
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses of four porous 3-D Cd(II) metal–organic frameworks from a new multidentate ligand 5-(imidazol-1-yl)-N′-(pyridin-4-ylmethylene) nicotinohydrazide and their characterization and adsorption properties†
Gui-Ying Zhu‡
a,
Zhenliang Liu‡b,
Jia Wanga,
Xue Niua,
Teng Wanga,
Guo-Xia Jin*a and
Jian-Ping Ma *a
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, P. R. China. E-mail: xxgk123@163.com; guoxiajin@sdnu.edu.cn
bSchool of Chemistry and Pharmaceutical Engineering, Shandong First Medical University & Shandong Academy of Medical Sciences, Taian 271016, P. R. China
First published on 18th August 2020
Abstract
Herein, a new multidentate ligand, 5-(imidazol-1-yl)-N′-(pyridin-4-ylmethylene) nicotinohydrazide (L), with an acylhydrazone group was synthesized and characterized. Subsequently, four porous Cd(II)-MOFs, i.e. [Cd(L)(NO3)]n (1), [Cd(L)Cl]n (2), [Cd(L)Br]n (3), and [Cd(L)I]n (4), were assembled using the ligand L by a solvothermal method and characterized by single-crystal X-ray diffraction, infrared spectroscopy, thermogravimetric analysis, and powder X-ray diffraction. Structural analysis shows that the coordination environments around Cd(II) in all the four compounds are different due to the different coordinated anions. Among them, the coordination geometries and the arrangement of five-coordinated groups of the compound 1 containing the coordinated NO3− anions are significantly different from those of the other three compounds containing halides. However, all the four MOFs have similar one-dimensional rhombic channels. In these channels, both the nitrate ions and the halide ions are attached to the inner walls of the pores. The CO2 adsorption properties of 1–4 were studied at 273 K, and the results showed that these compounds exhibit different adsorption capacities for CO2 due to the presence of different ions in their pores.
Introduction
In recent years, metal–organic frameworks (MOFs) have been extensively studied in the adsorption and separation of guest molecules, such as heavy metal ions,1,2 chiral molecules,3 dyes,4 water,5 and gases and volatile organic compounds,6–10 due to their advantages of high porosity, low density, large surface area, regular channels, and tunable pore diameter. As is well-known, the pore size and functional interior environment of MOFs are the key factors determining the type of guest molecules adsorbed. Therefore, it is important to design and synthesize MOFs with suitable pore size and chemistry for the specific adsorption and separation of guest molecules. The pore size and interior surface of MOFs can be tuned and modified by changing the organic or inorganic building units via various ways, for example, changing the length and geometry of ligands,11 introducing functional groups,12,13 shifting counteranions,9 using a mixed-ligand strategy,14 and increasing the unsaturation sites of metal centers and improving the symmetry of inorganic core clusters.15,16 Usually, the MOFs self-assembled from ligands containing multi-heteroatoms have better guest adsorption capacities due to weak host–guest interactions. For example, Zheng and co-workers have demonstrated that the MOFs synthesized from ligands containing acylamide groups have significantly improved CO2 selective adsorption due to their enhanced CO2 binding ability.17 Dong and co-workers have reported that a Cd(II)-MOF constructed using a 4-amino-1,2,4-triazole-bridged ligand exhibits excellent adsorption and separation ability for aromatic, chlorocarbon, and C2H2 molecules because of its host–guest hydrogen-bonding interactions.9,18,19 In this study, we designed and synthesized a new multidentate ligand, 5-(imidazol-1-yl)-N′-(pyridin-4-ylmethylene) nicotinohydrazide (L), with an acylhydrazide chelating group and two terminal coordination groups. Subsequently, four 3D porous Cd(II)-MOFs, [Cd(L)(NO3)]n (1), [Cd(L)Cl]n (2), [Cd(L)Br]n (3), and [Cd(L)I]n (4), containing one-dimensional rhombic channels were synthesized by the self-assembly of the ligand L under solvothermal conditions and characterized by single-crystal X-ray diffraction, infrared spectroscopy, thermogravimetric analysis, and powder X-ray diffraction. In addition, the CO2 adsorption properties of the MOFs 1–4 were preliminarily studied.Results and discussion
Crystal structure of the compound [Cd(L)(NO3)]n (1)
Single-crystal X-ray diffraction analysis shows that the compound 1 crystallizes in the monoclinic space group P21/c. Its asymmetric unit contains one Cd(II) atom, one ligand L, and one nitrate anion (Fig. 1). Cd(II) lies in an irregular [CdN4O3] pentagonal bipyramidal coordination environment. N6ii, O1i, N2i, O3, and O2 from the imidazolyl and acylhydrazone chelating groups of the two ligands and one nitrate anion lie in the equatorial plane [Cd1–N6ii = 2.289(3), Cd1–O1i = 2.291(2), Cd1–N2i = 2.387(3), Cd1–O3 = 2.408(3), and Cd1–O2 = 2.638(3) Å]. The bond angles of N6ii–Cd1–O1i, N2i–Cd1–O3, N6i–Cd1–O2, and O1i–Cd1–N2i are 82.62(10), 81.84(11), 76.80(10), and 69.37(10)°, respectively. Furthermore, two Npyridyl atoms from the terminal and di-substituted pyridyl groups of the other two ligands are located at the axial positions with the bond angles N1iii–Cd1–N4, N6ii–Cd1–N4, N6ii–Cd1–N1iii, O1i–Cd1–N4, and N6ii–Cd1–O1i being 169.46(10), 90.06(12), 99.51(12), 90.19(11), and 82.62(10)°, respectively. The bond lengths of Cd1–N1iii and Cd1–N4 are 2.332(3) and 2.425(3) Å, respectively. Moreover, two Cd(II) atoms are connected by the imidazolyl and acylhydrazone chelating groups of the two ligands [C7–O1 = 1.257(5), C7–N3 = 1.359(5), and C6–N2 = 1.277(5) Å] to form two kinds of 22-membered planar macrocycles with different axial positions (Fig. 2a). The dihedral angle between the two kinds of macrocyclic planes is ca. 77°. These two kinds of macrocyclic units are alternatively linked through Cd–N bonds between the Cd(II) atoms and di-substituted pyridyl nitrogen atoms to form a 2D network extending along the bc plane. The 2D nets are further parallelly connected by the Cd–N1 bonds (N1 from the terminal pyridyl groups), as viewed down the crystallographic c-axis (Fig. 2b), to produce a 3D framework with 4,4-connected net nodes and pts topology (Fig. 3). Note that there exist 1D rhombic channels (cross section 12.18 Å × 13.52 Å), as viewed along the crystallographic [101] direction. The coordinated nitrate anions are located on the interior surface of the channels. Based on our previous study,20 ligands containing monodentate and bidentate chelating binding sites can be very useful types of organic ligands for the construction of porous frameworks with high dimensionality. This result shows that the ligand L containing the imidazolyl and pyridyl terminal groups and acylhydrazone chelating binding sites can be an efficient organic linker for the construction of porous 3D frameworks.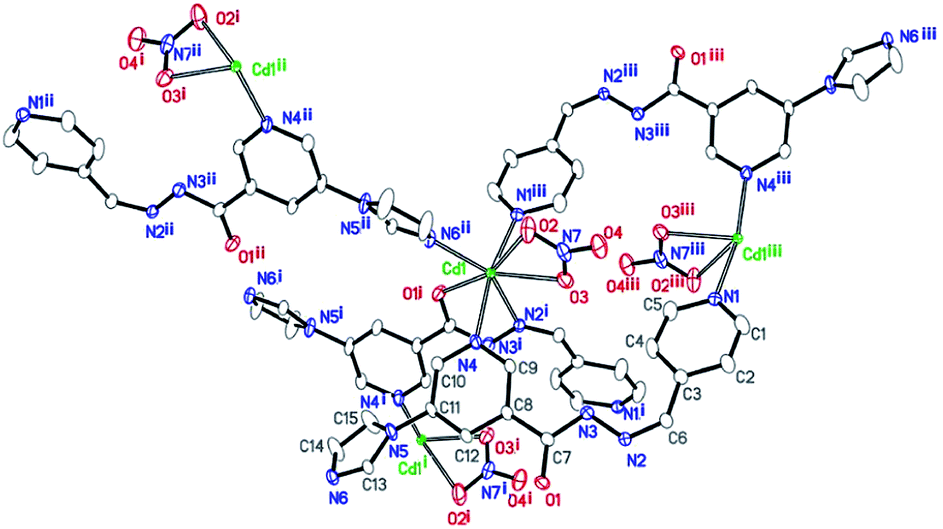 | ||
| Fig. 1 Coordination environment of the Cd(II) centers in 1, with displacement ellipsoids drawn at a 50% probability level [symmetry codes: (i): −x, −y + 1, −z + 2; (ii): −x + 1, y + 1/2, −z + 5/2; and (iii): x, −y + 1/2, z + 1/2]. | ||
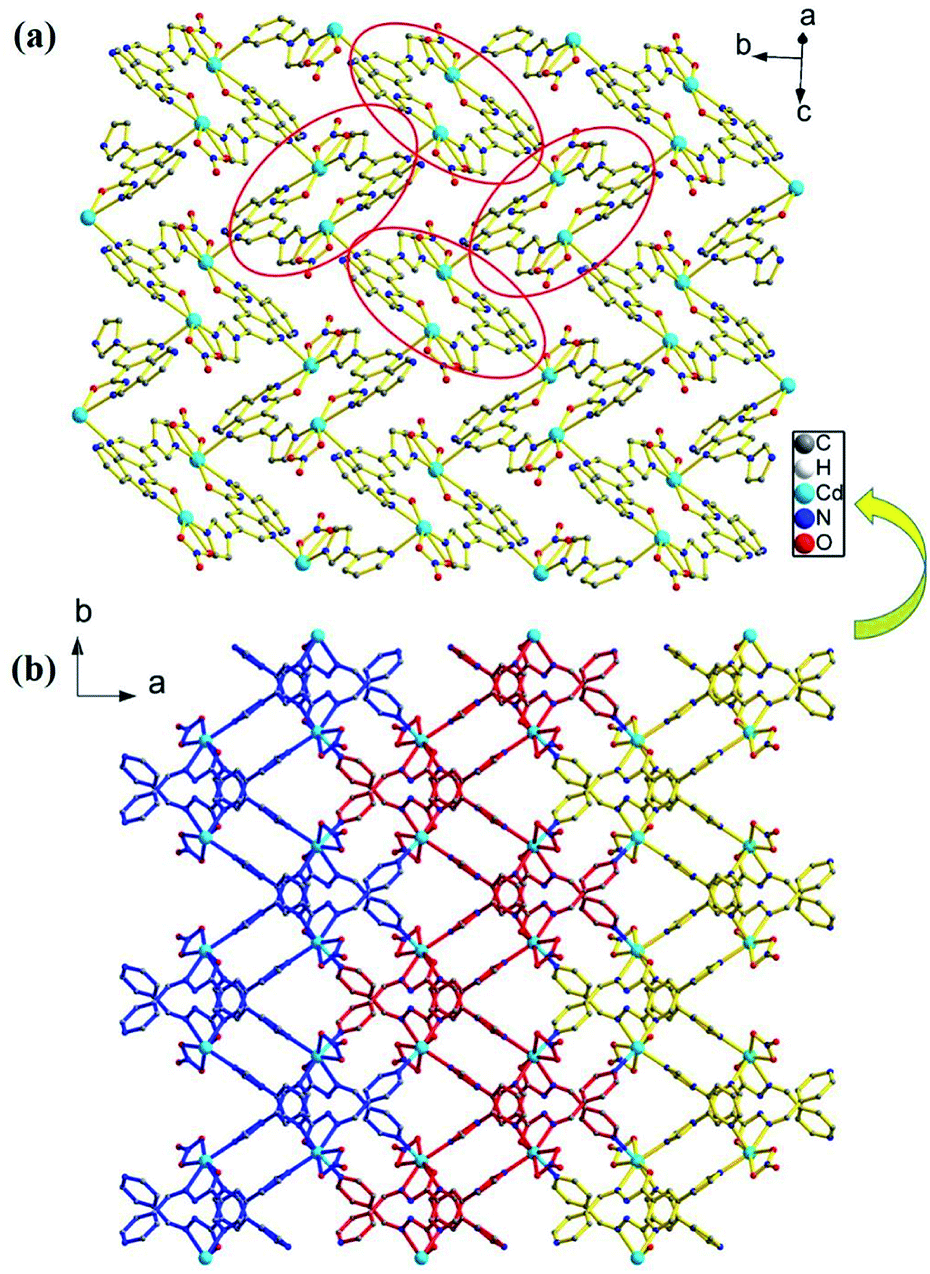 | ||
| Fig. 2 (a) A 2D network containing two kinds of macrocycles with different axial positions viewed along the [302] crystallographic direction. (b) Parallel arrangement of the 2D networks along the a-axis, connected through Cd–Npyridyl bonds to form a 3D framework in 1. | ||
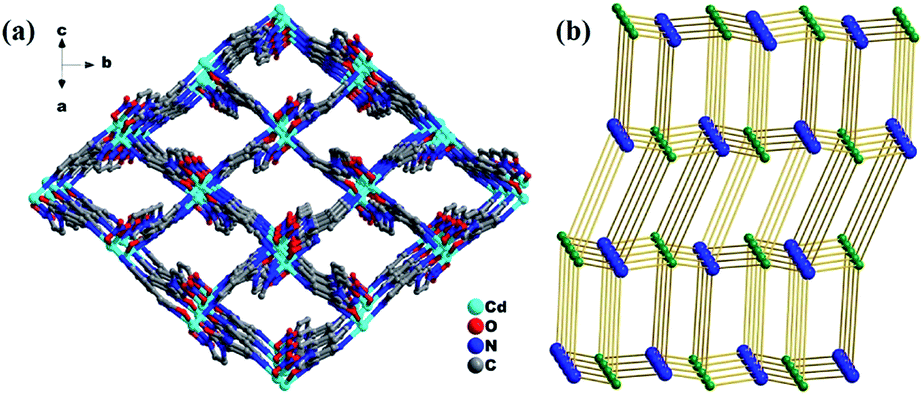 | ||
| Fig. 3 (a) 3D framework of 1 and the rhombic channels viewed along the [101] crystallographic direction and (b) schematic of the (4,4)-connected pts network. | ||
Crystal structures of the compounds [Cd(L)(X)]n (2: X = Cl; 3: X = Br; and 4: X = I).
Compound 2 also crystallizes in the monoclinic space group P21/c. Single-crystal analysis shows that the compound 2 has a similar 3D porous structure as 1. As shown in Fig. 4, its asymmetric unit consists of one cadmium atom, one ligand, and one coordinated chloride ion. The Cd(II) center lies in a distorted octahedral environment. N5iii, O1ii, and N2ii from the imidazolyl group and acylhydrazone chelating group of the two ligands and Cl1 lie in the equatorial plane [Cd1–N5iii = 2.291(3), Cd1–N2ii = 2.358(3), Cd1–O1ii = 2.287(2), and Cd1–Cl1 = 2.4997(8) Å]. The bond angles of O1ii–Cd1–N5iii, N5iii–Cd1–Cl1, O1ii–Cd1–N2ii, and N2ii–Cd1–Cl1 are 86.04(9), 100.15(7), 69.91(9), and 103.32(7)°, respectively. In addition, two Npyridyl atoms (N1i and N6) from the terminal and di-substituted pyridyl groups of the other two ligands are located at the axial positions with the bond angle N1i–Cd1–N6 being 176.71(9)°. The bond lengths of Cd1–N1i and Cd1–N6 are 2.403(3) and 2.500(3) Å, respectively. The bond angles of Cl1–Cd1–N6, N5iii–Cd1–N6, N2ii–Cd1–N1i, N5iii–Cd1–N1i, are 89.81(7), 92.48(10), 95.49(10), and 90.69(10)°, respectively.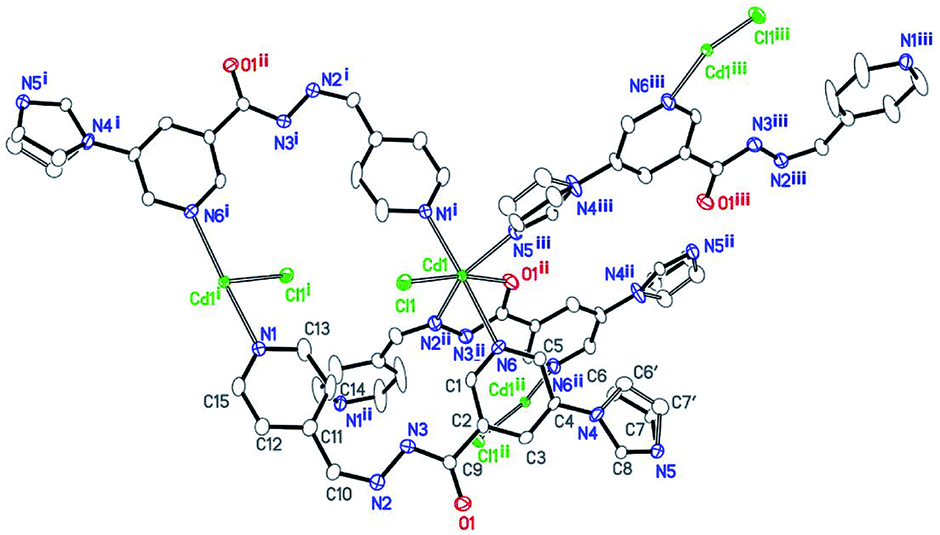 | ||
| Fig. 4 Coordination environment of the Cd(II) centers in 2, with displacement ellipsoids drawn at a 50% probability level [symmetry codes: (i): −x + 1, −y, −z; (ii): x, −y + 1/2, z + 1/2; and (iii): −x + 2, y − 1/2, −z + 1/2]. | ||
Similar to the case of 1, every two Cd(II) atoms are connected by the imidazolyl and acylhydrazone chelating groups of the two ligands [C9–O1 = 1.257(2), C9–N3 = 1.348(2), and C10–N2 = 1.287(2) Å] to form a 22-membered planar macrocycles with different axial positions (Fig. 5). The dihedral angle between these two kinds of macrocyclic planes is ca. 66°. These two kinds of macrocyclic units are alternatively linked through Cd–N6 bonds between the Cd atoms and di-substituted pyridyl nitrogen atoms to form a 2D network extending along the bc plane. The 2D nets are further parallelly connected by Cd–N1 (N1 from the terminal pyridyl groups) bonds to yield a 3D porous framework with rhombic channels (cross section 13.86 Å × 14.19 Å), as viewed along the [101] crystallographic direction (Fig. S1 and S2†). Compared to the case of 1, the coordinated chloride atoms are located on the interior surface of the channels in 2.
 | ||
| Fig. 5 2D network in 2 containing two kinds of macrocycles with different axial positions viewed along the [302] crystallographic direction. | ||
When CdBr2 and CdI2 were used in the assembly reactions with the ligand L, the porous MOFs [Cd(L)(Br)]n (3) and [Cd(L)(I)]n (4) were obtained, which were allomeric with 2 (Fig. S3–S6†). Single-crystal analysis indicated that the Cd(II) centers lie in a similar distorted octahedral environment as in the case of 2, except for the coordinated bromine and iodine atoms.
Further insights into the structures of 1–4 show that the coordination environments around Cd(II) in all the four compounds were different due to the different coordinated anions, as shown in Fig. 6. Among them, the coordination geometries and the arrangement of five-coordinated groups of the compound 1 containing coordinated NO3− are significantly different from those of the other three compounds containing halides. By comparison, the angles of N–Cd1–N located at the axial positions and O–Cd–N located at the equatorial positions decreased with an increase in the anion radius and size. The dihedral angles between the mono- and di-substituted pyridine rings also exhibited similar changes [2: 17.495°; 3: 18.126°; 4: 21.852°; and 1: 21.973°]. Due to the difference in the Cd(II) coordination environments, the pore sizes of 1–4 also exhibit regular subtle changes; for example, the cross section areas are 12.18 Å × 13.52 Å (1), 13.86 Å × 14.19 Å (2), 13.72 Å × 14.15 Å (3), and 12.09 Å × 13.69 Å (4). Among them, the MOFs 1 and 4 with the biggest nitrate and iodine ions have the smallest channels, and the MOF 2 with the smallest chlorine ions has the biggest channels. Thus, in this study, the pore sizes and interior surfaces of the Cd(II)-MOFs could be tuned by changing the anions.
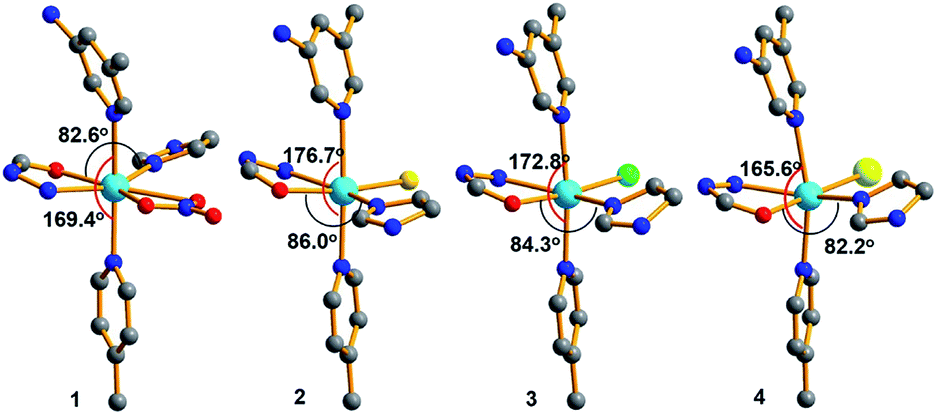 | ||
| Fig. 6 Coordination environment around the Cd(II) centers in 1–4. | ||
IR, TGA and PXRD
Infrared (IR) spectra of the compounds 1–4 (Fig. S7†) show strong absorption peaks around 1666 cm−1, which are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the acylhydrazone group; the peaks around 1590 cm−1 can be attributed to the CN stretching vibration of pyridyl groups; the peak around 1330 cm−1 corresponds to the N–N stretching vibration of the acyl hydrazone group.
O stretching vibration of the acylhydrazone group; the peaks around 1590 cm−1 can be attributed to the CN stretching vibration of pyridyl groups; the peak around 1330 cm−1 corresponds to the N–N stretching vibration of the acyl hydrazone group.
Thermogravimetric analysis (TGA) curves shown in Fig. 7 reveal that the frameworks of 1–4 have similar thermal stabilities. All the four MOFs were stable up to around 270 °C, after which they began to decompose. The compounds 1–3 underwent the first weight loss in the temperature range from 25 to 200 °C due to the loss of the guest DMF molecules, which were confirmed to be present in the MOFs by an 1H NMR spectrum (Fig. S8†), and the framework started to disintegrate at ca. 270 °C. In the case of the compound 4, these encapsulated DMF molecules were removed at temperatures ranging from 25 to 270 °C, and then, the framework began to decompose.
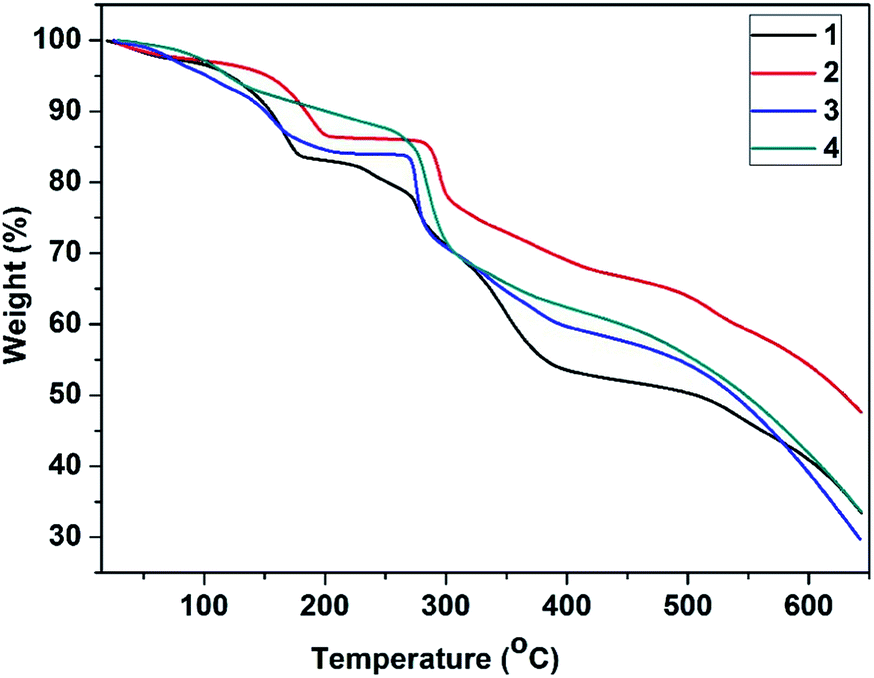 | ||
| Fig. 7 TGA curves of 1–4. | ||
The room-temperature powder X-ray diffraction (PXRD) patterns of 1–4 are shown in Fig. S9.† All the peaks in the obtained curves approximately match those in the simulated curves generated based on the single-crystal diffraction data; this confirms the phase purity of the as-prepared products.
CO2 adsorption properties of the MOFs 1–4
Considering the existence of 1D channels, the CO2 adsorption measurements were performed for the desolvated MOFs 1–4. Before measurement, the as-synthesized crystal samples of 1–4 were immersed in methanol to exchange the uncoordinated DMF molecules. Then, drying was conducted at 60 °C under high vacuum for 3 h to obtain activated samples.The adsorption isotherm curves of the MOFs 1–4 for CO2 were measured, and the MOFs 3 and 4 exhibited typical type-I isotherms and the adsorption curves were in agreement with the desorption curves at 273 K, as shown in Fig. 8. However, the other two MOFs, especially the MOF 1, displayed a sharp adsorption increase at one point upon increasing the pressure. This phenomenon indicated that the MOFs might undergo subtle structural changes and the windows of the channels suddenly open; this might also have resulted in the hysteretic phenomenon of their desorption curves. The adsorption isotherms suggest that the CO2 sorption capacity is 6.80 cm3 g−1 for 2, 17.67 cm3 g−1 for 3, 23.27 cm3 g−1 for 4, and 28.30 cm3 g−1 for 1, which are close to those of some of the reported MOFs such as ZIF-95 (19.7 cm3 g−1) and ZIF-8 (32.22 cm3 g−1).21,22 The BET surface areas of 1–4, calculated according to their CO2 adsorption isotherms, are 266.54, 164.10, 151.26, and 165.42 m2 g−1, respectively. In particular, the Cd-MOFs 1 and 4 with bigger nitrate and iodine ions are beneficial for the adsorption of CO2 than 2 with a smaller chloride ion. These results show that the anions attached to the frameworks have an effect on the adsorption ability of the Cd-MOFs. The CO2 uptake capacity gradually increases with an increase in the size of the anion. This is probably because different anions change the pore volumes differently such that the pores with larger anions better match the CO2 molecules. On the other hand, the I− anion has bigger polarizability than Cl− and Br−, and NO3− has more N–O polar bonds. Thus, the lone pairs of the more polarizable I atom or electronegative O atom may interact with the electropositive C atom of CO2; this would probably lead to better sorption capacity of the MOFs containing I− and NO3− anions than those of the MOFs with Cl− and Br−.23 Overall, the pore sizes are substantially large as compared to the adsorbed gas molecules; therefore, the sorption capacities of all the four MOFs are relatively low.
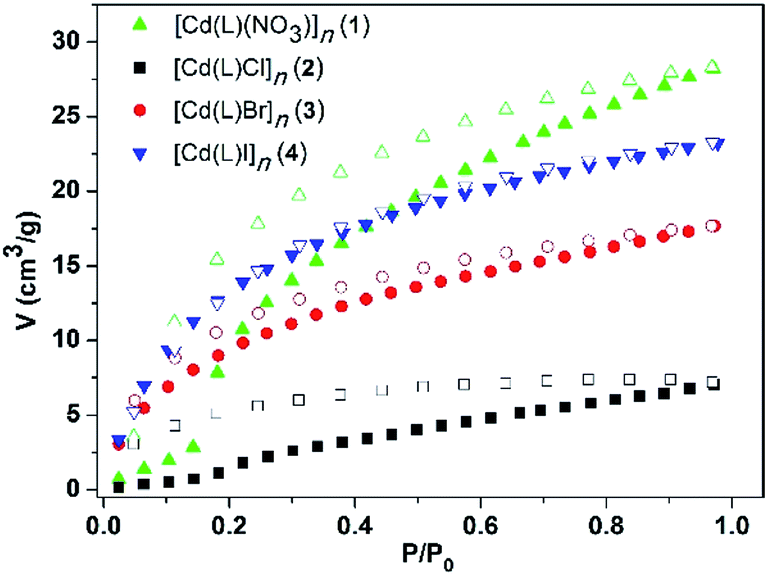 | ||
| Fig. 8 CO2 adsorption isotherm curves of the MOFs 1–4 at 273 K. | ||
Experimental
General methods
All reagents were used as purchased without further purification. IR spectra were obtained by a BIO-RAD FTS-40 Infrared Spectrometer (KBr pellet). TGA was conducted using a TA instrument, Q5 series thermogravimetric analyzer, at the heating rate of 10 °C min−1. Powder X-ray diffraction was carried out by a Bruker powder diffractometer with Cu Kα radiation as the X-ray source and the scanning angle of 2-theta = 5–50°. Photoluminescence was determined by an FLS-920 fluorescence analyzer.Synthesis of the ligand
Synthesis of the MOFs
Refinement
For 1: the hydrogen atoms attached to carbon atoms were placed in geometrically idealized positions and refined using a riding model with C–H = 0.93 (aromatic) and Uiso(H) = 1.2Ueq(C) for all H atoms. A large void space is present in the framework, which exhibits many significant electron density peaks. The species were too severely disordered to be modeled and were treated with SQUEEZE/PLATON. The solvent-accessible void volume calculated by this program was 812 Å3 (35.4% of the total unit cell volume (2291.8(2) Å3)), corresponding to 264 e− per cell. These species include some DMF and H2O molecules.For 2: the atoms C6 and C7 of the imidazole ring are disordered over two orientations, with refined site occupation factors of 0.46(5)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.54(5). A large void space is present in the framework, and the species were too severely disordered to be modeled. Therefore, they were treated with SQUEEZE/PLATON, and the solvent-accessible void volume calculated by this program was 758 Å3 (34.2% of the total unit cell volume (2219.14(9) Å3)), corresponding to 228 e− per cell. These species include some DMF and H2O molecules.
:0.54(5). A large void space is present in the framework, and the species were too severely disordered to be modeled. Therefore, they were treated with SQUEEZE/PLATON, and the solvent-accessible void volume calculated by this program was 758 Å3 (34.2% of the total unit cell volume (2219.14(9) Å3)), corresponding to 228 e− per cell. These species include some DMF and H2O molecules.
For 3: all non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms attached to carbon atoms were placed in geometrically idealized positions and refined using a riding model with C–H = 0.93 (aromatic) and Uiso(H) = 1.2Ueq(C) for all H atoms. The atoms C6 and C7 of the imidazole ring are disordered over two orientations, with the refined site occupation factors of 0.48(8):0.52(8). A large void space is present in the framework, which exhibits many significant electron density peaks. The species were too severely disordered to be modeled and were treated with SQUEEZE/PLATON. The solvent-accessible void volume calculated by this program was 721 Å3 (31.9% of the total unit cell volume (2262.9(2) Å3)), corresponding to 231 e− per cell. These species include some DMF and H2O molecules. The contribution of the disordered species was removed from the structure factor calculations. The tabulated F(000), MW, and density reflect known cell contents only.
For 4: The hydrogen atoms attached to carbon atoms were placed in geometrically idealized positions and refined using a riding model with C–H = 0.93 (aromatic) and Uiso(H) = 1.2Ueq(C) for all H atoms. A large void space is present in the framework, and the species were too severely disordered to be modeled and were treated with SQUEEZE/PLATON. The solvent-accessible void volume calculated by this program was 810 Å3 (35.4% of the total unit cell volume (2284.1(2) Å3)), corresponding to 281 e− per cell. These species include some DMF and H2O molecules.
Gas adsorption measurements
The CO2 sorption isotherms were measured using an automatic volumetric adsorption apparatus (Quantachrome Autosorb-IQ-MP) at 273 K. Before gas adsorption testing, the as-synthesized compounds 1–4 were placed in renewed methanol for one day (three times) in order to totally remove the DMF molecules. The samples were activated by drying in a vacuum oven at 90 °C for 30 min. Before measurement, the activated samples were dehydrated again via the “outgas” function of the adsorption analyzer for 3 h at 80 °C.Conclusions
In this study, a new multidentate ligand, 5-(imidazol-1-yl)-N′-(pyridin-4-ylmethylene) nicotinohydrazide (L), with an acylhydrazide group was synthesized and characterized. Subsequently, four porous Cd(II)-MOFs, [Cd(L)(NO3)]n (1), [Cd(L)Cl]n (2), [Cd(L)Br]n (3) and [Cd(L)I]n (4), were assembled using the ligand L by a solvothermal method and characterized by single-crystal X-ray diffraction, infrared spectroscopy, thermogravimetric analyses, and powder X-ray diffraction. Structural analysis shows that the coordination environments around Cd(II) in all the four compounds were different due to the different coordinated anions. Among them, the coordination geometries and the arrangement of five-coordinated groups of the compound 1 containing the coordinated NO3− anions are significantly different from those of the other three compounds containing halides. However, all four MOFs have similar one-dimensional rhombic channels. In these channels, both nitrate ions and halide ions are attached on the inner walls of the pores. The CO2 adsorption properties of 1–4 were studied at 273 K, and the results showed that these compounds exhibit different adsorption capacities for CO2 due to different ions in their pores. Further research on the adsorption properties of this type of Cd-MOFs for other guests is underway.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support provided by NSFC (Grant No. 21771120).Notes and references
- Z. H. Yang, J. Cao, Y. P. Chen, X. Li, W. P. Xiong, Y. Y. Zhou, C. Y. Zhou, R. Xu and Y. R. Zhang, Microporous Mesoporous Mater., 2019, 277, 277–285 CrossRef CAS.
- Y. Wu, Y. Ma, G. Xu, T. Xia, W. Liu, Z. Dong, Q. Yuan, C. Zhang and Q. J. Hu, Mater. Sci., 2019, 54, 2093–2101 CAS.
- D. Balestri, D. Capucci, N. Demitri, A. Bacchi and P. Pelagatti, Materials, 2017, 10, 727 CrossRef PubMed.
- D. Jiang, M. Chen, H. Wang, G. Zeng, D. Huang, M. Cheng, Y. Liu, W. Xue and Z. Wang, Coord. Chem. Rev., 2019, 380, 471–483 CrossRef CAS.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- X. Wang, Y. Wu, J. Peng, Y. Wu, J. Xiao, Q. Xia and Z. Li, Chem. Eng. J., 2019, 358, 1114–1125 CrossRef CAS.
- P. Q. Liao, N. Y. Huang, W. X. Zhang, J. P. Zhang and X. M. Chen, Science, 2017, 356, 1193–1196 CrossRef CAS PubMed.
- N. Thi, X. Huynh, V. Chihaia and D. N. Son, J. Mater. Sci., 2019, 54, 3994–4010 CrossRef.
- G. X. Jin, X. Niu, J. Wang, J. P. Ma, T. L. Hu and Y. B. Dong, Chem. Mater., 2018, 30, 7433–7437 CrossRef CAS.
- G. X. Jin, J. Wang, J. Y. Liu, J. P. Ma and Y. B. Dong, Inorg. Chem., 2018, 57, 6218–6221 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- Y. B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed.
- Z. Lu, F. Meng, L. Du, W. Jiang, H. Cao, J. Duan, H. Huang and H. He, Inorg. Chem., 2018, 57, 14018–14022 CrossRef CAS PubMed.
- B. Liu, H. F. Zhou, L. Hou and Y. Y. Wang, Dalton Trans., 2018, 47, 5298–5303 RSC.
- I. Bratsos, C. Tampaxis, I. Spanopoulos, N. Demitri, T. A. Steriotis and P. N. Trikalitis, Inorg. Chem., 2018, 57, 7244–7251 CrossRef CAS PubMed.
- J. Jiang, Z. Lu, M. Zhang, J. Duan, W. Zhang, Y. Pan and J. Bai, J. Am. Chem. Soc., 2018, 140, 17825–17829 CrossRef CAS PubMed.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed.
- Q. K. Liu, J. P. Ma and Y. B. Dong, J. Am. Chem. Soc., 2010, 132, 7005–7017 CrossRef CAS PubMed.
- Q. K. Liu, J. P. Ma and Y. B. Dong, Chem. Commun., 2011, 47, 12343 RSC.
- X. W. Wu, F. Pan, D. Zhang, G. X. Jin and J.-P. Ma, CrystEngComm, 2017, 19, 5864–5872 RSC.
- A. Phan, C. J. Doonan, F. J. Uribe, C. B. Knobler, M. O. Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- J. W. Maina, P. G. Cristina, J. A. Schütz, J. T. Wang and L. F. Dumee, Carbon, 2019, 148, 80–90 CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1951144–1951147. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05131d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |