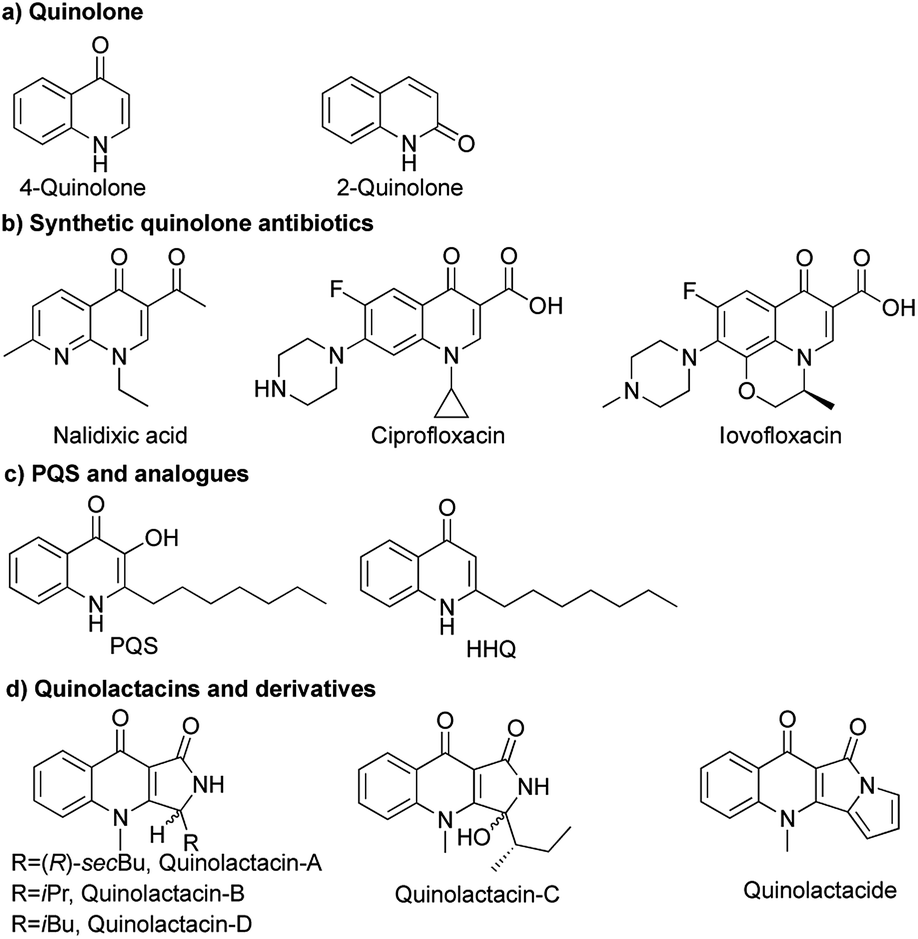 | ||
| Fig. 1 Structures of quinolones and derivative. | ||
Quinolactacins are the first reported pyrroloquinoline-type natural compounds and are considered to be novel quinolone antibiotics. The first example of quinolactacins was reported by Norihiro Kakinuma and co-workers in 2000, which was isolated from the cultured broth of an unidentified Penicillium sp. EPF-6. Quinolactacin A has inhibitory activity against tumor necrosis factor (TNF) production induced by murine peritoneal macrophages and macrophage-like J774.1 cell stimulated with lipopolysaccharide. At that moment, the stereochemistry of quinolactacin-A, -B and -C were unknow.8 In 2001, Kin and co-workers discovered (S)-quinolactacin-A and (R)-quinolactacin-A as a diastereomers from solid-state fermentation of Penicillium citrinum 90648.9 (S)-Quinolactacin-D and (R)-quinolactacin-D were isolated from a solid phase fermentation of an Australian P. citrinum strain in 2006.10 Abe and co-workers discovered a new compound named quinolactacide from P. citrinum F 1539 which showed 88% mortality against green peach aphids at 250 ppm (Fig. 1d).11
The particularity of the marine ecological environment has created vast biodiversity and unique chemical diversity, hence the formation of the secondary metabolites of miscellaneous, unique structure and its variations, and the activity of the high efficiency, caused the extensive attention of pharmacologist, chemists and biologists. Ocean source of raw materials has become an important source of new drug discovery. There has been a sharp upward swing in the number of new natural products reported from marine microorganisms. The number of reported new marine natural products has steadily grown from 332 in 1984 to 1340 in 2015.12 57% of brand new marine natural products reported in 2017 were from marine microorganisms.13
During screening for new bioactive compounds from marine microorganism, we successfully discovered a new quinolactacin named quinolactacin-H from the cultured of Penicillium sp. ENP701, which was originated from the water sample from the East China Sea. So far, all the quinolactacins were isolated from Penicillium sp. However, it is interesting that the new quinolactacin-H comes from a marine source of Penicillium for the first time. Different from other quinolactacins, the structure of the new compound consists of a molecular (R)-quinolactacin-H and a molecular (S)-quinolactacin-H coordinating to a central metal of magnesium ion. This special structure may be formed under the condition of microbial source. The absolute configuration of quinolactacin-H was determined by X-ray crystallographic analysis (Fig. 2). The structure kindled our interest in magnesium complex as research target for its total synthesis.
 | ||
| Fig. 2 Structures of the (S)-quinolactacin-H and (R)-quinolactacin-H. | ||
In 2003, Zhang and co-workers reported an enantioselective total synthesis of (S)-quinolactacins-A, (R)-quinolactacins-A and (R)-quinolactacins-B.14 The most important synthetic procedure is asymmetric Pictet–Spengler reaction.15 The β-carboline structures achieved by Pictet–Spengler reaction and quinolone skeleton could be assembled by Winterfeldt oxidation.16 The diastereoselective syntheses of chiral β-carboline by employing N,N-phthaloyl-protected tert-leucine chlorides as chiral auxiliaries.17 On this basis, we designed asymmetric total synthesis routes for (R)-quinolactacin-H (Scheme 1).
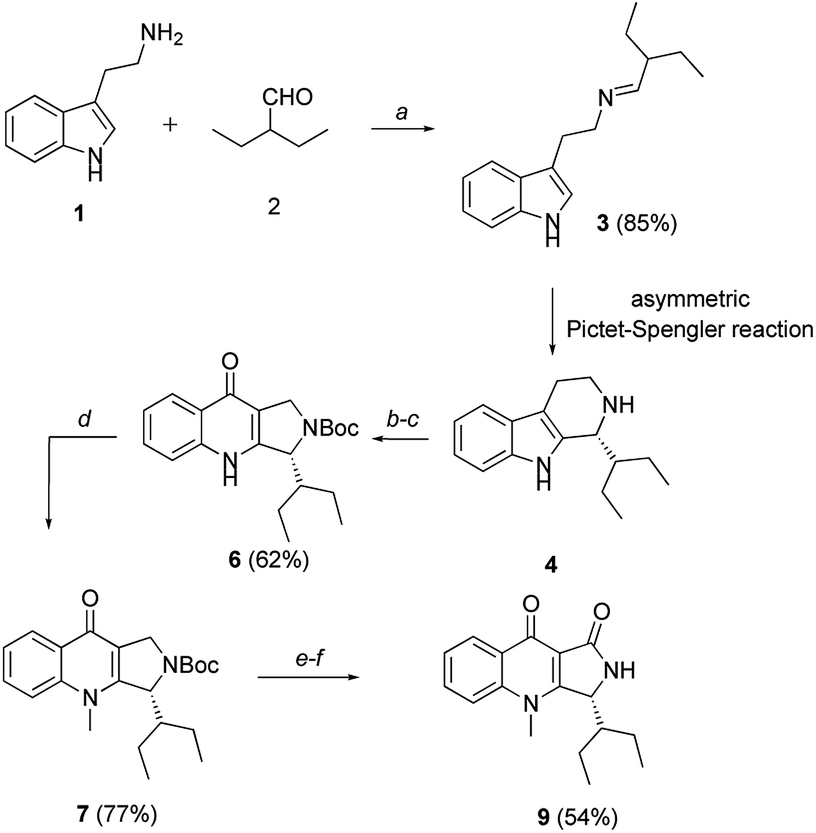 | ||
| Scheme 1 Total synthesis of (R)-quinolactacin-H. Reagents and conditions: (a) 1 (50 mmol), 2 (1.2 equiv.), DCM (200 mL), MgSO4 (10 g), r.t.; (b) 4 (4.1 mmol), Boc2O (1.2 equiv.), TEA (1.5 equiv.), DMF (15 mL), r.t.; (c) KO2 (4.0 equiv.), 18-Crown-6 (1.0 equiv.), DMF (15 mL), r.t., 62% over two steps; (d) 6 (1.4 mmol), MeI (2.0 equiv.), K2CO3 (2.0 equiv.), DMF (8 mL), r.t.; (e) 7 (0.5 mmol), CuBr (0.2 mmol, 40 mol%), t-BuOOH (2.0 equiv, 5.5 M in decane), PhMe (5 mL), 50 °C to r.t.; (f) TFA (4.0 equiv.), DCM (4 mL), r.t., 54% over two steps. | ||
Based on the same synthetic route, we synthesized the marine natural products (R)-quinolactacin-H by applying the asymmetric Pictet–Spengler reaction to get chiral β-carboline intermediates. N-protected β-carboline 12b was obtained from tryptamine-derived imine 10 by Waldmann's asymmetric Pictet–Spengler reaction using N,N-phthaloyl-protected t-leucine chlorides as chiral auxiliary (Scheme 2).15 Diastereomers of carboline 12a and 12b were fully separated by silica gel column chromatography and crystallization in 80% yield in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The chiral auxiliary on carboline 12b was removed by LiAlH4 in dry THF, obtaining tetrahydro-β-carboline in 36% yield. We also investigated other methodology to get (S)-quinolactacin-H in high yield and high enantioselectivity. We screened a series of organocatalysts, which were successfully applied in asymmetric Pictet–Spengler reaction18 previously. Hence, we were attracted by asymmetric Pictet–Spengler reaction catalysed by chiral thiourea catalyst. Jacobsen et al. developed a highly enantioselective catalytic Pictet–Spengler reaction to synthesize a range of N-acetyl β-carbolines in high enantioselectivity.19 Asymmetric acetyl-Pictet–Spengler cyclization of imine 10 was demonstrated by using chiral thiourea catalyst A in the presence of AcCl and 2,6-lutidine in diisopropyl ether (DIPE) at −30 °C to afford the N-acetyl tetrahydro-β-carboline 10 in 63% yield and 99.5% enantiomeric excess (ee) (Scheme 2). Tetrahydro-β-carboline could be obtained by reductive deacetylation of N-acetyl tetrahydro-β-carboline (S)-4 with LiH2NBH3 in THF in 46% yield. We then total synthesized (S)-quinolactacin-H via the same strategy.
:1 ratio. The chiral auxiliary on carboline 12b was removed by LiAlH4 in dry THF, obtaining tetrahydro-β-carboline in 36% yield. We also investigated other methodology to get (S)-quinolactacin-H in high yield and high enantioselectivity. We screened a series of organocatalysts, which were successfully applied in asymmetric Pictet–Spengler reaction18 previously. Hence, we were attracted by asymmetric Pictet–Spengler reaction catalysed by chiral thiourea catalyst. Jacobsen et al. developed a highly enantioselective catalytic Pictet–Spengler reaction to synthesize a range of N-acetyl β-carbolines in high enantioselectivity.19 Asymmetric acetyl-Pictet–Spengler cyclization of imine 10 was demonstrated by using chiral thiourea catalyst A in the presence of AcCl and 2,6-lutidine in diisopropyl ether (DIPE) at −30 °C to afford the N-acetyl tetrahydro-β-carboline 10 in 63% yield and 99.5% enantiomeric excess (ee) (Scheme 2). Tetrahydro-β-carboline could be obtained by reductive deacetylation of N-acetyl tetrahydro-β-carboline (S)-4 with LiH2NBH3 in THF in 46% yield. We then total synthesized (S)-quinolactacin-H via the same strategy.
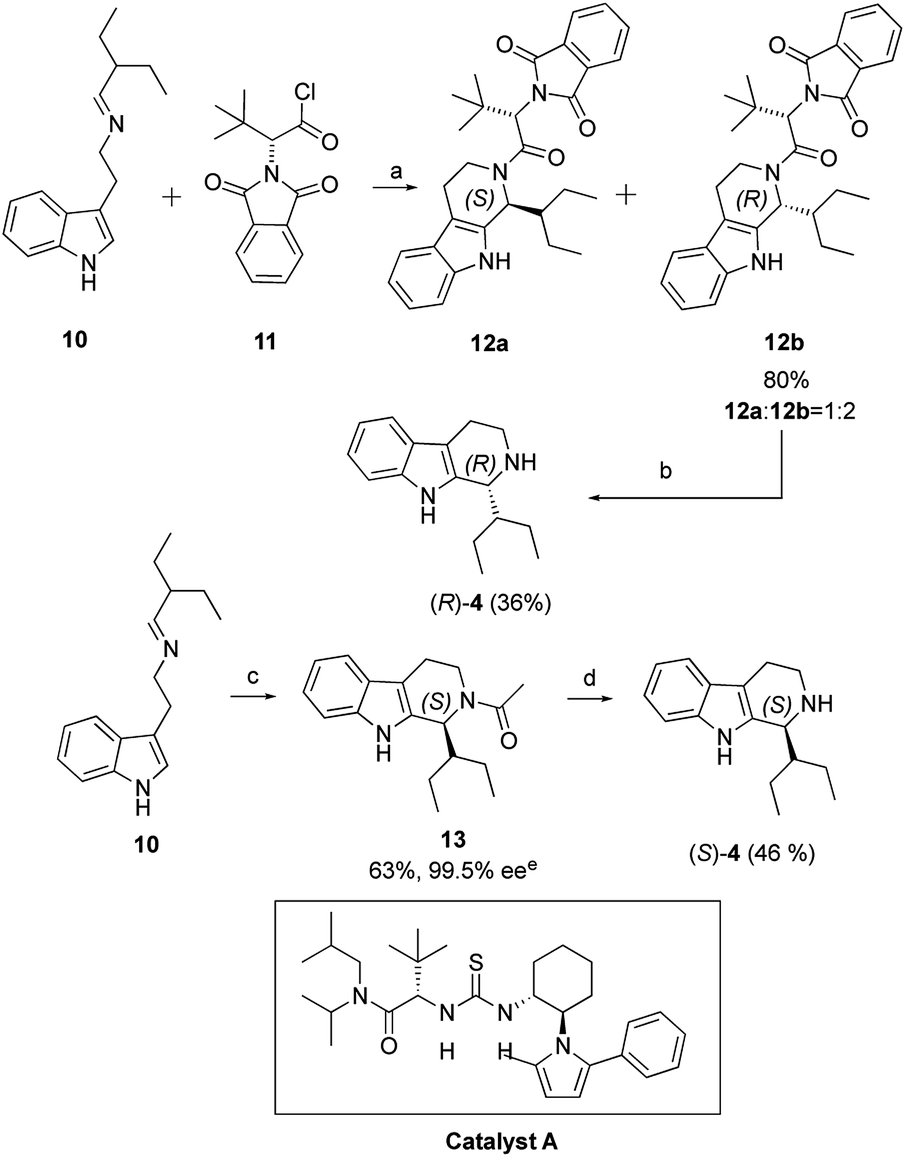 | ||
| Scheme 2 Asymmetric Pictet–Spengler reaction. Reagents and conditions: (a) 10 (24.8 mmol), 11 (1.2 equiv.), Ti(O-n-Pr)4 (1.2 equiv.), dry DCM (200 mL), r.t., (b) 12b (2.06 mmol), H4AlLi (LAH) (2.0 equiv.), Dry THF (20 mL), reflux, (c) 10 (2.0 mmol), Catalyst A (0.1 mmol, 5 mol%), AcCl (1.05 equiv.), 2,6-lutidine (1.05 equiv.), DIPE (40 mL), −80 °C to −30 °C, (d) LiH2NBH3, Dry THF. (e) The ee was determined by HPLC analysis on the Chiralcel OD-H column. | ||
With the enantiopure (S)-quinolactacin-H and (R)-quinolactacin-H in hand, we turned towards determination of their in vitro antimicrobial activity. Unfortunately, both enantiomers of quinolactacin-H showed weak antimicrobial activity. However, in the meantime, we accidentally found that biofilm growth of microorganisms was inhibited. Biofilms are extremely difficult to be cleared because they are encased in a protective, impermeable extracellular matrix that makes biofilm-related bacteria resistant to both the antibiotic and host's immune response.20 In fact, biofilm clearance typically requires about 1000-fold more conventional antibiotics than planktonic bacteria.21 Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that poses a threat to immunocompromised patients, and is a cause of death in patients with cystic fibrosis, AIDS and cancer.22 It is also one of the most studied models of biofilm formation.
Herein we tested the ability of (S)-quinolactacin-H and (R)-quinolactacin-H to inhibit biofilm growth in a wild-type P. aeruginosa strain (PAO1) using standard static biofilm growth assays. Biofilms were grown in a Luria-Bertani (LB) media in 96-well microtiter plates, and quantified biofilm growth after 24 hours by using crystal violet staining of the surface-associated biomass.23 Dose–response curves of crystal-violet-stained biofilms in the presence of (S)-quinolactacin-H and (R)-quinolactacin-H are shown in Fig. 3, benzimidazole used as positive control.24 Follow-up dose–response experiments revealed that (S)-quinolactacin-H had half-maximal inhibitory concentration (IC50) values of 16.7 μM against P. aeruginosa PAO1. Moreover, (R)-quinolactacin-H also showed strong inhibitory activity against P. aeruginosa biofilm growth (IC50 = 24.5 μM).
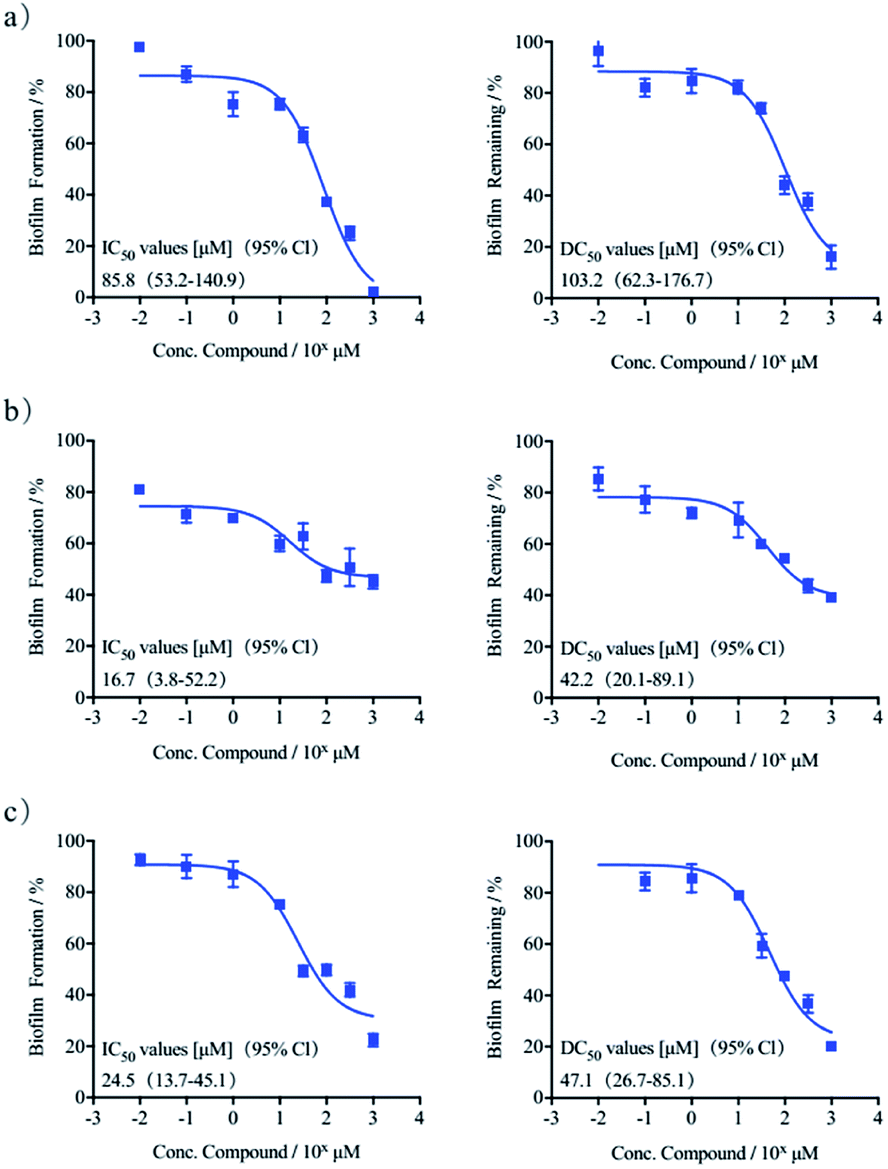 | ||
| Fig. 3 Dose–response curves of crystal violet biofilm inhibitory and dispersion in P. aeruginosa: (a) benzimidazole, (b) (S)-quinolactacin-H, (c) (R)-quinolactacin-H. | ||
The new natural products quinolactacins are capable of not only inhibiting bacteria biofilm growth, but also dispersing preformed biofilms. We used the crystal violet staining assay to test the ability of (S)-quinolactacin-H and (R)-quinolactacin-H to disperse 24 hour-old P. aeruginosa biofilms. The results showed that (S)-quinolactacin-H and (R)-quinolactacin-H strongly disperse P. aeruginosa biofilms, with half-maximal dispersion (DC50) values of 42.2 and 47.1 μM, respectively (Fig. 3).
Conclusions
In conclusion, we discovered the new natural product named quinolactacin-H, isolated from a Penicillium which came from the east China sea. The absolute configuration was determined by X-single crystal diffraction. Total synthesis (S)-quinolactacin-H and (R)-quinolactacin-H by asymmetric Pictet–Spengler reaction as one of the most important steps in the whole route. The preliminary biological results obtained for this series of quinolactacin-H suggests that quinolactacins as potent antibiofilm agents in P. aeruginosa. Both (S)-quinolactacin-H and (R)-quinolactacin-H show strong activity in inhibiting the growth of biofilms and dispersing preformed biofilms. This is the first report that quinolactacins strongly inhibit and disperse bacterial biofilm. Ongoing work in our group is directed towards the study of other quinolactacins and derivatives, as well as assessing their biofilm inhibitory activities across an expanded set of bacterial species and investigate the mechanism by which the quinolactacins elicits its biofilm inhibitory activity in P. aeruginosa.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (No. 2018YFC0311002) and National Natural Science Foundation of China (No. 81773628).Notes and references
- (a) G. F. Zhang, S. Zhang, B. Pan, X. Liu and L. S. Feng, Eur. J. Med. Chem., 2018, 144, 710 CrossRef PubMed; (b) S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415 CrossRef CAS PubMed; (c) E. Fernández-Álvaro, W. D. Hong, G. L. Nixon, P. M. O'Neill and F. Calderón, J. Med. Chem., 2016, 59, 5587 CrossRef PubMed.
- (a) Z. Xu, S. J. Zhao, Z. S. Lv, F. Gao, Y. Wang, F. Zhang, L. Bai and J. L. Deng, Eur. J. Med. Chem., 2019, 162, 396 CrossRef CAS PubMed; (b) Y. Q. Hu, C. Gao, S. Zhang, L. Xu, Z. Xu, L. S. Feng, X. Wu and F. Zhao, Eur. J. Med. Chem., 2017, 139, 22 CrossRef CAS; (c) Y. L. Fan, X. W. Cheng, J. B. Wu, M. Liu, F. Z. Zhang, Z. Xu and L. S. Feng, Eur. J. Med. Chem., 2018, 146, 1 CrossRef CAS.
- F. Gao, P. Wang, H. Yang, Q. Miao, L. Ma and G. Lu, Eur. J. Med. Chem., 2018, 157, 1223 CrossRef CAS PubMed.
- (a) K. C. Sekgota, S. Majumder, M. Isaacs, D. Mnkandhla, H. C. Hoppe, S. D. Khanye, F. H. Kriel, J. Coates and P. T. Kaye, Bioorg. Chem., 2017, 75, 310 CrossRef CAS PubMed; (b) Z. G. Luo, J. J. Tan, Y. Zeng, C. X. Wang and L. M. Hu, Mini-Rev. Med. Chem., 2010, 10, 1046 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423 CrossRef CAS PubMed.
- H. A. A. Ezelarab, S. H. Abbas, H. A. Hassan and G. E. D. A. Abuo-Rahma, Arch. Pharm., 2018, 351, 1 CrossRef PubMed.
- (a) E. Deziel, F. Lepine, S. Milot and L. G. Rahme, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1339 CrossRef CAS PubMed; (b) S. P. Diggle, S. Matthijs, V. J. Wright, M. P. Fletcher, S. R. Chhabra, I. L. Lamont, X. Kong, R. C. Hider, P. Cornelis, M. Camara and P. Williams, Chem. Biol., 2007, 14, 87 CrossRef CAS PubMed; (c) H. Huse and M. Whiteley, Chem. Rev., 2011, 111, 152 CrossRef CAS.
- (a) N. Kakinuma, H. Iwai and A. Nakagawa, J. Antibiot., 2000, 53, 1247 CrossRef CAS PubMed; (b) S. Takahashi, N. Kakinuma and A. Nakagawa, J. Antibiot., 2000, 53, 1252 CrossRef CAS.
- W. G. Kim, N. K. Song and I. D. Yoo, J. Antibiot., 2010, 33, 831 Search PubMed.
- B. Clark, R. J. Capon and E. Lacey, Org. Biomol. Chem., 2006, 4, 1512 RSC.
- (a) M. Abe, T. Imai, N. Ishii, M. Usui, T. Okuda and T. Oki, Biosci., Biotechnol., Biochem., 2005, 69, 1202 CrossRef CAS PubMed; (b) M. Abe, T. Imai, N. Ishii and M. Usui, Biosci., Biotechnol., Biochem., 2006, 70, 303 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers and M. H. G. Munro, Nat. Prod. Rep., 2015, 32, 116 RSC; J. W. Blunt, B. R. Copp, R. A. Keyzers and M. H. G. Munro, Nat. Prod. Rep., 2016, 33, 382 RSC.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2019, 36, 122 RSC.
- X. Q. Zhang, W. Q. Jiang and Z. H. Sui, J. Org. Chem., 2003, 68, 4523 CrossRef CAS PubMed.
- H. Waldmann, G. Schmidt, H. Henke and M. Burkard, Angew. Chem., Int. Ed., 1955, 34, 2402 CrossRef.
- W. Jiang, X. Zhang and Z. Sui, Org. Lett., 2003, 5, 43 CrossRef CAS PubMed.
- G. Schmidt, H. Waldmann, H. Henke and M. Burkard, Chem. - Eur. J., 1996, 2, 1565 CrossRef.
- (a) L. Qi, H. Hou, F. Ling and W. H. Zhong, Org. Biomol. Chem., 2018, 16, 566 RSC; (b) D. Huang, F. X. Xu, X. F. Lin and Y. G. Wang, Chem. - Eur. J., 2012, 18, 3148 CrossRef CAS PubMed.
- M. S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 10558 CrossRef CAS PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623 CrossRef CAS PubMed.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114 CrossRef CAS PubMed.
- (a) R. C. Boucher, Eur. Respir. J., 2004, 23, 146 CrossRef CAS PubMed; (b) R. B. Moss, Clin. Infect. Dis., 1995, 21, 839 CrossRef CAS PubMed.
- (a) G. D. Christensen and W. A. Simpson, J. Clin. Microbiol., 1985, 22, 996 CrossRef CAS PubMed; (b) G. A. O'Toole and R. Kolter, Methods Enzymol., 1999, 310, 91 Search PubMed.
- R. Frei, A. S. Breitbach and H. E. Blackwell, Angew. Chem., Int. Ed., 2012, 51, 5226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC crystallographic data of quinolactacin-H have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the reference number CCDC 1517245. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05244b |
| This journal is © The Royal Society of Chemistry 2020 |