
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopographical and compositional engineering of core–shell Ni@Pt ORR electro-catalysts†
Gerard M. Leteba ab,
David R. G. Mitchellc,
Pieter B. J. Levecquea,
Eric van Steena and
Candace I. Lang*b
ab,
David R. G. Mitchellc,
Pieter B. J. Levecquea,
Eric van Steena and
Candace I. Lang*b
aCatalysis Institute, Department of Chemical Engineering, University of Cape Town, Cape Town 7700, South Africa. E-mail: gerard.leteba@uct.ac.za
bSchool of Engineering, Macquarie University, Sydney, NSW 2109, Australia
cElectron Microscopy Centre, Innovation Campus, University of Wollongong, Wollongong, NSW 2517, Australia
First published on 7th August 2020
Abstract
Complex faceted geometries and compositional anisotropy in alloy nanoparticles (NPs) can enhance catalytic performance. We report on the preparation of binary PtNi NPs via a co-thermolytic approach in which we optimize the synthesis variables, which results in significantly improved catalytic performance. We used scanning transmission electron microscopy to characterise the range of morphologies produced, which included spherical and concave cuboidal core–shell structures. Electrocatalytic activity was evaluated using a rotating disc electrode (1600 rpm) in 0.1 M HClO4; the electrocatalytic performance of these Ni@Pt NPs showed significant (∼11-fold) improvement compared to a commercial Pt/C catalyst. Extended cycling revealed that electrochemical surface area was retained by cuboidal PtNi NPs post 5000 electrochemical cycles (0.05–1.00 V, vs. SHE). This is attributed to the enclosure of Ni atoms by a thick Pt shell, thus limiting Ni dissolution from the alloy structures. The novel synthetic strategy presented here results in a high yield of Ni@Pt NPs which show excellent electro-catalytic activity and useful durability.
Introduction
Metal nanoparticles (NPs) with novel, well-designed surface functionalities are of particular interest in the field of catalysis,1–8 as NPs with unusual morphologies have the potential to exhibit improved catalytic performance.1,9–12 Platinum (Pt) is the most active catalytic material used in fuel cell applications; however, it exhibits sluggish kinetics towards the oxygen reduction reaction (ORR). Furthermore, it has a very high cost and is a scarce resource, which hinders fuel cell commercialization.1,13,14 From both catalytic and economic perspectives, the incorporation of secondary non-noble metals into the Pt NPs will not only reduce the cost of the catalyst, but can also improve the catalytic activity.1,15–17 In this regard, dilution of Pt with 3d-transition metals such as nickel has been successful.16–19 Disordered PtNi systems are thought to be key for the development of high performance electrocatalysts: surface defects, terraces, steps and curvatures in these NPs can influence catalytic performance by exposing crystal facets with under-coordinated atoms.6,10,16,20–23Common methods to produce metal NPs are wet chemical, vapour deposition, impregnation, solvothermal, pyrolysis, etc.16,21,24–26 Of these, wet-chemical synthetic approaches are widely used to prepare monodisperse alloy NPs because they yield uniform sizes, well-defined morphologies and good dispersability in organic solvents.4,18,19,21,27–29 These structural properties are vital for the utilization of NPs in a wide range of industrial applications.16,18,21,28,30,31 Optimising the surface structure of NPs for electrocatalysis requires an understanding of the effect of each step in the solution-based synthetic process used.21,27,32 Simultaneous reduction of different metal precursors in liquid organic media can result in the evolution of core–shell, hetero-structured, alloyed or segregated NPs due to differences in the standard reduction potentials of the components.30,33 Thermolytic decomposition of organometallic precursors in the presence of organic surfactants (which can serve as solvents, surface coordinating agents and reductants) represents a powerful approach for the manipulation of NP structure. The result can be the synthesis of monodisperse alloy NPs with crystallographically complex surface structures and compositional anisotropy within the particles.4,16,18,19,21,29
Herein, we describe the optimization of several synthesis variables. The outcome of this process is the production of high quality, binary core–shell Ni@Pt NPs from organometallic precursors, reduced by high temperature and by the use of co-surfactants. The resulting NP nanostructures are dominated by asymmetric rough surfaces and display compositional zoning, being predominantly Pt-rich in the outer shell. These electrocatalysts exhibited excellent performance (both in terms of mass- and area-specific activities) towards ORR when compared with a commercial Pt/C catalyst. They also showed good durability even after extended electrochemical cycling (5000 cycles). Catalyst degradation resulted from preferential dissolution of Ni. These NPs have significant potential in fuel cell applications, with considerable scope for further improvements in performance and durability.
Results and discussion
Thermolytic decomposition and characterization of binary PtNi systems
The nature of precursor salts, reduction temperature, reducing agents and surfactants can determine the final surface structural properties of NPs, by changing the nucleation and growth kinetics of the NPs. In particular, the binding affinity (adsorption) of two or more surfactants on growing crystal surfaces within the organic synthetic system can differ, depending on the nature of each surfactant's functional group.28,29,34–36 Here, we explore the effect of systematically varying the surfactant components, Pt precursor salt and reducing agent (reductant), on the formation of the solid NPs. The experimental approach to synthesis is shown in Table 1.| Synthesis | Precursors salts | Surfactants | Solvent | Reductant (s) | Temperature (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|
| H2PtCl6 (mmol) | Ni(Ac)2·4H2O (mmol) | Pt(acac)2 (mmol) | OAm (mL) | ODA (g) | OA (mL) | 1-OD (mL) | Temperature + surfactants or TBAB | ||
| 1 | 0.82 | 0.24 | — | 15 | 4.4 | — | 20 | Temperature + surfactants | 310 |
| 2 | 0.82 | 0.24 | — | 15 | 4.4 | 5 | 20 | Temperature + surfactants | 310 |
| 3 | 0.24 | 0.82 | 15 | 4.4 | — | 20 | Temperature + surfactants | 310 | |
| 4 | 0.24 | 0.82 | 15 | 4.4 | 5 | 20 | Temperature + surfactants | 310 | |
| 5 | 0.82 | 0.24 | — | 15 | 4.4 | — | 20 | TBAB | 310 |
| 6 | 0.82 | 0.24 | — | 15 | 4.4 | 5 | 20 | TBAB | 310 |
| 7 | 0.24 | 0.82 | 15 | 4.4 | — | 20 | TBAB | 310 | |
| 8 | 0.24 | 0.82 | 15 | 4.4 | 5 | 20 | TBAB | 310 | |
All experiments used a reduction temperature of 310 °C in a high-boiling point solvent, 1-octadecene (1-OD, boiling point: 315 °C). We note here the rapid formation of a dark organic growth medium and subsequent flocculation of black precipitates during synthesis (suggesting the growth of dense nanoparticle products). This precluded investigation of NP nucleation and growth as a function of reaction time. The synthesis using platinic acid without additional reductant (syntheses #1 and #2) resulted in core–shell NPs with a high surface area (details of materials synthesis are given in the ESI† Experimental section). The various surfactant mixtures and temperature influenced the co-thermolytic decomposition of precursor salts (Ni(Ac)2·4H2O) and (H2PtCl6 (8 wt% in water (H2O))). The differences in reduction kinetics of metal precursors could be key determinants for the creation of these nanostructures with different core and heteroepitaxial shell configurations. In these experiments, it is therefore reasonable to conclude that the evolution of these complex compositionally-zoned structures proceeded via the formation of Ni near-spherical clusters, followed by preferential Pt deposition on the vertices of preformed Ni surfaces, as illustrated in Scheme 1.
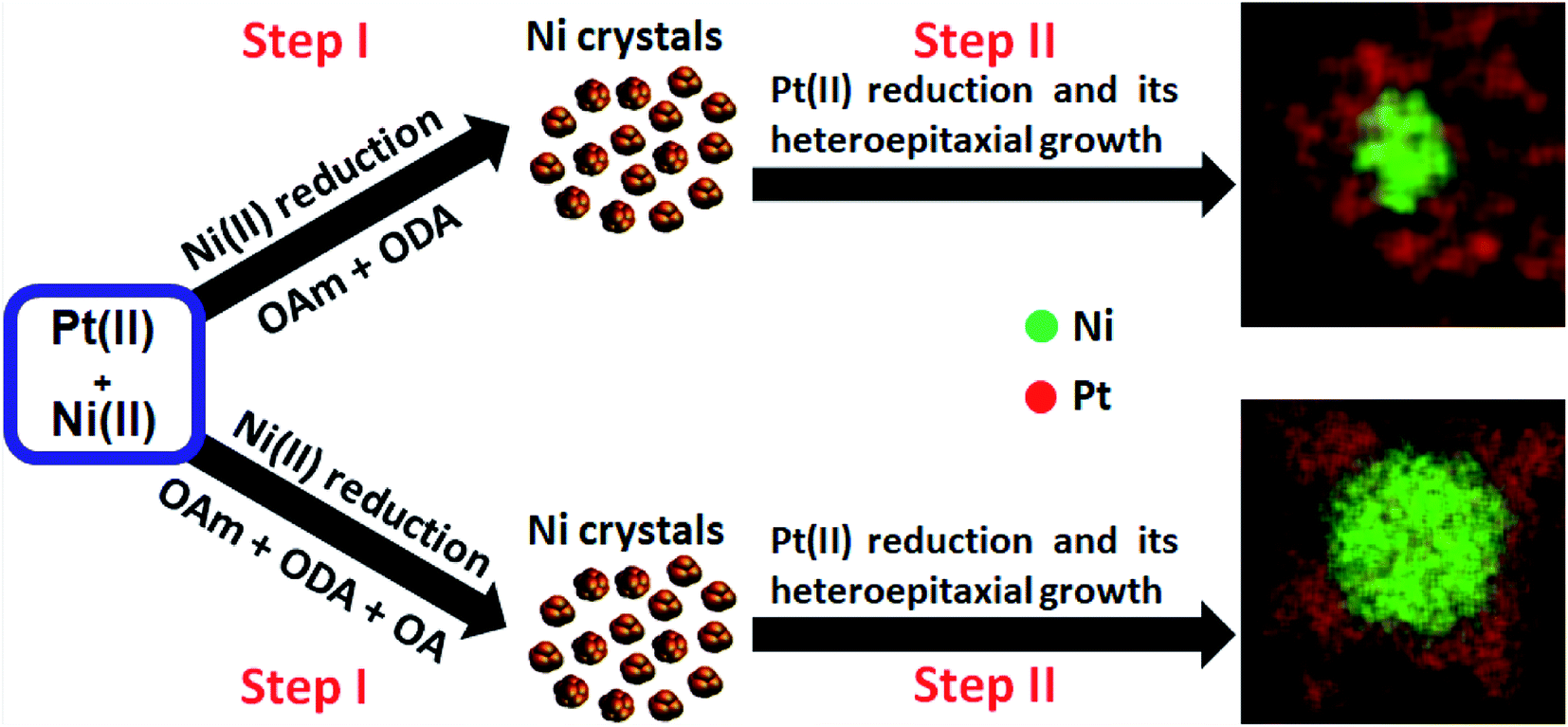 | ||
| Scheme 1 Scheme illustrating topographical evolution of selective Pt epitaxial growth on preformed Ni crystal substrates when using binary and ternary surfactant systems. Figures at the right are STEM-EDS elemental maps of NPs and image diameter = 50 nm. | ||
The sizes and morphologies of the NPs synthesized using binary and ternary surfactants, were analysed by scanning transmission electron microscopy (STEM). Nanostructures prepared using dual surfactants (OAm and ODA, synthesis #1) are referred to hereafter as PtNi(#1) while those prepared in ternary surfactants (OAm, ODA and OA, synthesis #2) are referred to as PtNi(#2). Fig. 1a–g shows PtNi(#1) NPs: a bright-field (BF) STEM micrograph (Fig. 1a), high-angle annular dark-field STEM (HAADF) micrographs (Fig. 1b–d), energy dispersive X-ray spectroscopy (EDS) elemental maps (Fig. 1e and f) and a line scan profile extracted through regions along the white arrow of (d) (Fig. 1g). Fig. 1a (and the corresponding HAADF-STEM image, Fig. S1a†) shows a collection of roughly spherical, branched, dendritic NPs with an average particle diameter of 69.5 ± 5.4 nm, from measurement of 300 NPs (Fig. S2a†). The HAADF images in Fig. 1b–d display bright Pt-rich overlayers associated with a concentration of Pt and its preferential deposition along the vertices of the crystal facets. The atomic resolution HAADF image, Fig. 1c (acquired from the boxed region in Fig. 1b) reveals highly crystalline particles with lattice fringes (marked on Fig. 1c): the measured interplanar spacings (d) and crystal facets are shown. The insert is the corresponding fast Fourier transform (FFT) with rings of spots, indicating that the NPs are individually polycrystalline. The chemical compositions and distribution of PtNi NPs were determined using energy dispersive X-ray spectroscopy (EDS) combined with HAADF imaging. The elemental maps (Fig. 1e and f, S4a†) indicate a core@shell NP morphology comprising a Ni-rich core decorated with Pt. This surface atomic arrangement/distribution on single NPs was further confirmed by cross-sectional line profiles derived from the spectrum images of these NPs (Fig. 1e and f). The corresponding intensity profiles extracted from the spectrum images reveal chemical zoning within single NPs (Fig. 1g). These NPs exhibit a Ni-rich core, suggesting that the final structures are core@shell nanostructures with the majority of the outer layer enriched in Pt due to heteroepitaxial growth. EDS analyses extracted from 300 NPs yielded an average composition (at%) of Ni12Pt88 for these PtNi NPs.
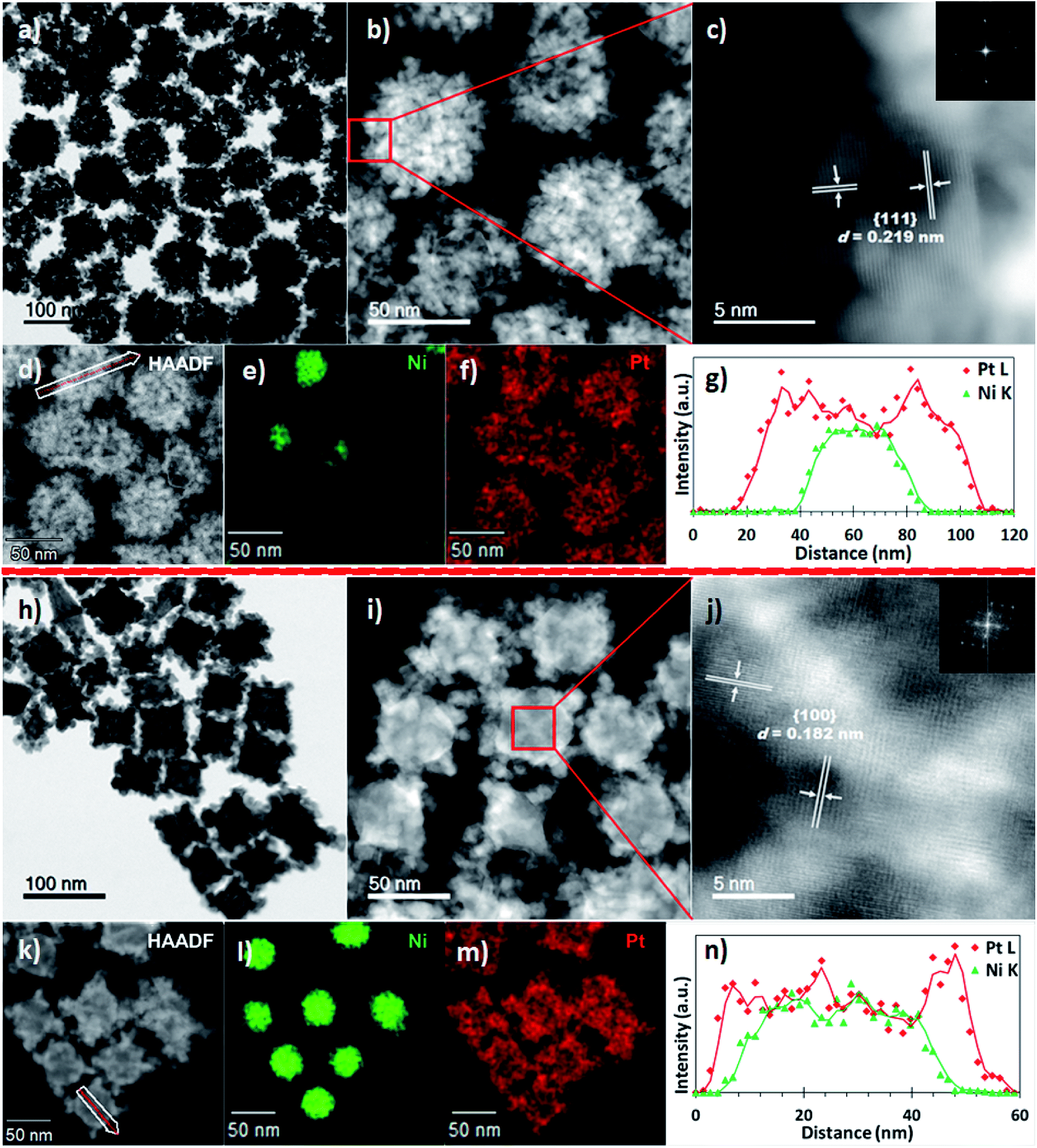 | ||
| Fig. 1 Syntheses #1 and #2: (a) BF-STEM, (b) and (d) HAADF-STEM, (c) atomic resolution detail of the boxed region in (b) (d-spacings indicated with the associated FFT inset), (e) Ni (Kα) map (green), (f) Pt (Lα) map (red) images, and (g) EDS line scan profile along the white arrow in (d) showing PtNi chemical sector zoning. These PtNi(#1) NPs showed EDS-derived average atomic compositions of 12% Ni and 88% Pt. (h) BF-STEM of PtNi(#2), (i and k) HAADF-STEM, (j) atomic resolution detail of the core region boxed in (i) (d-spacings indicated with the associated FFT inset), (l) Ni (Kα) map (green), (m) Pt (Lα) map (red) images, and (n) EDS line scan profiles along the white arrow in (k) showing compositional zoning in PtNi. These NPs showed STEM-EDS average atomic compositions of 21% Ni and 79% Pt. | ||
Fig. 1(h–n) shows BF-STEM micrographs of PtNi(#2) NPs, i.e. those synthesized by adding a third surfactant component (OA). A BF-STEM image is shown in Fig. 1h (the corresponding HAADF-STEM image is shown in Fig. S1b†), with HAADF micrograph (Fig. 1i–k), STEM-EDS elemental distribution (Fig. 1l and m, S4b†) and line scan profile extracted through regions along the line path (white) of (k) (Fig. 1n). PtNi(#2) NPs are seen to have a similar compositional arrangement to PtNi NPs (shown in Fig. 1a–g), of Ni-rich core decorated with Pt–shell. The average particle diameter of PtNi(#2) NPs was 75.4 ± 4.5 nm, from measurement of 300 NPs (Fig. S2b†). Atomic resolution HAADF image (acquired from a boxed region Fig. 1i centre) reveals crystalline particles with lattice fringes (marked on Fig. 1j): the measured interplanar spacings (d) and crystal facets are indicated. The insert is the corresponding FFT, typical of a single particle. The composition (at%) extracted from the STEM-EDS of 300 NPs gave an average of Ni21Pt79 for these PtNi(#2) NPs.
A key difference between the roughly spherical PtNi(#1,S where S = spherical) NPs seen in Fig. 1a and the cuboidal PtNi(#2,C where C = cuboidal) NPs in Fig. 1h, lies in latter's preferential Pt crystal overgrowth along the edges and vertices of the Ni crystal facets. This results in the well-defined concave cuboidal (95%) surface structures exhibited by PtNi(#2,C) NPs. The corresponding secondary electron image (Fig. S3†) of Fig. 1h shows the topographical/compositional contrast present, along the facet apices. Compared to the dendritic multi-layered PtNi(#1,S) NPs, the PtNi(#2,C) NPs exhibit cores that are richer in Ni. In addition, the PtNi(#2,C) NPs showed more compact surface nanostructures than the PtNi(#1,S) NPs, with closely spaced and interconnected Pt nanocrystals forming porous surface structures on PtNi(#2,C) NPs. X-ray diffraction (XRD) was used to characterize the crystal structure of PtNi NPs, as shown in Fig. S5.† Diffraction lines were characteristic of a face-centred cubic (fcc) platinum phase. The slight shift of the peak positions (relative to pure platinum) towards higher angles indicates a reduced lattice parameter (d), due to partial dilution of Pt by Ni resulting in a smaller lattice parameter than metallic Pt. These PtNi(#1,S) NPs showed some splitting of the diffraction peaks, most notably of the prominent (111) diffraction peak (Fig. S5,† green plot). This secondary diffraction peak is at the position of Ni(111) suggesting that Ni-rich cores were present in these PtNi(#1,S) NPs. This split diffraction line may be associated with segregated Ni crystals or incomplete Pt NP coverage/interatomic diffusion with Ni–core crystal facets. In contrast, the XRD pattern for PtNi(#2,C) displayed no extra diffraction peaks. Composition assessments of the PtNi(#1,S) particles from the split {111} diffraction peaks using Vegard's rule suggest Ni7Pt93 and Ni49Pt51 (versus Ni12Pt88) respectively whereas the PtNi(#2,C) NPs contain Ni24Pt76 (versus Ni21Pt79).
The co-thermolytic synthetic approach in synthesis #1 and synthesis #2, was aimed at fast reduction kinetics of different metal precursors (Ni(Ac)2·4H2O and H2PtCl6 (8 wt% in H2O)). This resulted in multi-layered to decorated NPs of core@shell nanostructures with the majority of the outer layer enriched in Pt heteroepitaxial growth. Our experiments indicate that the evolution and growth of alloy nanostructures is rapid at 310 °C, with the resulting binary structures obtained within 20 min of reaction. The formation of a dark-black organic growth medium and subsequent flocculation of black precipitates during synthesis, suggested the reduction of precursor salts and growth of dense nanoparticle products. The use of dual amine (OAm and ODA) surfactants resulted in Ni crystals with random Pt nanocrystal deposition on their surfaces, forming a collection of roughly spherical dendritic multi-layered PtNi(#1,S) NPs, a result of preferential amine adsorption on the {111} crystal facets. The inclusion of OA, as the third surfactant component, appeared to alter the growth kinetics of the expected multi-layered dendritic-like structures: the resultant NPs were a mixture of concave cuboidal (95%) and elongated polyhedral (≤5%) core@shell PtNi(#2,C) NPs, with non-uniform heteroepitaxial Pt crystal growth occurring preferentially along the sharper edges and vertices of the core Ni crystal facets. This is expected as the lattice mismatch accommodates Pt shell enrichment better near edges and corners, due to a lower energy cost. The evolution of both concave cuboidal and elongated polyhedral structures indicate that there was selective binding of OA on the {100} and {111} crystal facets, permitting anisotropic growth along the <111> direction. This bears comparison with a report of pure Pt concave nanocubes, bounded by high-index {720}-facets, which showed improved catalytic performance towards ORR, relative to a commercial Pt/C material.37 Weak selective passivation on the {111} crystal facets is reported to favour the formation of concave high-index {511}-faceted elongated bipyramidal morphologies.29
Syntheses #3 and #4 resulted in NPs with a random atomic arrangement and high surface area. The influence of the Pt precursor salt on NP morphology, was investigated by replacing H2PtCl6 with Pt(acac)2. The remainder of the thermolytic conditions in the NP synthesis were the same as syntheses #1 and #2. In syntheses #3 and #4, the use of either dual surfactants (OAm and ODA) or a ternary mixture of surfactants (OAm, ODA and OA), resulted in the formation of hyperbranched PtNi nanoparticle structures with ∼100% yields (Fig. S6a and b,† first column). The inclusion of OA did not result in any structural changes. Selected area electron diffraction (SAED) patterns acquired from these nanoparticle populations (Fig. S6a and b,† second column), confirm the expected fcc lattice structures. FFTs (last column), inserts in BF-STEM images, reveal arcs of spots characteristic of polycrystalline structures displaying well-resolved lattice fringes (the measured d-spacings are indicated on the figures). The lattice spacing of 0.219 nm (PtNi(#3)) and 0.212 nm (PtNi(#4)) further confirms that both types of binary alloys were encased by {111} crystal facets. The evolution of these facets arises from the deposition of small single crystals on high surface energy facets, from the core outward, which results in the formation of multiply exposed crystal facets. This demonstrates that the NPs exhibit a narrow range of orientations, suggesting that templating has occurred: the alignment of new crystals is guided by growth on pre-existing surfaces in a controlled manner.19 Although low energy {111} crystal facets encase these multiply branched binary alloys, they are high surface area nanostructures. Multiple edges, steps, corners and kinks in addition to densely packed centres dominated the observed symmetrically branched morphologies. These results may suggest that the evolution of these highly branched alloy NPs occurred through kinetic-growth control. The growth mechanism of pure Pt into dendritic structures (using OAm as both surfactant and reductant) has been reported to occur anisotropically along the <111> orientation at low temperatures (≤150 °C), requiring extended reaction time.38 Increasing the dwell temperature to 250 °C resulted in the formation of spherical Pt NPs (7.9 nm).38 However, our findings have suggested that the formation of these large branched NPs could arise from the destabilized binding affinity of the surfactants, triggering rapid particle coarsening (Ostwald ripening) at the elevated reduction temperature (310 °C).19 STEM imaging and mapping show a solid solution with disordered atomic distributions within the individual NPs (Fig. S7a and b†). The corresponding STEM-EDS spectra are shown in Fig. S7c and d.† The estimated atomic compositions were Ni42Pt58 for PtNi(#3) and Ni30Pt70 for PtNi(#4) NPs. The recorded XRD patterns of these alloys reveal fcc solid solution phases (Fig. S8†). These observations suggest that complete shape-selectivity, in uniform-sized alloy NP systems, can be achieved by a suitable choice of Pt precursor complex.
The inclusion of an additional reductant in the form of tetrabutylammonium borohydride (TBAB = C16H36N(BH4); syntheses #5, #6, #7 and #8) resulted in small NPS with a lower surface area. Good control of the nucleation and subsequent growth of solution-derived NPs often requires the use of some specific reducing agents or metal precursors.39,40 The TBAB-assisted co-reduction of Ni(Ac)2·4H2O and H2PtCl6 precursors produced cuboidal particles (Fig. 2a) while simultaneous reduction of organometallic Ni(Ac)2·4H2O and Pt(acac)2 precursors produced predominantly rounded particles (Fig. 2b), in dual (OAm and ODA) surfactants. Similar spherical morphologies were obtained when the same precursors were used with ternary (OAm, ODA and OA) surfactants (Fig. S9a and b†). We thus observed no significant effect of the third surfactant component on the morphological and size evolution. Our results indicate that TBAB-assisted reduction of metal precursors is a versatile synthetic technique for the formation of monodisperse small-sized nanoparticles with a narrow size distribution (Fig. S10a–d†). Unlike the large particles obtained via the thermolytic reduction approach (which precipitated during synthesis and were afterwards resuspended in chloroform), these small-sized alloy NPs remained as stable colloids. Based on these experiments, it is therefore reasonable to conclude that the reduction of precursors proceeds faster in the presence of TBAB and nucleation of NPs thus occurs faster.
 | ||
| Fig. 2 Syntheses #5 and #7: BF-STEM images of (a) PtNi(#5) and (b) PtNi(#7) synthesized in co-surfactants (OAm and ODA), with the inclusion of TBAB as the additional reductant. Inserts are enlarged BF-STEM images. | ||
Electrochemical surface area, ORR activity and durability
NPs synthesized using syntheses #1 and #2, which exhibited a core@shell structure with high surface area, were tested electrochemically. The synthesised NPs were dispersed onto a commercial carbon support using chloroform and sonication (details in Experimental section†). Fig. S11a and b† show STEM micrographs of the carbon-supported binary core@shell PtNi(#1,S) and PtNi(#2,C) NPs, respectively, prior to electrochemical evaluations. The particles were well dispersed on the support and did not agglomerate. The surface electrochemical properties of these nanoalloys were then investigated using a rotating disk electrode (RDE). Cyclic voltammograms for the three catalysts, recorded after 100 electrochemical cycles to activate the surface, are shown in Fig. 3a. These curves exhibit both the hydrogen desorption/adsorption current density peaks (0.05–0.35 V, vs. SHE) and the oxide formation/reduction current density peaks (0.7–1.0 V, vs. SHE).15,41 The magnitude of these peaks (in mA cm−2) scaled as: PtNi(#2,C)/C > PtNi(#1,S)/C ≫ commercial Pt/C electrocatalysts. The prominent oxide formation/reduction current density peaks of both PtNi(#2,C)/C and PtNi(#1,S)/C materials are associated with the presence of oxophilic Ni atoms on the alloys. The ECSAs of these materials (60 wt% NP loading) and the commercial Pt/C electrocatalyst (HiSPEC on carbon, 60 wt% loading) were estimated by integrating the charges from adsorption of the underpotential-deposited hydrogen (ECSAHupd, using 210 μC cm−2)42,43 and carbon monoxide (CO) adsorbed (COads, using 420 μC cm−2).44 The calculated ECSAHupd values, scaled as PtNi(#2,C)/C (78.4 m2 gPt−1) > commercial Pt/C (74.0 m2 gPt−1) ≫ PtNi(#1,S)/C (69.5 m2 gPt−1) and the ECSACO values as PtNi(#2,C)/C (80.3 m2 gPt−1) > commercial Pt/C (84.4 m2 gPt−1) ≫ PtNi(#1,S)/C (67.2 m2 gPt−1) as presented in Table S1.† The ECSACOads/ECSAHupd ratios determined for all the NPs were very similar, approximating unity (Table S2†). Previous reports suggest that ECSACOads/ECSAHupd values close to unity are representative of either pure Pt, Pt-rich surface, thicker Pt–shell or homogeneous Pt–Ni solute–solvent atomic arrangements whereas values around 1.5 represent Pt–skin type surfaces.45,46 The calculated charge ratio values of these binary alloy electrocatalysts are therefore characteristic of thicker Pt-atomic shell overlayers, as confirmed by the line scan profiles extracted from EDS maps (Fig. 1g and n). The CO stripping current peaks of these Pt alloy electrocatalysts displayed a negative potential shift relative to a commercial Pt/C electrocatalyst. This indicates that the adsorbed CO is stripped at lower electrode potentials on the alloy electrocatalysts as a result of lower binding energy or weaker interaction between CO and the Pt surface facilitated by the presence of oxophilic Ni oxide species. The result is improved CO tolerance on the alloy materials. The reduced CO coverage is a consequence of changed electronic properties induced by incorporated Ni in the Pt crystal structure.47,48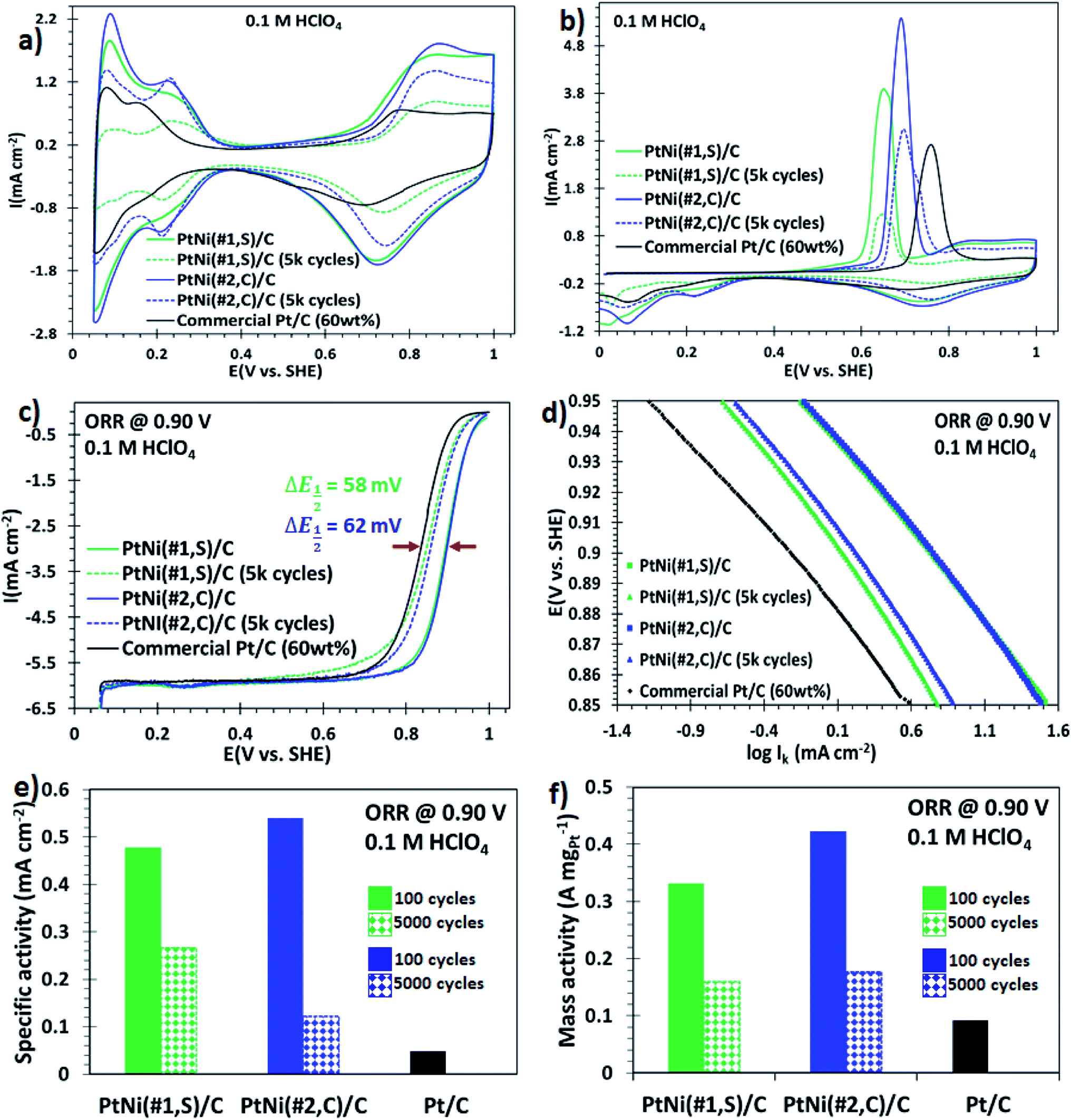 | ||
| Fig. 3 Catalytic performance comparisons per unit Pt surface area between PtNi(#1,S)/C, PtNi(#2,C)/C and commercial Pt/C electrocatalysts. (a) Cyclic voltammograms of PtNi(#1,S)/C (green), PtNi(#2,C)/C (blue) and commercial Pt/C (black) after 100 electrochemical cycles of surface activation and 5000 potential cycles of durability tests. (b) Carbon monoxide (CO) stripping curves of PtNi(#1,S)/C, PtNi(#2,C)/C and commercial Pt/C. (c) ORR polarization curves recorded at 1600 rpm and (d) the corresponding Tafel plots in the potential range of 0.85–0.95 V (vs. SHE). (e) and (f) Are histograms of ORR intrinsic area-specific activities and mass-specific activities at +0.9 V (vs. SHE) after 100 electrochemical cycles of catalyst activation and post 5000 accelerated durability cycles, respectively. No durability measurements were conducted for the commercial Pt/C electrocatalyst. | ||
The ORR catalytic performance of these heterogeneous alloy electrocatalysts was evaluated by acquiring the polarization curves in O2-saturated 0.1 M HClO4 solution, using a rotating disk electrode, at the rotation speed of 1600 rpm. The ORR polarization curves in Fig. 3c display two observable potential regimes: a mixed, kinetic-diffusion-controlled region in the potential window of 0.8–1.0 V (vs. SHE) and a diffusion-limited current density regime (∼0.05–0.8 V, vs. SHE).49 Furthermore, these ORR polarization curves of the electrocatalysts, including the commercial Pt/C, reached their diffusion limited-current at ∼6.0 mA cm−2 (Fig. 3c), consistent with the reported theoretical values (5.8–6.02 mA cm−2).50,51 The half-wave potentials (E1/2) in the ORR polarization plots for PtNi(#1,S)/C (0.89 V, vs. SHE) and PtNi(#2,C)/C (0.90 V, vs. SHE) electrocatalysts show 58 mV and 62 mV positive shifts (indicated in Fig. 3c) respectively, compared to commercial Pt/C (0.84 V, vs. SHE) catalyst. These shifts to higher potentials correspond to improved ORR electrocatalytic activities for the alloy electrocatalysts. The measured currents were corrected for mass transport to acquire the true kinetic currents. The corresponding ORR Tafel plots (Fig. 3d) obtained from the potentials in the range of 0.85–0.95 V (vs. SHE), exhibit catalytic activities which follow this trend: PtNi(#2,C)/C > PtNi(#1,S)/C ≫ commercial Pt/C. The mass-specific and area-specific activities at 0.9 V (vs. SHE) were calculated by normalizing the kinetic currents (Ik) with the ECSAHupd and the Pt catalyst mass immobilized on the glassy carbon, respectively. The kinetic current (Ik) can be calculated using the Koutecky–Levich equation:51 plots of the present work indicate that PtNi(#2,C)/C displayed the most positive onset potential of all the electrocatalysts, suggesting enhanced catalytic performance of these binary core–shell NPs. The area-specific activities and Pt mass-specific activities of the binary NPs were compared with the commercial Pt/C electrocatalysts. The area-specific activities showed that the absolute value of the electrocatalyst intrinsic activity of PtNi(#2,C)/C and PtNi(#1,S)/C exhibited ∼10- and ∼11-fold electrocatalytic improvement, respectively, relative to a commercial Pt/C electrocatalyst (Fig. 3e). In conjunction with the area-specific activity evaluations, the Pt mass activities for the effective Pt utilization at 0.9 V (vs. SHE) of PtNi(#2,C)/C and PtNi(#1,S)/C exhibit ∼4- and ∼5-fold enhanced ORR electrocatalytic activity, respectively, compared with the commercial Pt/C (Fig. 3f). Previous reports have shown that heteroatom or nitrogen-doped carbon (NC) supports display enhanced catalytic activity and durability of supported metal nanoparticles for ORR. The nitrogen atoms interact with surface Pt atoms at a catalyst/support interface, thereby improve the dispersability and catalytic performance of the immobilized nanocatalysts.52–56 In this study, electronic interactions between the support and the metal particles cannot be excluded. However, across all the electrocatalysts the support material was kept the same and is comparable to the support used for the commercial catalyst. It is therefore likely that the differences we observe in catalytic activity are not related to changes in metal–support interaction. We conclude that the complex anisotropic shapes of the particles with protruding surface defects such as branched edges/corners, stepped interfaces, roughness as well as distinct crystal facets of these alloy NPs provide a large area of surface active sites for the ORR.
The cyclic voltammograms (Fig. 3a, dashed graphs) and CO stripping voltammograms (Fig. 3b, dashed plots) after 5000 potential cycles (0.05–1.00 V, vs. SHE, at 100 mV s−1) showed a decrease in ECSA. The prominent decline in the hydrogen desorption/adsorption and the oxide formation/reduction current density peaks with potential cycling is correlated with Ni oxide dissolution and atomic restructuring on the surface of electrocatalysts.57 The ECSAHupd decay after 5000 electrochemical cycles, relative to the initial values, was: 66% (PtNi(#1,S)/C) and 18% (PtNi(#2,C)/C) (Table S1†). The noticeable deterioration in CO current peaks (PtNi(#1,S)/C (67% loss) and PtNi(#2,C)/C (23% loss)) is similar to the ECSAHupd loss. These ECSAHupd losses could arise from any of the following: Ni oxide dissolution; metal alloy oxide formation due to prolonged exposure to an oxidizing environment and morphological transformations. This leads to attenuated CO tolerance of the alloy NPs and the appearance of insignificant double peaks with extended potential cycling (Fig. 3b, dashed plots). The ECSACOads/ECSAHupd ratios were PtNi(#1,S)/C (∼0.92) and PtNi(#2,C)/C (∼0.96) (Table S2†). Our results show that these ECSACOads/ECSAHupd ratios are representative of a high density of surface defects for PtNi(#1,S)/C electrocatalysts whereas the PtNi(#2,C)/C electrocatalyst reflects a thicker and distorted Pt-overlayer shell. These observations are in good agreement with previous reports.58
In addition, these binary alloys showed a significant decrease in the ORR activity, with the polarization curves shifting from higher (positive) to lower (negative) potentials (Fig. 3c). The PtNi(#1,S)/C system shows a negative half-wave potential shift of 39.2 mV whereas for the PtNi(#2,C)/C electrocatalyst this is 36.1 mV, after 5000 cycles. These performance losses may be associated with oxide formation on both Ni and Pt surfaces in addition to atomic restructuring at the alloy surfaces, thereby suppressing the catalytic activity of Pt atoms.57,59 Furthermore, impurities or metal oxides are more likely to interfere with the surface adsorption of molecular O2 species to the working electrode interface, thus limiting ORR performance. In terms of area-specific activity the catalysts presented here still show ∼5 (PtNi(#1,S)/C, 44% loss) and ∼3 (PtNi(#2,C)/C, 77% loss) improvement factors (Fig. 3e), while in terms of mass-specific activity, PtNi(#1,S)/C and PtNi(#2,C)/C still display ∼2 (52% loss) and ∼2 (58% loss) better performance, respectively, compared with pure commercial Pt/C (Fig. 3f and Table S1†). BF-TEM micrographs show no agglomeration of individual NPs before electrochemical measurements (Fig. 4a and b). However, with cycling the nanocrystals making up each nanoparticle densified as shown in Fig. 4c and d (Fig. S12a and b†). The relatively large particle size of the NPs conferred resistance to agglomeration due to restricted mobility on the support material. PtNi(#1,S)/C catalyst shows substantial morphological changes, becoming more porous during cycling when compared with the PtNi(#2,C)/C electrocatalysts. Fig. 4e shows the decay of ECSAHupd of the binary NPs whereas Fig. 4f exhibits the dissolution of Ni, with extended cycling. The PtNi(#1,S)/C system suffered severe Ni pitting corrosion (∼100%) possibly because of its lower density, with closely spaced and interconnected Pt nanocrystals forming porous surface structures (as observed in Fig. 1a–g) whereas the more compact PtNi(#2,C)/C electrocatalyst (Fig. 1h–n) experienced ∼26% Ni leaching, post 5000 electrochemical cycles. The observed acid leaching of oxophilic Ni atoms from the PtNi(#1,S)/C system during extended electrochemical cycling is in line with previous reports.24,29,60 The compositional stabilities of the PtNi(#2,C)/C electrocatalyst could be correlated with Pt shell restructuring, suppressing Ni disintegration and thus leading to more induced lattice compression.61–63 Nonetheless, the core@shell nanomaterials obtained here display enhanced catalytic performance relative to a commercial catalyst, even after 5000 electrochemical cycles. Furthermore, they also showed good durability after accelerated durability cycling. Extended cycling shows some performance degradation due to compositional and microstructural changes.
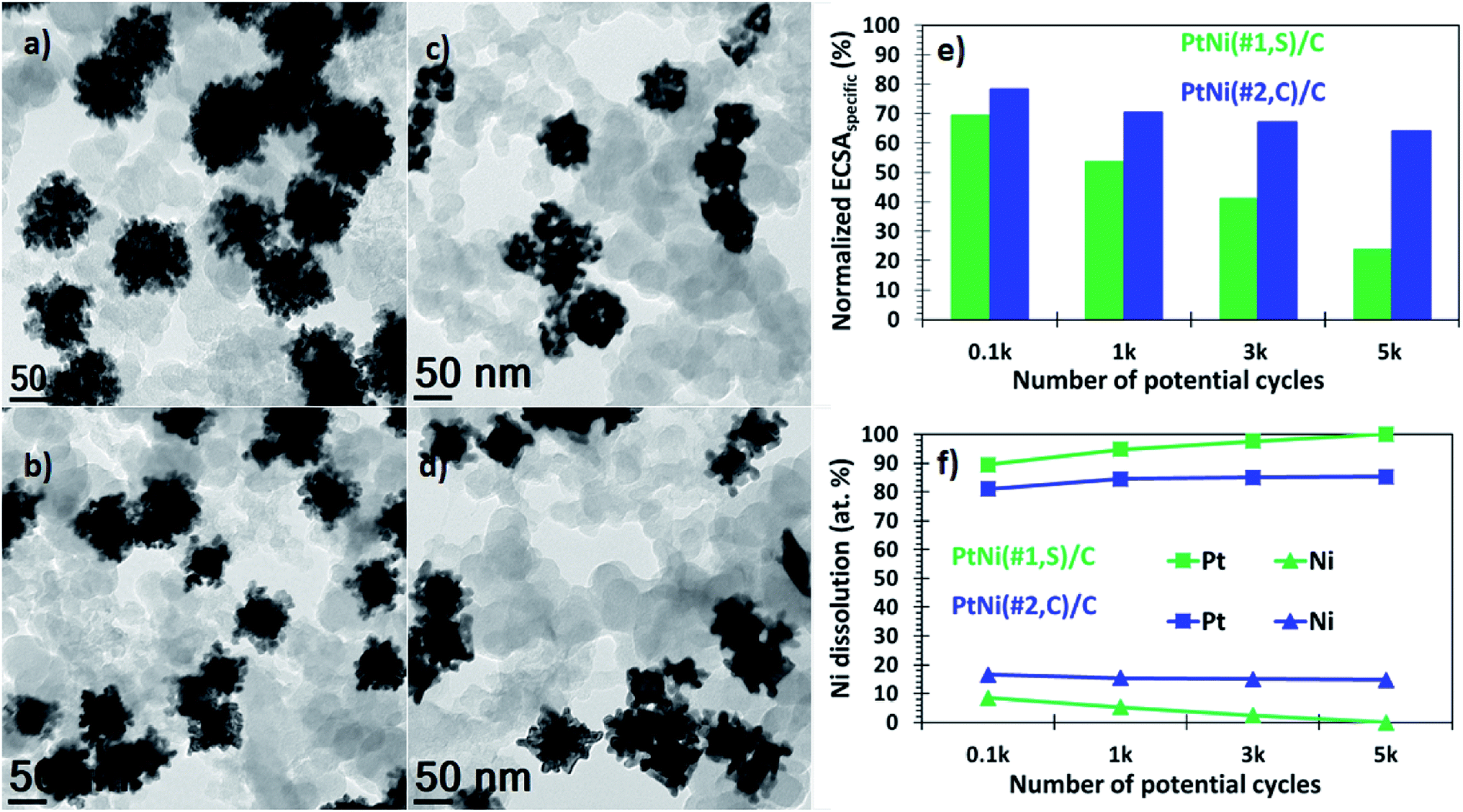 | ||
| Fig. 4 Representative BF-TEM micrographs of as-deposited (a) PtNi(#1,S)/C and (b) PtNi(#2,C)/C nanomaterials. (c) and (d) Are respective BF-TEM images of PtNi(#1,S)/C and PtNi(#2,C)/C electrocatalysts post 5000 extended electrochemical cycling, showing the morphology changes. (e) ECSA deterioration and (f) compositional change as a function of potential cycling (up to 5000 cycles) of PtNi(#1,S)/C (green) and PtNi(#2,C)/C (blue) electrocatalysts. | ||
Conclusions
We report the outcome of an optimization of synthesis variables, for the production of PtNi NPS. A high temperature, rapid thermolytic approach produced topographically- and compositionally varied PtNi NPs. Synthesis methods #1 and #2 resulted in dendritic-like multi-layered NPs and NPs with a Ni–core decorated with Pt nanocrystals, respectively. The significant microstructural variations between the two types of binary nanostructures are associated with the influence of the third surfactant (OA) component used during synthesis, yielding predominantly high-index (cuboidal and elongated polyhedral) core@shell PtNi(#2,C) NPs. These dendritic-like multi-layered and concave decorated (cuboidal and elongated polyhedral) core@shell PtNi electrocatalysts exhibited approximately 11-fold catalytic improvements toward ORR relative to the commercially available Pt/C electrocatalyst, in perchloric acid (0.1 M HClO4) solution and at a rotation speed of 1600 rpm. Post 5000 electrochemical cycles (0.05–1.00 V, vs. SHE, at 100 mV s−1), the alloy electrocatalysts showed activity decay to a value comparable to the initial commercial Pt/C electrocatalyst measurements; it also resulted in Ni/Ni oxide dissolution and particle shape change. The reported novel synthetic strategy for shape-dependent core@shell PtNi(#1 and #2) NPs has the potential for high-yield NP production as high performance ORR electrocatalysts. The focus of further work will be directed at further improvements to performance and durability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Macquarie University and the University of Cape Town. The contribution of the Electron Microscopy Centre, University of Wollongong and the Electron Microscope Unit, University of Cape Town, is gratefully acknowledged. This research used equipment funded by the Australian Research Council (ARC), Linkage, Infrastructure, Equipment and Facilities (LIEF) grant (LE120100104): JEOL JEM-ARM200F, located at the University of Wollongong Electron Microscopy Centre. G. M. L and P. B. J. L acknowledge funding from the Hydrogen South Africa (HySA) Catalysis Centre of Competence, and E. V. acknowledges funding from the South African Research Initiative (NRF grant number 114606).References
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- J. Snyder, K. Livi and J. Erlebacher, Adv. Funct. Mater., 2013, 23, 5494–5501 CrossRef CAS.
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904 CrossRef CAS PubMed.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765 CrossRef CAS.
- C.-J. Zhong, J. Luo, P. N. Njoki, D. Mott, B. Wanjala, R. Loukrakpam, S. Lim, L. Wang, B. Fang and Z. Xu, Energy Environ. Sci., 2008, 1, 454 RSC.
- K. Jiang, Q. Shao, D. Zhao, L. Bu, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27, 1700830 CrossRef.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- J. Lim, H. Shin, M. Kim, H. Lee, K.-S. Lee, Y. Kwon, D. Song, S. Oh, H. Kim and E. Cho, Nano Lett., 2018, 18, 5343 CrossRef CAS PubMed.
- G. Wang, M. A. Van Hove, P. N. Ross and M. I. Baskes, Prog. Surf. Sci., 2005, 79, 28–45 CAS.
- C. Wang, L. Zhang, H. Yang, J. Pan, J. Liu, C. Dotse, Y. Luan, R. Gao, C. Lin, J. Zhang, J. P. Kilcrease, X. Wen, S. Zou and J. Fang, Nano Lett., 2017, 17, 2204–2210 CrossRef CAS PubMed.
- S. Kühl, M. Gocyla, H. Heyen, S. Selve, M. Heggen, R. E. Dunin-Borkowski and P. Strasser, J. Mater. Chem. A, 2019, 7, 1149–1159 RSC.
- W. Gao, Z. D. Hood and M. Chi, Acc. Chem. Res., 2017, 50, 787–795 CrossRef CAS PubMed.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen, J. Rossmeisl and I. Chorkendorff, J. Am. Chem. Soc., 2011, 133, 5485–5491 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547 CrossRef CAS PubMed.
- Y.-C. Wang, T. J. A. Slater, G. M. Leteba, A. M. Roseman, C. P. Race, N. P. Young, A. I. Kirkland, C. I. Lang and S. J. Haigh, Nano Lett., 2019, 19, 732–738 CrossRef CAS.
- G. M. Leteba, D. R. G. Mitchell, P. B. J. Levecque and C. I. Lang, Nanomaterials, 2018, 8, 462 CrossRef.
- L. Bu, J. Ding, S. Guo, X. Zhang, D. Su, X. Zhu, J. Yao, J. Guo, G. Lu and X. Huang, Adv. Mater., 2015, 27, 7204–7212 CrossRef CAS.
- Z. Niu, N. Becknell, Y. Yu, D. Kim, C. Chen, N. Kornienko, G. A. Somorjai and P. Yang, Nat. Mater., 2016, 15, 1188–1194 CrossRef CAS PubMed.
- A. S. Bandarenka, H. A. Hansen, J. Rossmeisl and I. E. L. Stephens, Phys. Chem. Chem. Phys., 2014, 16, 13625–13629 RSC.
- F. Calle-Vallejo, M. D. Pohl, D. Reinisch, D. Loffreda, P. Sautet and A. S. Bandarenka, Chem. Sci., 2017, 8, 2283–2289 RSC.
- R. M. Arán-Ais, F. Dionigi, T. Merzdorf, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, M. Gliech, J. Solla-Gullón, E. Herrero, J. M. Feliu and P. Strasser, Nano Lett., 2015, 15, 7473–7480 CrossRef.
- C. Cui, L. Gan, H.-H. Li, S.-H. Yu, M. Heggen and P. Strasser, Nano Lett., 2012, 12, 5885–5889 CrossRef CAS.
- V. Di Noto, E. Negro, S. Polizzi, F. Agresti and G. A. Giffin, ChemSusChem, 2012, 5, 2451–2459 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef CAS.
- G. M. Leteba, D. R. G. Mitchell, P. B. J. Levecque, L. Macheli, E. van Steen and C. I. Lang, ACS Appl. Nano Mater, 2020, 3, 5718–5731 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- I. McCue, E. Benn, B. Gaskey and J. Erlebacher, Annu. Rev. Mater. Res., 2016, 46, 263–286 CrossRef CAS.
- S. Shan, J. Luo, L. Yang and C.-J. Zhong, Catal. Sci. Technol., 2014, 4, 3570–3588 RSC.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, Z. Zhang, W. Niu, C. Li, N. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409–6455 CrossRef CAS PubMed.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- H.-T. Zhang, J. Ding and G.-M. Chow, Langmuir, 2008, 24, 375–378 CrossRef CAS PubMed.
- F. Dumestre, Science, 2004, 303, 821–823 CrossRef CAS.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808–4812 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS.
- T. R. Ralph, G. A. Hards, J. E. Keating, S. A. Campbell, D. P. Wilkinson, M. Davis, J. St-Pierre and M. C. Johnson, J. Electrochem. Soc., 1997, 144, 3845–3857 CrossRef CAS.
- J. I. Shui, C. Chen and J. C. M. Li, Adv. Funct. Mater., 2011, 21, 3357–3362 CrossRef CAS.
- F. Maillard, S. Schreier, M. Hanzlik, E. R. Savinova, S. Weinkauf and U. Stimming, Phys. Chem. Chem. Phys., 2005, 7, 385–393 RSC.
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2012, 51, 3139–3142 CrossRef CAS.
- N. Becknell, Y. Kang, C. Chen, J. Resasco, N. Kornienko, J. Guo, N. M. Markovic, G. A. Somorjai, V. R. Stamenkovic and P. Yang, J. Am. Chem. Soc., 2015, 137, 15817–15824 CrossRef CAS PubMed.
- E. Christoffersen, P. Liu, A. Ruban, H. L. Skriver and J. K. Nørskov, J. Catal., 2001, 199, 123–131 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 CrossRef CAS PubMed.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- B. Dembinska, A. Zlotorowicz, M. Modzelewska, K. Miecznikowski, I. A. Rutkowska, L. Stobinski, A. Malolepszy, M. Krzywiecki, J. Zak, E. Negro, V. Di Noto and P. J. Kulesza, Catalysts, 2020, 10, 689 CrossRef CAS.
- P. J. Kulesza, J. K. Zak, I. A. Rutkowska, B. Dembinska, S. Zoladek, K. Miecznikowski, E. Negro, V. Di Noto and P. Zelenay, Curr. Opin. Electrochem., 2018, 9, 257–264 CrossRef CAS.
- L. Perini, C. Durante, M. Favaro, V. Perazzolo, S. Agnoli, O. Schneider, G. Granozzi and A. Gennaro, ACS Appl. Mater. Interfaces, 2015, 7, 1170–1179 CrossRef CAS PubMed.
- S. Zhang, T. Fu, J. Li, Y. Peng and J. Zhao, ACS Appl. Energy Mater., 2018, 1, 6198–6207 CrossRef.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M.-G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef CAS PubMed.
- R. Chattot, I. Martens, M. Scohy, J. Herranz, J. Drnec, F. Maillard and L. Dubau, ACS Energy Lett., 2020, 5, 162–169 CrossRef CAS.
- M. Ahmadi, F. Behafarid, C. Cui, P. Strasser and B. R. Cuenya, ACS Nano, 2013, 7, 9195–9204 CrossRef CAS PubMed.
- X. Zhao, S. Takao, K. Higashi, T. Kaneko, G. Samjeskè, O. Sekizawa, T. Sakata, Y. Yoshida, T. Uruga and Y. Iwasawa, ACS Catal., 2017, 7, 4642–4654 CrossRef CAS.
- X. Yan, S. Yu, Y. Tang, D. Sun, L. Xu and C. Xue, Nanoscale, 2018, 10, 2231–2235 RSC.
- Y. Chen, Z. Liang, F. Yang, Y. Liu and S. Chen, J. Phys. Chem. C, 2011, 115, 24073–24079 CrossRef CAS.
- V. Di Noto, E. Negro, A. Nale, P. J. Kulesza, I. A. Rutkowska, K. Vezzù and G. Pagot, Electrocatalysis, 2020, 11, 143–159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05195k |
| This journal is © The Royal Society of Chemistry 2020 |