DOI:
10.1039/D0RA05188H
(Paper)
RSC Adv., 2020,
10, 28674-28679
The geometrical structure and electronic properties of trivalent Ho3+ doped Y2O3 crystals: a first-principles study
Received
12th June 2020
, Accepted 29th July 2020
First published on 4th August 2020
1. Introduction
Rare-earth (RE) doped laser materials have attracted enormous interests because of their abundant transition channels and sharp luminescence bands.1–5 The potential applications have been widely investigated in a diversity of fields, such as optical imaging, quantum cascade lasers, high-density optical data storage and biophotonic areas.6–9 A recent study reveals that the directly pumped Ho3+-doped silica microsphere may be an excellent candidate for fabricating 2 μm laser, which can serve as laser-emitting source for mid-infrared telecommunications.10
Trivalent holmium ion (4f10 configuration) is a greatly promising laser ion due to the substantial transition channels at various wavelengths in the UV, visible and infrared regions.11,12 A well-known emission transition 5I7 → 5I8 with wavelength near 2 μm of Ho3+ can serve as the so called “eye safe” solid-state laser system.13 Yttrium oxide (Y2O3) is a typical cubic phase crystal structure with Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group, which possesses low phonon energy and desirable physical properties including low thermal expansion, high melting point and photochemical stability.14–16 The Y3+ ions of yttrium oxide crystal are six-fold coordinated to nearest O2− ligands, forming a [YO6]9− local unit with C2 site symmetry.17 After being doped with appropriate rare-earth ions, Y2O3 crystals can serve as excellent laser host materials because of their high thermal conductivity and low phonon energy.18 In recent years, Ho3+-doped Y2O3 (Y2O3
space group, which possesses low phonon energy and desirable physical properties including low thermal expansion, high melting point and photochemical stability.14–16 The Y3+ ions of yttrium oxide crystal are six-fold coordinated to nearest O2− ligands, forming a [YO6]9− local unit with C2 site symmetry.17 After being doped with appropriate rare-earth ions, Y2O3 crystals can serve as excellent laser host materials because of their high thermal conductivity and low phonon energy.18 In recent years, Ho3+-doped Y2O3 (Y2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ho) crystal has been the subject of intensive investigations as a great promising laser material.19 Laversenne et al. first demonstrated the growth of Ho3+-doped Y2O3 single crystal by using the Laser Heated Pedestal Growth (LHPG) technique.20 In addition, they especially analyzed the dynamical laser resonant characteristics of Y2O3:Ho. Qin et al. studied the luminescence spectra of Ho3+-doped Y2O3 under the excitation of a 532 nm continuous-wave laser.21 The results indicate that Ho3+ ion possesses several fluorescence transitions in the ultraviolet and violet region (306, 390 and 428 nm) which are assigned to the transitions of 3D3 → 5I8, 5G4 → 5I8 and 5G5 → 5I8, respectively. Wang et al. reported a high output laser operation at around 2.1 μm of Y2O3:Ho with low scattering loss and excellent optical quality.22 Their results revealed that Ho3+-doped Y2O3 system shows attractive prospect in high-power and efficient laser applications as a laser gain medium. Although numerous investigations have been widely reported on Y2O3:Ho, there is no systematic study to elucidate its microstructure and electronic properties.
:Ho) crystal has been the subject of intensive investigations as a great promising laser material.19 Laversenne et al. first demonstrated the growth of Ho3+-doped Y2O3 single crystal by using the Laser Heated Pedestal Growth (LHPG) technique.20 In addition, they especially analyzed the dynamical laser resonant characteristics of Y2O3:Ho. Qin et al. studied the luminescence spectra of Ho3+-doped Y2O3 under the excitation of a 532 nm continuous-wave laser.21 The results indicate that Ho3+ ion possesses several fluorescence transitions in the ultraviolet and violet region (306, 390 and 428 nm) which are assigned to the transitions of 3D3 → 5I8, 5G4 → 5I8 and 5G5 → 5I8, respectively. Wang et al. reported a high output laser operation at around 2.1 μm of Y2O3:Ho with low scattering loss and excellent optical quality.22 Their results revealed that Ho3+-doped Y2O3 system shows attractive prospect in high-power and efficient laser applications as a laser gain medium. Although numerous investigations have been widely reported on Y2O3:Ho, there is no systematic study to elucidate its microstructure and electronic properties.
In this paper, we perform extensive structure searches to obtain the ground-state structure of Y2O3:Ho based on the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization)23–27 method coupled with the DFT (density functional theory). Furthermore, we calculate and analyze the band structure, density of states and the ELF (electron localized function) to gain deeper insights into the electronic properties of Ho3+-doped Y2O3 system. The outline of this paper is organized as follows. We exhibit a brief description of the calculation method in Section 2. In Section 3, we present our results and discussion. A conclusion is finally given in Section 4.
2. Computational methods
We have explored the structural evolution of Y2O3:Ho crystal by using the unbiased CALYPSO23–27 method. The CALYPSO is a reliable structure prediction method which has been validated by a large variety of crystal structures.28–32 We perform an evolutionary variable-cell structure prediction with 80 atoms per simulation cell at ambient pressure. To determine the most stable structure of Y2O3:Ho system, we optimized the all lowest-lying candidate structures by using the density functional theory in VASP (Vienna Ab Initio Simulation Package) code.33–35 The frozen-core all electron projector-augmented wave (PAW) method has been adopted, with 4f115s25p66s2, 4d15s2 and 2s22p4 treated as valence electrons for Ho, Y and O, respectively. For describing the influence of the correlation effect introduced by 4f electrons of Ho atoms, we employ the local density approximation (LDA) with an onsite Coulomb repulsion parameter U36 to determine the electronic band structure of Y2O3:Ho. The U value of Ho has been determined to be 6.8 eV by Min et al.37 Phonon dispersion curve have been calculated by the PHONOPY code.38
3. Results and discussion
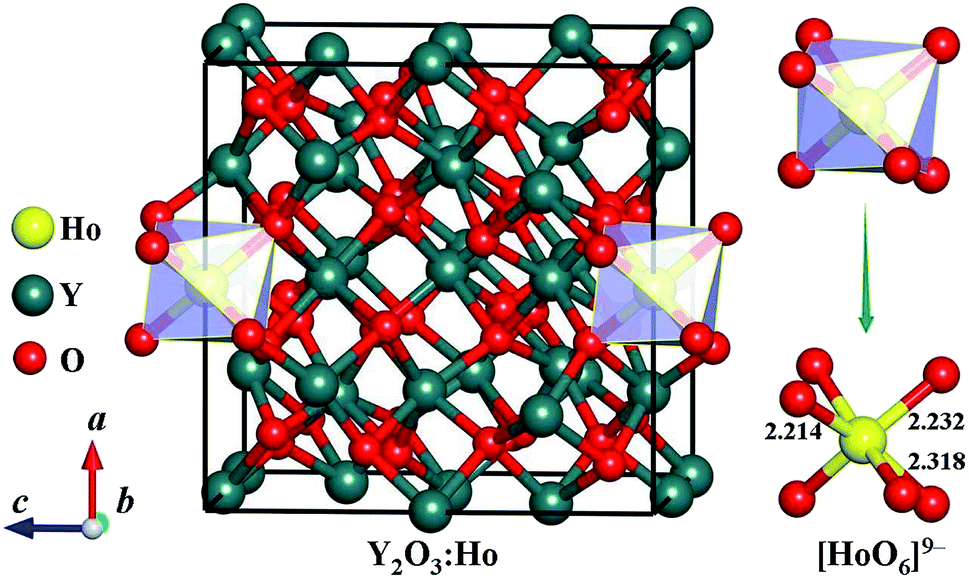
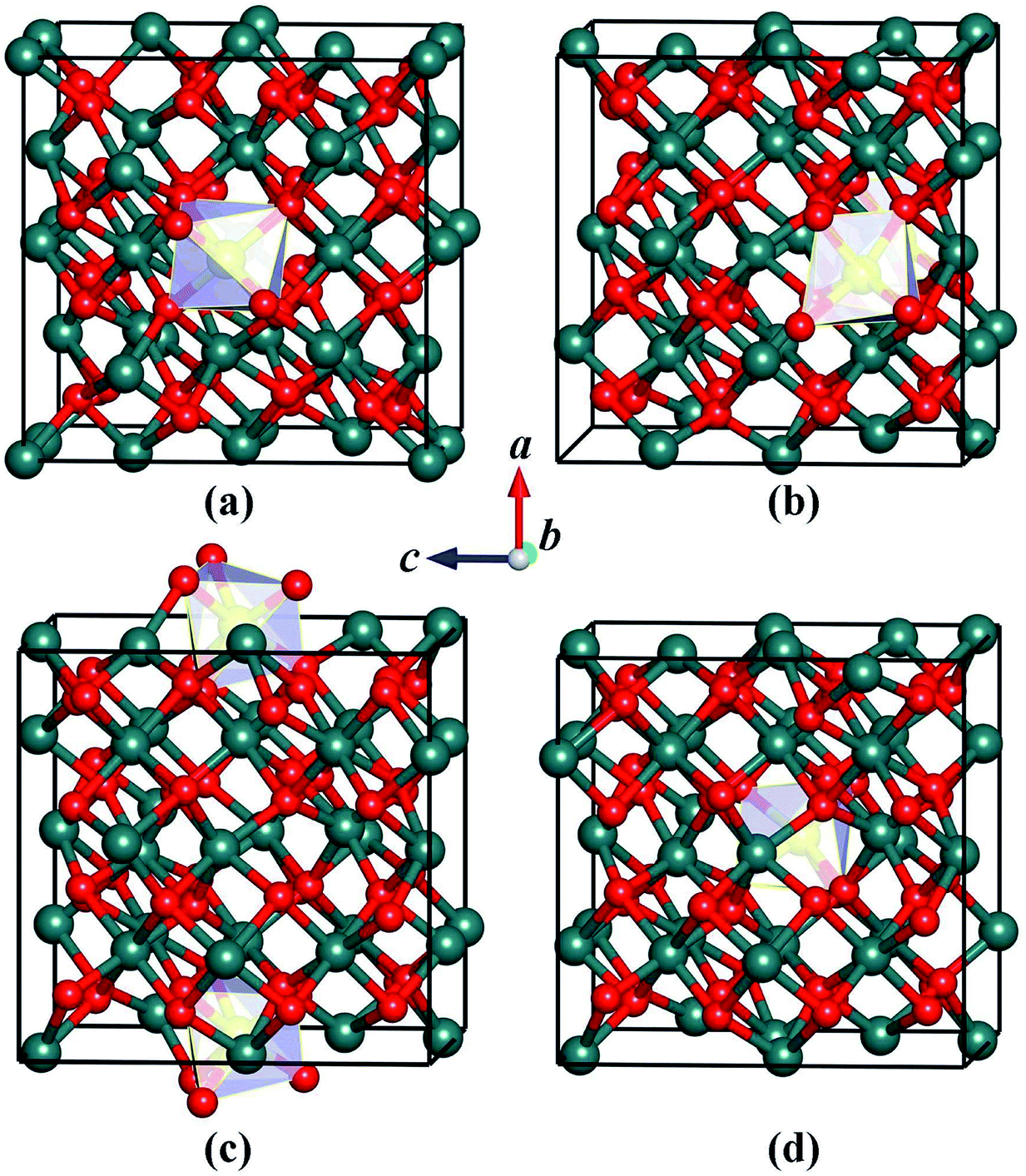
We carefully examine the ground-state crystal structure of Ho3+-doped Y2O3 by using the unbiased CALYPSO structure search method with the stoichiometric ratio of Ho:Y:O = 1:31:48 under ambient conditions. The lowest-energy structure of Y2O3:Ho is successfully identified and displayed in Fig. 1. It can be seen from Fig. 1 that the ground-state Y2O3:Ho crystal possesses the monoclinic configuration with the Ho3+ ion (0.901 Å) substitute for Y3+ ion (0.900 Å) in the Y2O3 host. The concentration of the impurity Ho3+ is equal to 3.125%, which is in excellent agreement with the result measured by Atabaev et al.15 The site symmetry of [HoO6]9− local structure are calculated to be C2 and Ho3+ position are six-coordinated by oxygen atoms. The three different bond lengths between Ho–O bonds are calculated to be 2.214, 2.232, and 2.318 Å, respectively. The Ho3+ doped Y2O3 crystal structure belongs to the standardized P2 symmetry and the calculated lattice constants are a = b = c = 10.524 Å, β = 90°. The coordinates of all atoms for the ground state Y2O3:Ho are summarized in Table 1 for further investigations. Moreover, our structure searches also predict many metastable structures of Ho3+-doped Y2O3 which can play important roles to explore the structural evolutions. The first four optimized low-lying structures (a), (b), (c) and (d) from low to high energy are exhibited in Fig. 2. It is found that the Y3+ ions of these isomers are replaced by Ho3+ ions at different sites in the host crystals. Interestingly, the isomer (a) possesses the same P2 monoclinic configuration with the ground-state structure while the isomers (b), (c) and (d) exhibit the P1 space group. In these metastable structures, we find that the impurity Ho3+ ions tend to occupy the crystal face site positions of the Y3+.
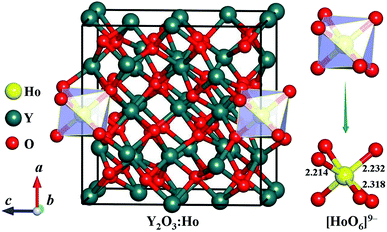 |
| | Fig. 1 Crystal structure and [HoO6]9− local unit of the ground-state Ho3+-doped Y2O3. The bond lengths are in the unit of Å. | |
Table 1 Coordinates of all atoms for the ground state Ho3+-doped Y2O3
| Atom |
x |
y |
z |
Wyckoff site symmetry |
| Ho |
0.50000 |
0.03244 |
−0.00000 |
1c |
| O(1) |
−0.09805 |
0.37926 |
0.39177 |
2e |
| O(2) |
0.40264 |
0.87998 |
0.89255 |
2e |
| O(5) |
0.40205 |
0.12070 |
0.60831 |
2e |
| O(6) |
−0.09818 |
0.62076 |
0.10808 |
2e |
| O(9) |
0.14194 |
0.15190 |
0.37921 |
2e |
| O(10) |
0.64186 |
0.65190 |
0.87934 |
2e |
| O(11) |
−0.14176 |
0.84802 |
0.87927 |
2e |
| O(12) |
0.35819 |
0.34791 |
0.37925 |
2e |
| O(17) |
0.12928 |
0.39175 |
0.15192 |
2e |
| O(18) |
0.62931 |
0.89190 |
0.65180 |
2e |
| O(19) |
0.62868 |
0.10740 |
0.84899 |
2e |
| O(20) |
0.12931 |
0.60822 |
0.34801 |
2e |
| O(25) |
0.59794 |
0.62079 |
0.60830 |
2e |
| O(26) |
0.09816 |
0.12070 |
0.10813 |
2e |
| O(29) |
0.09808 |
0.87932 |
0.39175 |
2e |
| O(30) |
0.59805 |
0.37924 |
0.89184 |
2e |
| O(33) |
0.35820 |
0.84806 |
0.62099 |
2e |
| O(34) |
−0.14195 |
0.34792 |
0.12077 |
2e |
| O(35) |
0.64100 |
0.15153 |
0.12014 |
2e |
| O(36) |
0.14182 |
0.65199 |
0.62075 |
2e |
| O(41) |
0.37080 |
0.60848 |
0.84803 |
2e |
| O(42) |
−0.12934 |
0.10816 |
0.34796 |
2e |
| O(43) |
−0.12943 |
0.89189 |
0.15191 |
2e |
| O(44) |
0.37080 |
0.39172 |
0.65211 |
2e |
| Y(1) |
0.00004 |
0.24998 |
0.24998 |
2e |
| Y(2) |
0.49982 |
0.75036 |
0.24967 |
2e |
| Y(3) |
0.50011 |
0.24976 |
0.75024 |
2e |
| Y(4) |
0.00005 |
0.75003 |
0.75003 |
2e |
| Y(9) |
−0.25020 |
0.24979 |
0.96795 |
2e |
| Y(10) |
−0.25006 |
0.75002 |
0.53211 |
2e |
| Y(13) |
0.71753 |
0.00010 |
0.24967 |
2e |
| Y(15) |
−0.21791 |
0.50004 |
0.24995 |
2e |
| Y(21) |
−0.25026 |
0.75030 |
0.03217 |
2e |
| Y(22) |
0.25002 |
0.24997 |
0.53226 |
2e |
| Y(25) |
−0.21803 |
0.00001 |
0.75019 |
2e |
| Y(26) |
0.28218 |
0.49999 |
0.24999 |
2e |
| Y(17) |
0.00000 |
0.96781 |
0.00000 |
1a |
| Y(30) |
0.00000 |
0.46784 |
0.00000 |
1a |
| Y(18) |
0.50000 |
0.53219 |
−0.00000 |
1c |
| Y(19) |
0.00000 |
0.03214 |
0.50000 |
1b |
| Y(29) |
0.00000 |
0.53220 |
0.50000 |
1b |
| Y(20) |
0.50000 |
0.46778 |
0.50000 |
1d |
| Y(31) |
0.50000 |
0.96786 |
0.50000 |
1d |
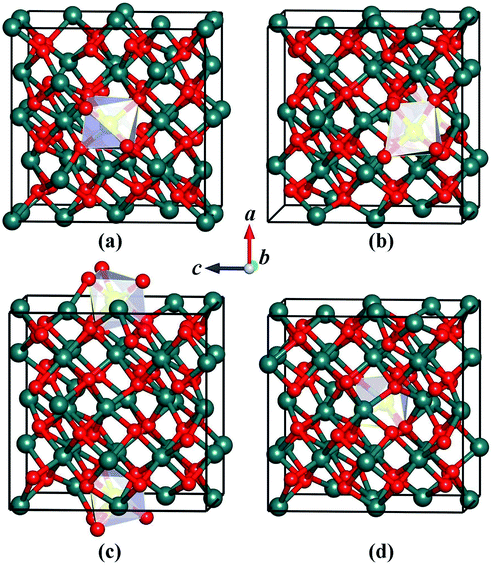 |
| | Fig. 2 Coordination structures of the metastable (a–d) for Y2O3:Ho. | |
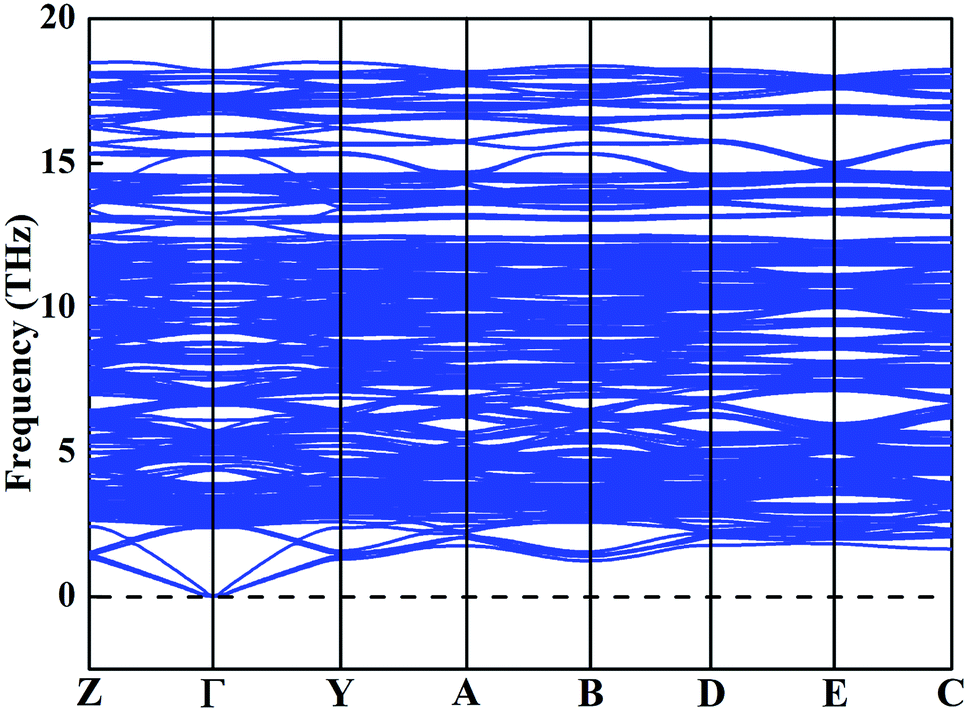
To clarify the true structure of the ground-state Y2O3:Ho, as shown in Fig. 3, we calculate the X-ray diffraction (XRD) patterns of the ground state structure. We can clearly see from Fig. 3 that the simulated spectrum of Y2O3:Ho are in good accordance with the observations in experiment.15 In addition, the XRD patterns of the four metastable structures are calculated and the results are also plotted in Fig. 3. It can be seen from Fig. 3 that the overall distribution of the peaks is similar, suggesting that the structural parameters of the four metastable structures are close to each other. To further validate the dynamical stability of Ho3+-doped Y2O3 system, we have calculated the phonon dispersion curves in Fig. 4 and no imaginary phonon frequencies can be seen over the entire Brillouin zones. The result indicates that the determined ground-state structure of Ho3+-doped Y2O3 crystal is dynamically stable. These theoretical results provide great support for the reliability of our structural prediction methodology.
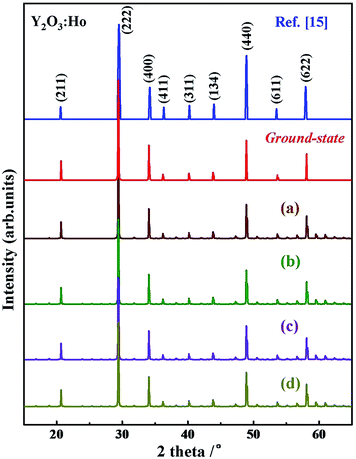 |
| | Fig. 3 Comparison of the simulated XRD spectrum for the ground-state and metastable (a–d) Y2O3:Ho with experimental patterns. | |
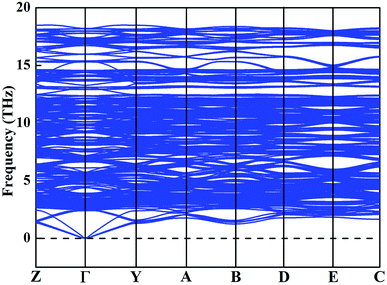 |
| | Fig. 4 Calculated phonon dispersion curve for the ground-state Y2O3:Ho. | |
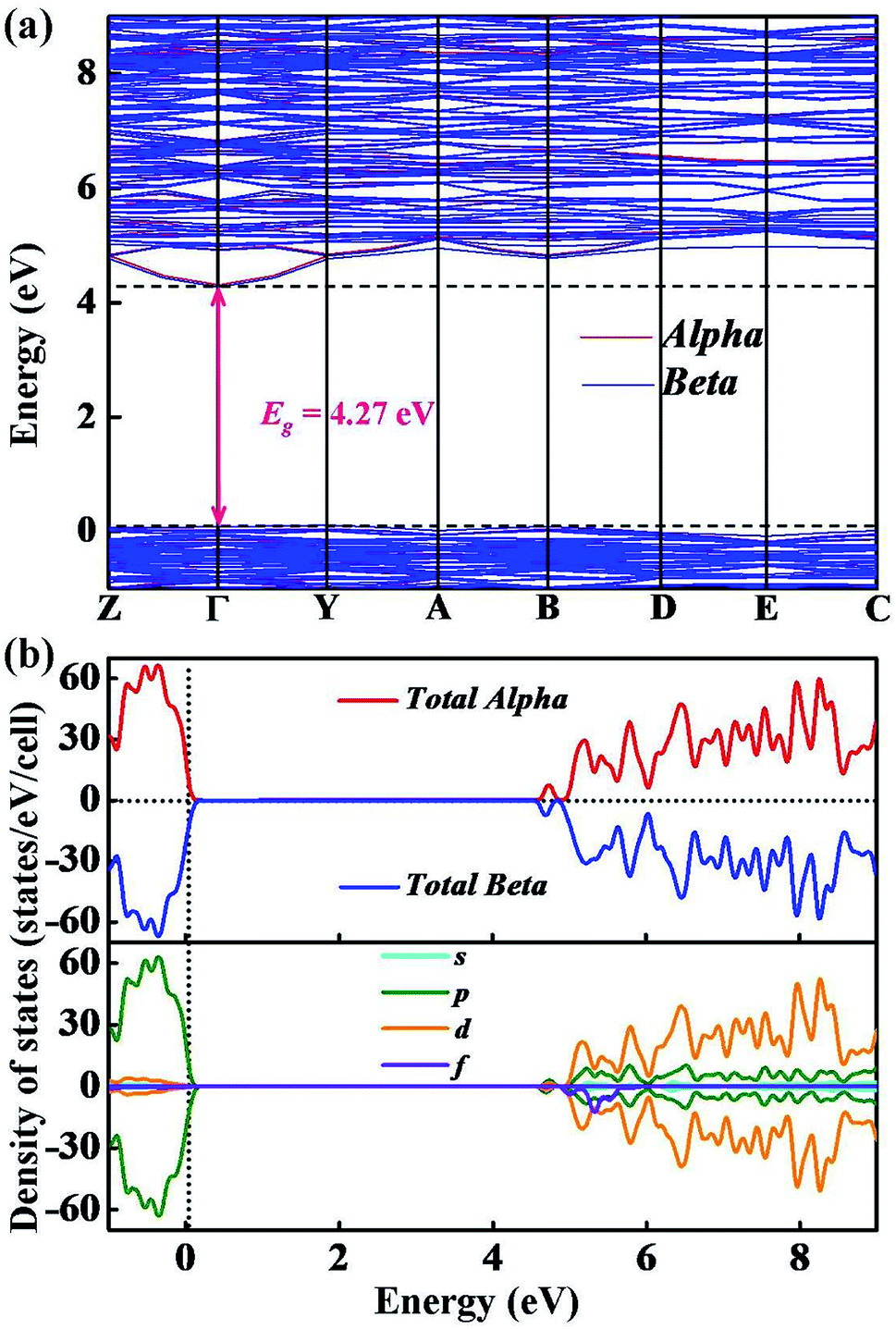
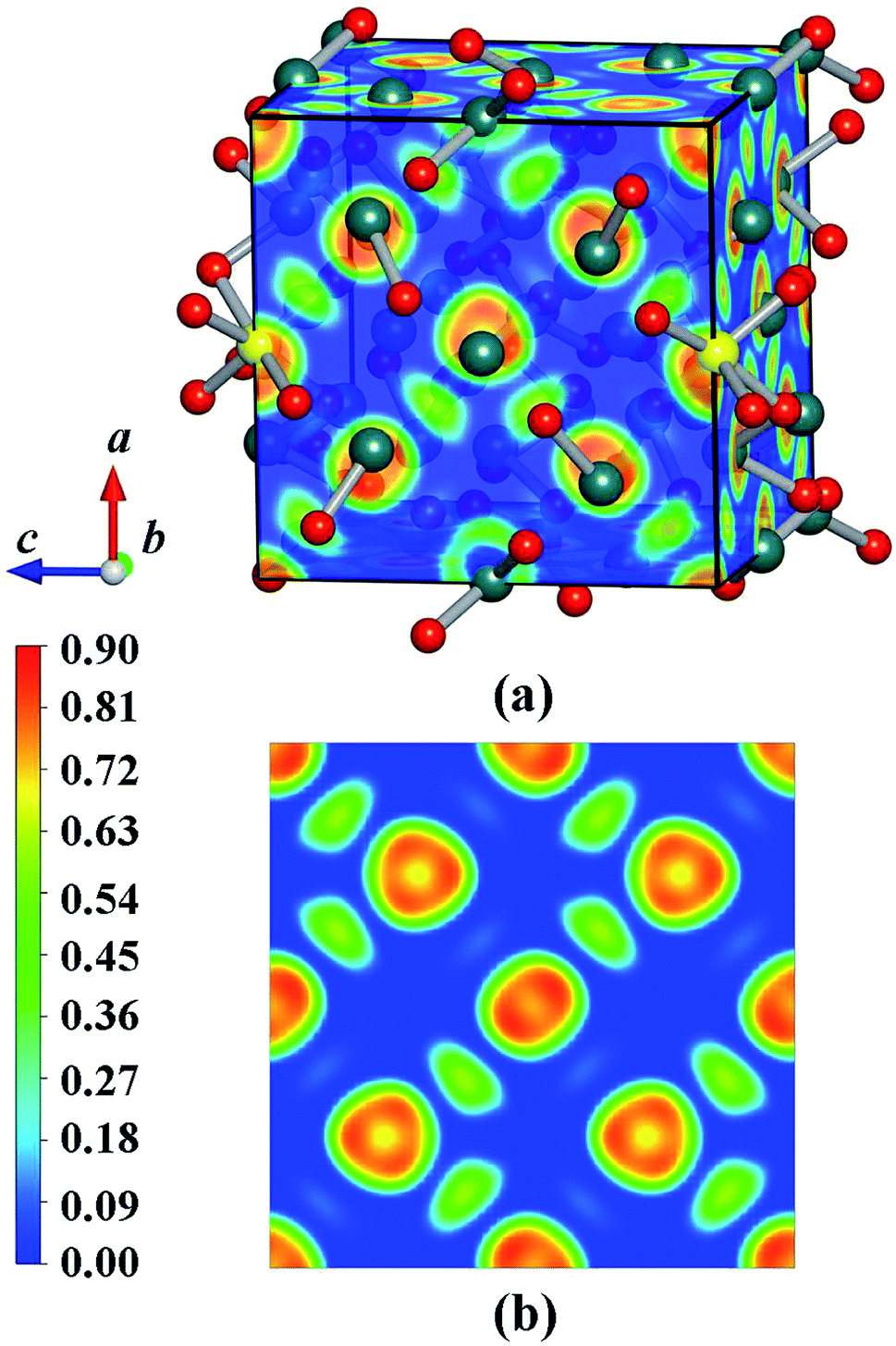
We have calculated the electronic band structure and the total as well as partial DOS for Y2O3:Ho. As illustrated in Fig. 5(a), the direct band gap value for Ho3+-doped Y2O3 is about 4.27 eV at the Γ point, which is approximately 2/3 of the experimental value (Eg = 6.2 eV) determined by Wallace and Wilk.39 This result can be ascribed to the general underestimation of band gap value by the first-principle calculations. The result indicates that the Ho3+ impurity ion remains the insulating character of Ho3+-doped Y2O3 crystal. From Fig. 5(b), we can clearly see that the low valence band region is mainly composed of p states with the smaller contributors of d states ranging from −1 eV to 0 eV, and the dominant contributions of the high conduction band between 4.3 eV to 9 eV are mainly occupied by p, d and f states. It should be noted that the s states is very weak from −1 eV to 9 eV. In addition, we have calculated the electron localized function (ELF) to visualize the chemical bonding character in Y2O3:Ho crystal. The ELF in crystal structure and the ELF of the (100) plane are presented in Fig. 6. It is shown that the ELF near the Y and Ho atoms value is close to 0.9, which suggests that the electrons are extremely localized around the Y and Ho atoms.
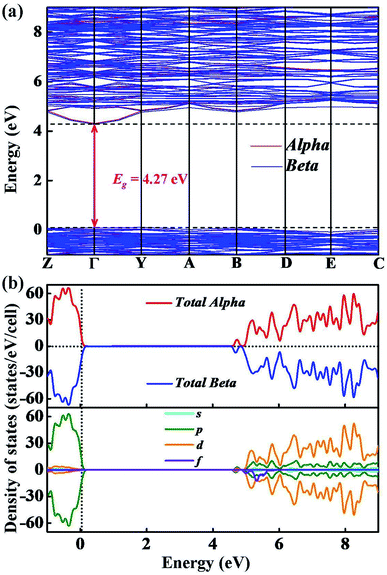 |
| | Fig. 5 The calculated (a) electronic band structure and (b) total as well as partial densities of states of Y2O3:Ho. | |
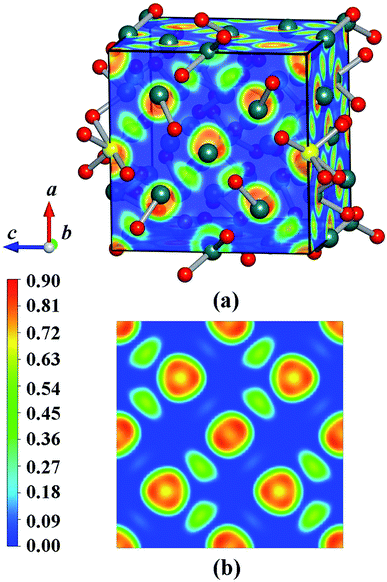 |
| | Fig. 6 ELF maps of (a) the structure and (b) 〈100〉 plane for the ground-state Y2O3:Ho. | |
4. Conclusions
In summary, we have explored the ground-state crystal structure of Ho3+-doped Y2O3 by means of the unbiased CALYPSO method combined with first-principle calculations. It is shown that the ground-state Y2O3:Ho structure possesses a novel P2 phase with the monoclinic symmetry. We carry out a systematical investigation to the microstructure evolutions for the ground-state Y2O3:Ho crystal. The results indicate that the impurity Ho3+ ion substitutes the positions of Y3+ ions in the host crystal lattice, forming the [HoO6]9− local structure. We find that the impurity Ho3+ ions tend to occupy the crystal face positions of the Y3+ ions from the structural features of the ground-state and metastable structures. We further calculate the band structure and density of states by LDA + U method for Y2O3:Ho. Our result reveals that the electronic band gap of Ho3+-doped Y2O3 is 4.27 eV. We hope that these findings can provide valuable guidance for future experiment research of Y2O3:Ho.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11904297, 11747139 and 11804031) and the Fundamental Research Funds for the Central Universities (SWU118055), the Scientific Research Project of Education Department of Hubei Province (No. Q20191301), the Youth Fund of Yangtze University (No. 2016cqn, Y. Y. J.).
References
- P. S. J. Bharadwaj, S. Kundu, V. S. Kollipara and K. B. R. Varma, Synergistic effect of trivalent (Gd3+, Sm3+) and high-valent (Ti4+) co-doping on antiferromagnetic YFeO3, RSC Adv., 2020, 10, 22183–22195 RSC.
- J. Bai, P. Duan, X. Wang, G. Han, M. Wang and G. Diao, Upconversion luminescence enhancement by Fe3+ doping in CeO2:Yb/Er nanomaterials and their application in dye-sensitized solar cells, RSC Adv., 2020, 10, 18868–18874 RSC.
- Monika, R. S. Yadav, A. Bahadur and S. B. Rai, Concentration and pump-mediated color tenability, optical heating and temperature sensing via TCLs of red emission in an Er3+/Yb3+/Li+ co-doped ZnGa2O4 phosphor, RSC Adv., 2019, 9, 40092–40108 RSC.
- N. Choudhary, M. K. Verma, N. D. Sharma, S. Sharma and D. Singh, Correlation between magnetic and transport properties of rare earth doped perovskite manganites La0.6R0.1Ca0.3MnO3 (R = La, Nd, Sm, Gd, and Dy) synthesized by Pechini process, Mater. Chem. Phys., 2020, 242, 122482 CrossRef CAS.
- M. P. Ghosh, S. Sharma, H. K. Satyapal, K. Tanbir, R. K. Singh and S. Mukherjee, Tuning the microstructural, optical and superexchange interactions with rare earth Eu doping in nickel ferrite nanoparticles, Mater. Chem. Phys., 2020, 241, 122383 CrossRef CAS.
- S. D. Jackson, Towards high-power mid-infrared emission from a fibre laser, Nat. Photonics, 2012, 6, 423–431 CrossRef CAS.
- I. Cieslik, T. Bolek, M. J. Wozniak, A. Majchrowski, S. Hirano and A. Budzianowski, Potassium gadolinium tungstate nanocrystals doped with holmium ions as candidates for optical imaging, Appl. Surf. Sci., 2018, 446, 139–144 CrossRef CAS.
- Y. R. Fang and M. T. Sun, Nanoplasmonic waveguides: towards applications in integrated nanophotonic circuits, Light: Sci. Appl., 2015, 4, e294 CrossRef CAS.
- X. Y. Wang, Y. L. Wang, S. Wang, B. Li, X. W. Zhang, L. Dai and R. M. Ma, Lasing Enhanced Surface Plasmon Resonance Sensing, Nanophotonics, 2017, 6, 472–478 CAS.
- A. Z. Li, M. Zhang, X. Wang, S. B. Wang, B. G. Guo, E. Lewis and P. F. Wang, Directly pumped Ho3+-doped microspheres lasing at 2.0 μm, IEEE Photonics Technol. Lett., 2019, 31, 1366–1368 CAS.
- S. Balaji, G. Gupta, K. Biswas, D. Ghosh and K. Annapurna, Role of Yb3+ ions on enhanced ∼2.9 μm emission from Ho3+ ions in low phonon oxide glass system, Sci. Rep., 2016, 6, 29203 CrossRef CAS PubMed.
- M. Eichhorn, Quasi-three-level solid-state lasers in the near and mid infrared based on trivalent rare earth ions, Appl. Phys. B, 2008, 93, 269–316 CrossRef CAS.
- M. C. Pujol, J. Massons, M. Aguiló, F. Díaz, M. Rico and C. Zaldo, Emission Cross Sections and Spectroscopy of Ho3+ Laser Channels in KGd(WO4)2 Single Crystal, IEEE J. Quantum Electron., 2002, 38, 93–100 CrossRef CAS.
- A. Ciric and S. Stojadinovic, Structural and photoluminescence properties of Y2O3 and Y2O3:Ln3+ (Ln = Eu, Er, Ho) films synthesized by plasma electrolytic oxidation of yttrium substrate, J. Lumin., 2020, 217, 116762 CrossRef CAS.
- T. S. Atabaev, H. H. T. Vu, Y. D. Kim, J. H. Lee, H. K. Kim and Y. H. Hwang, Synthesis and luminescence properties of Ho3+ doped Y2O3 submicron particles, J. Phys. Chem. Solids, 2012, 73, 176–181 CAS.
- B. M. Walsh, J. M. McMahon, W. C. Edwards, N. P. Barnes, R. W. Equall and R. L. Hutcheson, Spectroscopic characterization of Nd:Y2O3: application toward a differential absorption lidar system for remote sensing of ozone, J. Opt. Soc. Am. B, 2002, 19, 2893–2903 CrossRef CAS.
- M. Ju, Y. Xiao, W.
G. Sun, C. Lu and Y. Y. Yeung, In-depth determination of the microstructure and energy transition mechanism for Nd3+-doped yttrium oxide laser crystals, J. Phys. Chem. C, 2020, 124, 2113–2119 CrossRef CAS.
- V. Singh, V. K. Rai, B. Voss, M. Haase, R. P. S. Chakradhar, D. T. Naidu and S. H. Kim, Photoluminescence study of nanocrystalline Y2O3:Ho3+ phosphor, Spectrochim. Acta, Part A, 2013, 109, 206–212 CrossRef CAS PubMed.
- G. A. Newburgh, A. W. Daniels, A. Michael, L. D. Merkle, A. Ikesue and M. Dubinskii, Resonantly diode-pumped Ho3+:Y2O3 ceramic, Opt. Express, 2011, 19, 3604–3611 CrossRef CAS PubMed.
- L. Laversenne, C. Goutaudier, Y. Guyot, M. T. Cohen-Adad and G. Boulon, Growth of rare earth (RE) doped concentration gradient crystal fibers and analysis of dynamical processes of laser resonant transitions in RE-doped Y2O3 (RE = Yb3+, Er3+, Ho3+), J. Alloys Compd., 2002, 341, 214–219 CrossRef CAS.
- F. Qin, Y. D. Zheng, Y. Yu, Z. M. Chen, P. S. Tayebi, W. W. Cao and Z. G. Zhang, Ultraviolet and violet upconversion luminescence in Ho3+-doped Y2O3 ceramic induced by 532-nm CW laser, J. Alloys Compd., 2011, 509, 1115–1118 CrossRef CAS.
- F. Wang, J. W. Tang, E. H. Li, C. F. Shen, J. Wang, D. Y. Tang and D. Y. Shen, Ho3+:Y2O3 ceramic laser generated over 113 W of output power at 2117 nm, Opt. Lett., 2019, 44, 5933–5936 CrossRef CAS PubMed.
- Y. C. Wang, J. Lv, L. Zhu and Y. M. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. C. Wang, J. Lv, L. Zhu and Y. M. Ma, CALYPSO: a method for crystal structure prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. C. Wang, M. S. Miao, J. Lv, L. Zhu, K. T. Yin, H. Y. Liu and Y. M. Ma, An effective structure prediction method for layered materials based on 2D particle swarm optimization algorithm, J. Chem. Phys., 2012, 137, 224108 CrossRef PubMed.
- S. H. Lu, Y. C. Wang, H. Y. Liu, M. S. Miao and Y. M. Ma, Self-assembled ultrathin nanotubes on diamond (100) surface, Nat. Commun., 2014, 5, 3666 CrossRef CAS PubMed.
- L. Zhu, H. Y. Liu, C. J. Pickard, G. T. Zou and Y. M. Ma, Reactions of xenon with iron and nickel are predicted in the Earth's inner core, Nat. Chem., 2014, 6, 644–648 CrossRef CAS PubMed.
- J. Lv, Y. C. Wang, L. Zhu and Y. M. Ma, Particle-swarm structure prediction on clusters, J. Chem. Phys., 2012, 137, 084104 CrossRef PubMed.
- Y. Xiao, M. Ju, X. Y. Kuang and Y. Y. Yeung, A systematic study of the microstructure and laser characteristics of Pr3+-doped lithium lutetium fluoride, J. Alloys Compd., 2018, 749, 391–398 CrossRef CAS.
- G. Bo, P. Gao, S. Lu, J. Lv, Y. Wang and Y. Ma, Interface structure prediction via CALYPSO method, Sci. Bull., 2019, 64, 301 CrossRef.
- C. Lu and C. Chen, Indentation-strain stiffening in tungsten nitrides: mechanisms and implications, Phys. Rev. Mater., 2020, 4, 043402 CrossRef CAS.
- C. Lu and C. Chen, Structure-strength relations of distinct MoN phases from first-principles calculations, Phys. Rev. Mater., 2020, 4, 044002 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA + U Method, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- B. I. Min, H. J. F. Jansen, T. Oguchi and A. J. Freeman, Local density total energy description of ground and excited state properties of the rare earth metals, J. Magn. Magn. Mater., 1986, 61, 139–150 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- R. M. Wallace and G. Wilk, Alternative gate dielectrics for microelectronics, Bulletin, 2002, 27, 186 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Lu Pana,
Chuanzhao Zhang
ab,
Lu Pana,
Chuanzhao Zhang