
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of substituents and promoters on the Diels–Alder cycloaddition reaction in the biorenewable synthesis of trimellitic acid†
Tuhin Suvra Khan *a,
Shelaka Gupta*b,
Maaz Ahmadc,
Md Imteyaz Alamc and
M. Ali Haider*c
*a,
Shelaka Gupta*b,
Maaz Ahmadc,
Md Imteyaz Alamc and
M. Ali Haider*c
aNanocatalysis Area, Light Stock Processing Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, Uttarakhand, India. E-mail: tuhins.khan@iip.res.in; Tel: +91-135-2525915
bDepartment of Chemical Engineering, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, 502205, India. E-mail: shelaka@che.iith.ac.in
cRenewable Energy and Chemicals Laboratory, Department of Chemical Engineering, Indian Institute of Technology Delhi, Hauz Khas, Delhi, 110016, India. E-mail: haider@iitd.ac.in; Tel: +91-11-2658-2037 Tel: +91-11-26591016
First published on 18th August 2020
Abstract
An efficient route to produce oxanorbornene, a precursor for the production of bio-based trimellitic acid (TMLA) via the Diels–Alder (DA) reaction of biomass-derived dienes and dienophiles has been proposed by utilizing density functional theory (DFT) simulations. It has been suggested that DA reaction of dienes such as 5-hydroxymethyl furfural (HMF), 2,5-dimethylfuran (DMF), furan dicarboxylic acid (FDCA) and biomass-derived dienophiles (ethylene derivatives e.g., acrolein, acrylic acid, etc.) leads to the formation of an intermediate product oxanorbornene, a precursor for the production of TMLA. The activation barriers for the DA reaction were correlated to the type of substituent present on the dienes and dienophiles. Among the dienophiles, acrolein was found to be the best candidate showing a low activation energy (<40 kJ mol−1) for the cycloaddition reaction with dienes DMF, HMF and hydroxy methyl furoic acid (HMFA). The FMO gap and (IPdiene + EAdienophile)/2 were both suggested to be suitable descriptors for the DA reaction of electron-rich diene and electron-deficient dienophile. Further solvents did not have a significant effect on the activation barrier for DA reaction. In contrast, the presence of a Lewis acid was seen to lower the activation barrier due to the reduction in the FMO gap.
Introduction
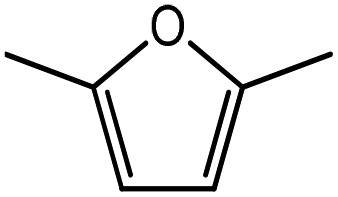
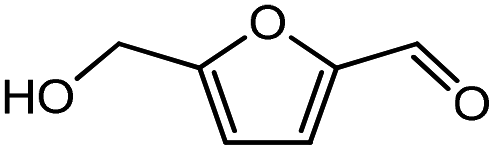
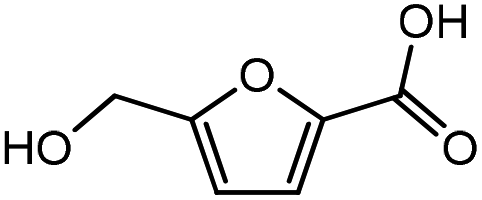
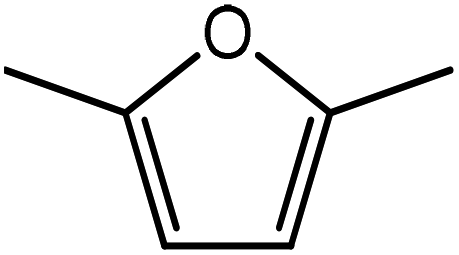
Advances in the field of bio-renewable chemicals have created many opportunities to produce fossil-based chemicals from renewable carbon resources and limit the dependence on fossil reserves.1–8 Since aromatics, predominantly procured from petroleum reserves, are the critical building blocks of the chemical industry for the production of a wide range of products, there is a need to develop bio-based processes for their production.9,10 As a result, multiple approaches have been investigated for the production of aromatics from biomass that includes biological,11 chemocatalytic12,13 and thermochemical14,15 routes. However, the selectivity of aromatics remains a challenge in all these processes. On the other hand, Diels–Alder (DA) reactions which involve the coupling of a conjugated diene and a dienophile, offer a robust route to combine biomass-derived intermediates, particularly furan and furan derivatives, to obtain cyclic products.16–18 Advances in the field of metabolic engineering have expanded the number of biomass-derived intermediates that can be used for DA reactions and will open up avenues for producing aromatic monomers with diverse functionalities.19–21 Following the DA reaction, the cyclic compounds undergo either dehydration,22 decarboxylation,20 or dehydrogenation19 to produce aromatic compounds.Davis et al.17 showed that para-terephthalic acid (PTA) could be produced by reacting 5-hydroxymethylfurfural (HMF) and its derivatives with ethylene, to form an oxanorbornene intermediate, which can undergo dehydration over a solid acid catalyst to form its aromatic equivalent. The aromatic intermediates were subsequently oxidized under mild condition, over low-cost oxide catalyst to obtain PTA. A similar study has been performed by Do et al.23 to obtain p-xylene from DA reaction between 2,5-dimethylfuran (DMF) and ethylene. Moreover, Dauenhauer et al.24 reacted 2-methylfuran (MF) with ethylene to obtain toluene. Furthermore, Toste et al. used DA reaction between DMF and acrolein to obtain p-xylene after oxidative dehydration and decarboxylation.18 However, all DA reactions using DMF have a major obstacle of hydrogenation of HMF to DMF which necessarily requires precious metal catalysts like Pd.25,26 Notably, a higher cost of the catalyst can be a crucial and big hindrance in process commercialization. To overcome the above limitation, Davis et al.17 proposed DA reactions by using HMF and its oxidative derivatives like hydroxy methyl furoic acid (HMFA) and furan dicarboxylic acid (FDCA). Gong et al.27 performed the DA reaction between FDCA and ethylene in water, but the yield of the product was only in traces. The presence of two electron-withdrawing carboxylic groups reduces the electron density on the diene making the normal demand DA reaction unfavourable. Changing the substituent on the furan substituted diene or ethylene substituted dienophile would alter the reaction rate, hence choice of best diene and dienophile combination is essential.
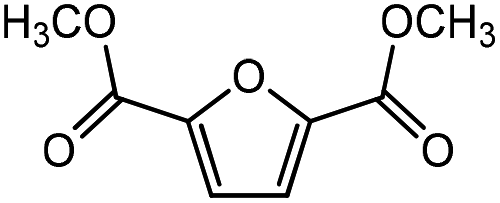
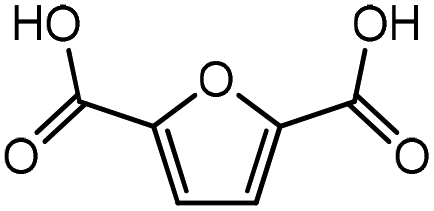
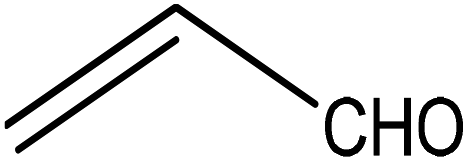
Trimellitic acid (TMLA), a large scale commodity chemical, so far, is produced from fossil fuel-based sources.28 The TMLA and its derivatives find wide applications in the synthesis of resins, paints, coatings, dental adhesives, anti-cancer drugs, plasticizers etc.28–33 A renewable pathway for the production of TMLA can have a broad impact on the chemical value chain. To the best of our knowledge, the possibility of producing TMLA from biomass is yet to be explored. In the present study, an alternative pathway for the production of biobased TMLA has been proposed (Fig. 1(b)). DA reaction between biomass-derived dienes and ethylene based dienophile molecules (Fig. 1(a)) have been proposed to form oxanorbornene, a precursor for the bio-based production of TMLA. This study is particularly focused on studying the effect of substituents present on the dienes and dienophiles (Fig. 1) on the activation barriers for the formation of oxanorbornene via DA reaction using density functional theory (DFT) simulations. DFT simulation have been utilized to study the effect of the type of the substituents present in biomass derived dienes and dienophiles on the rate of formation of the oxanorbornene adduct.
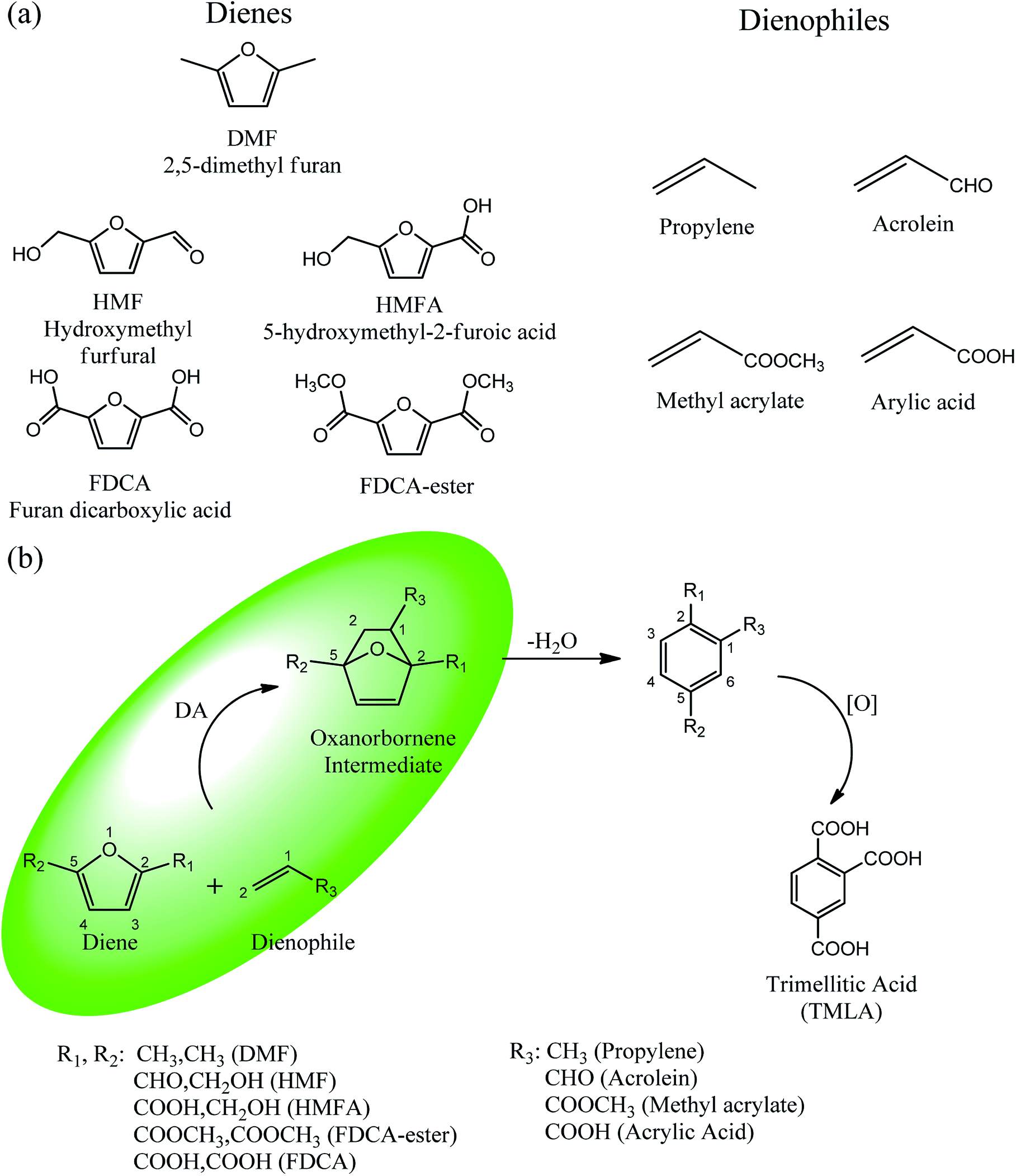 | ||
| Fig. 1 (a) Molecular structure of the dienes and dienophiles studied under the scope of our study. (b) The reaction mechanism of the DA reaction to produce trimellitic acid (TMLA) after dehydration and oxidation of the oxanorbornene intermediate. The highlighted portion in the reaction schematic is the focus of our study. | ||
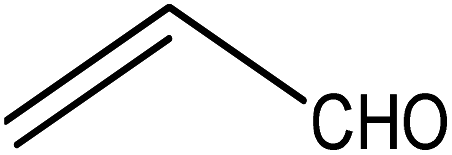
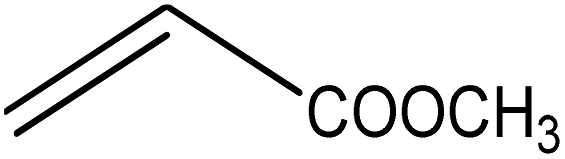
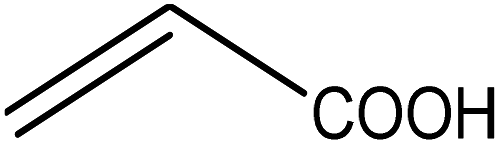
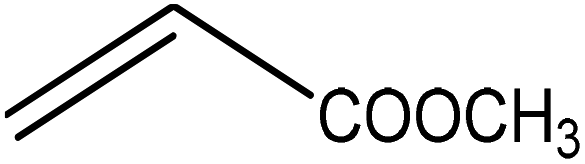
Five different furan based dienes (DMF, HMF, HMFA, FDCA, FDCA-ester) and four ethylene based dienophiles (propylene, acrolein, methyl acrylate and acrylic acid) with different substituents were considered to study the reaction pathway for the formation of the oxanorbornene intermediate (Fig. 1(a)). The DA reactions involving HMF as the diene followed by DMF and then, FDCA-ester was discussed to compare the electronic effects of the substituents present in HMF (electron-withdrawing in FDCA or electron-donating in DMF) on the overall reactivity. The dienophiles discussed in this study are focused mainly on methyl acrylate, acrylic acid, both of which have electron-withdrawing groups attached to the C1 of ethylene and propylene which has a mild electron-donating methyl group at the same position. The above-mentioned set of dienes and dienophiles would cover up all combinations of molecules with varying electronic effects. DFT calculations were performed to calculate the activation and reaction energies for all combinations of the DA reactions between the chosen dienes and dienophiles. The oxanorbornene adduct obtained from the DA reaction can be dehydrated over an acidic catalyst34 to produce aromatic intermediate,35 which upon oxidation will produce TMLA, as shown in the scheme in Fig. 1(b). In this paper, we have presented the kinetic feasibility of the DA reaction by altering the diene and the most optimum pathway for the formation of the oxanorbornene intermediate, a precursor for bio-based production of TMLA.
Methods
DFT calculations were studied based on the Double Numerical Plus Polarization function (DNP) basis set using the DMol3 module in Material Studio 8 (Biovia, San Diego, USA).36 Generalized Gradient Approximation (GGA) with BLYP was used to describe the exchange and correlation energy in the DFT calculations.37 GGA based functional has also been previously used to obtain good correlation with experimental trends in the retro Diels–Alder (rDA) reaction of partially saturated 2-pyrones.38–40 The dispersion effects in the DA reaction were included using Grimme (G06) DFT-D method.41 Convergence criteria were set to 0.0001 eV, 0.05 eV Å−1 and 0.005 Å with respect to energy, force and atom displacement respectively for the geometry optimization. Transition state (TS) search was performed with a linear synchronous transit/quadratic synchronous transit (LST/QST) method using 0.05 eV Å−1 as force convergence criteria.42 During LST, single-point calculations were carried out on linearly interpolated structure on the minimum energy path joining the reactant and product states. The transition state is one which has the maximum energy along this minimum energy path. The structure obtained was taken as an intermediate for QST optimization, resulting in a more refined structure closer to the TS geometry. The obtained TS geometries for different reaction steps were further iterated using the ‘TS optimization’ module in DMol3 to achieve refined TS structures. The vibrational frequencies of TS were analysed and confirmed to be all positive except one imaginary in the direction of the reaction coordinate. The activation energy, Ea was calculated as the energy difference between the transition and initial states,| Ea = ETS − EIS |
The reaction energies, Er for the reactions were calculated as
| Er = EFS − EIS |
B3LYP43 hybrid exchange–correlation functional along with Grimme (G06) DFT-D correction was also applied in order to check the change in the activation barriers of DA reaction. The hybrid B3LYP functional has been shown to be one of the best exchange–correlation functional for studying the reactivity and properties of organic molecules.44,45 The comparison between the result obtained for the activation energies calculated from BLYP functional with Grimme (G06) vdW correction result in a much lower value as compared to B3LYP functional with Grimme (G06) vdW correction (Table SI-1†). However, the general trend in the activation barrier between the diene + dienophile combination remains unchanged, as can be seen in the Table SI-1.†
The energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for the dienes and dienophiles were obtained by checking the orbitals property tab during the geometry optimization in DMol3.
The values of ionization potential and electron affinity for the dienes and dienophiles was calculated by the formula given below,
| IP = Ecation − Eneutral |
| EA = Eneutal − Eanion |
Energies of the cations and anions were obtained by assigning a +ve and −ve charge, respectively, during the geometry optimization of the diene and dienophiles. The solvent environment was simulated using conductor-like screening model (COSMO)46 in which solvent was represented by their respective dielectric constant, ε; water (ε = 78.54), ethanol (ε = 24.3), acetone (ε = 20.7), diethylether (ε = 4.335), CCl4 (ε = 2.238) and n-hexane (ε = 1.89). COSMO based solvent model is based on the dielectric constant of the solvent and is known to capture the electrostatic contributions of the solvent environment towards the reaction energy profile. COSMO based solvent model has been previously used by us to successfully capture the effect of polar and non-polar solvent in the rDA reaction of partially saturated 2-pyrones.39,40
Results and discussions
DA reactions can either proceed via a normal electron demand or an inverse demand path, which primarily depends on the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the diene and dienophile.47,48 In normal demand DA reactions, diene donates the electron to the dienophile during the reaction. Thus, the difference between the HOMO of diene and LUMO of dienophile gives the frontier molecular orbital (FMO) gap.49 Notably, for reactions to proceed via normal demand path, the diene should be electron-rich and the dienophile should be electron deficient in driving the reaction in the forward direction. On the contrary, in inverse demand DA reactions, the dienophile donates the electron to the diene and hence the FMO gap is measured by the difference between the HOMO of dienophile and LUMO of the diene. This situation typically arises when the diene is electron-poor and the dienophile is electron-rich. Hence, the type of substituents (electron-donating or withdrawing) present on the diene and dienophile play an essential role in determining the course of the DA reactions. With the aim to produce TMLA from substituted furan and ethylene derivatives, DFT simulations were used to study the effect of the type of substituents present on the dienes and dienophiles on the rate of formation of oxanorbornene intermediate formed as shown by the reaction scheme in Fig. 1(b). The DFT calculated values for the FMO gap, activation barrier and reaction energy are mentioned in Tables 1 and 2.| Diene + dienophile |
|
|
|
|
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | I | N | I | N | I | N | I | N | I | |
 |
4.16 | 6.06 | 4.46 | 5.61 | 1.84 | 5.55 | 2.52 | 5.95 | 2.22 | 6.13 |
 |
5.13 | 3.69 | 5.43 | 3.24 | 2.81 | 3.19 | 3.49 | 3.58 | 3.19 | 3.77 |
 |
5.34 | 4.06 | 5.64 | 3.61 | 3.02 | 3.55 | 3.70 | 3.95 | 3.4 | 4.14 |
 |
5.57 | 3.56 | 5.87 | 3.10 | 3.25 | 3.05 | 3.93 | 3.44 | 3.64 | 3.63 |
 |
5.89 | 3.22 | 6.20 | 2.77 | 3.58 | 2.71 | 4.26 | 3.11 | 3.96 | 3.29 |
| Diene + dienophile |
|
|
|
|
|
|---|---|---|---|---|---|
 |
83.8 (−11) | 90.0 (−5.9) | 36.9 (−0.8) | 54.6 (−2.1) | 47 (−7.5) |
 |
78.3 (6.7) | 75.5 (11.8) | 39.2 (11.5) | 60.2 (16.8) | 48.1 (3.2) |
 |
74.1 (−7.9) | 69.5 (−5.1) | 39.3 (6.9) | 58.2 (9.3) | 46.1 (−15.7) |
 |
91.4 (22.7) | 90 (25.7) | 68.2 (37.3) | 81.6 (38.6) | 68.9 (11.7) |
 |
93.9 (23.6) | 94.6 (27.5) | 84 (44) | 87.3 (45.3) | 80.8 (32.5) |
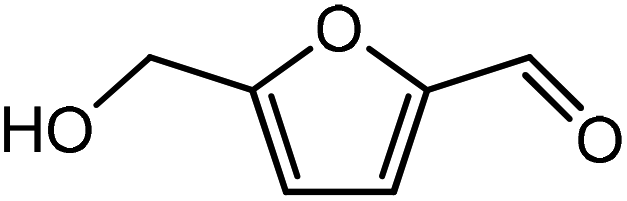
HMF is a versatile biomass derived platform molecule which has been included in the top-10 list of value-added biomass-based chemicals by the US Department of Energy (DOE).50 HMF is obtained from the cellulosic part of lignocellulosic biomass51 by catalytic conversion of carbohydrates, monosaccharides, disaccharides and polysaccharides.52,53 Cellulose upon acid hydrolysis breaks into its sugar monomers i.e., glucose which subsequently in the presence of Lewis acid catalyst undergoes isomerization reaction to produce fructose followed by dehydration in the presence of a Brønsted acid catalyst to produce HMF.54
To begin with, the reaction between HMF and ethylene under the solvent-less condition was studied. The reaction followed the inverse demand path with an FMO gap of 3.69 eV (Table 1). The DFT calculated activation barrier and reaction energy for the reaction between HMF and ethylene was found to be 78.3 and 6.7 kJ mol−1 respectively (Table 2). However, the absence of substituents at C2 position on ethylene implies that ethylene would lead to the production of PTA after dehydration and oxidation of the oxanorbornene intermediate. It cannot be utilized for the production of TMLA unless the aromatic intermediate is methylated and oxidized at a later stage. For this study, ethylene has been used as a reference to study the substituent effect on the dienophiles. On the addition of an electron-withdrawing group on ethylene like an ester, acid or aldehyde, the nature of DA reaction reversed and followed a normal demand path rather than inverse demand.
The result was consistent with the theoretical explanation that electron withdrawing groups in dienophiles make them electron-poor and hence, it is the diene that would transfer an electron to the dienophile during the reaction. The reactant, transition and product states for each of the reactions were also studied, where the bond distance between the carbon atoms of the diene and dienophile involved in bond formation was calculated. For the reaction between HMF and methyl acrylate, the bond distance between the two pair of C atoms i.e. C5 of diene & C2 of dienophile (C5–C2) and C2 of diene & C1 of dienophile (C2–C1) was measured to be 3.29 Å and 3.16 Å in the geometrically optimized reactant state structure (Fig. 2(a)). The transition state was found to be product like as the bond distance between the same pair of atoms reduced to 2.09 Å and 2.15 Å in the transition state structure. In contrast, in the oxanorbornene product, the corresponding bond distances were calculated to be 1.57 Å and 1.60 Å (Fig. 2(a)). The FMO gap for the same reaction was 3.49 eV (Table 1), while the activation barrier was calculated to be 60.2 kJ mol−1 (Table 2). When acrylic acid was chosen as the dienophile, the FMO gap reduced further to 3.19 eV (Table 1) due to a stronger electron-withdrawing acidic group than in methyl acrylate; whereas, the activation barrier reduced to 48.1 kJ mol−1 (Table 2). The bond distances between the bond forming C atoms in the reactant state were 3.58 Å and 3.47 Å which decreased to 2.28 Å and 1.92 Å in the transition state structure and further reduced to 1.58 Å and 1.64 Å in the product (Fig. 2(b)). With an even stronger electron-withdrawing group in acrolein, the FMO gap further reduced to 2.81 eV and similarly, the activation barrier reduced to 39.2 kJ mol−1, thereby making acrolein the most favorable dienophile for the DA reaction with HMF as diene. For the HMF and acrolein DA reaction, the bond distances between the bond forming C atoms in the reactant state were 3.18 Å and 3.61 Å, which were decreased to 1.73 Å and 2.41 Å in the transition state structure and further reduced to 1.57 Å and 1.65 Å in the product (Fig. 2(c)). However, when propylene was chosen as the dienophile which has a mild electron donating methyl group, the reaction proceeded with an inverse path with the FMO gap of 3.24 eV (Table 1). The activation barrier for this reaction was calculated to be around 75.5 kJ mol−1 similar to the one obtained in reaction with ethylene (∼78.3 kJ mol−1). In comparison, the reaction energy was 11.8 kJ mol−1, as shown in Table 2. The bond distances between C5–C2 and C2–C1 were measured to be 3.25 Å and 3.62 Å in reactant state, which decreased to 2.08 Å and 2.27 Å in the transition state and further to 1.57 Å and 1.61 Å in the product structure (Fig. 2(d)). Reactions with electron deficient dienophiles such as methyl acrylate, acrylic acid or acrolein with HMF serve as better reactant constituents with respect to the ease of oxanorbornene formation.
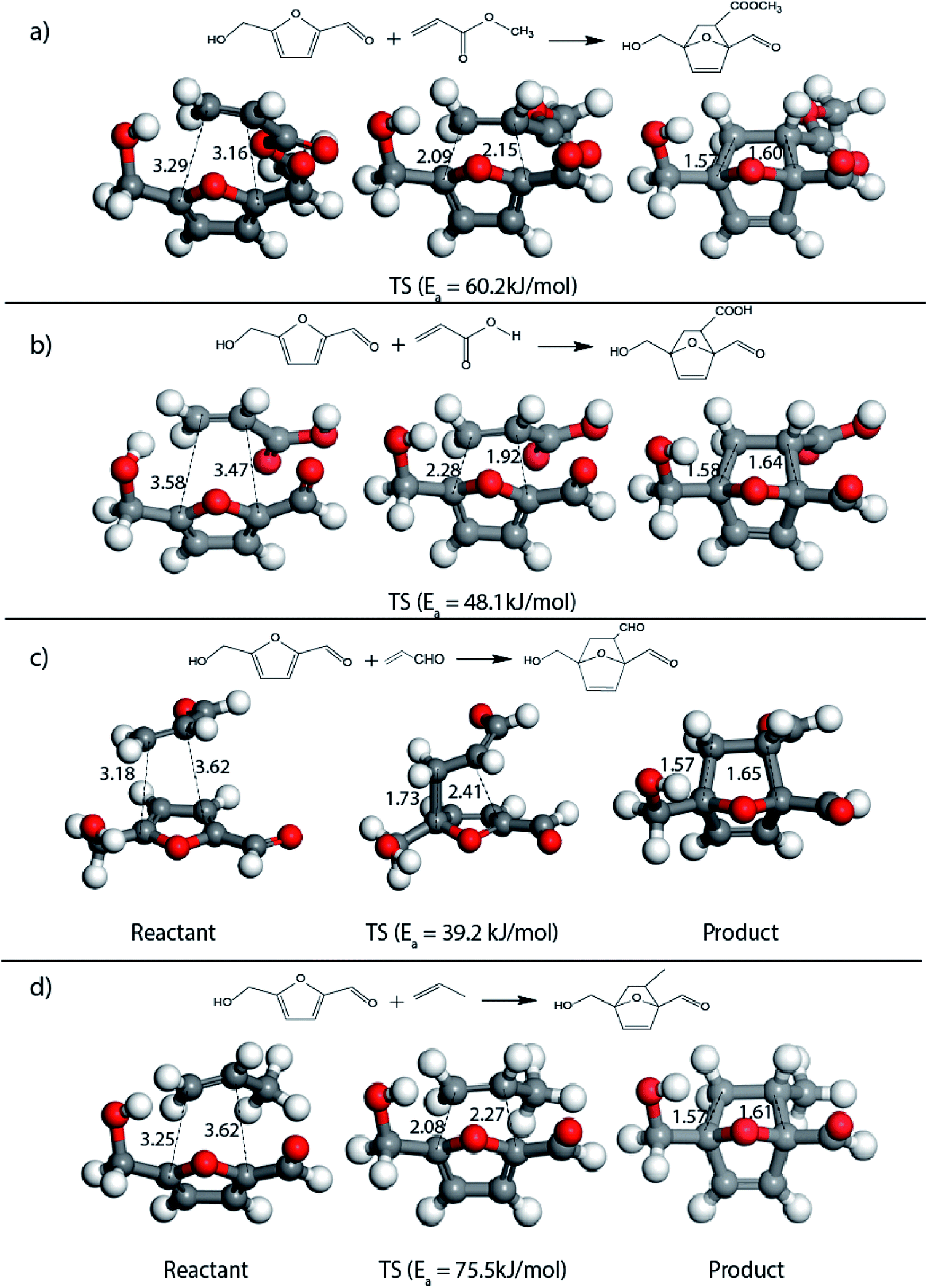 | ||
| Fig. 2 Reactant, transition state and product structures for reaction between (a) HMF + methyl acrylate, (b) HMF + acrylic acid, (c) HMF + acrolein, (d) HMF + propylene. All bond lengths are in Å. | ||
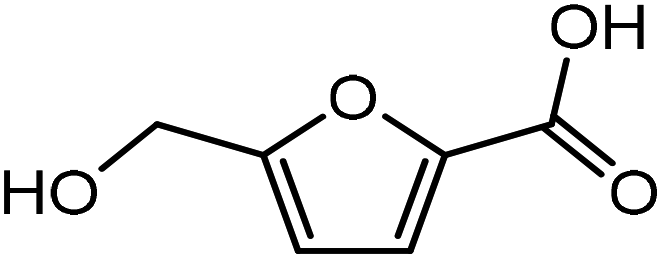
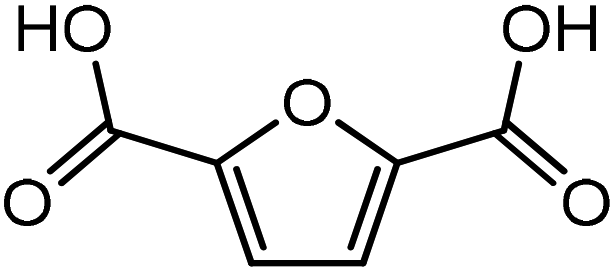
It was observed that when acrolein was used as the dienophile, the corresponding activation barriers for all the dienes chosen in this study, reduced considerably with respect to other dienophiles like acrylic acid, methyl acrylate and propylene, as can be seen in Table 2. As the nature of the diene became more nucleophilic, i.e. more electron-rich, the activation barrier for its reaction with acrolein decreased further. One possible reason could be an easier overlap of molecular orbitals formed during the reaction due to the lower FMO gap. The barrier for the reaction between FDCA and acrolein was calculated to be 84 kJ mol−1 (Table 2). The C5–C2 and C2–C1 distances were measured to be 3.72 Å and 3.57 Å in the reactant state, 1.84 Å and 2.28 Å in the transition state and 1.58 Å and 1.59 Å in product state (Fig. 3(a)). On replacing FDCA with electronically similar diene, FDCA-ester, the barrier reduced to 68.2 kJ mol−1 while the corresponding C5–C2 and C2–C1 distances were measured to be 3.33 Å and 3.26 Å in the reactant state, 1.73 Å and 2.34 Å in the transition state and 1.62 Å and 1.59 Å in product state (Fig. 3(b)). However, a significant reduction in the activation barriers was observed when HMFA and HMF were used as dienes. With HMFA, the activation barrier dropped to 39.3 kJ mol−1. The bond distances between C5–C2 and C2–C1 were calculated to be 3.16 Å and 3.15 Å in the reactant state, 1.71 Å and 2.52 Å in the transition state and 1.58 Å and 1.63 Å in product state (Fig. 3(c)). The lowest barrier of the reaction was calculated for electron-rich diene, DMF, wherein the barrier was calculated to be around 36.9 kJ mol−1. The corresponding C5–C2 and C2–C1 distances were measured to be 3.04 Å and 3.14 Å in the reactant state, 1.68 Å and 2.44 Å in the transition state and 1.58 Å and 1.63 Å in product state (Fig. 3(d)).
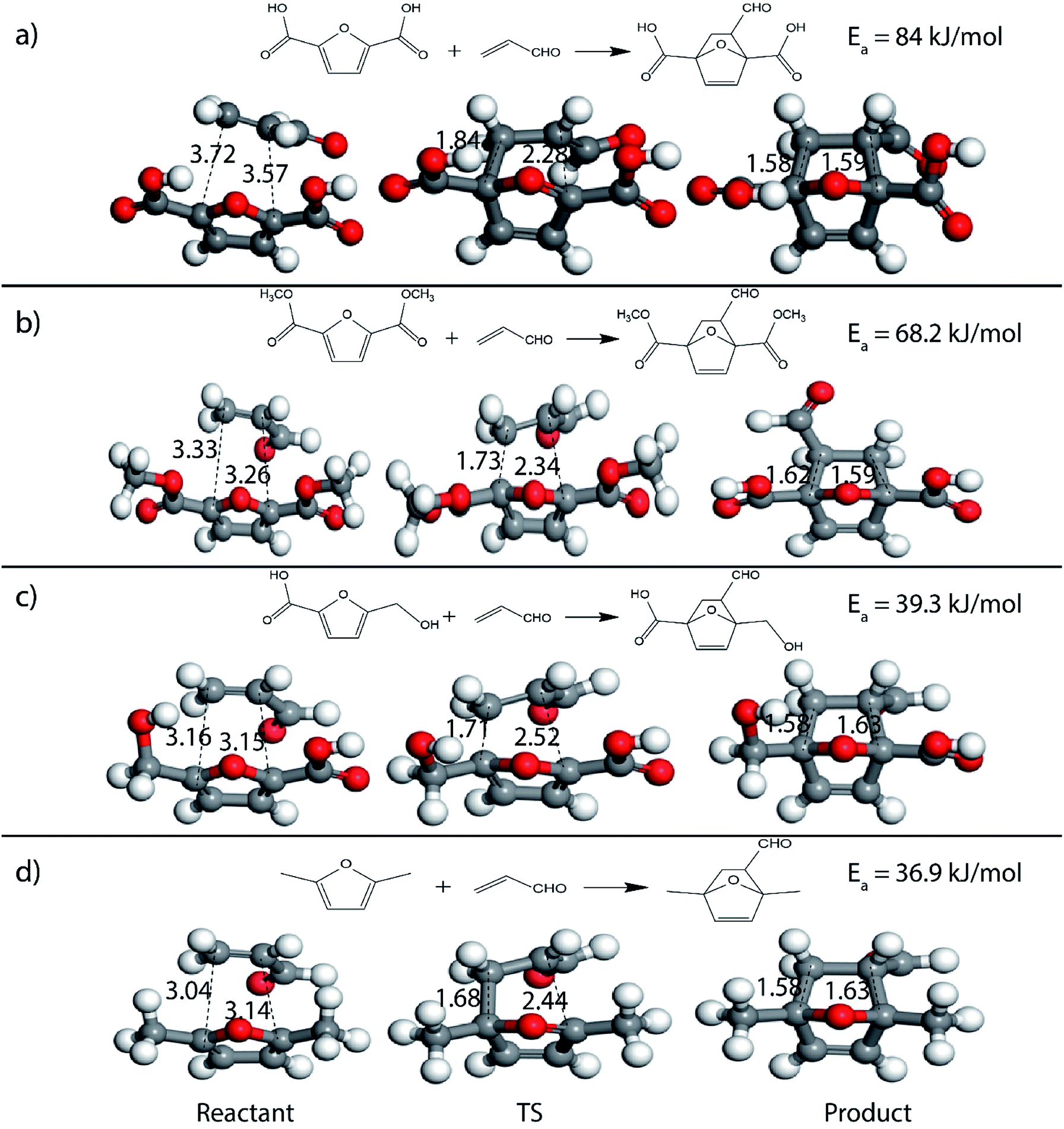 | ||
| Fig. 3 Reactant, transition state and product structures for reaction between (a) FDCA + acrolein, (b) FDCA-ester + acrolein, (c) HMFA + acrolein, (d) DMF + acrolein. All bond lengths are in Å. | ||
Further, to study the effect of electron-donating substituents present on the dienes, reaction with DMF were explored. Notably, the hydrodeoxygenation of HMF yields DMF. Unlike HMF with an electron-withdrawing group at the C2 position, DMF has two mild electron-donating methyl groups at C2 and C5 positions (Fig. 1). DMF, being an electron rich diene due to the presence of the methyl groups preferably proceed via normal demand reaction pathway on reacting with a dienophile, as shown in Table 1. The activation barrier for the reaction with ethylene was calculated to be 83.8 kJ mol−1 (Table 2), while the FMO gap for the same reaction was calculated to be 4.16 eV as shown in Table 1. When electron withdrawing groups were substituted to ethylene, lowering of activation barrier with respect to ethylene was observed. The results were in accordance with the decrease in the FMO gap for normal DA reactions when dienophile becomes electron poorer. Thus, for a reaction involving methyl acrylate, the activation barrier was calculated to be 54.6 kJ mol−1 as the FMO gap dropped down to 2.52 eV. The bond distances between C5–C2 and C2–C1 were 3.13 Å and 3.10 Å in the reactant state, 2.08 Å and 2.18 Å in the transition state and 1.58 Å and 1.61 Å in the product structures (Fig. 4(a)). When acrylic acid was used as the dienophile, the activation barrier further reduced to 47 kJ mol−1 while the FMO gap reduced to 2.22 eV. The corresponding bond distances were obtained as 3.06 Å and 3.06 Å in reactant state, 2.05 Å and 2.18 Å in the transition state and 1.58 Å and 1.62 Å in product state structures (Fig. 4(b)). With an even stronger electron withdrawing acrolein, the activation barrier was calculated to be only 36.9 kJ mol−1 with the FMO gap as 1.84 eV, as has been previously shown in Fig. 3(d). This decreasing trend of the activation barrier is consistent with the explanation provided by the FMO gap descriptor, where a normal demand DA reaction is favoured by electron-rich diene and electron-poor dienophile. As the strength of the electron-withdrawing group increases from methyl acrylate, acrylic acid to acrolein, the dienophile becomes more deficient in electron and hence, the FMO gap reduces, leading to a reduction in activation barrier. Whereas, for DA reaction with propylene as the dienophile, due to the presence of electron-donating methyl group, resulted in an increase in the FMO gap (∼4.46 eV, Table 1), makes the DA reaction less favorable. The activation barrier for DA reaction of propylene with DMF was calculated as high as 90 kJ mol−1 (Table 2), comparable to the reaction involving ethylene (83.8 kJ mol−1, Table 2). The bond distances measured here were found to be 3.23 Å and 3.19 Å in reactant state, 2.15 Å and 2.14 Å in transition state and 1.58 Å and 1.59 Å in the product state (Fig. 4(c)).
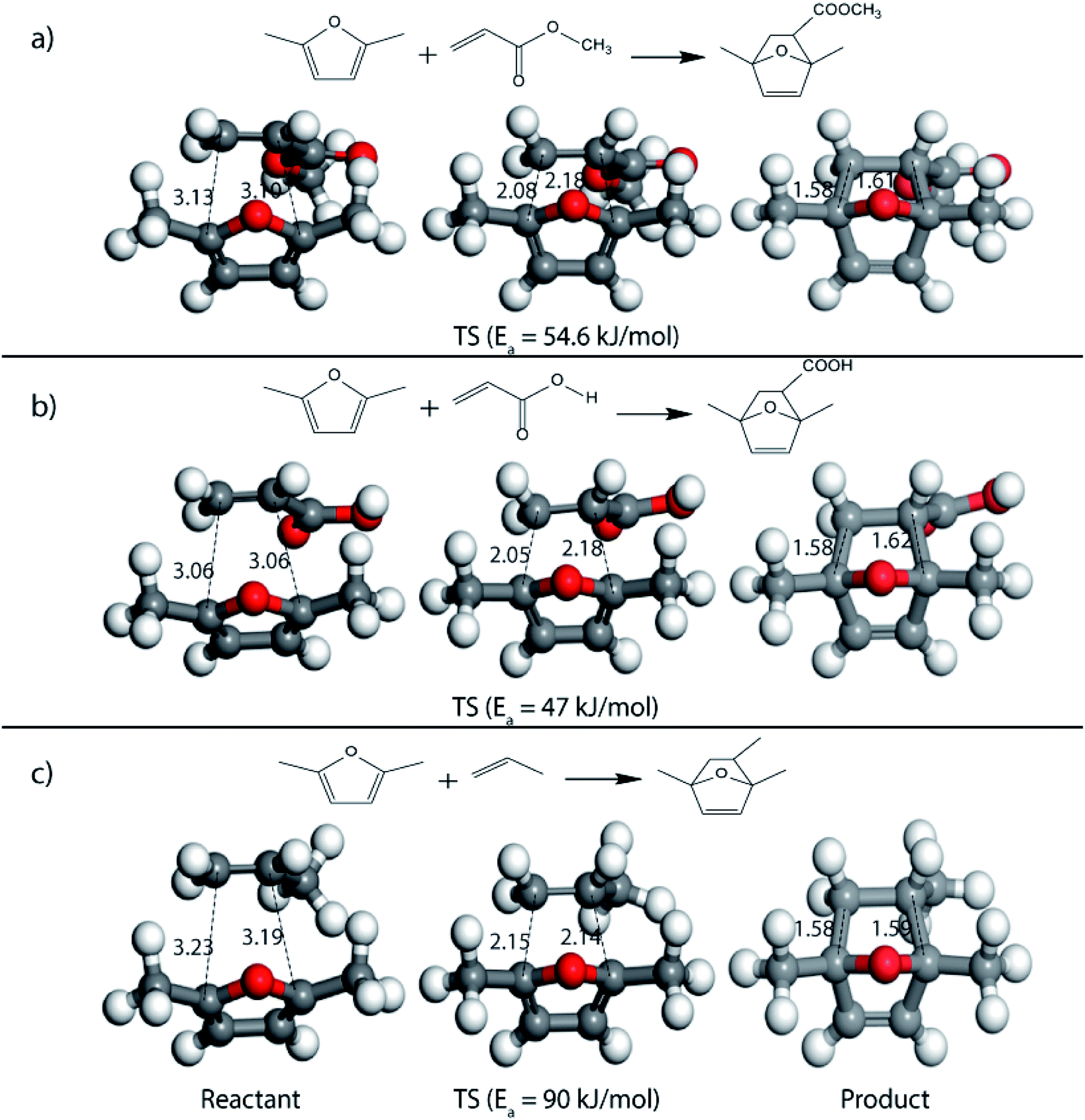 | ||
| Fig. 4 Reactant, transition state and product structures for reaction between (a) DMF + methyl acrylate, (b) DMF + acrylic acid, (c) DMF + propylene. All bond lengths are in Å. | ||
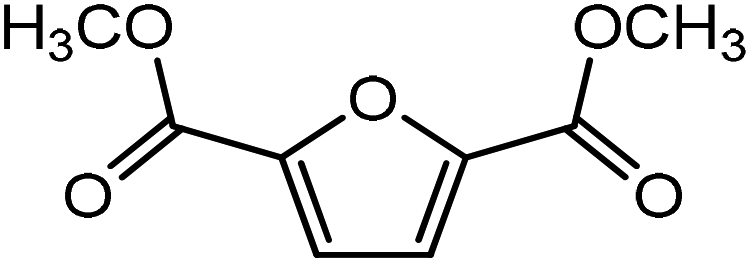
On replacing the substituents at C2 and C5 position of HMF with methyl ester groups, FDCA-ester, an electrophilic diene, is obtained, which can be viewed as the oxidized derivative of HMF. HMF can be easily oxidized to compounds like HMFA, FDCA-ester, or FDCA under mild conditions by using cheap oxide catalysts, as studied by Davis et al.17 An added advantage of using HMF or its oxidized derivatives is that the oxanorbornene intermediate obtained by the DA reaction could be readily converted to TMLA as the intermediate already contains oxidative substituents at C2 and C5 positions unlike for DMF, which contains 2 methyl groups as substituents. Here, the focus is on FDCA-ester as the diene and its DA reactions with different dienophiles. The methyl ester group attached to the C2 and C5 position of the furan molecule makes FDCA-ester an electron-poor diene. Consequently, it was observed by DFT simulations that all DA reactions involving FDCA-ester as the diene made the reaction proceed via an inverse demand path, irrespective of the dienophile used in the reaction. The FMO gap for the reaction of FDCA-ester with methyl acrylate was 3.44 eV while the corresponding activation barrier was 81.6 kJ mol−1. The bond distance between C5–C2 and C2–C1 was measured as 3.13 Å and 3.15 Å in the reactant state, 2.03 Å and 2.13 Å in the transition state and 1.59 Å and 1.62 Å in the product state structures (Fig. 5(a)). The effect of an even electron poorer dienophile, acrylic acid, was reflected in further increase of FMO gap to 3.63 eV (Table 1) due to the reaction following inverse demand path, and a high activation barrier of 68.9 kJ mol−1 (Table 2). The C5–C2 and C2–C1 bond distances were measured to be 3.42 Å and 3.07 Å in the reactant state, 2.14 Å and 2.09 Å in the transition state and 1.58 Å and 1.61 Å in product structure (Fig. 5(b)). When propylene was used as the dienophile, the FMO gap reduced to 3.04 eV as compared to much higher values for methyl acrylate and acrylic acid. It was expected as all these reactions proceed via the inverse demand path and hence, an electron-rich dienophile would favor the electron donation from the dienophile to the diene. The corresponding C5–C2 and C2–C1 bond distances were calculated to be 3.16 Å and 3.16 Å in reactant state, 2.11 Å and 2.13 Å in transition state and 1.59 Å and 1.60 Å in the product state (Fig. 5(c)). The activation barrier for the reaction of FDCA-ester with propylene was calculated to be high ∼90 kJ mol−1.
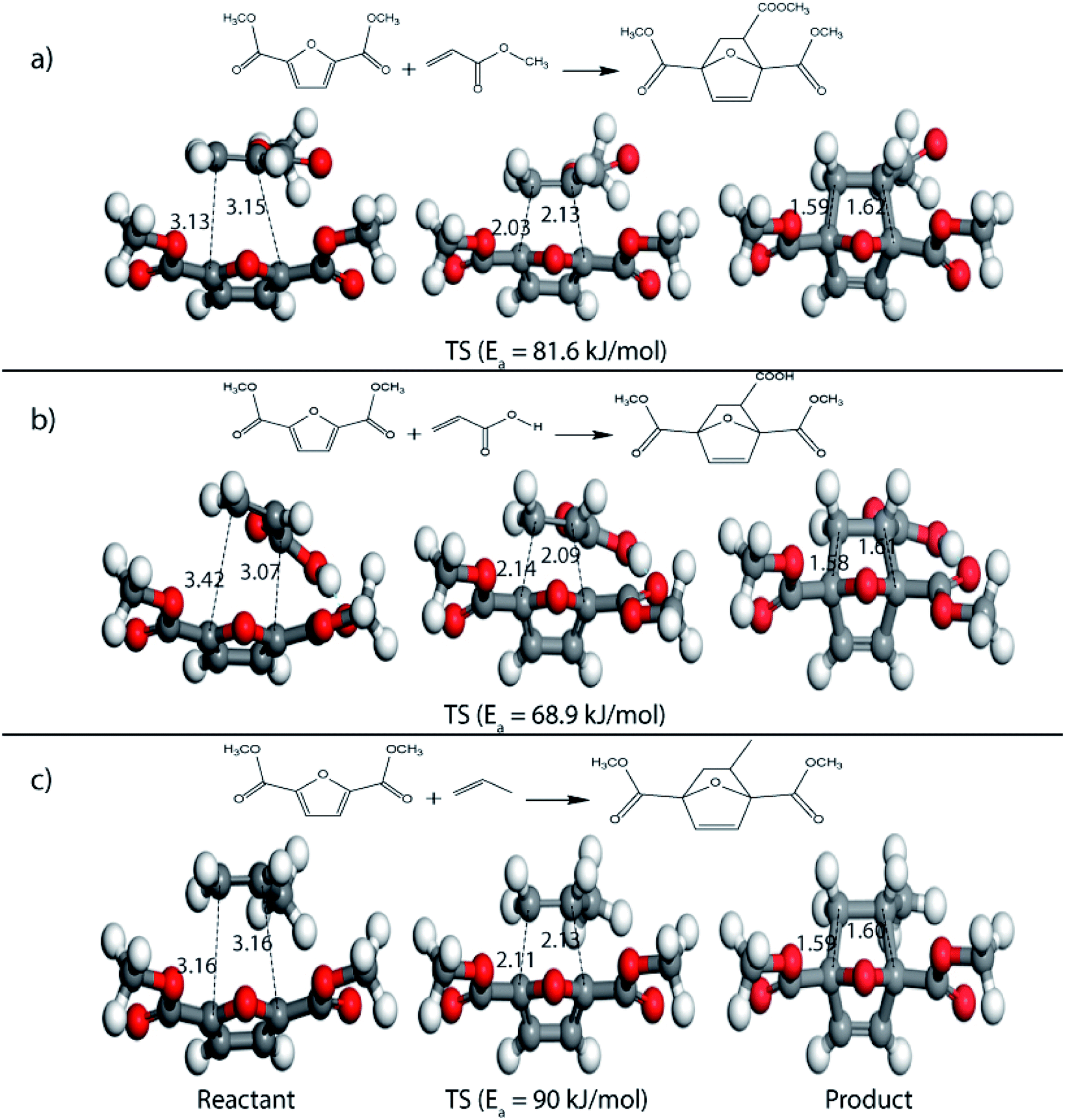 | ||
| Fig. 5 Reactant, transition state and product structures for reaction between (a) FDCA-ester + methyl acrylate, (b) FDCA-ester + acrylic acid, (c) FDCA-ester + propylene. All bond lengths are in Å. | ||
The activation energy for the DA reaction were found to scale linearly with the normal FMO gap, as shown in Fig. 6. The normal FMO gap was found to be a better descriptor than the conventional FMO gap (Fig. SI-1(a)†) and inverse FMO gap (Fig. SI-1(b)†), as can be seen from the fitting parameters shown in Table SI-2.† The activation energy for the DA reaction increases linearly with the increase in the normal FMO gap. Similar trend has been previously observed for rDA reaction of partially saturated 2-pyrones by Gupta et al.40 and Khan et al.55 Among the furan-based dienes, the variance of activation barrier with normal FMO gap is highest for DMF, followed by HMF and HMFA, as can be seen in Fig. 6. The activation barrier for DA reaction of furan-based carboxylic substituted dienes FDCA and FDCA-ester are much less susceptible to the change in the normal FMO gap (Fig. 6).
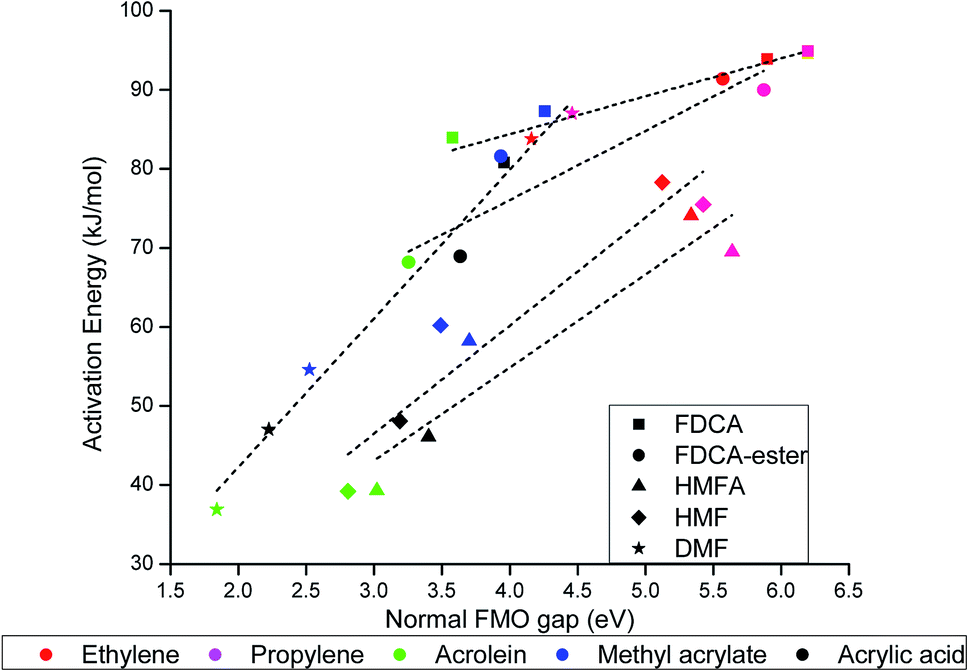 | ||
| Fig. 6 Activation energy (kJ mol−1) versus normal FMO gap (eV) for DA reaction of the molecules included in our scope of study. | ||
The ‘FMO gap’ has been used for decades to understand the reactivity of diene and dienophiles undergoing DA reactions. In our previous articles39,40 the ‘FMO gap’ was shown to be a good descriptor in explaining the reactivity of various partially saturated 2-pyrones undergoing rDA reaction. However, the use of ‘FMO gap’ as an universal descriptor for DA reactivity have several drawbacks as noted by Dewar,56 Fisher et al.57 and I. Fernández et al.58 Michael J. S. Dewer in in his study56 explained that the FMO theory was based on simple assumptions and did not have a good basis in fundamental quantum mechanics and failed to provide satisfying accuracy in determining the chemical reactivity. Fernandez et al.58 studied the DA reaction between substituted butadienes and ethylenes and suggested that both the frontier orbital interactions and deformation energy in the transition states determines the activation barriers and focus on only one of them may lead to a wrong prediction of activation barriers and reactivity trends. Souza et al.59 showed that the DA reaction can follow different pathways, the conventional synchronous concerted pathways, the asynchronous concerted pathway involving a short-lived biradicaloid and a stepwise pathway having a stable biradical intermediate. The dynamics (quasi-classical trajectories) approaches were shown to be better suited to describe the reactivity trends of the asynchronous concerted DA reactions compared to the conventional static transition state search or the intrinsic reaction coordinate (IRC) approaches. Similarly, Wang et al. in their study60 concluded that the DA reactions are complex in nature and the dynamic effects play a central role in determining the DA selectivity.
The DA reaction studied here follow an asynchronous concerted mechanism as suggested by Souza et al.59 The Diels–Alder reactions are single step with no stable intermediate in between, however it can be seen in the transition states that one of the newly formed C–C bond was shorter compared to the other C–C bond, resulting in an asymmetric transition state structure. We would also like to mention that the normal FMO gap, the smaller of normal and inverse FMO gap, is not a good descriptor as can be seen in the Fig. SI-1(a).† We think the DA reactions studied here are following the short-lived biradicaloid mechanism as suggested by Souza et al.59 not fully synchronous concerted [4 + 2] cycloaddition.
To look for an alternative descriptor for DA reactions, the descriptor proposed by Khan et al.55 in their work on rDA reaction was used. The activation barrier, Ea, was plotted against the proposed descriptor (IPdiene + EAdienophile)/2, and it followed a linear scaling relationship with all the reactions chosen in our scope of the study (Fig. 7). The ionization potential of the diene (IPdiene) and electron affinity of dienophile (EAdienophile) could be related to a bond forming potential between the diene and dienophile; IPdiene indicating the ease with which the diene can donate electrons while EAdienophile indicates the ease of accepting electron by the dienophile, needed for the formation of a bond. As the descriptor value increased, the activation barrier for the reactions also followed an increasing trend. Due to less ambiguity in the descriptor (IPdiene + EAdienophile)/2 compared to the FMO gap, this descriptor can be proposed as a suitable alternative to the FMO descriptors commonly utilized for describing the DA reaction.
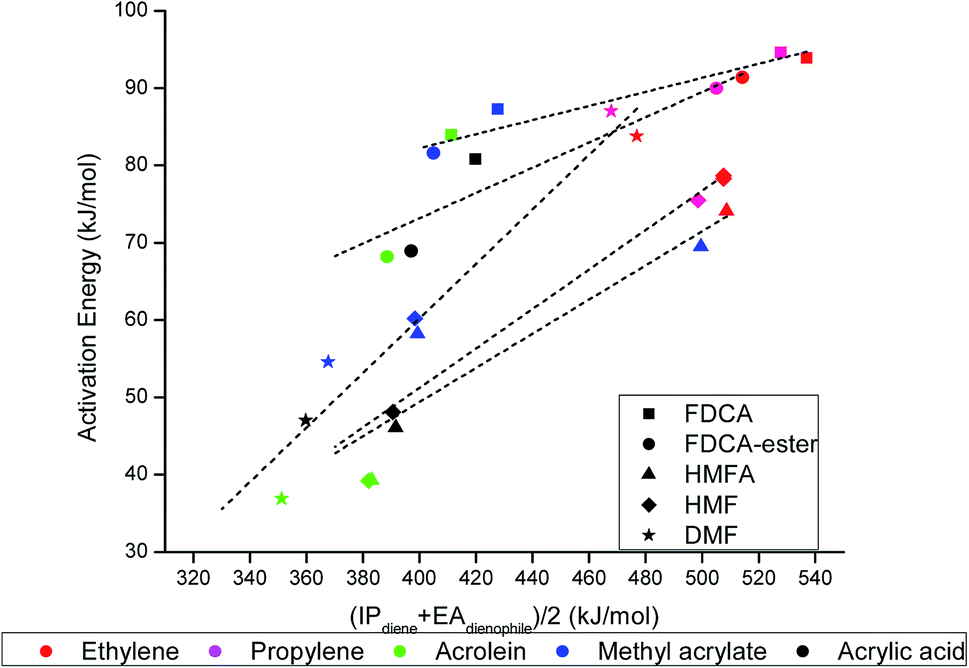 | ||
| Fig. 7 Linear scaling relationship between the activation barrier and proposed descriptor, (IPdiene + EAdienophile)/2 for all DA reactions chosen under the scope of our study. | ||
Further, the effect of solvent on the activation barrier and reaction energy was studied for all the DA cycloaddition reactions. In our previous studies for rDA reactions of partially saturated 2-pyrone molecules, solvents are observed to play a significant role in reduction of activation barrier through stabilization of polar transition state.40,55,61 For our reaction between FDCA-ester and ethylene in two different solvents, water (ε = 78.54) and dioxane (ε = 10.42), we found that the reaction in both the solvents yielded the same value of 26.84 kJ mol−1 for the difference in solvation energy in transition and reactant state. Hence, the activation barrier would remain unaffected largely due to change in solvents, as was observed for the reactions in our study. The reaction involving DMF and ethylene was studied in solvents having different dielectric constants such as acetone (ε – 20.7), acetonitrile (ε – 37.5), CCl4 (ε – 2.24), diethylether (ε – 4.34), ethanol (ε – 24.3), hexane (ε – 1.89) and water (ε – 78.54). DFT calculations provided similar activation barrier of 85 kJ mol−1 for the same reaction in presence of all the above-mentioned solvents, as given in Table SI-3.† Similar observation was made by Cinzia et al.62 and Taha et al.63 where the authors found is no significant change in the activation barrier for uncatalyzed DA reaction in the presence of different solvents.62,63 Taha et al.63 studied the solvent effect on the DA reaction between two hydrocarbon reactants, DMF and maleic anhydride and found that there were only a few kilocalories per mole drop in the activation barrier for the uncatalyzed reaction in the presence of varying solvents. The authors stated that variation in the activation barrier in different solvents varies in proportion to the difference in solvation energy between the transition state and the reactant state in the solvents. In general, the simple COSMO model fails to capture the effect of hydrogen bonding, nature of hydrophobic and hydrophilic interactions, effect of solvent cavity and the contribution of excess entropy in the reaction. Ab initio based CPMD method with explicit solvent molecules are known to be better suited to capture these contributions.64,65 However, the ab initio CMPD simulations are hugely computationally extensive calculations and was out of scope for this study involving many combinations of diene and dienophiles undergoing DA reactions.
Furthermore, many studies have shown that in the presence of Lewis acids the rate of DA reaction is enhanced due to the formation of a complex with the diene or the dienophile.63,66–68 In the present study, Na+ cation was chosen to model the Lewis acid environment for all the DA reaction as has been previously proposed by Taha et al. in their theoretical study on DA reaction between DMF and maleic anhydride (MA).63 When HMF reacts with the chosen dienophiles in the presence of Na+ as Lewis acid environment, Na+ binds preferably to the HMF molecule as compared to the dienophiles studied here, as has been shown in Fig. 8. When HMF reacts with acrolein in Na+ environment, the C5–C2 and C2–C1 bond distances were calculated to be 3.31 Å and 3.38 Å in reactant state, 1.73 Å and 2.41 Å in transition state and 1.58 Å and 1.66 Å in the product state (Fig. 8(a)). The activation barrier for the above-mentioned reaction was calculated as 31 kJ mol−1, the lowest for reaction of HMF with all chosen dienophiles. Same reaction in the absence of Na+ proceeded with an activation barrier of 39.2 kJ mol−1 (Fig. 2(c)). On reaction of HMF–Na+ with methyl acrylate, the C5–C2 and C2–C1 bond distances were calculated to be 4.33 Å and 4.24 Å in reactant state, 1.71 Å and 2.36 Å in transition state and 1.57 Å and 1.60 Å in the product state (Fig. 8(b)). The barrier of activation was measured to be 53.4 kJ mol−1 for the acid catalysed reaction, a drop of nearly 7 kJ mol−1 in the absence of Lewis acid. HMF–Na+ reacted with acrylic acid with an activation barrier of 47.6 kJ mol−1 and the corresponding C5–C2 and C2–C1 bond distances were calculated to be 3.26 Å and 3.12 Å in reactant state, 1.73 Å and 2.37 Å in transition state and 1.58 Å and 1.64 Å in the product state structures (Fig. 8(c)). The barrier for the DA reaction between HMF and propylene reduced from 75.5 kJ mol−1 in the absence to 35.8 kJ mol−1 in the presence of Lewis acid. The C5–C2 and C2–C1 bond distances were calculated to be 3.36 Å and 3.75 Å in reactant state, 1.88 Å and 2.35 Å in transition state and 1.57 Å and 1.61 Å in the product state (Fig. 8(d)). Similar reduction in activation barrier was also observed by Engberts et al.69 in their study of Lewis acid effect of Cu2+, Ni2+, Co2+ ions on DA reaction of 3-(p-nitrophenyl)-1-(2-pyridyl)-2-propene-1-one with cyclopentadiene. Since, Na+ was used as the Lewis acid to observe changes in the energetics of the reactions, there is no possibility of above-mentioned complexation, but considerable reduction in activation barrier for the reaction was noticed. The effect of Lewis acid (Na+) on DA reaction between HMFA and propylene is shown in Fig. 9(a), where the reactant, transition state and product structures in the uncatalyzed and catalysed environments has been shown.
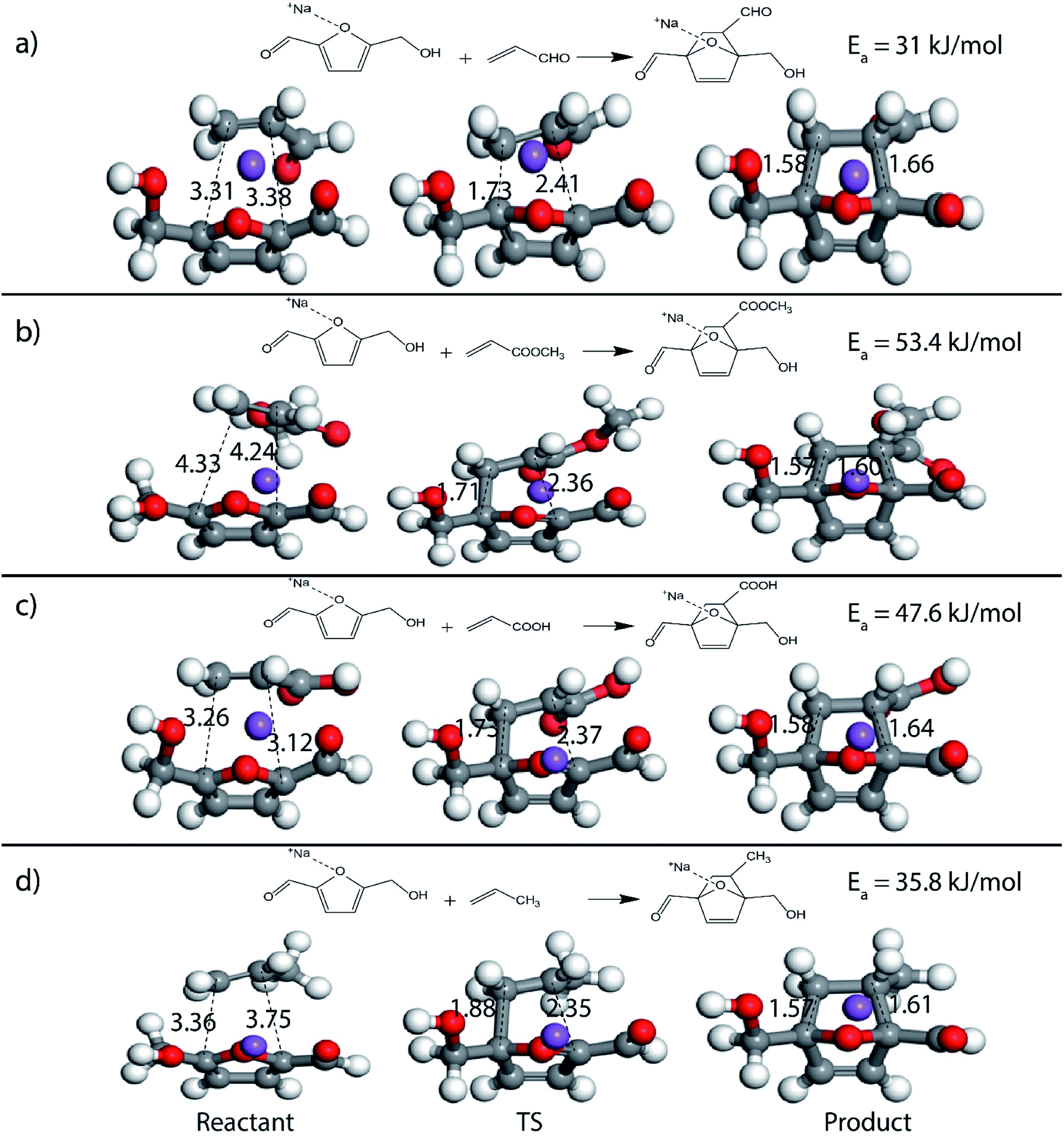 | ||
| Fig. 8 Reactant, transition state and product structures for reaction between (a) HMF and acrolein (b) HMF and methyl acrylate (c) HMF and acrylic acid (d) HMF and propylene, in the presence of Na+. All bond lengths are in Å. | ||
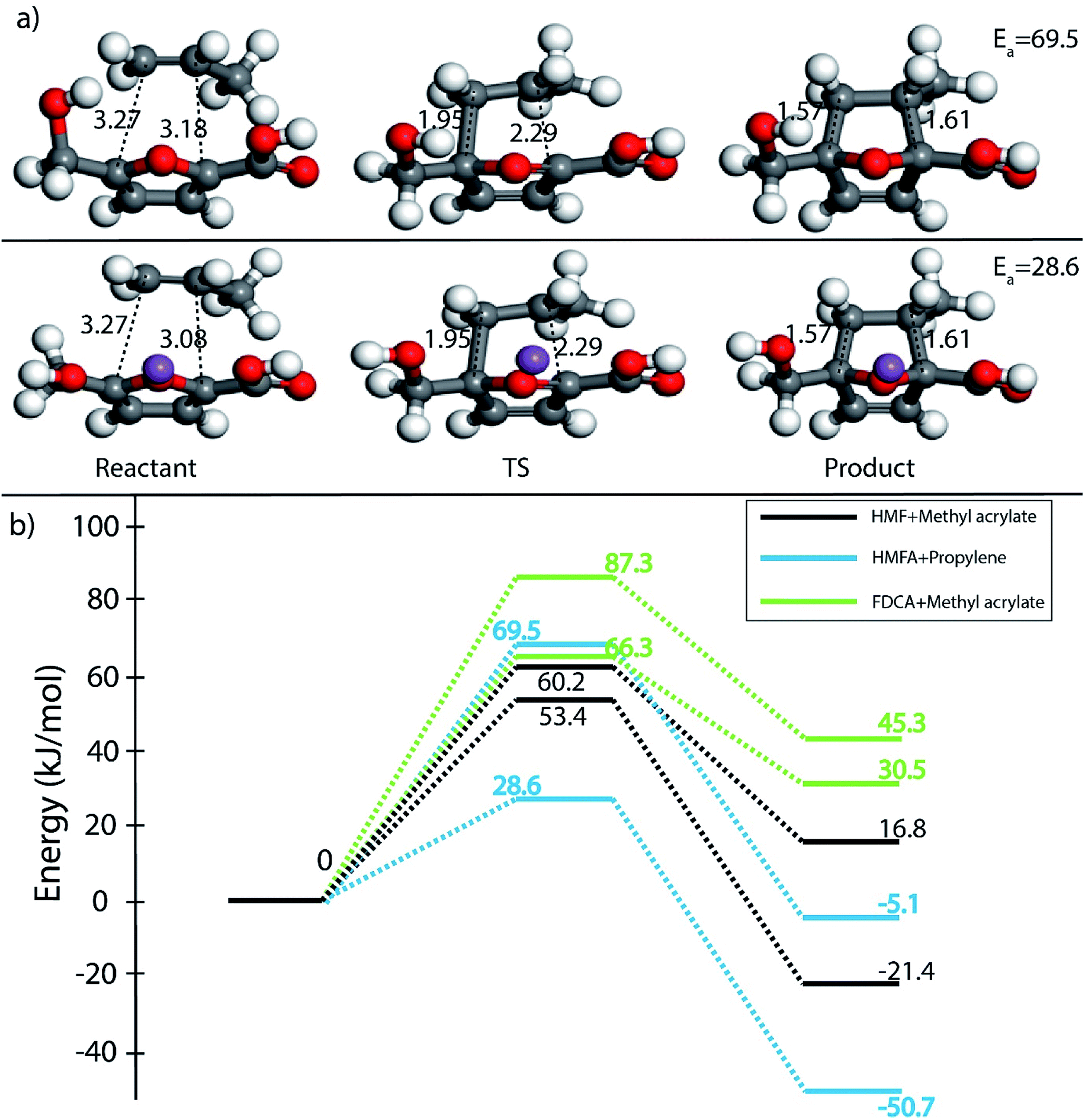 | ||
| Fig. 9 (a) Reactant, transition state and product structure for reaction between HMFA and propylene in non-catalysed environment (top) and Lewis acid catalysed (Na+) environment (below). All bond lengths are in Å. (b) Reaction diagram for DA reactions, HMF + methyl acrylate, HMFA + propylene and FDCA + methyl acrylate in the presence and absence of Na+ as Lewis acid. All energy values are in kJ mol−1. | ||
Effect of Na+ on three different DA reactions, HMF + methyl acrylate, HMFA + propylene and FDCA + methyl acrylate has been elucidated in reaction diagram shown in Fig. 9(b), which compares the Na+ catalysed reactions with the uncatalyzed ones. For the reaction with HMF and methyl acrylate, the activation barrier dropped from 60.2 kJ mol−1 to 53.4 kJ mol−1. Similarly, for reaction between HMFA and propylene, the barrier dropped from 69.5 kJ mol−1 to 28.6 kJ mol−1, while for the reaction between FDCA and methyl acrylate, the barrier for the reaction decreased from 87.3 kJ mol−1 to 66.3 kJ mol−1 (Fig. 9(b)). Such significant reduction in the activation barrier is consistent with previous theoretical and experimental studies done on numerous DA reaction as discussed.63,67 The drop in the activation barrier lies in the ability of Lewis acid to bind to the diene or the dienophile and decreasing the FMO gap.
The oxanorbornene intermediate produced directly by DA cycloaddition needs to be converted to the aromatic product to produce TMLA. This can be achieved by dehydration of oxanorbornene intermediate followed by oxidation of the substituents at C1, C2 and C4 positions of the substituted aromatic benzene ring. The dehydration of oxanorbornene can be carried over solid acid catalyst or activated carbon as catalyst to form its aromatic equivalent as shown by Davis et al.,17 and Williams et al.70 The resulting aromatic product could be oxidized to TMLA in the presence of multivalent catalyst containing cobalt and promoted by a source of bromine in a two-step process.71–73
Conclusion
In this study the DA reaction between furan substituted dienes and ethylene substituted dienophiles was studied theoretically using DFT simulations with the objective of producing the oxanorbornene cyclo-adduct, a precursor for the production of trimellitic acid (TMLA) as the end product. TMLA is a potential platform molecule for the production of value added chemicals which finds wide applications. A number of biomass derived furanic molecules as dienes and substituted ethylene as dienophiles were explored using DFT simulations to find out the most feasible pathway for the formation of oxanorbornene an intermediate for TMLA production via DA reaction. DFT simulations were used to explain the normal and inverse demand DA reactions for the oxanorbornene intermediate formation. The FMO gap theory successfully described the normal demand DA reactions, wherein low activation barrier and hence, high reaction rates were observed for reactions involving electron rich diene and electron poor dienophile. Another proposed descriptor, (IPdiene + EAdienophile)/2 was used to explain the reactivity trend for the reactions included in this study. The electronic effect of the substituents in the dienes and dienophiles were explained well by both the descriptors. The effect of solvent on the rate of DA reactions was also studied. Theoretically, no significant change in the activation barriers was observed for any of the reactions in the presence of different solvents. In order to understand the effect of Lewis acid on the DA reaction simulations were carried out in presence of Na+ cation. The activation energy reduced considerably for all the reactions in presence of Na+ implying that Lewis acid catalysed reactions can increase the rate of these reactions manifold. The proposed process in this study would open up alternative gateways for the production of TMLA through biomass derived platform molecules. Finally, the computational study presented here on the reactivity trend of dienes and dienophiles for the DA reaction using the linear scaling relationship, shows the effectiveness of simple computational DFT methods in understanding common chemical reactions. The study shows how trends in chemical reactions can be understood and explained through already known descriptors. In designing a novel reaction, the two proposed descriptors can be used for predicting reactivity of an unknown molecule of similar type, undergoing DA reaction in a range of solvents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge financial assistance from the Department of Science and Technology (Government of India, Grant No. CRG/2019/006176). Initial seed grant from ICAR-IITD joint MFIRP project (MI02031G) is appreciated to pursue this research. Computational resources are provided by the high performance computing (HPC) facility of IIT Delhi.References
- W. Zha, Z. Shao, J. W. Frost and H. Zhao, J. Am. Chem. Soc., 2004, 126, 4534–4535 CrossRef CAS PubMed.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- S. K. Bardhan, S. Gupta, M. E. Gorman and M. A. Haider, Renewable Sustainable Energy Rev., 2015, 51, 506–520 CrossRef CAS.
- S. Gupta, T. S. Khan and M. A. Haider, ACS Sustainable Chem. Eng., 2019, 7, 10165–10181 CrossRef CAS.
- M. I. Alam, S. Gupta, E. Ahmad and M. A. Haider, in Sustainable Catalytic Process, ed. B. Saha and M. Fan, Elsevier, 2015, pp. 157–177 Search PubMed.
- I. Alam, S. De, T. S. Khan, M. A. Haider and B. Saha, Ind. Crops Prod., 2018, 123, 629–637 CrossRef.
- M. I. Alam, S. Gupta, A. Bohre, E. Ahmad, T. S. Khan, B. Saha and M. A. Haider, Green Chem., 2016, 18, 6399–6696 RSC.
- M. I. Alam, M. A. Ali, S. Gupta and M. Ali Haider, in Microbial Applications, Springer International Publishing, 2017, vol. 2. pp. 153–166 Search PubMed.
- A. Maneffa, P. Priecel and J. A. Lopez-sanchez, ChemSusChem, 2016, 9, 1–14 CrossRef PubMed.
- X. Li, J. Li, G. Zhou, Y. Feng, Y. Wang and G. Yu, Appl. Catal., A, 2014, 481, 173–182 CrossRef CAS.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp and G. B. Hunsinger, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CAS.
- H. Zhang, Y. Cheng, T. P. Vispute and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 RSC.
- J. S. Luterbacher, Green Chem., 2019, 21, 2801–2809 RSC.
- A. Ã. Effendi, H. Gerhauser and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2008, 12, 2092–2116 CrossRef CAS.
- D. Shen, G. Liu, J. Zhao, J. Xue, S. Guan and R. Xiao, J. Anal. Appl. Pyrolysis, 2015, 112, 56–65 CrossRef CAS.
- M. J. S. Dewar and A. B. Pierini, J. Am. Chem. Soc., 1984, 106, 203–208 CAS.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Chemistry, 2011, 17, 12452–12457 CrossRef CAS PubMed.
- T. Pfennig, J. M. Carraher, A. Chemburkar, R. L. Johnson, A. T. Anderson, J. P. Tessonnier, M. Neurock and B. H. Shanks, Green Chem., 2017, 19, 4879–4888 RSC.
- T. Pfennig, R. L. Johnson and B. H. Shanks, Green Chem., 2017, 19, 3263–3271 RSC.
- J. J. Lee, G. R. Pollock, D. Mitchell, L. Kasuga and G. A. Kraus, RSC Adv., 2014, 4, 45657–45664 RSC.
- Y. Philip, I. Kristianto, H. Lee and J. Jae, Fuel, 2016, 182, 588–596 CrossRef.
- P. T. M. Do, J. R. McAtee, D. A. Watson and R. F. Lobo, ACS Catal., 2013, 3, 41–46 CrossRef CAS PubMed.
- S. K. Green, R. E. Patet, N. Nikbin, C. L. Williams, C. C. Chang, J. Yu, R. J. Gorte, S. Caratzoulas, W. Fan, D. G. Vlachos and P. J. Dauenhauer, Appl. Catal., B, 2016, 180, 487–496 CrossRef CAS.
- B. Saha, C. M. Bohn and M. M. Abu-Omar, ChemSusChem, 2014, 7, 3095–3101 CrossRef CAS PubMed.
- S. Gupta, T. S. Khan, B. Saha and M. A. Haider, Ind. Eng. Chem. Res., 2019, 58, 16153–16163 CrossRef CAS.
- W. H. Gong, US Pat., 20090124829A1, 2009, vol. 1–6.
- A. Frankhauser-Noti and K. Grob, Food Addit. Contam., 2004, 21, 711–718 CrossRef PubMed.
- D. F. Cadogan, Ullmann’s Encycl. Ind. Chem., 2012, pp. 35–154 Search PubMed.
- P. J. Andrulis, J. Biswas, A. Troy and P. Andrulis III, in Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy, 1988 Search PubMed.
- H. T. Aung, T. Nikai, M. Niwa and Y. Takaya, Bioorg. Med. Chem., 2011, 19, 7000–7002 CrossRef CAS PubMed.
- N. Manuja, R. Nagpal and I. Pandit, J. Clin. Pediatr. Dent., 2012, 36, 223–234 CrossRef CAS PubMed.
- I. Kavianinia, P. G. Plieger, N. G. Kandile and D. R. K. Harding, Mater. Today Commun., 2015, 3, 78–86 CrossRef CAS.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- G. A. Russell and S. Weiner, J. Org. Chem., 1966, 31, 248–251 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS.
- L. W. Wang and M. P. Teter, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13196–13220 CrossRef CAS PubMed.
- M. Chia, M. A. Haider, G. Pollock, G. a. Kraus, M. Neurock and J. a. Dumesic, J. Am. Chem. Soc., 2013, 135, 5699–5708 CrossRef CAS PubMed.
- T. S. Khan, S. Gupta, I. Alam and M. A. Haider, RSC Adv., 2016, 6, 101697–101706 RSC.
- S. Gupta, M. I. Alam, T. S. Khan, N. Sinha and M. A. Haider, RSC Adv., 2016, 6, 60433–60445 RSC.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414–205424 CrossRef.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys., 1959, 167, 378 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- N. Mardirossian and M. Head-gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- J. Tirado-rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- A. Klamt, V. Jonas, T. Burger and J. C. Lohrenz, J. Phys. Chem. A, 2011, 13, 146–152 Search PubMed.
- K. Fukui, Angew. Chem., Int. Ed., 1988, 27, 5–39 CrossRef.
- J. Chandrasekhar, S. Shariffskul and W. L. Jorgensen, J. Phys. Chem. B, 2002, 106, 8078–8085 CrossRef CAS.
- Y. Chung, B. F. Duerr, T. A. McKelvey, P. Nanjappan and A. W. Czarnik, J. Org. Chem., 1989, 54, 1018–1032 CrossRef CAS.
- G. Chemistry, J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- C. Li, Z. Zhang and Z. K. Zhao, Tetrahedron Lett., 2009, 50, 5403–5405 CrossRef CAS.
- P. Bhaumik, H. J. Chou, L. C. Lee and P. W. Chung, ACS Sustainable Chem. Eng., 2018, 6, 5712–5717 CrossRef CAS.
- P. Daorattanachai, S. Namuangruk, N. Viriya-empikul, N. Laosiripojana and K. Faungnawakij, J. Ind. Eng. Chem., 2012, 18, 1893–1901 CrossRef CAS.
- Y. Zhao, S. Wang, H. Lin, J. Chen and H. Xu, RSC Adv., 2018, 8, 7235–7242 RSC.
- T. S. Khan, S. Gupta, M. I. Alam and M. A. Haider, RSC Adv., 2016, 6, 101697–101706 RSC.
- M. J. S. Dewar, J. Mol. Struct.: THEOCHEM, 1989, 200, 301–323 CrossRef.
- G. Fisher, Found. Chem., 2016, 18, 241–262 CrossRef CAS.
- I. Fernández and G. Frenking, Eur. J. Org. Chem., 2019, 478–485 CrossRef.
- M. A. F. De Souza, E. Ventura, S. A. Do Monte, J. M. Riveros and R. L. Longo, J. Comput. Chem., 2016, 37, 701–711 CrossRef CAS PubMed.
- Z. Wang, J. S. Hirschi and D. A. Singleton, Angew. Chem., 2009, 48, 9156–9159 CrossRef CAS PubMed.
- G. Shrivastav, T. S. Khan, M. Agarwal and M. A. Haider, J. Phys. Chem. C, 2018, 122, 11599–11607 CrossRef CAS.
- C. Chiappe, M. Malvaldi and C. S. Pomelli, Green Chem., 2010, 12, 1330–1339 RSC.
- T. Salavati-Fard, S. Caratzoulas and D. J. Doren, J. Phys. Chem. A, 2015, 119, 9834–9843 CrossRef CAS PubMed.
- G. Shrivastav, T. S. Khan, M. Agarwal and M. A. Haider, J. Phys. Chem. C, 2018, 122, 11599–11607 CrossRef CAS.
- G. Shrivastav, T. S. Khan, M. Agarwal and M. A. Haider, React. Chem. Eng., 2020, 5, 651–662 RSC.
- J. B. F. N. Engberts, Pure Appl. Chem., 1995, 67, 823–828 CAS.
- A. Sbai, V. Branchadell, R. M. Ortuño and A. Oliva, J. Org. Chem., 1997, 62, 3049–3054 CrossRef CAS PubMed.
- D. M. Gelman, C. M. Forsyth and P. Perlmutter, Org. Lett., 2009, 11, 4958–4960 CrossRef CAS PubMed.
- S. Otto and J. B. F. N. Engberts, Tetrahedron Lett., 1995, 36, 2645–2648 CrossRef CAS.
- C. L. Williams, C. C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- J. K. Darin and A. G. Bemis, US Pat., 489597819A, 1985, vol. 19.
- M. R. Green and W. P. Schammel, US Pat., 4948921A, 1985, vol. 19.
- W. P. Schammel and J. K. Darin, US Pat., 4755622A, 1985, vol. 19.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04318d |
| This journal is © The Royal Society of Chemistry 2020 |