
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed highly regioselective hydroformylation to linear aldehydes by employing porous organic polymer as a ligand†
Zhaozhan Wanga and
Yong Yang *ab
*ab
aCAS Key Laboratory of Bio-Based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: yangyong@qibebt.ac.cn
bDalian National Laboratory for Clean Energy, Dalian 116023, China
First published on 7th August 2020
Abstract
In this work, we developed a new structural porous organic polymer containing biphosphoramidite unit, which can be used as a solid bidentate phosphorous ligand for rhodium-catalyzed solvent-free higher olefins hydroformylation. The resultant catalyst demonstrated unprecedently high regioselectivity to linear aldehydes and could be readily recovered for successive reuses with good stability in both catalytic activity and regioselectivity.
Introduction
Hydroformylation, also known as the oxo process, is a highly atom-economical synthetic method to prepare aldehydes by the addition of syngas (H2/CO) into olefins.1 To date, homogeneous ligand-modified rhodium or cobalt complexes have been successfully used in industry, and more than 10 million tons of aldehydes are produced annually.2 In particular, rhodium compounds generally give rise to high reaction rates and good selectivity to the desired products. However, industrial applications of these homogeneous catalysts remain a challenge because they are expensive, cannot be recycled, and are difficult to separate from the product mixture. In contrast, the heterogeneous catalysts hold significant advantages, such as easy of catalyst separation and recyclability, compared with homogeneous analogues. More importantly, heterogeneous catalysts can often present different or even unexpected catalytic performance, due to the surface support properties, metal–support interactions, and microenvironments of catalytically active sites.3Due to the commercial interest, the linear aldehydes, as versatile intermediates for the production of plasticizer alcohols, detergents, surfactants and other useful fine chemicals, have been the most desired products for hydroformylation of higher olefins (>C5).4 To this end, considerable efforts have been devoted in designing new types of phosphorous ligands for rhodium-catalyzed hydroformylation processes. A family of ligands, e.g., xantphos,5 bisbi,6 naphos,7 biphephos,8 calix[4]arene bisphosphate,9 spiroketal-based bisphosphoramidite ligands,10 pyrrole-based tri- and tetra-phosphoramidite ligands,11 have been developed and demonstrated high regioselectivities to linear aldehydes. Among them, pyrrole-based phosphoramidites developed firstly by van Leeuwen,10a followed by Ding,10b,c Zhang,11a,b and Chen11c respectively, gave unprecedentedly high regioselectivity due to the strong π-acceptor ability of phosphorous. Nonetheless, all cases are still limited to homogeneous catalytic system. Therefore, the development of a heterogeneous catalyst with excellent regioselectivity to linear aldehydes is highly desirable.
Over the past few years, porous organic polymers (POPs) have attracted considerable attention due to their high specific surface areas, porous channels, and adjustable functionalities in their structures.12 Specifically, some P or N-containing molecule units, serving as coordination sites to ligate with metal species to form catalytically active sites, can be effectively incorporated into the POPs structures. As such, POPs can be employed as solid supports and porous organic ligands (POLs) in catalysis. In this context, Xiao, Ding and co-workers developed POLs bearing a PPh3 unit to coordinate Rh species to form a heterogeneous supported Rh catalyst (denoted as Rh/POL-PPh3), which exhibited superior activity and excellent recyclability for 1-octene hydroformylation.13 Later on, two groups have independently developed several different POPs-based metal catalysts containing various coordination molecule units for organic transformations.14 Very recently, POPs-based heterogeneous catalysts have also been expanded for hydrosilylation,15 CO2 fixation,16 and others.17
Herein, we report a new porous organic polymers (named as CPOL-BPa&1VB) with biphosphoramidite molecule unit (Scheme 1), which has large surface area with hierarchical pores and can coordinate with Rh species to form a heterogeneous catalyst. The resultant catalyst shows superior activity and excellent regioselectivity to linear aldehydes in hydroformylation of higher olefins. In addition, the catalyst can be readily recovered for successive reuse without appreciable loss of catalytic performance.
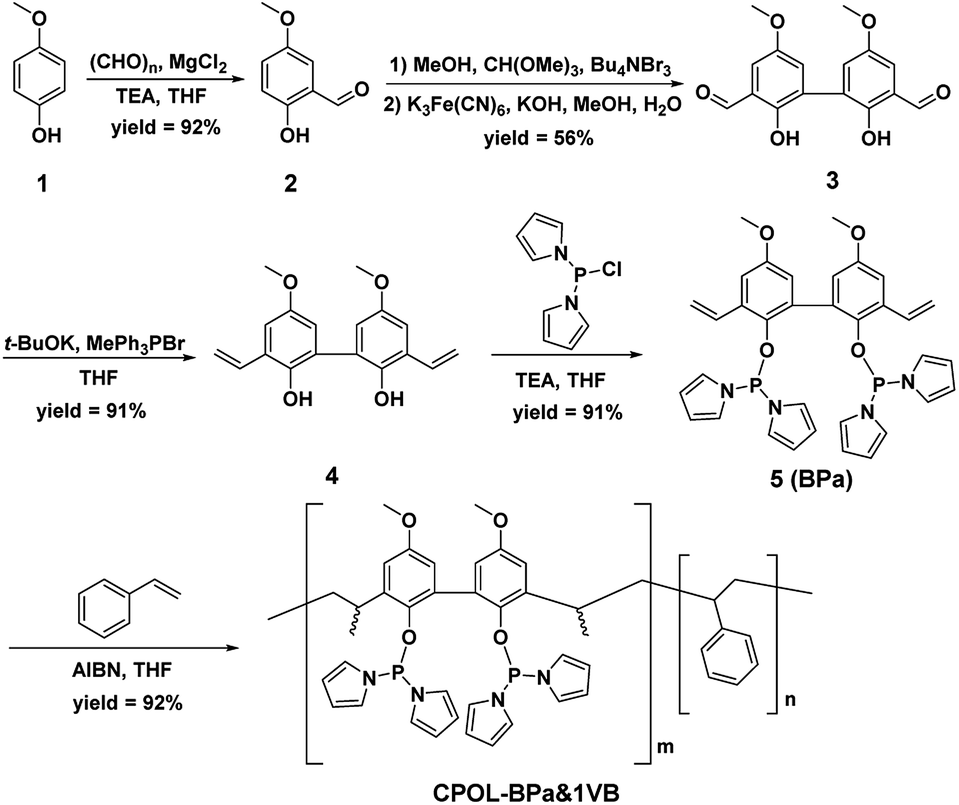 | ||
| Scheme 1 Synthesis of CPOL-BPa&1VB. | ||
Results and discussion
The CPOL-BPa&1VB was prepared via copolymerization of bivinyl-functionalized pyrrole-based biphosphoramidite (5, BPa) with styrene as the monomers under solvothermal conditions, as shown in Scheme 1 (see details in the ESI†). Divinyl diphenol 4 was synthesized from 4-methoxyphenol 1 as starting material followed by sequential formylation, coupling, and Witting reactions. Divinyl-functionalized pyrrole-based biphosphoramidite 5 was prepared by treatment of 4 with chlorodipyrrolyphosphine in the presence of Et3N as a HCl scavenger. CPOL-BPa&1VB was obtained in 92% yield by copolymerization of 5 with styrene (St) under solvothermal conditions in tetrahydrofuran (THF) at 100 °C for 48 h in the presence of azobisisobutyronitrile (AIBN). For comparison, copolymerization of 5 with other different structure monomers, such as divinylbenzene (2VB), tris(phenylene)vinylene (3VTPB), and tri(4-vinphenyl)phosphane (3VPPh3), tris(4-vinylphenyl)phosphite (3VTPPi), were also prepared under similar conditions, denoted as CPOL-BPa&2VB, CPOL-BPa&3VTPB, and CPOL-BPa&3VPPh3, CPOL-BPa&3VTPPi, respectively.Comprehensive characterizations were carried out to investigate the local chemical structure of the obtained CPOL-BPa&1VB. 13C MAS NMR spectrum (Fig. 1A) demonstrates that an additional peak at around 42 ppm with strong intensity appears, assignable to the polymerized vinyl groups, accompanying with the appearance of the characteristic peaks attributed to the pyrrole-based biphosphoramidite upon copolymerization. This result strongly suggests the success of copolymerization of 5 and styrene monomer. A single peak at 105.4 ppm without appearance of oxidized phosphorous species was observed in 31P MAS NMR spectrum (Fig. 1B), which is in well consistent with that of the monomer 5 (see the ESI†), indicating that P species are stable during the copolymerization. N2 sorption isotherm (Fig. 1C) shows a sharp uptake of N2 at low relative pressure (P/P0 < 0.1) and a hysteresis loop at higher relative pressure (0.4 < P/P0 < 1.0), indicating the existence of hierarchical micro-, meso-, and macropores in the framework, which was further confirmed by analysis of pore-size distribution (Fig. 1D). The total pore volume and BET surface area derived from N2 adsorption data were as high as 0.4 cm3 g−1 and 140 m2 g−1, respectively, which should be beneficial for its application as a support to immobilize metal species and in turn facilitate the reaction effectively. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 1E and F) display a quite rough surface morphology with presence of hierarchically porous structures in the framework. The thermogravimetric analysis (TGA) (Fig. S1†) shows that the CPOL-BPa&1VB has good thermal stability. Upon reaction of Rh(acac)(CO)2 with CPOL-BPa&1VB, a new peak at 129.8 ppm was observed in the solid state 31P MAS NMR spectrum (Fig. S2†), assignable to the P species coordinated with Rh with a possible structure of HRh(BPa)2(CO),10a indicating the success of the coordination.
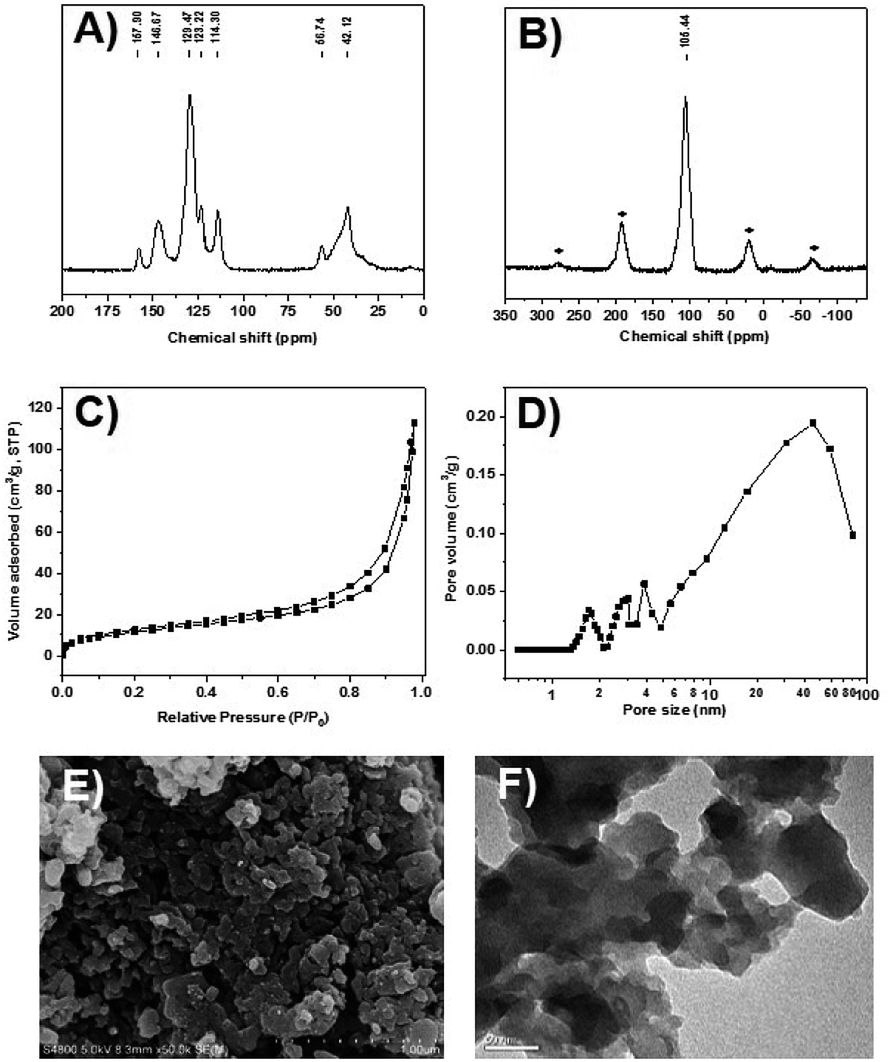 | ||
| Fig. 1 (A) 13C MAS NMR spectrum, (B) 31P MAS NMR spectrum, (C) N2 sorption isotherm, (D) pore size distribution calculated by nonlocal density functional theory (NLDFT), (E) SEM and (F) TEM image of CPOL-BPa&1VB. | ||
Subsequently, we treated the CPOL-BPa&1VB with Rh(acac)(CO)2 (acac = acetylacetonate) to afford its corresponding Rh catalyst for solvent-free 1-hexene hydroformylation as a benchmark reaction to investigate its catalytic performance, the results are compiled in Table 1. Firstly, a set of parameters, including reaction temperature, reaction time, pressure of syngas, and substrate/catalyst (S/C) ratios were screened intensively. Higher temperature gave an increased reaction efficiency, while lower reaction temperatures dramatically improved the regioselectivity to linear aldehyde together with a reduced product selectivity to hydrogenation and isomerization of 1-hexene (entries 1–3). To our surprise, unprecedented regioselectivity to linear 1-heptanal with l/b ratio up to 158.5 and almost complete conversion of 1-hexene was achieved when the reaction was performed at 80 °C and 2.0 MPa pressure of syngas after 8 h (entry 2). To the best of our knowledge, this is the highest value among all obtained results catalyzed by a heterogeneous Rh catalyst so far. A decrease or increase of pressure of syngas led to relatively lower regioselectivity to linear 1-heptanal with comparable reaction efficiency, while a considerable poorer selectivity to aldehyde was afforded at lower pressure under otherwise identical conditions (entries 5 and 6). Survey of different substrate/catalyst (SC) ratios ranging from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 30000 for the reaction reveals no appreciable changes in regioselectivity to linear 1-heptanal, while a slight longer reaction time was required to guarantee complete conversion, higher selectivity, and excellent regioselectivity to 1-haptanal for the reaction with higher S/C ratio (entries 2, 7–10).
000 to 30000 for the reaction reveals no appreciable changes in regioselectivity to linear 1-heptanal, while a slight longer reaction time was required to guarantee complete conversion, higher selectivity, and excellent regioselectivity to 1-haptanal for the reaction with higher S/C ratio (entries 2, 7–10).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Olefin | S/C | Temperature (°C) | Time (h) | PH2+CO (MPa) | Conversionb (%) | l/bb | Selectivityb (%) | ||
| Aldehyde | Isomerization | Alkane | ||||||||
| a Reaction conditions: 1-hexene (3 mL), CPOL-BPa&1VB (8 mg), Rh(acac)(CO)2 (0.49 mg), H2/CO = 1/1.b Determined by GC and GC-MS using decane as an internal standard sample and confirmed with their corresponding authentic samples.c 1-Octene (2 mL), toluene (2 mL).d 2-Octene (3 mL).e Styrene (3 mL). | ||||||||||
| 1 | 1-Hexene | 12750 |
60 | 8 | 2 | 79.5 | 175.7 | 90.6 | 5.7 | 3.6 |
| 2 | 1-Hexene | 12750 |
80 | 8 | 2 | 98.5 | 158.5 | 87.8 | 6.8 | 5.4 |
| 3 | 1-Hexene | 12750 |
100 | 8 | 2 | 99.3 | 35.7 | 69.7 | 6.2 | 23.9 |
| 4 | 1-Hexene | 12750 |
80 | 5 | 2 | 91.5 | 134.9 | 82.9 | 10.1 | 6.9 |
| 5 | 1-Hexene | 12750 |
80 | 8 | 1 | 96.5 | 95.7 | 65.8 | 16.0 | 18.2 |
| 6 | 1-Hexene | 12750 |
80 | 8 | 4 | 94.1 | 89.0 | 90.0 | 6.3 | 3.7 |
| 7 | 1-Hexene | 10000 |
80 | 8 | 2 | 99.2 | 164.0 | 88.4 | 7.3 | 4.3 |
| 8 | 1-Hexene | 20000 |
80 | 8 | 2 | 91.7 | 157.0 | 83.9 | 9.0 | 7.1 |
| 9 | 1-Hexene | 30000 |
80 | 8 | 2 | 70.1 | 123.0 | 59.6 | 22.8 | 17.6 |
| 10 | 1-Hexene | 30000 |
80 | 12 | 2 | 96.4 | 155.3 | 82.8 | 9.6 | 7.6 |
| 1c | 1-Octene | 6710 | 80 | 8 | 2 | 99.6 | 104.3 | 73.7 | 24.7 | 1.5 |
| 12d | 2-Octene | 10107 |
100 | 8 | 2 | 66.0 | 8.9 | 66.8 | 32.6 | 0.4 |
| 13e | Styrene | 13740 |
80 | 15 | 2 | 85.0 | 0.1 | 95.8 | — | 4.2 |
Under the optimized conditions, we next investigated other olefins hydroformylation. Longer chain 1-octene could be efficiently hydroformylated, affording excellent regioselectivity to linear aldehyde with l/b ratio of 104.3 in full conversion (entry 11). Internal 2-octene was also suitable for this reaction conditions, while a considerably lower l/b ratio (8.9) of aldehyde was achieved in this case (entry 12). Besides, styrene, as a functionalized olefin, was also compatible, giving the branched aldehyde as major product (l/b = 0.10) in excellent selectivity with TON as high as 11680 (entry 13).
For comparison, 5 was used as a ligand for the homogeneous hydroformylation under otherwise identical conditions. It turns out that nearly equal reaction efficiency to that of Rh/CPOL-BPa&1VB was achieved, but with a considerably lower regioselectivity to linear 1-heptanal (l/b = 33.0 vs. 158.5) (Table 2, entry 1). Such a result not only clearly indicates that CPOL-BPa&1VB can achieve equal level of reaction efficiency as a homogeneous counterpart, but also showcases that the created microenvironment of catalytically active sites can unexpectedly tune the catalytic performance. However, in the absence of ligand, 1-hexene was converted into hexane as major product with poor selectivity to 1-heptanal (Table 2, entry 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Conversionb (%) | l/bb | Selectivityb (%) | ||
| 1 + 2 | 3 | 4 | ||||
| a Reaction conditions: 1-hexene (3 mL), Rh(acac)(CO)2 (0.49 mg), H2/CO pressure (2 MPa, H2/CO = 1/1), 80 °C, CPOL-BPa&1VB (8 mg), CPOL-BPa&2VB (18.2 mg), CPOL-BPa&3VTPB (42.4 mg), CPOL-BPa&3VPPh3 (19.1 mg), CPOL-BPa&3VTPPi (21.4 mg).b Determined by GC and GC-MS using decane as an internal standard sample and confirmed with their corresponding authentic samples.c In the absence of ligand. | ||||||
| 1 | 5 (BPa) | 99.4 | 33.0 | 85.1 | 2.5 | 12.4 |
| 2c | — | 98.5 | 1.2 | 25.7 | 13.7 | 60.6 |
| 3 | CPOL-BPa&1VB | 98.5 | 158.5 | 87.8 | 6.8 | 5.4 |
| 4 | CPOL-BPa&2VB | 99.4 | 82.9 | 55.7 | 13.4 | 30.9 |
| 5 | CPOL-BPa&3VTPB | 89.5 | 31.1 | 86.6 | 6.8 | 6.6 |
| 6 | CPOL-BPa&3VPPh3 | 86.3 | 13.7 | 88.2 | 6.0 | 5.8 |
| 7 | CPOL-BPa&3VTPPi | 97.5 | 10.1 | 90.7 | 4.1 | 5.2 |
Furthermore, several different structural CPOLs (e.g., CPOL-BPa&2VB, CPOL-BPa&3VTPB, CPOL-BPa&3VPPh3, CPOL-BPa&3VTPPi) were employed as solid ligands derived from copolymerization of 5 with varying vinyl-functionalized molecules as monomers (Table 2). The combination of Rh(acac)(CO)2 with these solid ligands demonstrated nearly comparable catalytic reactivity but with huge differences in regioselectivity to linear 1-heptanal under the optimized conditions. Among all investigated CPOLs, the CPOL-BPa&1VB stood out, affording the highest regioselectivity to linear 1-heptanal.
Finally, recyclability of the Rh/CPOL-BPa&1VB was investigated using the hydroformylation of 1-hexene under the optimized conditions. After each reaction, the solid catalyst was separated by centrifugation, washed with methanol and dried for next reaction. As shown in Fig. 2, both the catalytic activity and regioselectivity to linear 1-heptanal was remained without significant change after 5 recycles, indicating its high stability of the catalyst.
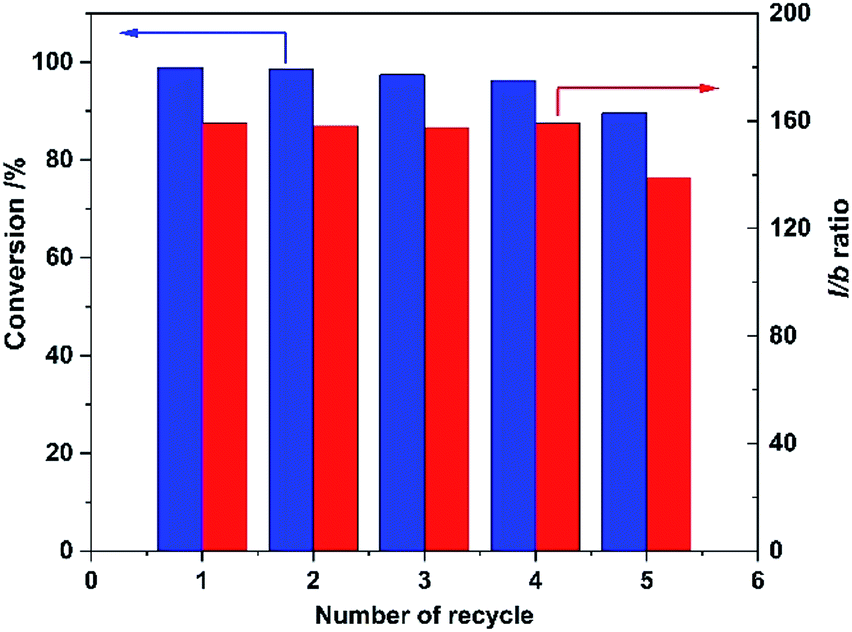 | ||
| Fig. 2 Recyclability of Rh/CPOL-BPa&1VB for 1-hexene hydroformylation under the optimized conditions. | ||
Conclusions
In conclusion, we developed a new porous organic polymer (POP) with large surface area and hierarchical pores via copolymerization of bivinyl-functionalized pyrrole-based biphosphoramidite with styrene under solvothermal conditions. The polymer contains biphosphoramidite molecule unit and can be used as a solid support and ligand for Rh-catalyzed solvent-free higher olefins hydroformylation, which demonstrates outstanding activity and unprecedentedly high regioselectivity to linear aldehydes. More importantly, the resultant catalyst can be readily recovered for successive reuse with good maintenance of both catalytic activity and regioselectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported DICP & QIBEBT (Grant No. DICP & QIBEBT UN201704, QIBEBT I201902), and the Dalian National Laboratory for Clean Energy (DNL) Cooperation Fund, Chinese Academy of Sciences (Grant No. DNL201904).Notes and references
- B. Breit, Top. Curr. Chem., 2007, 279, 139–172 CrossRef.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef PubMed.
- (a) J. Zhang, P. Sun, Z. Zhao, G. Gao and F. Li, Chin. Sci. Bull., 2019, 64, 1–15 CrossRef; (b) P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2011, 50, 396–400 CrossRef PubMed.
- E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 382–411 CrossRef CAS.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- T. J. Devon, G. W. Phillips, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, US Pat., 4694109, 1987.
- H. Klein, R. Jackstell, K. D. Wiese, C. Borgmann and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 3408–3411 CrossRef CAS.
- E. Billig, A. G. Abatjoglou and D. R. Bryant, US Pat., 4668651 (1987) and 4769498 (1988).
- R. Paciello, L. Siggel and M. Röper, Angew. Chem., Int. Ed., 1999, 38, 1920–1923 CrossRef CAS.
- (a) S. C. van der Slot, J. Duran, J. Luten, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2002, 21, 3873–3883 CrossRef CAS; (b) X. Jia, Z. Wang, C. Xia and K. Ding, Chem.–Eur. J., 2012, 18, 15288–15295 CrossRef CAS PubMed; (c) X. Ren, Y. Zheng, L. Zhang, Z. Wang, C. Xia and K. Ding, Angew. Chem., Int. Ed., 2017, 56, 310–313 CrossRef CAS PubMed.
- (a) S. Yu, Y. Chie, Z. Guan and X. Zhang, Org. Lett., 2008, 10, 3469–3472 CrossRef CAS PubMed; (b) C. Chen, Y. Qiao, H. Geng and X. Zhang, Org. Lett., 2013, 15, 1048–1051 CrossRef CAS PubMed; (c) W. Han, S. Qin, X. Shu, Q. Wu, B. Xu, R. Li, X. Zheng and H. Chen, RSC Adv., 2016, 6, 53012–53016 RSC.
- (a) L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS; (b) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS; (c) Y. Zhang and J. Y. Ying, ACS Catal., 2015, 5, 2681–2691 CrossRef CAS; (d) Q. Sun, Z. Dai, X. Meng, L. Wang and F. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS; (e) A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed; (f) G. Liu, Y. Wang, C. Shen, Z. Ju and D. Yuan, J. Mater. Chem. A, 2015, 3, 3051–3058 RSC; (g) Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- Q. Sun, M. Jiang, Z. Shen, Y. Jin, S. Pan, L. Wang, X. Meng, W. Chen, Y. Ding, J. Li and F.-S. Xiao, Chem. Commun., 2014, 50, 11844–11847 RSC.
- (a) Q. Sun, Z. Dai, X. Liu, N. Sheng, F. Deng, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204–5209 CrossRef CAS; (b) Q. Sun, B. Aguila, G. Verma, X. Liu, Z. Dai, F. Deng, X. Meng, F.-S. Xiao and S. Ma, Chem, 2016, 1, 628–639 CrossRef CAS; (c) C. Li, K. Xiong, L. Yan, M. Jiang, X. Song, T. Wang, X. Chen, Z. Zhan and Y. Ding, Catal. Sci. Technol., 2016, 6, 2143–2149 RSC; (d) C. Li, L. Yan, L. Lu, K. Xiong, W. Wang, M. Jiang, J. Liu, X. Song, Z. Zhan, Z. Jiang and Y. Ding, Green Chem., 2016, 18, 2995–3005 RSC; (e) Y. Wang, L. Yan, C. Li, M. Jiang, Z. Zhao, G. Hou and Y. Ding, J. Catal., 2018, 368, 197–206 CrossRef CAS; (f) C. Li, K. Sun, W. Wang, L. Yan, X. Sun, Y. Wang, K. Xiong, Z. Zhan, Z. Jiang and Y. Ding, J. Catal., 2017, 353, 123–132 CrossRef CAS; (g) Z. Ren, Y. Lium, Y. Lyu, X. Song, C. Zheng, S. Feng, Z. Jiang and Y. Ding, J. Catal., 2019, 369, 249–256 CrossRef CAS.
- (a) R.-H. Li, X.-M. An, Y. Yang, D.-C. Li, Z.-L. Hu and Z.-P. Zhan, Org. Lett., 2018, 20, 5023–5026 CrossRef CAS; (b) R.-H. Li, G.-L. Zhang, J.-X. Dong, D.-C. Li, Y. Yang, Y.-M. Pan, H.-T. Tang, L. Chen and Z.-P. Zhang, Chem.–Asian J., 2019, 14, 149–154 CrossRef CAS PubMed.
- W. Wang, Y. Wang, C. Li, L. Yan, M. Jiang and Y. Ding, ACS Sustainable Chem. Eng., 2017, 5, 4523–4528 CrossRef CAS.
- (a) R. Jana and J. A. Tunge, J. Org. Chem., 2011, 76, 8376–8385 CrossRef CAS; (b) Y.-B. Zhou, Y.-Q. Wang, L. C. Ning, Z.-C. Ding, W.-L. Wang, C.-K. Ding, R.-H. Li, J.-J. Chen, X. Lu, Y.-J. Ding and Z.-P. Zhan, J. Am. Chem. Soc., 2017, 139, 3966–3969 CrossRef CAS; (c) R. Cai, X. Ye, Q. Sun, Q. He, Y. He, S. Ma and X. Shi, ACS Catal., 2017, 7, 1087–1092 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04816j |
| This journal is © The Royal Society of Chemistry 2020 |