
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetically important ring opening reactions by alkoxybenzenes and alkoxynaphthalenes†
Ranadeep Talukdar
 *
*
Molecular Synthesis and Drug Discovery Laboratory, Centre of Biomedical Research, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow-226014, India. E-mail: ranadeep@chem.iitkgp.ernet.in
First published on 25th August 2020
Abstract
Alkoxybenzenes and alkoxynaphthalenes, as nucleophiles, have drawn great attention from organic chemists over the decades. Due to their high ring strain, those particular classes of molecules are often used in synthesis by utilizing their properties to undergo facile Friedel–Crafts alkylations. Different isomeric and low or densely substituted alkoxybenzenes are used for synthesis according to the structure of the target molecule. Isomeric methoxybenzenes, are the most commonly used molecule in this regard. This review aims to comprehensively cover the instances of different alkoxy-benzenes/naphthalenes used as nucleophiles for ring opening.
 Ranadeep Talukdar | Ranadeep Talukdar received his BSc in Honours Chemistry in 2007 from the University of Calcutta. After that, he went to the Indian Institute of Technology Kanpur to complete his Masters in 2009 and also got his PhD degree in organic synthesis working on domino-ring opening-cyclization of donor–acceptor cyclopropanes from the same institute. He completed his first post-doctoral research study on selenium chemistry and azetidine chemistry in 2017. He is currently pursuing research on organic photo-redox chemistry and N-heterocyclic carbenes as a National Post-Doctoral Fellow. |
1. Introduction
Alkylation of anisole is a popular undergraduate laboratory experiment. Such derivatization of electron-rich aromatic systems, notably, the Friedel–Crafts (FC) reaction, has been a boon to organic synthesis. The unique property of arenes, the nucleophilic character of their π-electron clouds, has been extensively utilized for their reaction against various ranges of electrophiles since then, to access a wide range of important motifs.1 This named reaction remains equally fashionable even after a century has passed because of its utter simplicity and the high regioselectivity displayed in the products.2There can be two scenarios for nucleophilic ring-opening functionalization by arenes. The neutral arenes (FC reaction) generally require pre-activation of the ring, preferably by a Lewis or a Brønsted acid, which attaches to a favourable “site” in the electrophilic ring, causing its activation.3 Metallo arenes can be another option, which do not require the prior activation of the electrophile ring, but require activation of the arene itself, caused by dissociation of the highly polarized sp2 carbon–metal bond, releasing an arene anion or an arene radical.4,5 As the second part of this story, i.e. metal-assisted ring opening of small strained rings by arenes, has been compiled in two excellent reviews by Wang5e and Yus,5f this review article is seriously aimed at the discussion of almost all ring-opening FC alkylations by neutral benzenes/naphthalenes reported to date.
2. “Neutral” arenes as nucleophiles
As the electron density along with electronic resonance increases with the addition of alkoxy groups in a certain ring, such substituted rings generally act more efficiently towards electrophilic substitutions.6 Moreover, the presence of alkoxy groups makes the reaction products more open to selective functionalization with phenolic derivatives further obtained by dealkylation.7 Other electron-rich arene species like dialkylamino,8 thioalkyl,9 simply alkyl benzenes10 or even unprotected phenols11 display similar nucleophilic properties, but this review will focus on the activities of the alkoxybenzene classes alone.2.1 Alkoxybenzenes
Alkoxybenzenes, on the other hand, in particular are treated as better nucleophiles, being much richer in electrons due to the +R effects of the substituents.6 The increase in the number of alkoxy groups not only increases the nucleophilicity but the specificity of the nucleophilic site as well. Considering this trend in regioselectivity there have been substantial efforts to utilize those classes of molecule as neutral nucleophiles en route to the synthesis of many important biomolecules.3The isomeric landscape of alkoxybenzenes is shown in Fig. 1 with an increase in the number of alkoxy groups in the ring (other substituents are omitted for clarity). Considering alkoxy groups as ortho/para directors only for upcoming electrophiles and depending on the number of alkoxy groups, they can have different nucleophilic sites (viz. for simple methoxybenzene, there might be ortho/para-selected products), although they react at only one site in a reaction (Fig. 1).
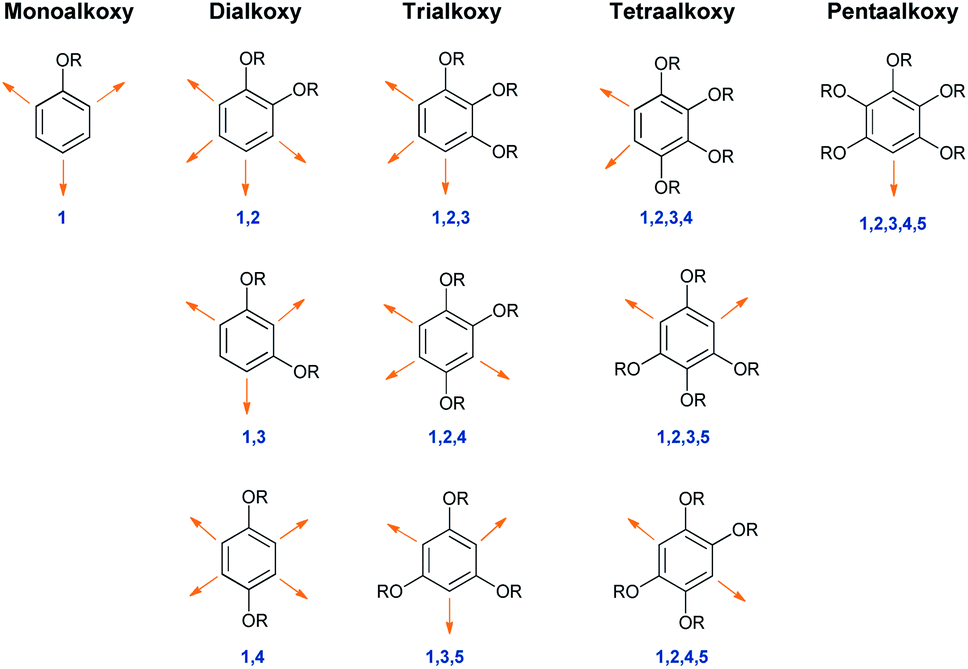 | ||
| Fig. 1 Alkoxybenzenes with number of possible regioisomeric nucleohilic sites (shown by the orange arrows). | ||
Small strained rings like cyclopropanes, aziridines, and epoxides, which are always planar molecules, are also electrophilic in nature for this reason. The tremendous angle strains inside them bend the bonds outwards (“banana” bonds) to increase the s-character in the bonds.12a Fig. 2 shows the actual angle difference of (109°28′′–104°) = 5°28′′ in cyclopropane leading to an angle strain of 27.5 kcal mol−1.12a,b This strain is gradually relieved as the ring size increases, with a greater degree of relaxation. Thus, any nucleophile that can “attack” this small ring can open it up and give vent to the strain. If the nucleophile is an electron-rich aromatic, then it will become alkylated in the process. As simple alkylations of alkoxy benzenes are abundant in the literature, this nucleophilic ring opening of small molecules turns out to be an interesting version of such alkylations. A point to be noted is that an aryl lithium or aryl Grignard addition is a prime competitor for Friedel–Crafts-type ring opening for the alkylation of aromatic ring alkylations.4 But, Grignard reactions generally require more inert although harsher conditions than the FC reaction to proceed. Added to this, they produce more side products, as the aryl Grignards are highly reactive. On the other hand, the FC reaction sometime produces regioisomeric mixtures of products and undergoes disproportionation.12b Therefore, considering all these constraints, the better practicability of the above two reactions still causes a great deal of debate, depending on the distribution of the desired products.
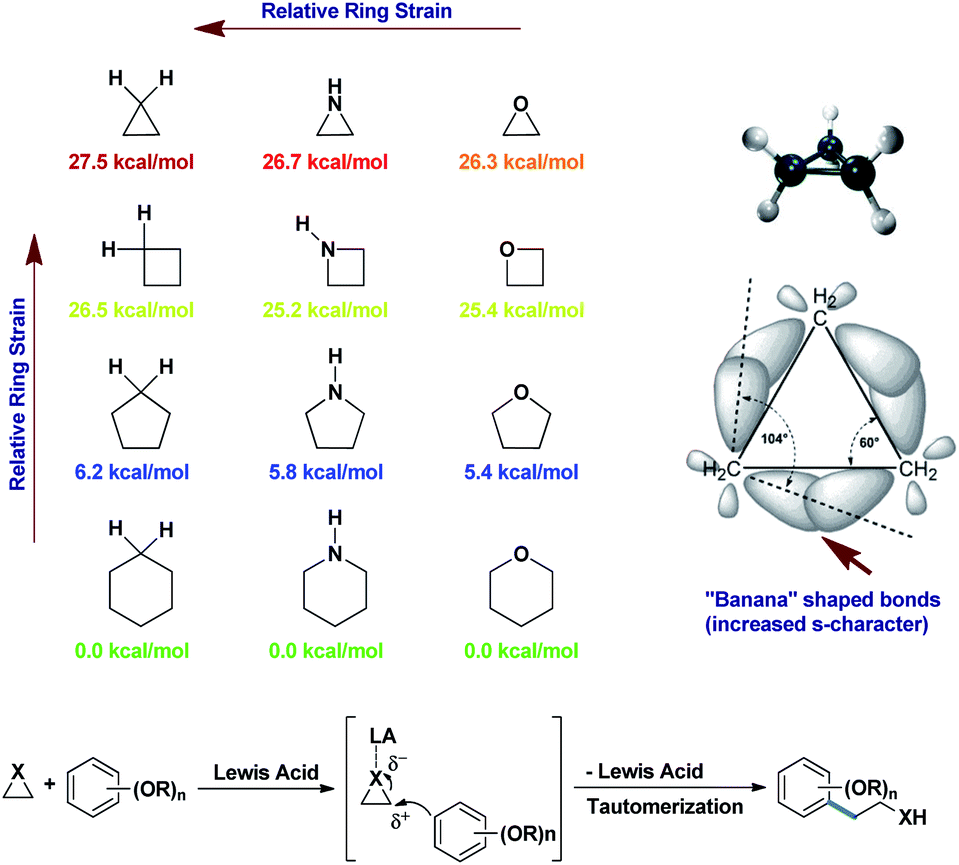 | ||
| Fig. 2 Release of angle strains in small carbo or heterocycles by ring opening.12a,b | ||
Such nucleophilic ring-opening derivatizations of alkoxybenzenes can be broadly classified depending on the ring size of the other electrophilic substrate in the reaction.
2.1.1.1 Ring opening of cyclopropanes. DA-cyclopropanes are activated further in the presence of catalytic amounts of Lewis/Brønsted acids to undergo ring-opening functionalization. Several methods are available in the literature for their opening by isomeric alkoxy benzenes. Ivanova et al. reported a 1,3-dimethoxybenzene-mediated nucleophilic ring opening of donor–acceptor cyclopropane diethyl diester.14 5 mol% lanthanide salt Lewis acid Yb(OTf)3 was loaded in refluxing chlorobenzene solvent to obtain a 73% yield of the alkylated 1,3-DMB product 2-(2,2-diarylethyl)malonate (Scheme 1).
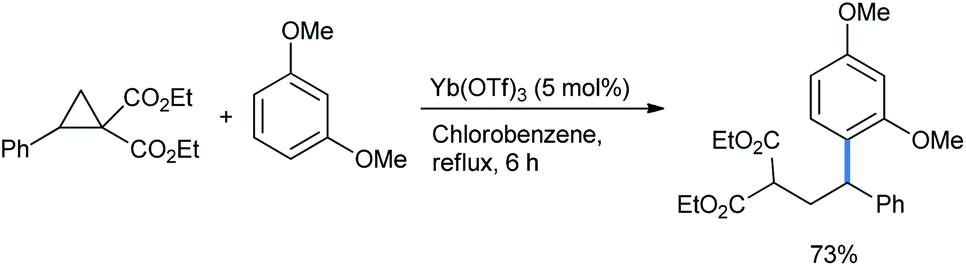 | ||
| Scheme 1 Ring-opening alkylation of 1,3-DMB by cyclopropane diester. | ||
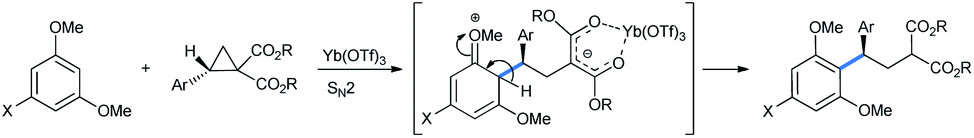
Ghorai and co-workers used 1,3,5-TMB for the stereoselective ring opening of DA-cyclopropane under the influence of a catalytic amount of Yb(OTf)3. Starting with an enantiopure substrate, excellent enantioselectivity was obtained with almost quantitative product yield (Scheme 2).15
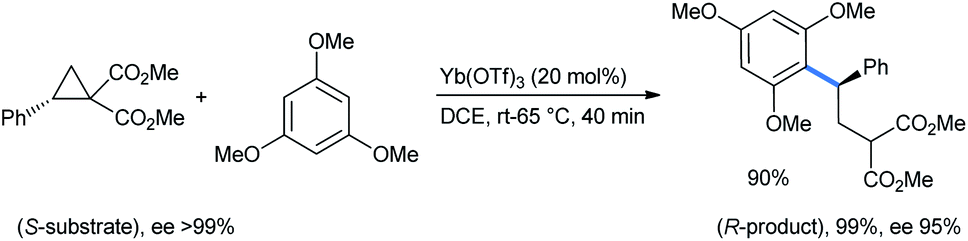 | ||
| Scheme 2 Stereoselective ring opening of DA-cyclopropanes by 1,3,5-TMB for the synthesis of 2-(2,2-diarylethyl)malonate. | ||
The above reactions follow a simple FC-type mechanism (Scheme 3). The catalytic oxophilic lanthanide(III) salt first coordinates with the ester carbonyl, making the benzyl position more electronically deficient. The multi-alkoxy benzene attacks the benzyl position of the ring in SN2 fashion. After ring opening, the aromaticity in the nucleophile is restored by proton transfer and tautomerization.
 | ||
| Scheme 3 Mechanism of ring opening of DA-cyclopropanes by electron-rich arenes. | ||
When the styryl substrate was used, the nucleophilic attack happened only in the ring carbon and the styryl double bond was absolutely untouched. The styryl-substituted product was then allylated and subjected to a ring-closing-metathesis (RCM) strategy by a Grubbs-2 catalyst for the facile synthesis of a trimethoxy phenyl substituted cyclohexene with 81% yield (Scheme 4). The RCM product structurally resembles the antimalarial agent (+)-methyllinderatin.16
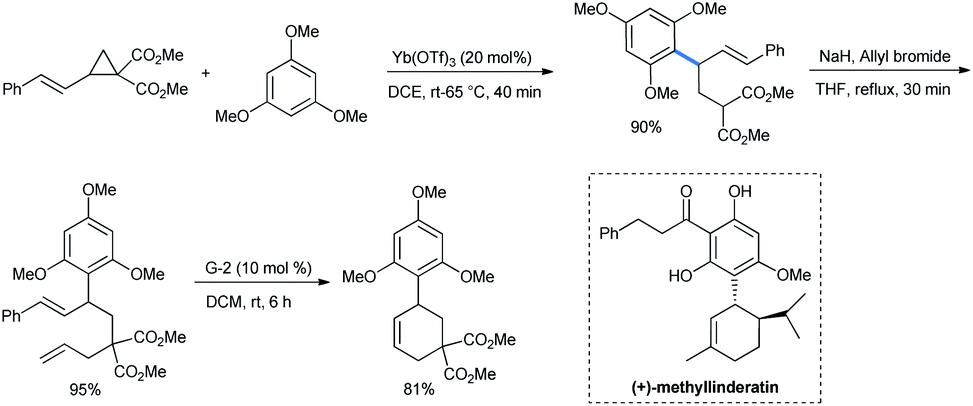 | ||
| Scheme 4 Ring opening of DA-cyclopropanes by 1,3,5-TMB. Synthesis of cyclohexenes by RCM. | ||
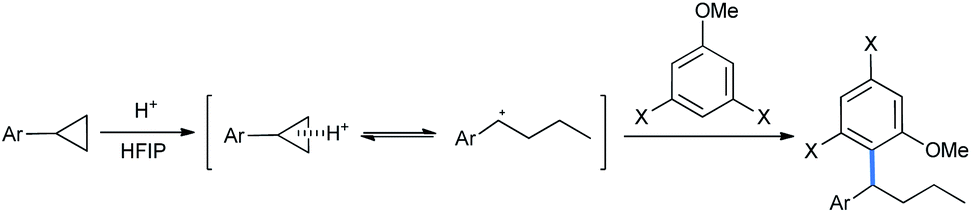
Moran's group claimed a Brønsted acid triflic acid catalyzed ring opening of DA-cyclopropanes in hexafluoroisopropanol medium. Using different substituted methoxy benzenes, a wide range of such 2-(2,2-diarylethyl)malonates were synthesized (Scheme 5). The bromo-substituted nucleophile was a suitable reactant for this reaction, giving 51% product yield.17
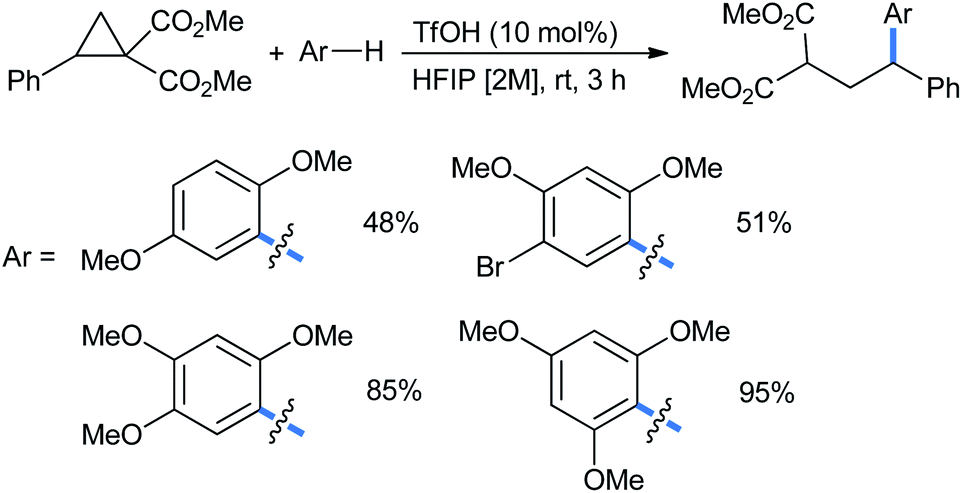 | ||
| Scheme 5 Ring opening of DA-cyclopropanes by Brønsted acid in HFIP solvent. | ||
The reaction scheme was extended to some chalcone-derived electrophiles to obtain the corresponding diaryl ketones (Scheme 6) in a regioselective homo-conjugate addition.17 The chloro-substituted dimethoxy nucleophile gave 72% of the ketone product.
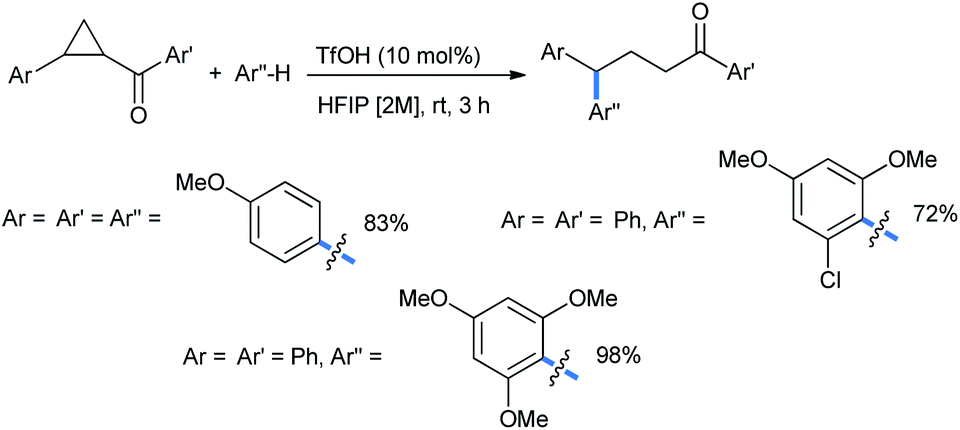 | ||
| Scheme 6 Synthesis of diaryl ketones from chalcone-derived DA-cyclopropanes. | ||
5 mol% Brønsted acid B(C6F5)3·H2O in HFIP medium was also suitable for this ring opening using 1,3,5-TMB (Scheme 7).17
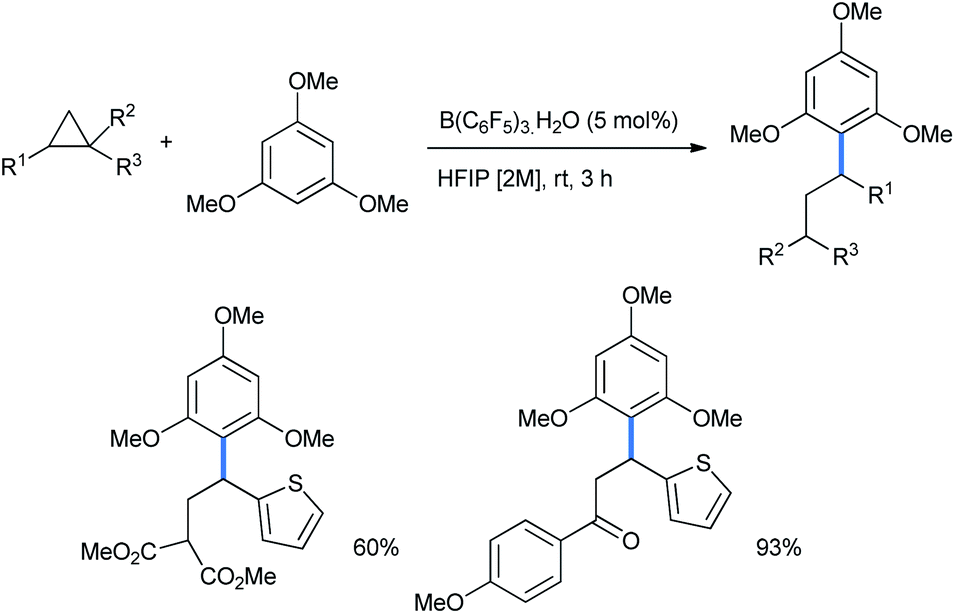 | ||
| Scheme 7 Ring opening of DA-cyclopropanes by Brønsted acid B(C6F5)3·H2O. | ||
Later on this strategy was taken to the functionalization of monosubstituted or gem-disubstituted cyclopropanes having either an electron-donor (aryl) or an electron-acceptor (keto or diester) group at a time. Although the reactions needed much higher temperature to proceed. Here 1,3-dimethoxybenzene displayed good ring-opening reactions with both diester or keto (benzoyl) containing rings (yields 77% and 87%, respectively).17 Fluoro-substituted dimethoxy benzene gave 82% of the ring-opened ketone (Scheme 8). 1,3,5-Trimethoxy benzene gave good yields of the corresponding products. Due to less activation of the ring by the solitary ester group, only 15% of product was obtained from methyl cyclopropanecarboxylate.
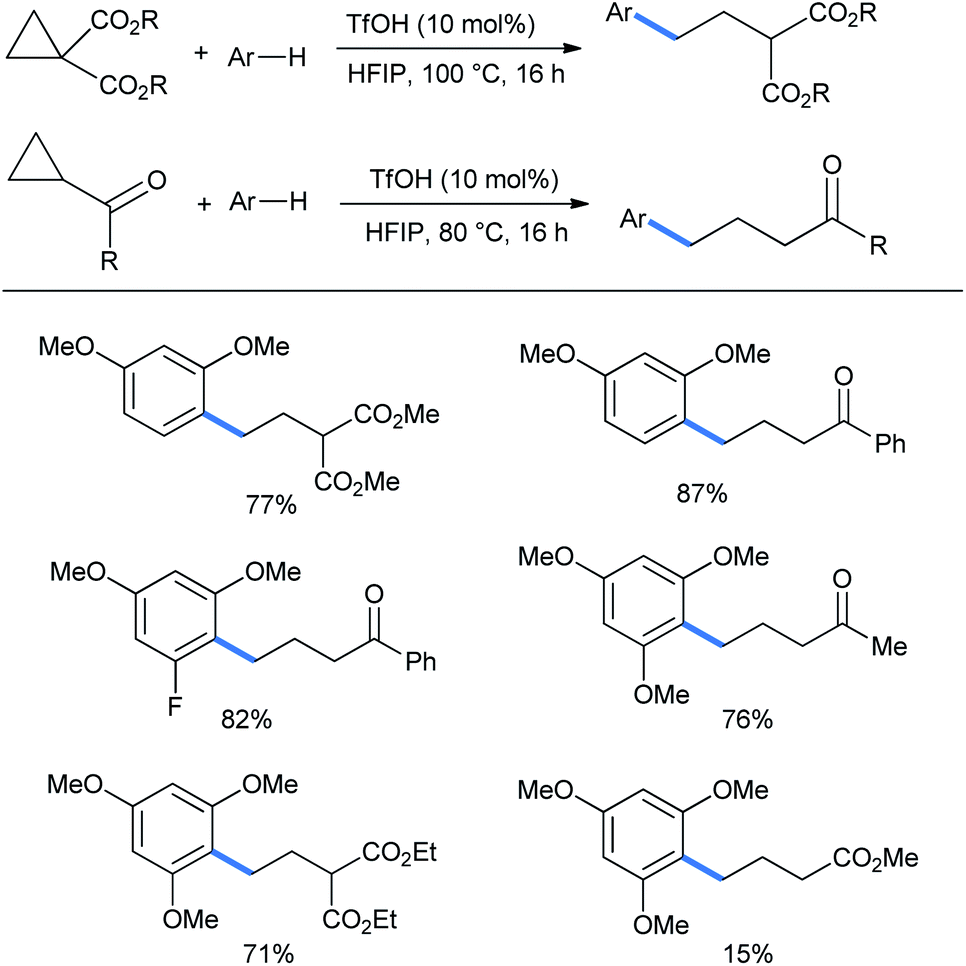 | ||
| Scheme 8 Ring opening of monosubstituted cyclopropanes by the Brønsted acid TfOH. | ||
The mechanism for carbonyls followed the same pathway as described, with triflic acid protonating the carbonyl oxygen which in turn polarizes the C–C bonds, making it easy for attack by methoxy benzene homologues (Scheme 9).17
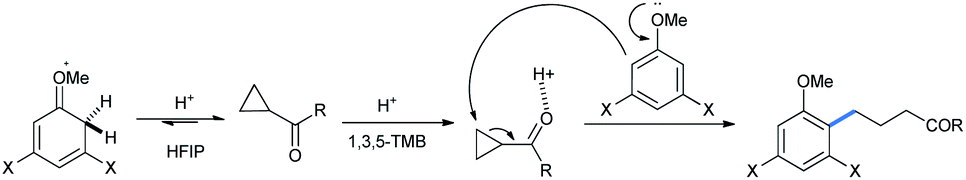 | ||
| Scheme 9 Mechanism of ring opening of cyclopropane carbonyls with alkoxy benzenes. | ||
Moran's group showed that the ring opening of simple aryl-substituted cyclopropanes can also be performed with each of the mono-, di- and trimethoxy benzene nucleophiles. The cyclopropanes lack the presence of any electron-withdrawing groups in the structures. The addition occurred at the benzylic position of cyclopropanes to give the corresponding geminal diaryl propanes in good to excellent yields (Scheme 10).18
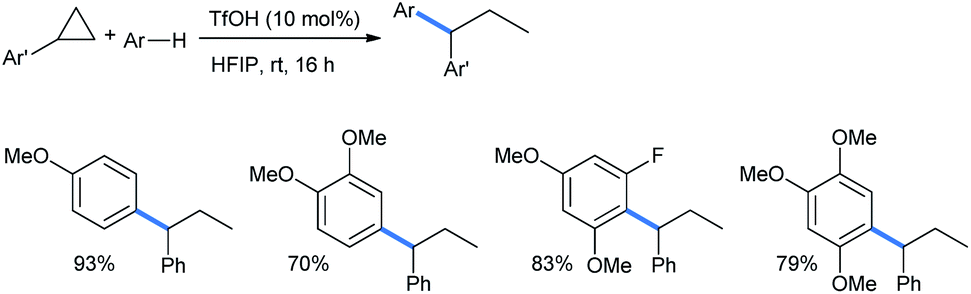 | ||
| Scheme 10 Ring opening of aryl cyclopropanes by methoxy benzenes in Brønsted acid TfOH. | ||
The non-benzyl C–C bond electrons first coordinate with the proton from triflic acid, in turn increasing the polarization on the benzyl carbon. Then it is attacked by the nucleophile to give the ring-opened product (Scheme 11).18
 | ||
| Scheme 11 Mechanism of ring opening of aryl cyclopropanes with no acceptor groups. | ||
2.1.1.2 Ring opening of aziridines. Aziridines are one of the most attractive building blocks in organic synthesis, especially because of the presence of a nitrogen atom. Great efforts have been made through the years for further ring-opening functionalization on this small and readily openable ring.19 Ghorai and co-workers synthesized 2,2-diarylethylamine via the regioselective ring opening of N-activated aziridine by 1,2,3-TMB. The reaction was catalyzed by a mixture of only 5% Lewis acids Zn(OTf)2 and Sc(OTf)3 in DCE solvent (Scheme 12).20
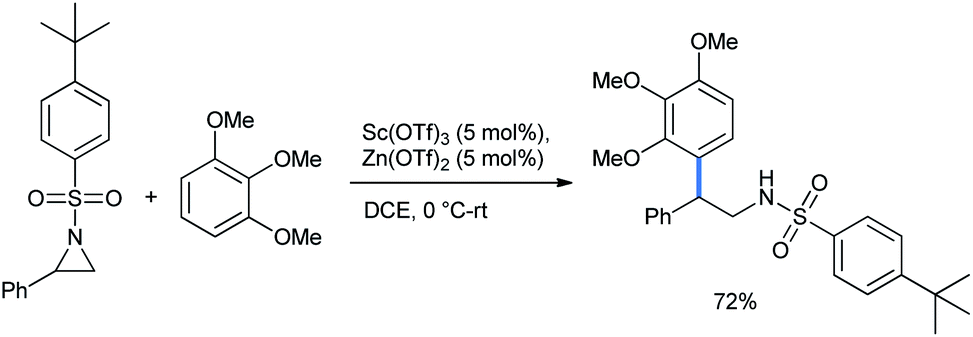 | ||
| Scheme 12 Ring opening of aziridines by 1,3,5-TMB. | ||
Similar results were obtained with the nucleophile 1,3,5-TMB under the same conditions to afford the stereo-enriched ring-opened 2,2-diarylethylamines (Scheme 13). The reaction proceeds via a similar Lewis acid catalyzed mechanism as described for the cyclopropane cases.20
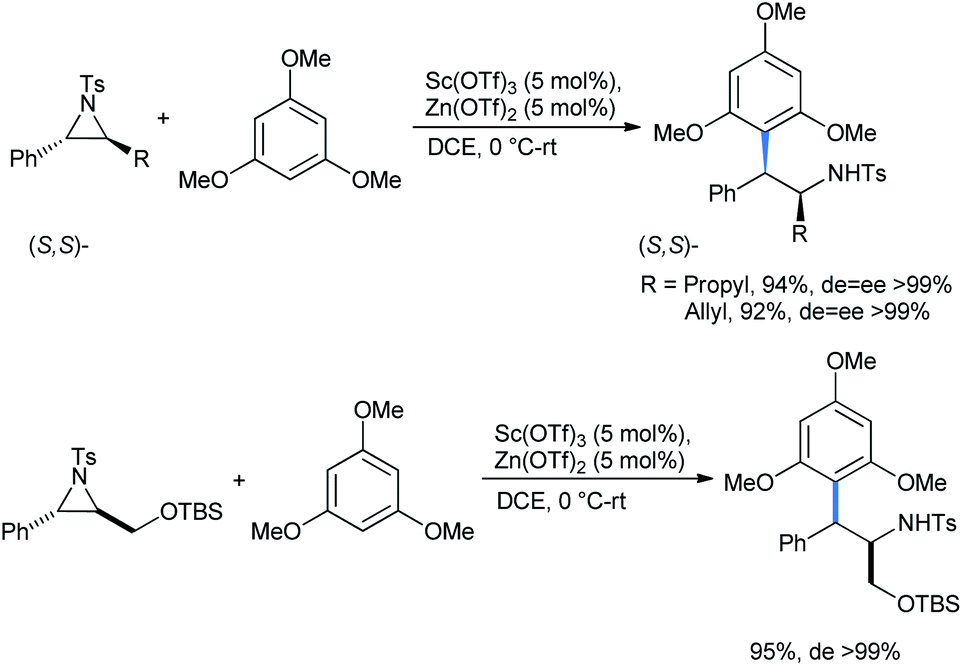 | ||
| Scheme 13 Ring opening of aziridines by 1,3,5-TMB. | ||
The Lewis acids are coordinated by tosyl oxygens, thus in turn weakening the benzyl C–N bond where the π-electrons of TMB could penetrate to give the desired product (Scheme 14).20
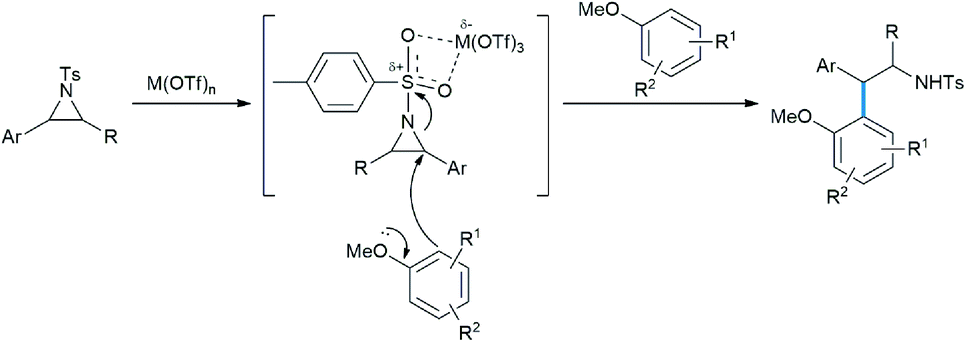 | ||
| Scheme 14 Mechanism of aziridine opening in Friedel–Crafts fashion. | ||
Takahashi et al. described an asymmetric formal synthesis of the antitumor agent (−)-podophyllotoxin.21a The key step was the synthesis of the aziridine ring opening by sesamol (Scheme 15).21b To check the feasibility and optimizing the Lewis acid concentration required for the reaction, the authors used less reactive (with fixed nucleophilic site) sesamol benzyl and allyl ethers with (±)-trans-arylaziridine-2-carboxylate in DCM solvent in the presence of catalytic InCl3.
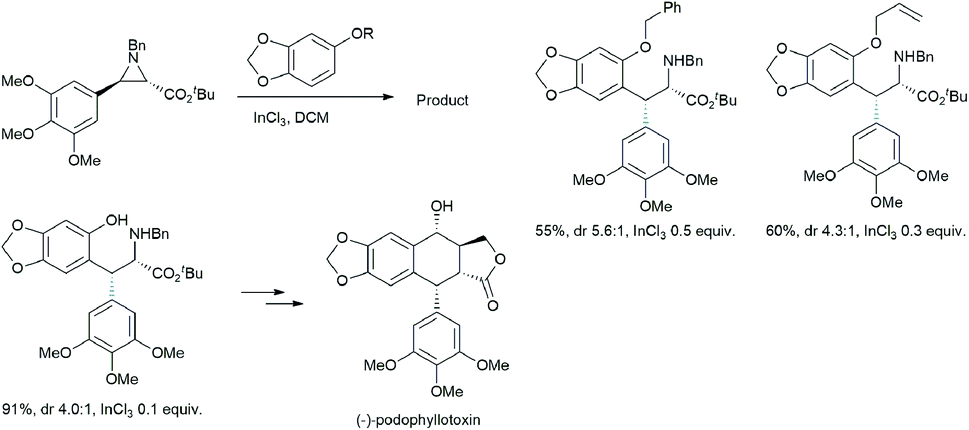 | ||
| Scheme 15 Total synthesis of (−)-podophyllotoxin from aziridine. | ||
Here, the metal coordinates with the aziridine ester oxygens for the ring activation followed by its cleavage (Scheme 16).21
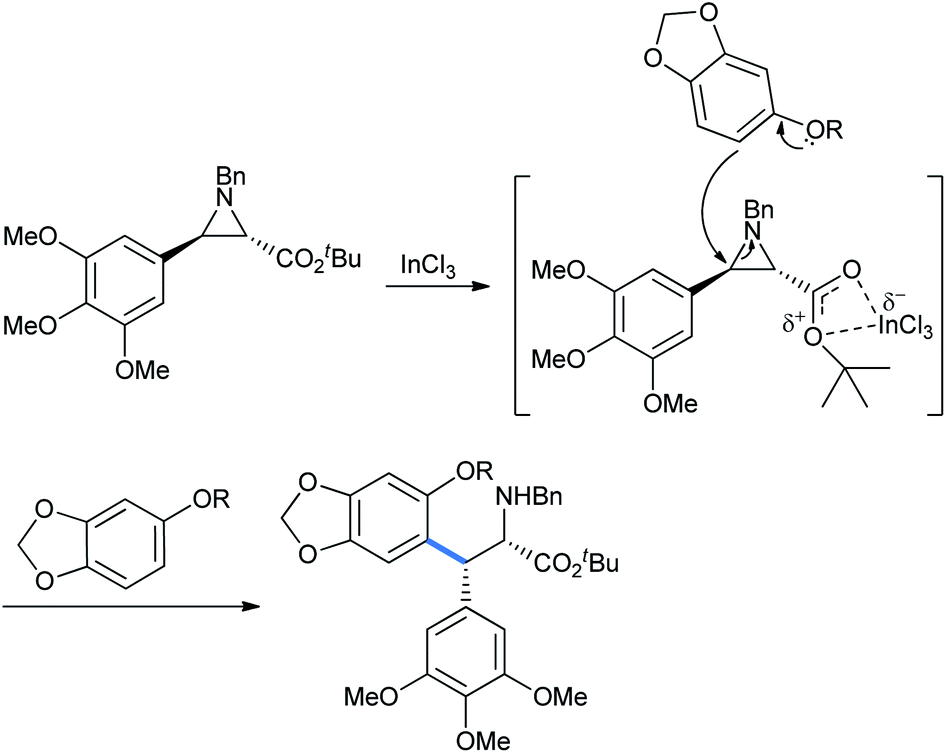 | ||
| Scheme 16 Synthesis of intermediate for (−)-podophyllotoxin. | ||
Hsueh described the ring opening of N-mesyl aziridine by 1,3,5-trimethoxy benzene in the presence of methanesulfonic acid as an activator.22 The reaction was carried out in a telescoped continuous flow process with both the formation and ring opening of aziridine to obtain 51% product yield (Scheme 17) with 40 minutes of residence time.
 | ||
| Scheme 17 Ring-opening reaction of N-mesyl aziridine under continuous flow. | ||
Xing and co-workers described a Lewis acid assisted synthesis of cis-1,4-disubstituted tetrahydroisoquinolines from N-activated aziridines by a tandem ring opening followed by a 3-component Pictet–Spengler reaction sequence in DCE medium (Scheme 18) under heating conditions.23
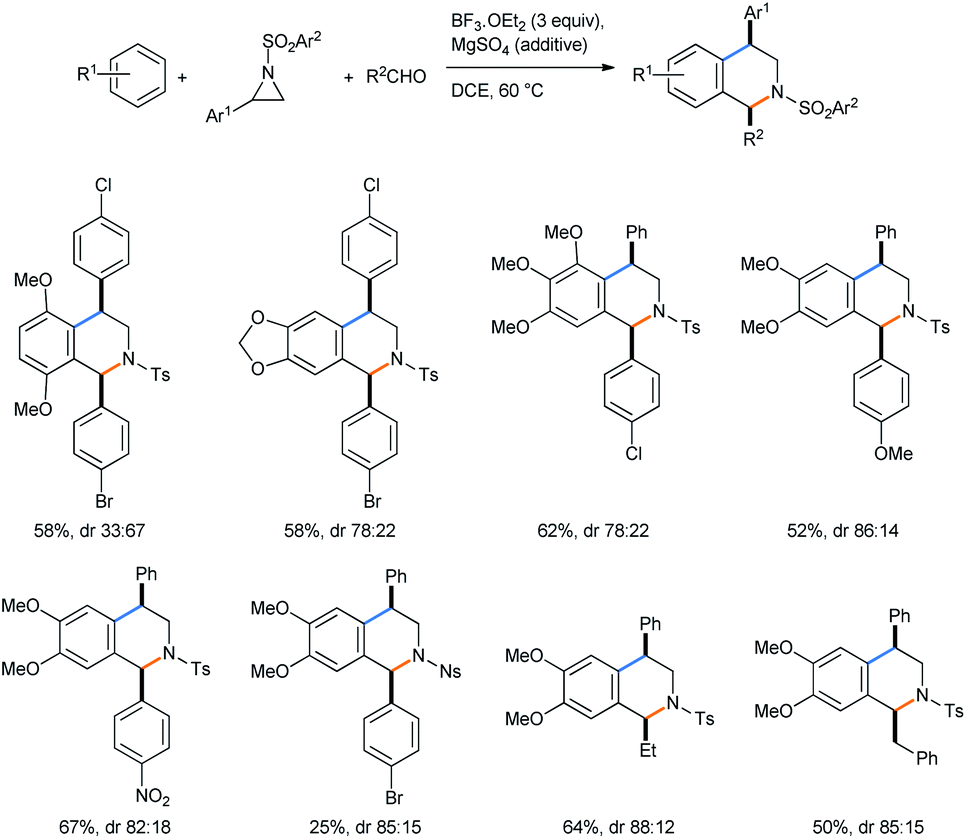 | ||
| Scheme 18 Synthesis of cis-1,4-disubstituted tetrahydroisoquinolines from N-activated aziridines. | ||
After the BF3·OEt2 catalyzed ring opening of aziridine, the free amine forms an iminium ion with aldehyde (Scheme 19). The iminium ion A is then reacted with electron-rich arene in Friedel–Crafts fashion.23 The second-step attack from the top face might result in steric repulsion between the aldehyde aryl group and the aziridine tosyl group (B), leaving the trans-diastereomer unfavorable. The approach of arene from the bottom face of imine only arranges an interaction between the tosyl and the small benzyl proton of the original aldehyde (C). This largely supports the formation of the cis-diasteromer of the tetrahydroisoquinoline product.
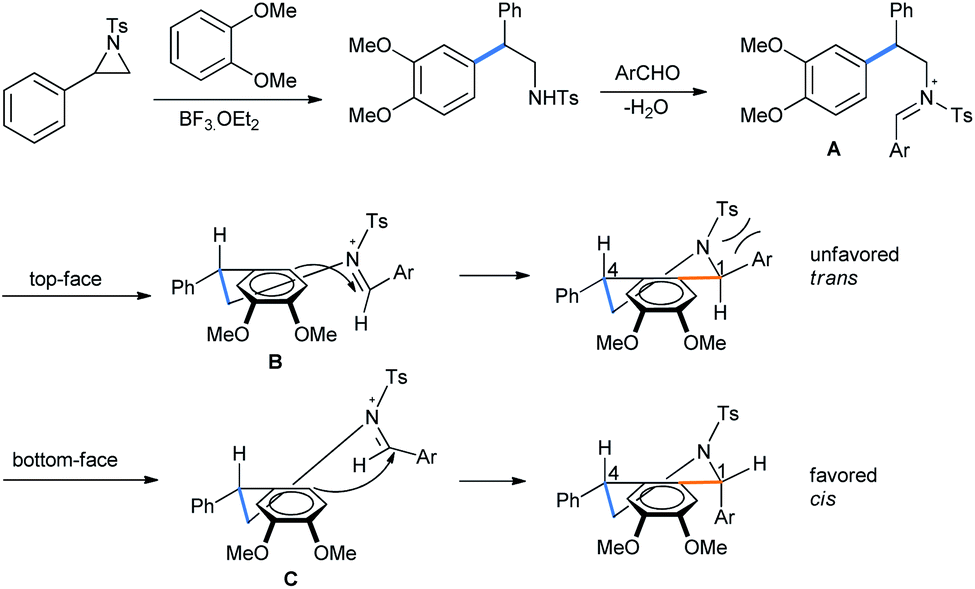 | ||
| Scheme 19 Mechanism of aziridine ring opening by dimethoxybenzene followed by diastereoselective Pictet–Spengler condensation. | ||
Acetals of the corresponding aldehydes also reacted in the same way, giving tetrahydroisoquinoline products. 3,4-Dimethoxy bromobenzene also acted in the same way as 1,2-DMB to afford the same product, proving that in the Pictet–Spengler process Br+ was liberated instead of a proton. Performing the reaction with a separately made ring-opened product with aldehyde for the Pictet–Spengler condensation gave similar yield and stereoselectivity in the final tetrahydroisoquinoline.
2.1.1.3 Ring opening of epoxides. Epoxides are important building blocks in organic syntheses.24 These highly strained and energetic small rings are sensitive to a wide range of nucleophiles, including moisture. They are easily opened up by alkoxy benzenes as well. Taylor and co-workers studied the ring opening of aromatic-substituted epoxides in the presence of 50% SnCl4 as a Lewis acid alkylation promoter (LAAP) to obtain a regioisomeric mixture of ring-open products with anisole as the solvent (Scheme 20).25 The yield increases with increasing electron density in the substrate.
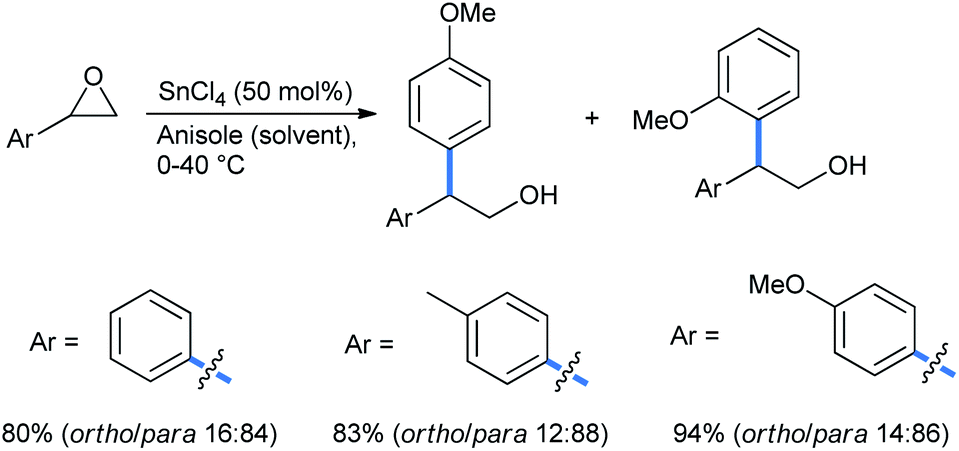 | ||
| Scheme 20 Ring opening of aryl epoxide with SnCl4. | ||
For the above reaction the yields were greatly improved over the previously reported benzene as solvent and AlCl3 as Lewis acid.26 Later on, Taylor et al. repeated their work with chiral epoxide to compare the stereochemical results with the Grignard reaction (Scheme 21).27 It is apparent from the specific rotation and ee values that both the FC and Grignard adduct from the ortho-position of anisole proceed via single inversion, which is also the case when the reaction is going through the para-position via the FC mode. The Grignard addition through the para-position of anisole only proceeds by double inversion. Although the sharp fall in enantioselectivity in all cases does not rule out a competitive SN1 pathway operating.
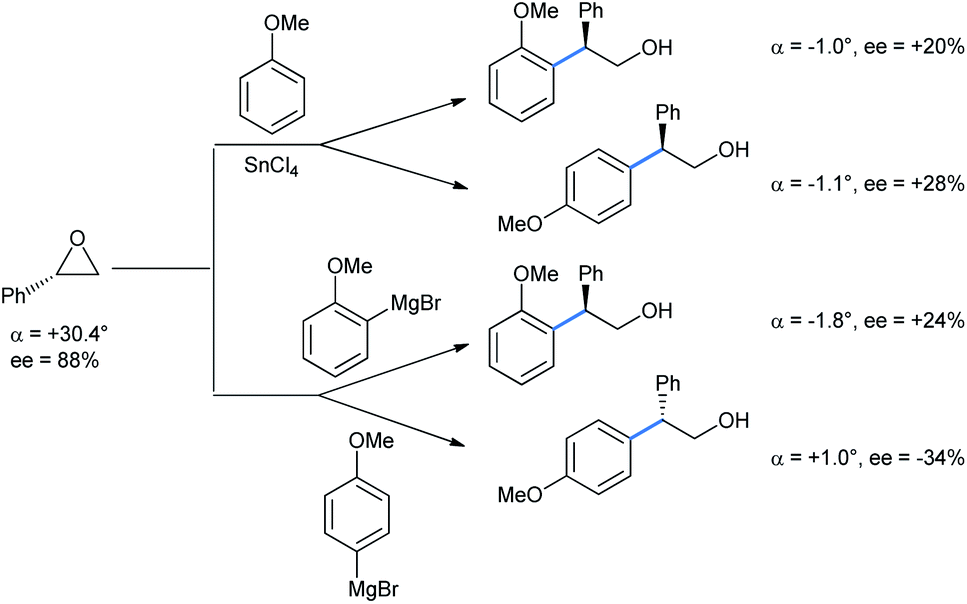 | ||
| Scheme 21 Ring opening of optically active phenyl oxirane: FC vs. Grignard reaction. | ||
As usual the epoxide oxygen first coordinates with the Sn4+ Lewis acid followed by ring opening at the benzyl position by anisole (Scheme 22).28
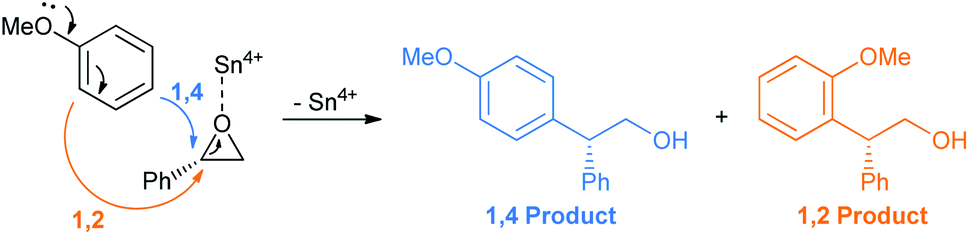 | ||
| Scheme 22 Proposed mechanism of epoxide opening with SnCl4 as the Lewis acid. | ||
Yang described a methodology for similar ring opening of styrene oxide in the presence of MoO2(acac)2 Lewis acid in CCl4 solvent (Scheme 23).29 If the para-position of anisole was blocked by a highly electron-withdrawing group, the relatively slower reaction gave only a trace amount of the ortho-ring-opened product.
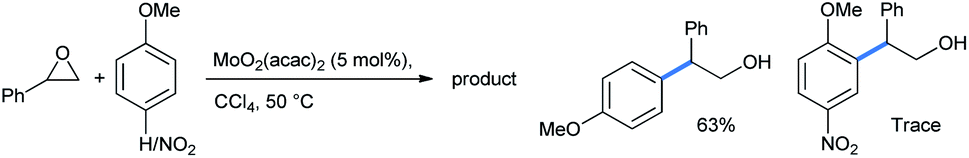 | ||
| Scheme 23 Ring opening of epoxide by anisole in the presence of MoO2(acac)2 Lewis acid. | ||
The catalytic MoIV interacts with the epoxy oxygen. The weakened benzyl carbon then reacts with methoxy benzene and the C–O bond is cleaved to give the ring-opened product (Scheme 24).29 It is noteworthy that similar Lewis acidic activity of MoO2(acac)2 is known previously.30
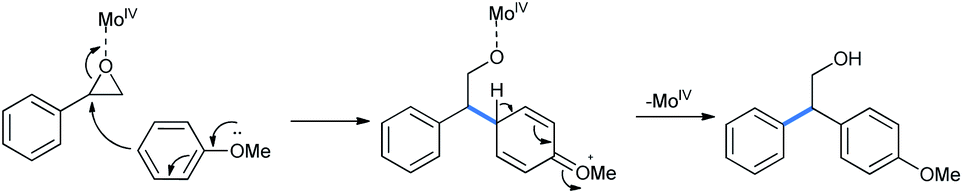 | ||
| Scheme 24 Mechanism of MoIV-assisted epoxide opening by anisole. | ||
An ingenious choice of oxirane substrates containing the alkoxy aryl groups lying just adjacent can undergo such intramolecular ring opening, giving rise to important molecular skeletons under suitable conditions, known as the intramolecular Friedel–Crafts epoxy–arene (IFCEA) cyclization strategy.31 In a such intramolecular fashion, attempting to synthesize the 7-membered ether tetrahydrobenzo[c]oxepin-4-ols from benzyl glycidyl ethers, Pericás tried different Lewis acid combinations.32 The 3,5-dimethoxy substrates gave the desired product, but the 4-methoxy analogues solely afforded 4-diarylmethyl-1,3-dioxolane (Scheme 25) with yields varying with the conditions used. Both sets of products were accompanied by high enantioselectivities.32a Condition D was chosen by the authors as the optimized one to keep the balance in yields between the 7-membered racemic hydroxyl ethers and the 1,3-dioxolane products.32b
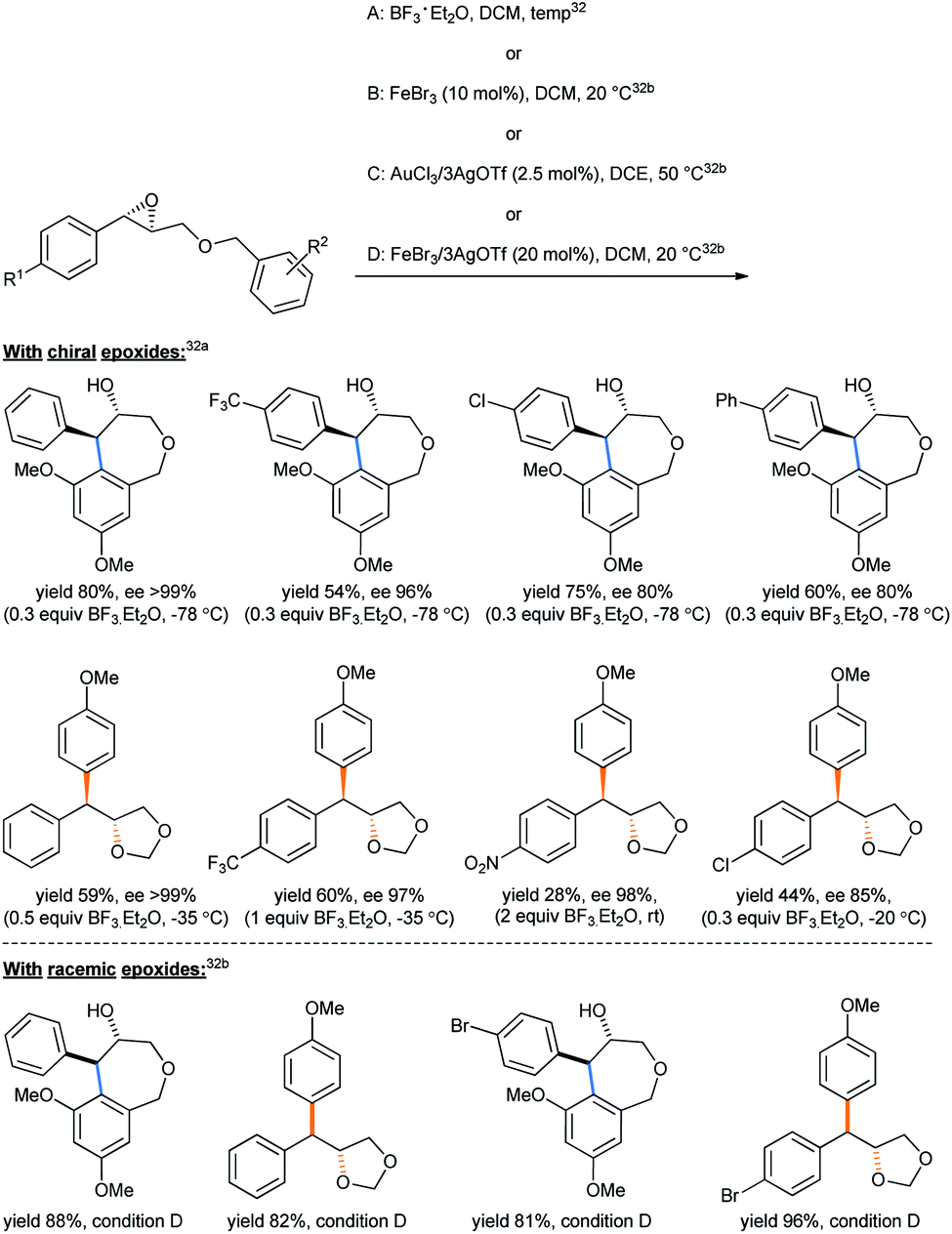 | ||
| Scheme 25 Synthesis of tetrahydrobenzo[c]oxepin-4-ols from benzyl glycidyl ether.32 | ||
The tetrahydrobenzo[c]oxepin-4-ol products arise from the usual Lewis acid activated ortho/para intramolecular ring opening of benzyl glycidyl ethers (Scheme 26).
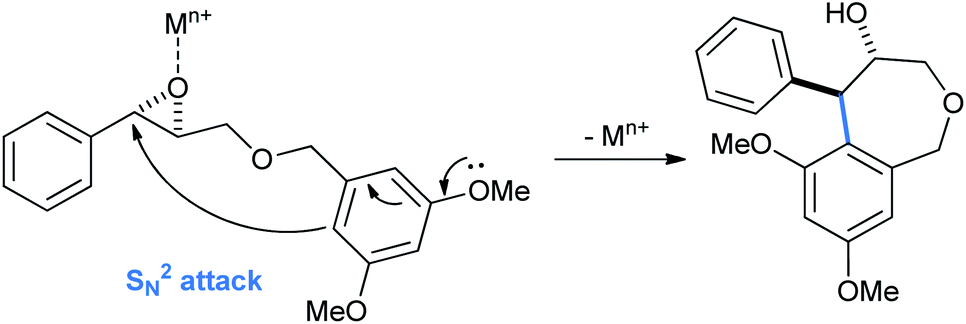 | ||
| Scheme 26 Mechanism of formation of tetrahydrobenzo[c]oxepin-4-ol from benzyl glycidyl ether. | ||
Interesting 4,4-diarylmethyl-1,3-dioxolane products arrive from an ipso attack from the electron-rich arene to the Lewis acid activated epoxide. The spiro-tetrahydropyran intermediate thus formed followed a fragmentation procedure, leading to the corresponding 4,4-diarylmethyl-1,3-dioxolane (Scheme 27).
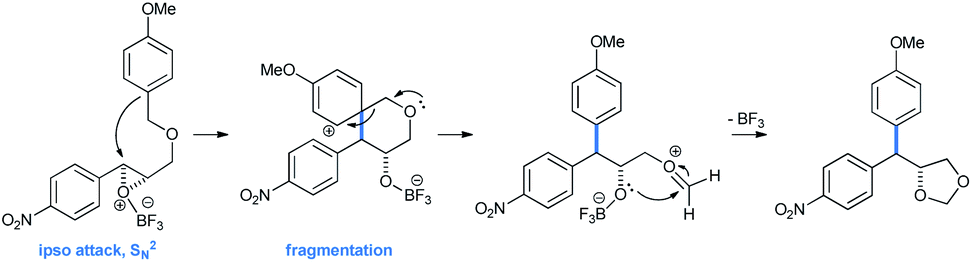 | ||
| Scheme 27 Mechanism of formation of 4,4-diarylmethyl-1,3-dioxolane. | ||
Qu reported an intramolecular regioselective ring opening of chiral epoxides with an adjacent methoxy benzene subunit under refluxing conditions in refluxing hexafluoroisopropanol as solvent to produce enantiopure 3-chromanol derivatives as trans-diastereomers (Scheme 28).33a This class of compounds are abundant in nature and largely have antioxidant properties.33b–d
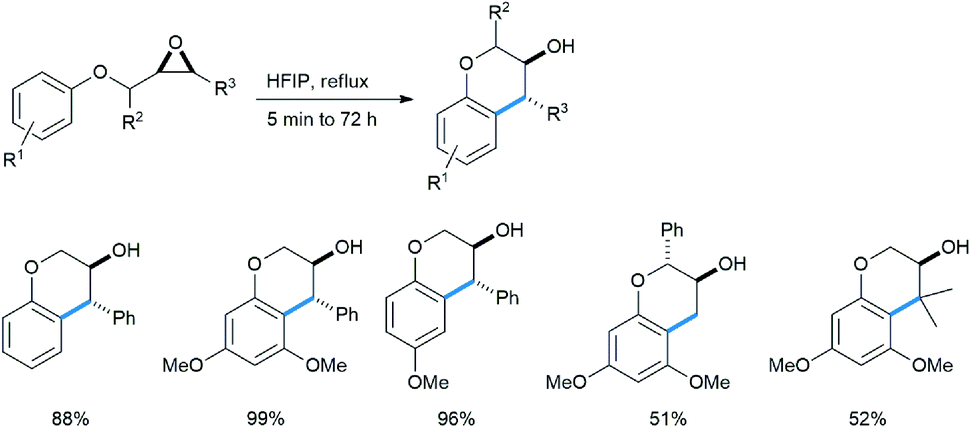 | ||
| Scheme 28 Synthesis of 3-chromanols from aryloxymethyl oxiranes in HFIP. | ||
This is a metal-free process and no Lewis acid was required for this protocol (Scheme 29). The “acidic” O–H proton of HFIP (pKa 9.3) activated the epoxy ring under refluxing conditions to open it in intramolecular fashion via “electrophilic solvent assistance (ESA)”. Apart from HFIP, 2,2,2-trifluoroethanol (pKa 12.4), water (pKa 15.7) and phenol (pKa 9.95) also helped ring opening by this method. The transition state shown in the mechanism is also stabilized by the highly ionizing solvent HFIP.
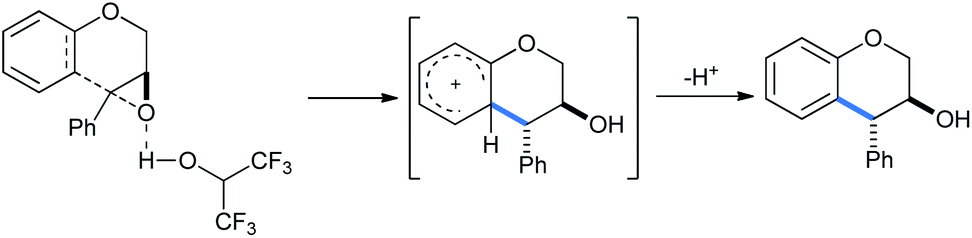 | ||
| Scheme 29 Mechanism of HFIP-assisted intramolecular ring opening of epoxides.33a | ||
The reaction scheme is well applicable to its intermolecular version (Scheme 30), also giving enantiopure product with 1,3,5-TMB. The yield increases with the number of methoxy groups. The only exception is the tetramethoxylated aromatic, probably for steric reasons. For the methoxy epoxide (the last product) a slow ring-opening reaction occurred at the least sterically hindered position.
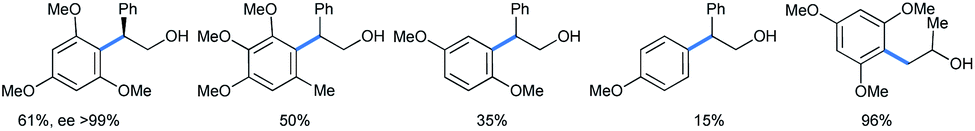 | ||
| Scheme 30 Intermolecular ring opening of epoxides with methoxy benzenes under HFIP.33a | ||
Devi used catalytically hydrated Brønsted acid TsOH·H2O for a cheap and metal-free intramolecular ring-opening reaction of 2-(aryloxymethyl)-3-aryloxiranes to obtain trans-4-arylchroman-3-ols in toluene solvent (Scheme 31).31a The reactions needed no inert atmosphere and the products have several biologically important activities as antioxidant, anticarcinogen, antiarteriosclerotic and antibacterial molecules. The yields ranged from 54 to 88% with the use of 20 mol% Brønsted acid. The use of stronger acids like trifluoro acetic acid, triflic acid or sulfuric acid lowered the yield, whereas weak acids like phenol or acetic acid failed to give any product.
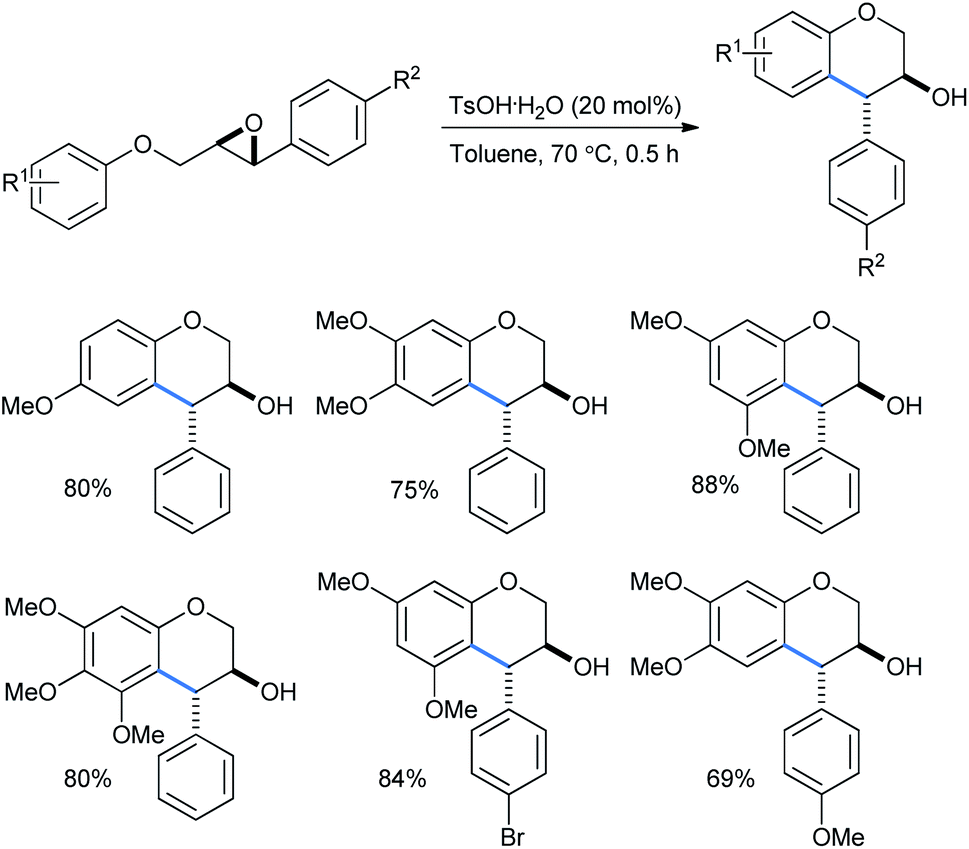 | ||
| Scheme 31 Brønsted acid-catalyzed intramolecular ring opening of 2-(aryloxymethyl)-3-aryloxiranes. | ||
Trans-isomers were exclusive in the products. According to Qu,33a a direct and concerted 6-endo-tet ring-opening cyclization of the epoxide can give rise to a trans-product (SN2, route a, Scheme 32). Otherwise if the reaction followed route b (SN1), the benzyl cation formed after acid-mediated cleavage of the epoxide, can stay in 4 different energetic transition states A–D. The TS C having the least steric requirement gave the final trans-product by a 6-exo-trig fashioned Friedel–Crafts attack from the alkoxy-bearing aryl group.
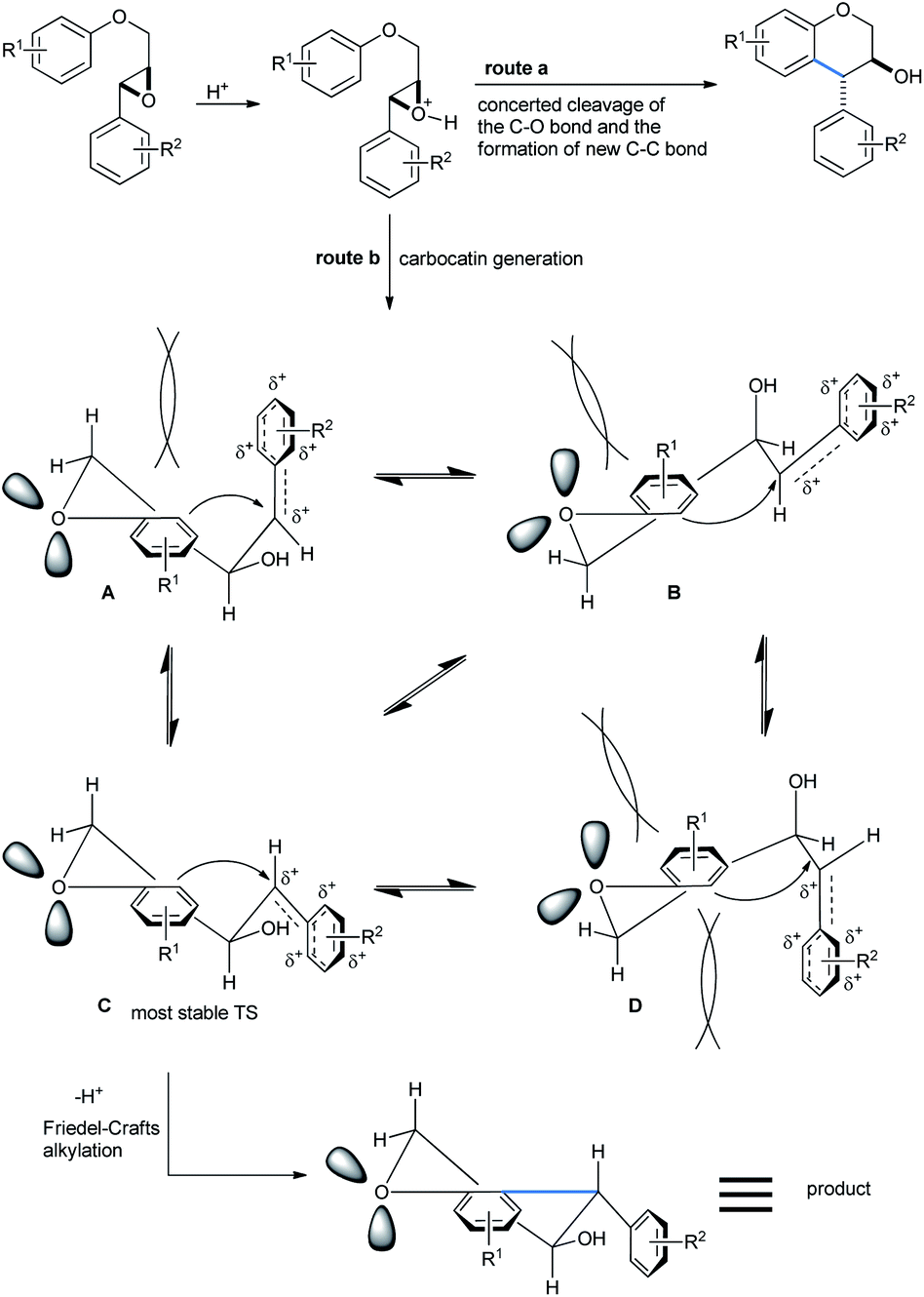 | ||
| Scheme 32 Mechanism of the formation of trans-4-arylchroman-3-ols by 6-exo-trig cyclization.31a | ||
Repeating the reactions under a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of toluene and acetonitrile, keeping other conditions the same, produced a comparable outcome for both alkoxy-substituted benzenes and naphthalenes.31b He and co-workers reported a gold(III)/silver(I) catalyzed ring-opening “cyclialkylation” of aryloxymethyl oxiranes in dichloroethane medium to synthesize 3-chromanols. A 2.5 mol% catalytic combination of AuCl3 and AgOTf catalysts was applied in a 1:3 ratio (Scheme 33).31c In some cases, the use of a 5 mol% catalyst afforded a better yield (given in parentheses). For 3,4-disubstituted chromanols, trans-diastereomers were the major product, indicating a possible SN1 mechanism. In the absence of gold(III) only trace amounts of 3-chromanols were obtained. Here Au(III) acted as the Lewis acid to coordinate with the epoxide oxygen and AgOTf was used as an additive to generate Au3+, probably by removing a Cl− ion from AuCl3.
:1 mixture of toluene and acetonitrile, keeping other conditions the same, produced a comparable outcome for both alkoxy-substituted benzenes and naphthalenes.31b He and co-workers reported a gold(III)/silver(I) catalyzed ring-opening “cyclialkylation” of aryloxymethyl oxiranes in dichloroethane medium to synthesize 3-chromanols. A 2.5 mol% catalytic combination of AuCl3 and AgOTf catalysts was applied in a 1:3 ratio (Scheme 33).31c In some cases, the use of a 5 mol% catalyst afforded a better yield (given in parentheses). For 3,4-disubstituted chromanols, trans-diastereomers were the major product, indicating a possible SN1 mechanism. In the absence of gold(III) only trace amounts of 3-chromanols were obtained. Here Au(III) acted as the Lewis acid to coordinate with the epoxide oxygen and AgOTf was used as an additive to generate Au3+, probably by removing a Cl− ion from AuCl3.
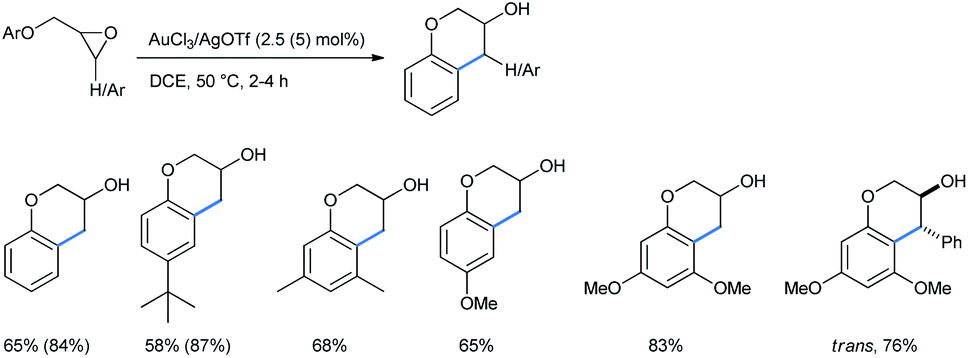 | ||
| Scheme 33 Gold(III)-catalyzed synthesis of 3-chromanols from aryloxymethyl oxiranes. | ||
When the reaction was performed with 1,3,5-TMB and propylene oxide, the sole regioisomer was obtained in 52% yield (Scheme 34).
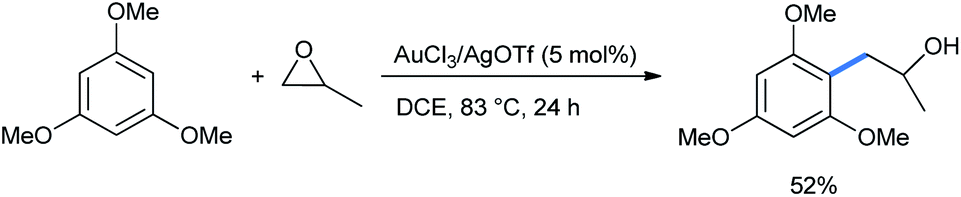 | ||
| Scheme 34 Ring opening of propylene oxide with 1,3,5-TMB. | ||
Adopting their previous strategy, Devi et al. described the parallel preparation of chroman-fused tetralins by ring opening of tetralin epoxy ethers in a cis-diastereoselective method.31d The products were B-ring-modified analogues of (±)-brazilin, a tetracyclic homoisoflavonoid natural red dye (Scheme 35).34
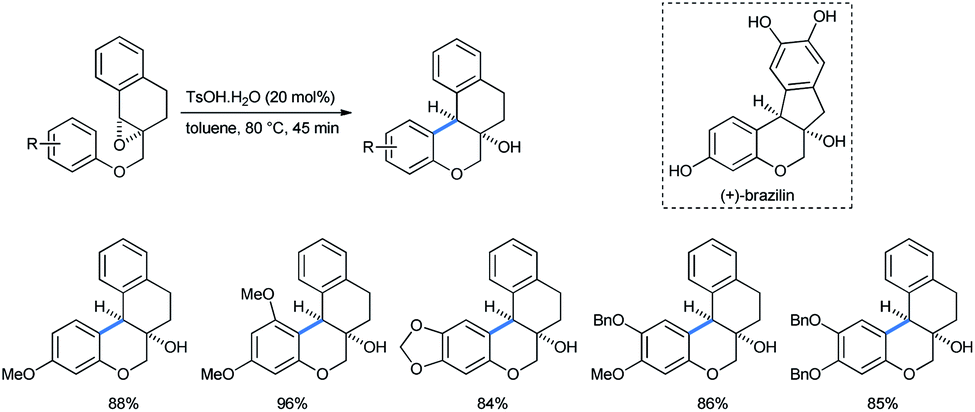 | ||
| Scheme 35 Synthesis of diastereoselective chroman-fused tetralins (cis exclusive). | ||
Later on using the same IFCEA strategy, they reported a 3-step synthesis of trans major tetrahydroquinoline-containing bridged benzothiaoxazepine-1,1-dioxides, starting from benzene sulfonamides via N-sulfonyl tetrahydroquinolines (Scheme 36).31e It is noteworthy that both N-arenesulfonyltetrahydroquinolines35a–d and benzothiaoxazepine-1,1-dioxides35e are important bioactive molecules.
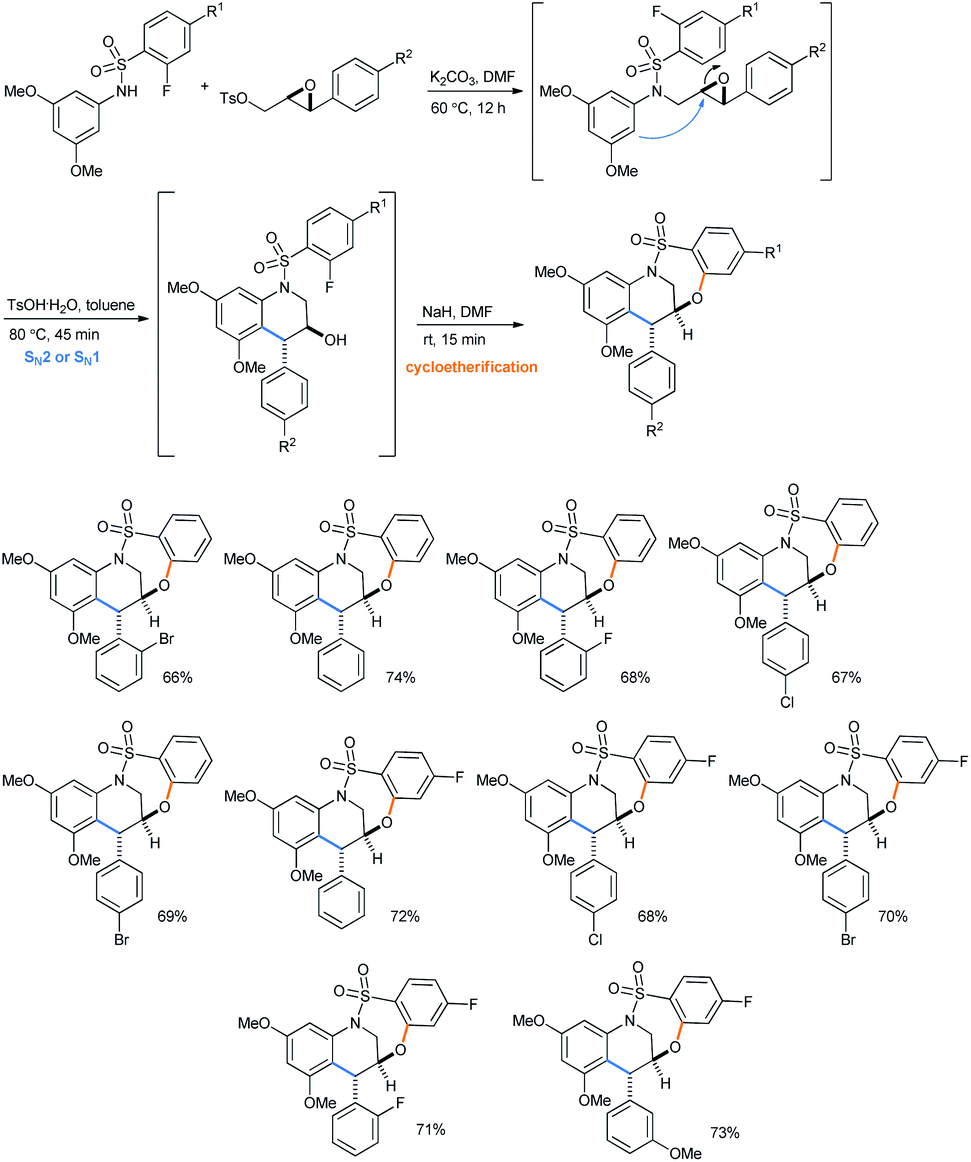 | ||
| Scheme 36 Synthesis of tetrahydroquinoline-containing bridged benzothiaoxazepine-1,1-dioxides from benzene sulfonamides.31e | ||
The enantioselective version was also made by starting from allyl alcohol using a chiral pool strategy by sharpless asymmetric epoxidation.36 The epoxide was then subjected to the optimized conditions to get (12S,13R)-tetrahydroquinoline with a 74% combined yield as the major enantiomer (Scheme 37).
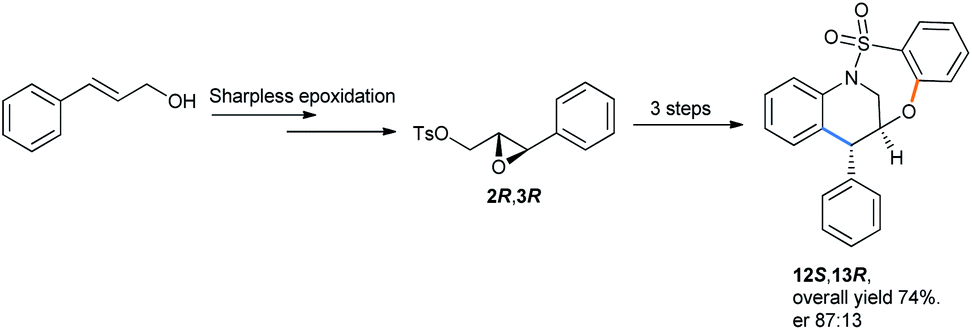 | ||
| Scheme 37 Synthesis of (12S,13R)-tetrahydroquinoline containing bridged benzothiaoxazepine-1,1-dioxide.31d | ||
The reaction intermediates need not be chromatographically purified to proceed to the next steps. The trans-selectivity in N-sulfonyl tetrahydroquinolines is attributed to the SN2 attack from the electron-rich arene section to the TsOH-activated epoxide for its ring opening followed by cycloetherification (Scheme 38). However, depending on the relative conformation of the transition state, an SN1 mechanism would give the same trans-product.
 | ||
| Scheme 38 Origin of trans-selectivity in products.31d | ||
2.1.1.4 Ring opening of thiiranes. Interesting 3-membered homologues of oxiranes are the thiiranes.37 The ring opening of 3-membered episulfonium salts was only reported by Ibragimov with electron-rich arene methoxy and 1,3-dimethoxy benzenes.38 This is an example of the β-arylthioalkylation strategy of arenes. For this purpose, olefin double bonds were sulfonated using arylsulfenyl halides to generate the episulfonium salts in situ in the presence of AgBF4.39 The reactions were carried out at −40 °C in a 1
:4 mixture of dichloroethane and dichloromethane solvents. The formation of thioethers resulted from the attack of a nucleophile on the sulfur atom instead of carbon (Scheme 39).
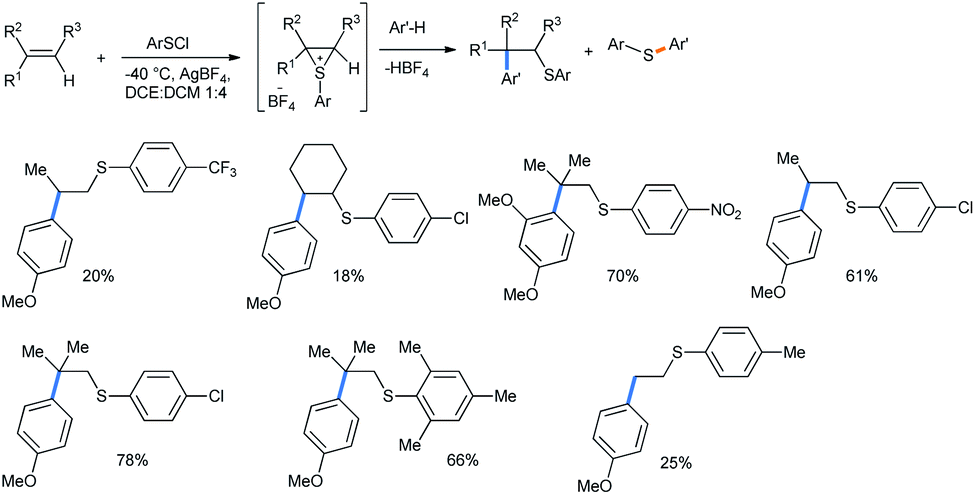 | ||
| Scheme 39 β-Arylthioalkylation of arenes using thiiranes. | ||
2.1.2.1 Ring opening of cyclobutanes. The simplest four-membered carbacycle is cyclobutane. Recently, the ring opening of its activated class, the donor–acceptor cyclobutanes, was studied by Kreft et al.9b The rings were further activated by stoichiometric use of AlCl3 Lewis acid (Scheme 40) to give the best yields. The use of a sub-stoichiometric amount (50 mol%) of Lewis acid afforded a lower product yield. The use of SnCl2, Sc(OTf)3, Yb(OTf)3 or In(OTf)3 needed a much longer reaction time than AlCl3.
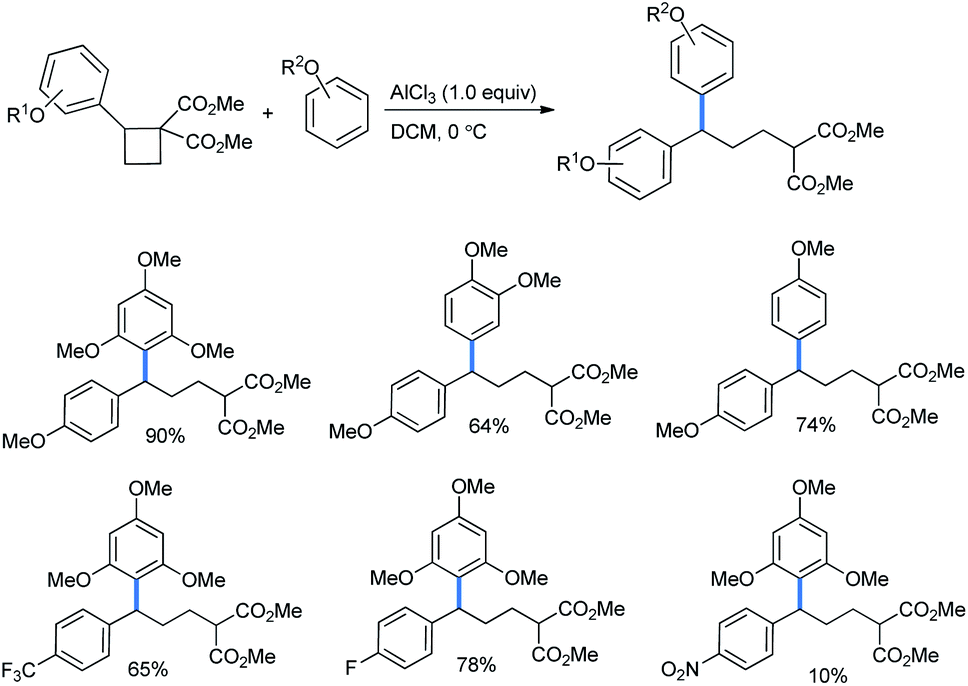 | ||
| Scheme 40 Ring opening of donor–acceptor cyclobutanes by methoxy benzenes. | ||
2.1.2.2 Ring opening of azetidines. The 4-membered homologue of aziridine, azetidine, behaves similarly if treated with a catalytic Lewis acid in the presence of alkoxy benzenes. Ghorai et al. have recently reported a catalytic Yb(OTf)3 assisted ring opening of N-activated azetidines with 1,3,5-TMB to synthesize 3,3-diarylpropylamines (Scheme 41).42 Dichloromethane was chosen as the best solvent for this reaction. The ring-opened products are H1-antihistaminic agents and are the chief composition of a number of marketed drugs.43
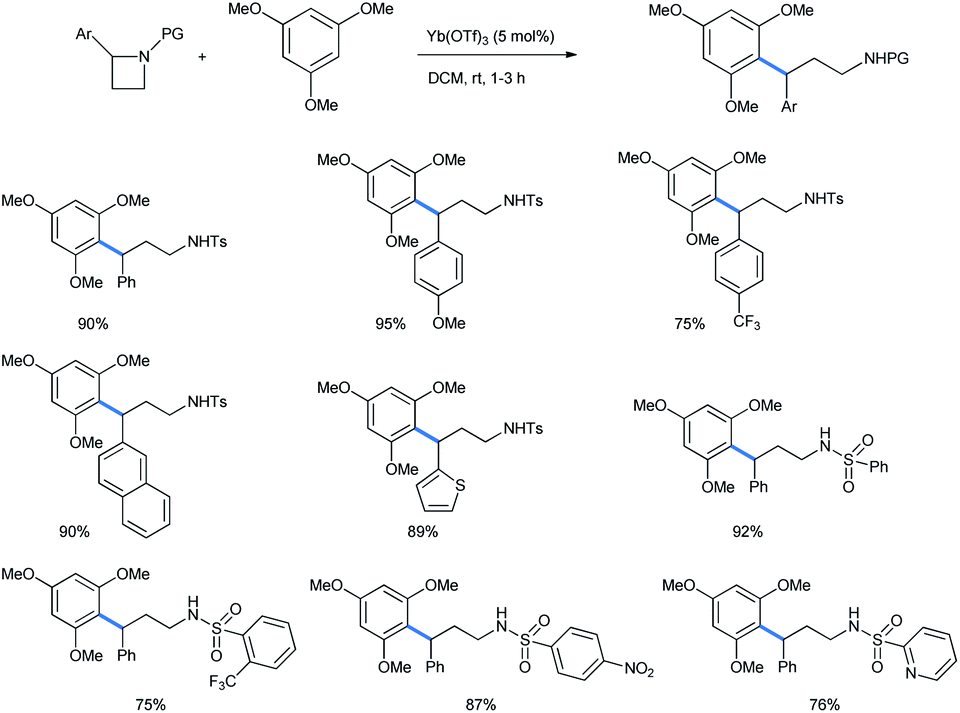 | ||
| Scheme 41 Yb(OTf)3 ring opening of azetidines with 1,3,5-trimethoxy benzene. | ||
Mono- and 1,2,4-trimethoxybenzene were also used as nucleophiles for this ring opening. The ring-opened products of the reaction were further utilized to synthesize the precursor of the urinary drug (±)-tolterodine in 4 further steps (Scheme 42). The target compound is a marketed antimuscarinic agent under the trade name Detrol.44
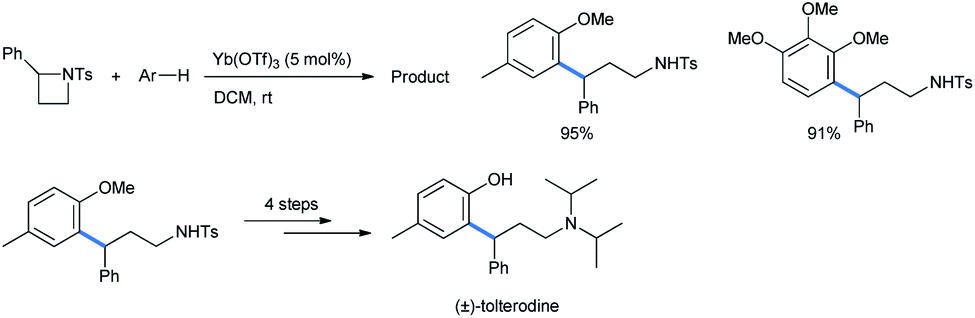 | ||
| Scheme 42 Synthesis of (±)-tolterodine from activated azetidine.42 | ||
The mechanism for the above reaction followed the same Lewis acid catalytic pathway, i.e. coordination of tosyl oxygens with Yb3+. As a result, the benzyl C–N bond is weakened, followed by attack by the methoxy benzene nucleophiles.
The Lewis acid assisted simple FC-ring-opening alkylation of alkoxy benzenes by oxetane systems is yet to be reported.
2.2 Alkoxy naphthalenes
Isomeric alkoxy naphthalenes are homologues of similar important electron-rich nucleophilic species like alkoxy benzenes, as discussed above. The nucleophilic sites of various mono, di- or trisubstituted naphthalenes are illustrated in Fig. 3. In all cases the alkoxy functionalities acted as either ortho- or para-directing groups.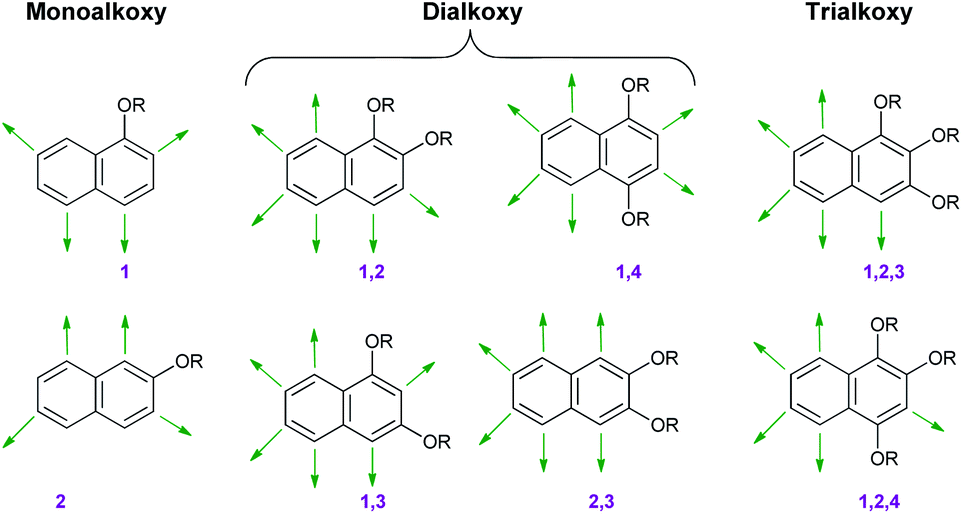 | ||
| Fig. 3 Nucleophilic sites in alkoxy naphthalenes (shown by green arrows). | ||
The use of alkoxy naphthalenes for FC ring opening is chiefly constrained to epoxides. Platt and co-workers synthesized methoxy anthanthrene by treating bisoxiranyl chrysene with BF3·OEt2 via Friedel–Crafts ring opening.45 Treatment of anthrathrene with boron tribromide gave the product hydroxy anthanthrene (Scheme 43).
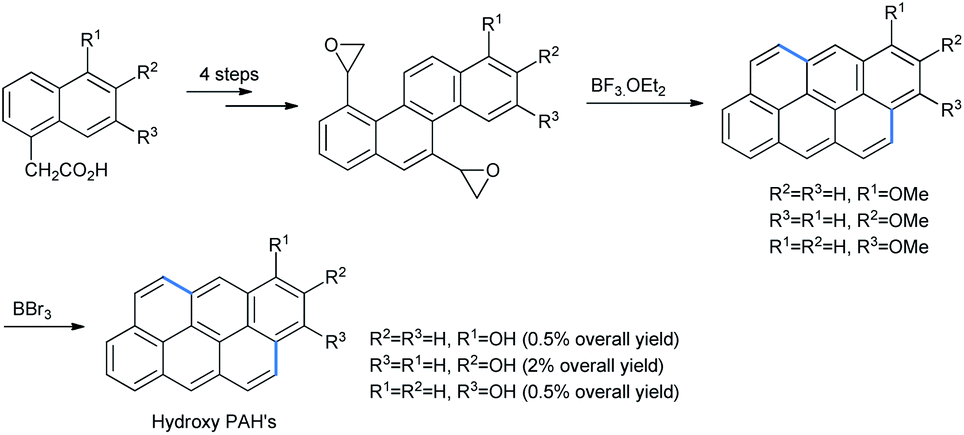 | ||
| Scheme 43 Synthesis of isomeric hydroxy anthanthrenes. | ||
These compounds constitute the class of hydroxy polyaromatic hydrocarbons (PAHs) which are carcinogenic in nature46a and possess bacterial mutagenicity in some histidine strains in Salmonella typhimurium.46b Later on a similar compound was synthesized in 94% yield by using InCl3 as a Lewis acid and starting with bis-epoxide by Ran and co-workers with radioactive 13C incorporated (shown by asterisks, Scheme 44).47 Further studies on the biological activities of the reduced product of benzopyran-7,8-dione, benzopyran-7,8-diol and epoxide anti-BPDE revealed their carcinogenic metabolite nature.48 Radioactive species were required as standards for mass spectrometry analysis of the patterns of benzopyrene (BP) metabolism and the structure of the DNA adduct in human cells.
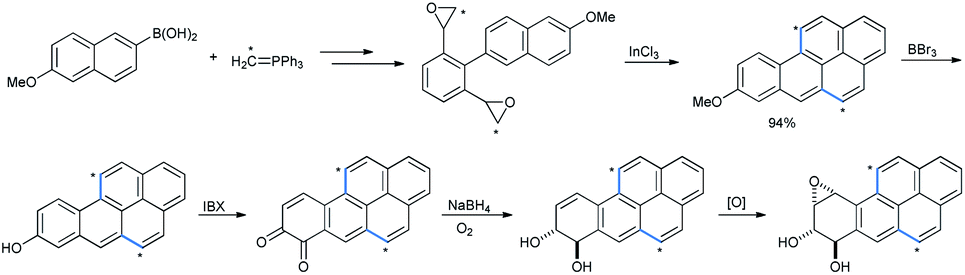 | ||
| Scheme 44 Synthesis of carcinogenic metabolites BP-7,8-dione, BP-7,8-diol and anti-BPDE. | ||
As described earlier, He's group also studied some electron-rich naphthalenes along with alkoxybenzenes for the regioselective ring opening of epoxides for the synthesis of 3-chromanols (Scheme 45). The products were in high yields and a diastereopure product was found for disubstituted epoxide.31c
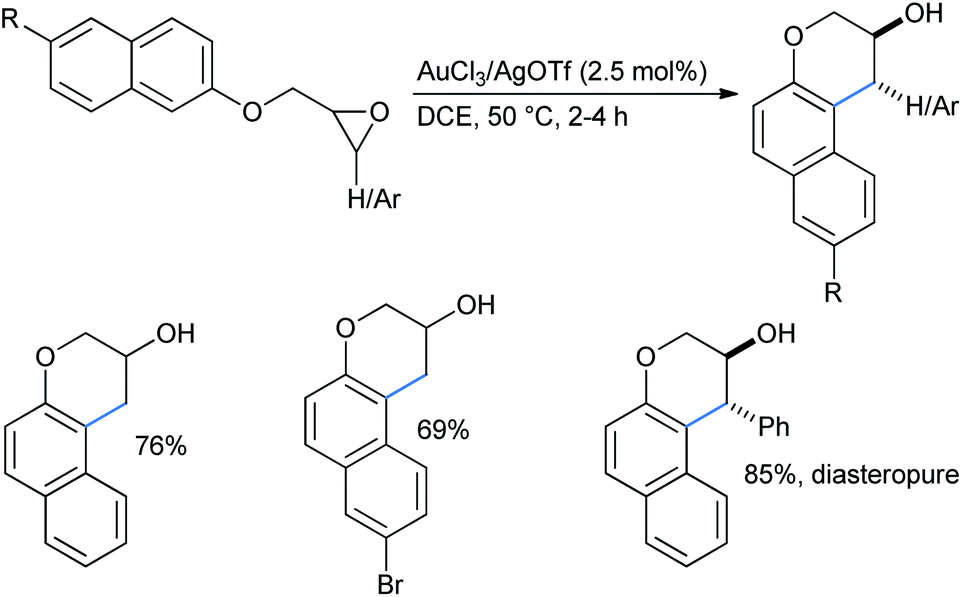 | ||
| Scheme 45 Gold–silver co-catalyzed synthesis of 3-chromanols. | ||
Qu also extended their work on intramolecular ring opening of epoxides with electron-rich naphthalenes. In HFIP medium 3-chromanols were synthesized in excellent yields (Scheme 46).33a
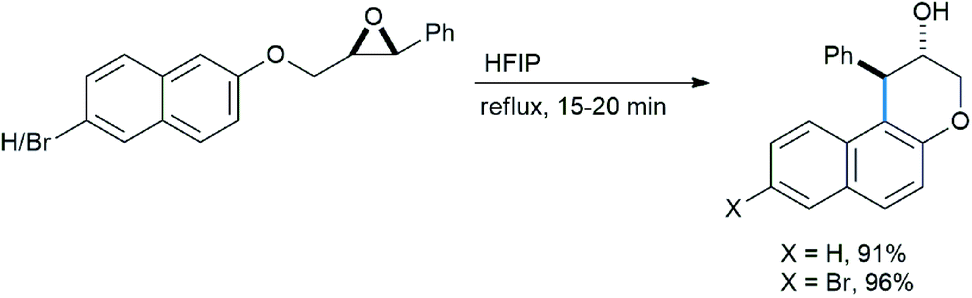 | ||
| Scheme 46 Synthesis of 3-chromanols in fluorinated alcohol solvent. | ||
The work done by Devi and co-workers when extended to naphthyl substrates provided the corresponding products, trans-4-arylchroman-3-ols, in good yields. No reaction was offered by the terminal epoxides, i.e., substrates had no direct aryl group attached even at 120 °C (Scheme 47).31a.b
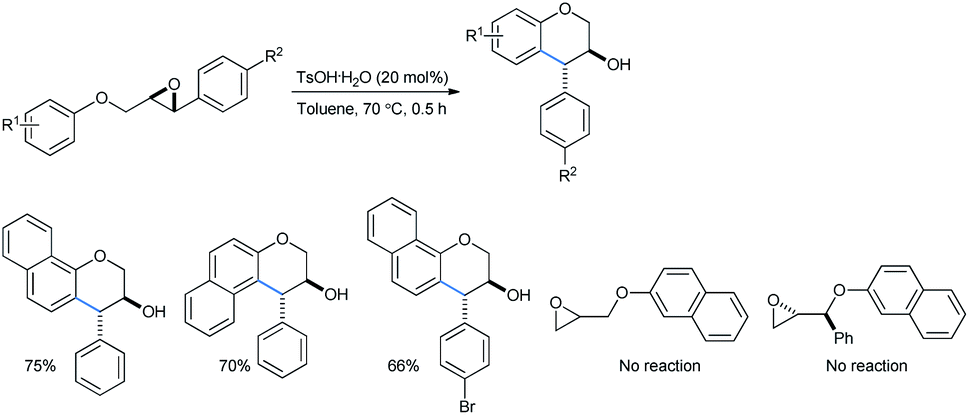 | ||
| Scheme 47 Intramolecular ring opening of naphthyl-substituted epoxides. | ||
When they performed the synthesis of cis-naphthochroman fused tetralins using the same intramolecular Friedel–Crafts epoxy–arene (IFCEA) cyclization strategy, it gave better yields with naphthalenes than with corresponding benzenes (Scheme 48).31d
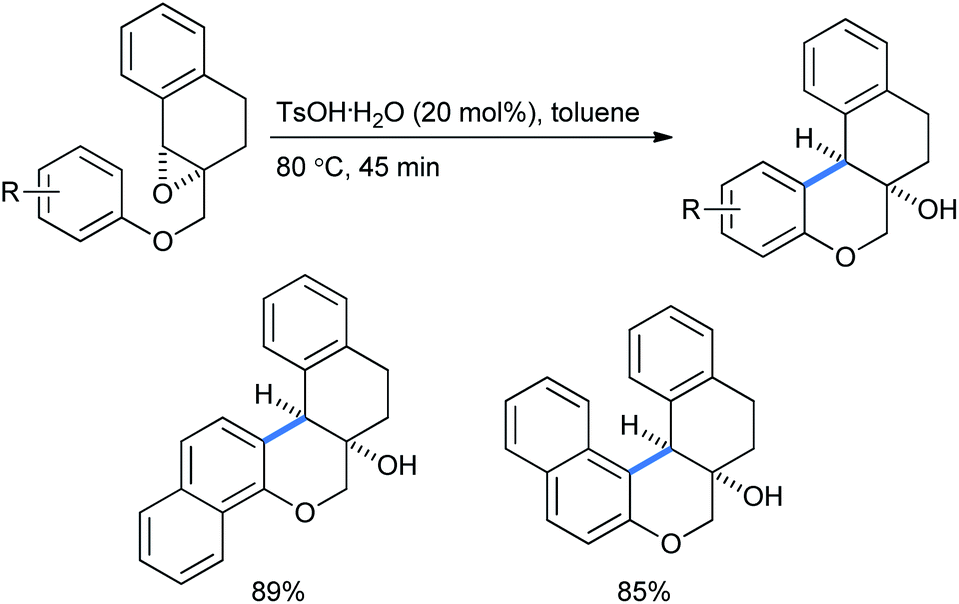 | ||
| Scheme 48 Syntheses of cis-naphthochroman fused tetralins using an intramolecular Friedel–Crafts epoxy–arene cyclization strategy. | ||
Using electron-rich 1- and 2-methoxy substituted naphthalenes, Moran and co-workers extended the ring opening with phenyl cyclopropane in excellent product yields using the same reaction conditions described in Scheme 10. For the 1-methoxy nucleophile, a 4:1 mixture of regioisomers was produced (Scheme 49).18
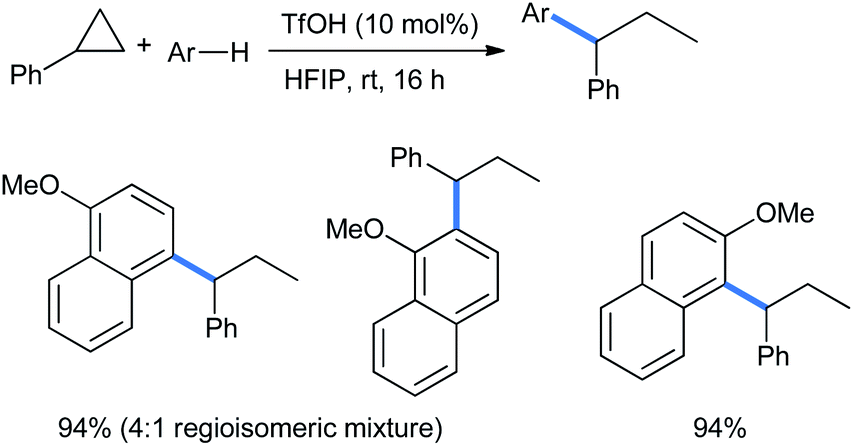 | ||
| Scheme 49 Ring opening of phenyl cyclopropane by methoxy naphthalenes. | ||
Nucleophilic ring opening by the higher homologues, i.e. alkoxy-anthracenes or phenanthrenes, has not been reported yet.
3. Conclusion and outlook
This concise review reports the ring-opening functionalization of small-sized molecular rings with alkoxybenzenes/naphthalenes in Friedel–Crafts fashion. Those “electrophilic” rings consist of different heteroatoms or simply of carbon. Sometimes the rings need subtle or considerable activation with the help of Lewis acid(s), followed by nucleophilic attack. As described, these are considered to be very important for synthesizing a diverse range of chemical commodities, obtained by single or multistep treatment(s) of the ring-opened products. The product spectrum stretches to biologically active natural products and drugs, important polymers, polyaromatic hydrocarbons, macrocycles, molecular motors, fluorescent sensors, photo-energy storage and more. The works cited in this article display a wide range of conditions and differently substituted substrates for target-oriented syntheses. But this is not the end of the story. The struggle of the electronically rich arenes towards the more challenging alkylative ring-opening functionalization of larger and more stable rings (five-membered ring onwards) to date poses a big problem to be solved. With its successful execution with non C–H activation pathways, this will definitely unleash an ocean of routes for chemists to enlighten the uncharted territories of Friedel–Crafts ring opening.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
The author would like to thank DST-SERB, India (grant number PDF/2018/000072/CS) for financial support.Notes and references
- For reviews check: (a) N. O. Calloway, Chem. Rev., 1935, 17, 327–392 CrossRef CAS; (b) T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed; (c) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC; (d) M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 1–24, DOI:10.3762/bjoc.6.6; (e) G. Sartori and R. Maggi, Chem. Rev., 2011, 111, PR181–PR214 CrossRef PubMed; (f) M. M. Heravi, V. Zadsirjan, P. Saedia and T. Momeni, RSC Adv., 2018, 8, 40061–40163 RSC; (g) Y. N. Nayak, S. Nayak, Y. F. Nadaf, N. S. Shetty and S. L. Gaonkar, Lett. Org. Chem., 2020, 17, 491–506 CrossRef.
- C. Friedel and J. M. Crafts, Compt. Rend., 1877, 84, 1392–1450 Search PubMed.
- (a) S. Stanković, M. D'hooghe, S. Catak, H. Eum, M. Waroquier, V. Speybroeck, N. D. Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643–665 RSC; (b) S. Ahmad, M. Yousaf, A. Mansha, N. Rasool, A. F. Zahoor, F. Hafeez and S. M. A. Rizvi, Synth. Commun., 2016, 46, 1397–1416 CrossRef CAS; (c) S. K. Dwivedi, S. Gandhi, N. Rastogi and V. K. Singh, Tetrahedron Lett., 2007, 48, 5375–5377 CrossRef CAS; (d) M. Karbalaei, M. Seifi and H. Sheibani, Res. Chem. Intermed., 2015, 41, 4679–4686 CrossRef CAS.
- Reports by non-transitional metallo-arenes: (a) V. G. Nenajdenko, A. S. Karpov and E. S. Balenkova, Tetrahedron: Asymmetry, 2001, 12, 2517–2527 CrossRef CAS; (b) M. Uemura, H. Yorimitsu and K. Oshima, Chem. Commun., 2006, 4726–4728 RSC; (c) B. Erdélyi, A. Szabé, G. Seres, L. Birincsik, J. Ivanics, G. Szatzkerd and L. Poppe, Tetrahedron: Asymmetry, 2006, 17, 268–274 CrossRef; (d) Y.-C. Wu and J. Zhu, Org. Lett., 2009, 11, 5558–5561 CrossRef CAS PubMed; (e) L. Miao, S. C. DiMaggio and M. L. Trudell, Synthesis, 2010, 91–97 CAS; (f) D.-H. Yoon, P. Kang, W. K. Lee, Y. Kim and H.-J. Ha, Org. Lett., 2012, 14, 429–431 CrossRef CAS PubMed; (g) E. Ertürk, M. A. Tezeren, T. Atalar and T. Tilki, Tetrahedron, 2012, 68, 6463–6471 CrossRef; (h) D. E. White, P. M. Tadross, Z. Lu and E. N. Jacobsen, Tetrahedron, 2014, 70, 4165–4180 CrossRef CAS PubMed; (i) D. Yang and N. Liang, Org. Biomol. Chem., 2014, 12, 2080–2086 RSC; (j) O. Dilek, M. A. Tezeren, T. Tilki and E. Ertürk, Tetrahedron, 2018, 74, 268–286 CrossRef CAS; (k) L. Liu, W. Lee, M. Yuan, C. Acha, M. B. Geherty, B. Williams and O. Gutierrez, Chem. Sci., 2020, 11, 3146–3151 RSC.
- Reports by transition metallo-arenes: (a) X. Tian, R. Maurya, K. Königsberger and T. Hudlicky, Synlett, 1995, 1125–1126 CrossRef CAS; (b) M. Asano, M. Inoue and T. Katoh, Synlett, 2005, 2599–2602 CAS; (c) Z. Wang, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 6140–6143 CrossRef CAS PubMed; (d) Y. Tan, Y. Yao, W. Yang, Q. Lin, G. Huang, M. Tan, S. Chen, D. Chen and D. Yanga, Adv. Synth. Catal., 2020, 362, 139–145 CrossRef CAS; (e) F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462–6477 RSC; (f) M. Yus, Pure Appl. Chem., 2003, 75, 1453–1475 CAS.
- (a) L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS; (b) L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley, New York, 2nd edn, 1991, pp. 14–17 Search PubMed; (b) C. B. Reese and R. E. Haslam, Protecting Groups in Organic Chemistry, ed. J. F. W. McOmie, Plenum Press, London, 1973, vol. 95, p. 145 Search PubMed; (c) A. Maercker, Angew. Chem., Int. Ed., 1987, 26, 972 CrossRef; (d) J. Oppenheimer, W. L. Johnson, M. R. Tracey, R. P. Hsung, P.-Y. Yao, R. Liu and K. Zhao, Org. Lett., 2007, 9, 2361–2364 CrossRef CAS PubMed; (e) M. V. Bhatt and S. U. Kulkarni, Synthesis, 1983, 249–282 CrossRef CAS; (f) P. Kraft and W. Eichenberger, Eur. J. Org. Chem., 2003, 3735–3743 CrossRef CAS.
- T. Manaka, S.-I. Nagayama, W. Desadee, N. Yajima, T. Kumamoto, T. Watanabe, T. Ishikawa, M. Kawahata and K. Yamaguchi, Helv. Chim. Acta, 2007, 90, 128–142 CrossRef CAS.
- (a) L. Changqing, H. Yukihiko, K. Kazuaki and S. Kazuhiko, Bull. Chem. Soc. Jpn., 1996, 69, 2095–2105 CrossRef; (b) A. Kreft, S. Ehlers, P. G. Jones and D. B. Werz, Org. Lett., 2019, 21, 6315–6319 CrossRef CAS PubMed.
- G. K. Surya Prakash, P. J. Linares-Palomino, K. Glinton, S. Chacko, G. Rasul, T. Mathew and G. A. Olah, Synlett, 2007, 1158–1162 CrossRef.
- (a) T. Kaicharla, T. Roy, M. Thangaraj, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 10061–10064 CrossRef CAS PubMed; (b) T. Kaicharla, A. Jacob, R. G. Gonnade and A. T. Biju, Chem. Commun., 2017, 53, 8219–8222 RSC.
- (a) E. V. Anslyn and D. E. Dougherty, Modern Physical Organic Chemistry, Edwards Brothers, Inc., 2006, p. 850 Search PubMed; (b) H. T. Lacey, Ind. Eng. Chem., 1954, 46, 1827–1835 CrossRef CAS.
- Some important reviews on DA-cyclopropanes: (a) W. Wu, Z. Lina and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315–7329 RSC; (b) P. Gopinath and S. Chandrasekaran, Curr. Org. Chem., 2019, 23, 276–312 CrossRef CAS; (c) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed; (d) M. Yu and B. L. Pagenkopf, Tetrahedron, 2005, 61, 321–347 CrossRef CAS; (e) M. Ya. Mel'nikov, E. M. Budynina, O. A. Ivanova and I. V. Trushkov, Mendeleev Commun., 2011, 21, 1–10 CrossRef; (f) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC.
- O. A. Ivanova, E. M. Budynina, Y. K. Grishin, I. V. Trushkov and P. V. Verteletskii, Eur. J. Org. Chem., 2008, 5329–5335 CrossRef CAS.
- R. Talukdar, A. Saha, D. P. Tiwari and M. K. Ghorai, Tetrahedron, 2016, 72, 613–624 CrossRef CAS.
- B. Portet, N. Fabre, V. Roumy, H. Gornitzka, G. Bourdy, S. Chevalley, M. Sauvain, A. Valentin and C. Moulis, Phytochemistry, 2007, 68, 1312–1320 CrossRef CAS PubMed.
- E. Richmond, V. D. Vuković and J. Moran, Org. Lett., 2018, 20, 574–577 CrossRef CAS PubMed.
- E. Richmond, J. Yi, V. D. Vuković, F. Sajadi, C. N. Rowley and J. Moran, Chem. Sci., 2018, 9, 6411–6416 RSC.
- (a) X. E. Hu, Tetrahedron, 2004, 60, 2701–2743 CrossRef CAS; (b) S. Sabir, G. Kumar, V. P. Verma and J. L. Jat, ChemistrySelect, 2018, 3, 3702–3711 CrossRef CAS; (c) R. Akhtar, S. A. R. Naqvi, A. F. Zahoor and S. Saleem, Mol. Diversity, 2018, 22, 447–501 CrossRef CAS PubMed.
- M. K. Ghorai, D. P. Tiwari and N. Jain, J. Org. Chem., 2013, 78, 7121–7130 CrossRef CAS PubMed.
- (a) D. B. Berkowitz, S. Choi and J.-H. Maeng, J. Org. Chem., 2000, 65, 847–860 CrossRef CAS PubMed; (b) M. Takahashi, N. Suzuki and T. Ishikawa, J. Org. Chem., 2013, 78, 3250–3261 CrossRef CAS PubMed.
- N. Hsueh, G. J. Clarkson and M. Shipman, Org. Lett., 2015, 17, 3632–3635 CrossRef CAS PubMed.
- S. Xing, J. Ren, K. Wang, H. Cui, W. Li and H. Yan, Tetrahedron, 2015, 71, 6290–6299 CrossRef CAS.
- (a) S. Faiz and A. F. Zahoor, Mol. Diversity, 2016, 20, 969–987 CrossRef CAS PubMed; (b) F. A. Saddique, A. F. Zahoor, S. Faiz, S. A. R. Naqvi, M. Usman and M. Ahmad, Synth. Commun., 2016, 46, 831–868 CrossRef CAS.
- S. K. Taylor, D. L. Clark, K. L. Heinz, S. B. Schramm, C. D. Westermann and K. K. Barnell, J. Org. Chem., 1983, 48, 592–596 CrossRef CAS.
- R. K. Summerbell and M. J. Kland-English, J. Am. Chem. Soc., 1955, 77, 5095–5098 CrossRef CAS.
- S. K. Taylor, W. C. Haberkamp, D. W. Brooks and D. N. Whittern, J. Heterocycl. Chem., 1983, 20, 1745–1747 CrossRef CAS.
- Y. Kita, S. Matsuda, R. Inoguchi, J. K. Ganesh and H. Fujioka, Tetrahedron Lett., 2005, 46, 89–91 CrossRef CAS.
- H. Yang, L. Xu, C. Luo, C. Lu and G. Cheng, Synth. Commun., 2015, 45, 1433–1441 CrossRef CAS.
- T. J. Korstanje, E. Folkertsma, M. Lutz, J. T. B. H. Jastrzebski and R. J. M. K. Gebbink, Eur. J. Inorg. Chem., 2013, 2013, 2195–2204 CrossRef CAS.
- (a) R. Devi, T. Kalita and S. K. Das, RSC Adv., 2015, 5, 39692–39696 RSC; (b) R. Devi, D. Gogoi, P. Bora and S. K. Das, Tetrahedron, 2016, 72, 4878–4888 CrossRef CAS; (c) Z. Shi and C. He, J. Am. Chem. Soc., 2004, 126, 5964–5965 CrossRef CAS PubMed; (d) D. Gogoi, R. Devi, P. Pahari, B. Sarma and S. K. Das, Beilstein J. Org. Chem., 2016, 12, 2816–2822 CrossRef CAS PubMed; (e) H. Borgohain, R. Devi, D. Dheer, B. J. Borah, R. Shankar and S. K. Das, Eur. J. Org. Chem., 2017, 6671–6679 CrossRef CAS.
- (a) G. Islas-González, J. Benet-Buchholz, M. A. Maestro, A. Riera and M. A. Pericàs, J. Org. Chem., 2006, 71, 1537–1544 CrossRef PubMed; (b) R. Marcos, C. Rodríguez-Escrich, C. I. Herrerías and M. A. Pericàs, J. Am. Chem. Soc., 2008, 130, 16838–16839 CrossRef CAS PubMed.
- (a) G.-X. Lia and J. Qu, Chem. Commun., 2010, 46, 2653–2655 RSC; (b) Q. Jiang, Free Radical Biol. Med., 2014, 72, 76–19 CrossRef CAS PubMed; (c) W. Gregor, G. Grabner, C. Adelwöhrer, T. Rosenau and L. Gille, J. Org. Chem., 2005, 70, 3472–3483 CrossRef CAS PubMed; (d) R. F. Bosch, R. Gaspo, A. E. Busch, H. J. Lang, G.-R. Li, S. Nattel, R. F. Bosch, R. Gaspo, A. E. Busch, H. J. Lang, G.-R. Li and S. Nattel, Cardiovasc. Res., 1998, 38, 441–450 CrossRef CAS PubMed.
- R. W. Dapson and C. L. Bain, Biotech. Histochem., 2015, 90, 401–423 CrossRef CAS PubMed.
- (a) V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed; (b) P. Bendale, S. Olepu, P. K. Suryadevara, V. Bulbule, K. Rivas, L. Nallan, B. Smart, K. Yokoyama, S. Ankala, P. R. Pendyala, D. Floyd, L. J. Lombardo, D. K. Williams, F. S. Buckner, D. Chakrabarti, C. L. M. J. Verlinde, W. C. Van Voorhis and M. H. Gelb, J. Med. Chem., 2007, 50, 4585–4605 CrossRef CAS PubMed; (c) L. Chen, P. T. Wilder, B. Drennen, J. Tran, B. M. Roth, K. Chesko, P. Shapiro and S. Fletcher, Org. Biomol. Chem., 2016, 14, 5505–5510 RSC; (d) C. Z. Ding, J. T. Hunt, C. Ricca and V. Manne, Bioorg. Med. Chem. Lett., 2000, 10, 273–275 CrossRef CAS PubMed; (e) T. G. Davies, W. E. Wixted, J. E. Coyle, C. Griffiths-Jones, K. Hearn, R. McMenamin, D. Norton, S. J. Rich, C. Richardson, G. Saxty, H. M. Willems, A. J. Woolford, J. E. Cottom, J. P. Kou, J. G. Yonchuk, H. G. Feldser, Y. Sanchez, J. P. Foley, B. J. Bolognese, G. Logan, P. L. Podolin, H. Yan, J. F. Callahan, T. D. Heighman and J. K. Kerns, J. Med. Chem., 2016, 59, 3991–4006 CrossRef CAS PubMed.
- T. Katsuki and B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- (a) M. Sander, Chem. Rev., 1966, 66, 297–339 CrossRef; (b) W. Chew and D. N. Harpp, Sulfur Rep., 1993, 15, 1–39 CrossRef CAS.
- M. A. Ibragimov, V. A. Smit, S. Gybin and M. Z. Krimer, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1983, 32, 137–141 CrossRef.
- W. A. Smit, M. Z. Krimer and E. A. Vorob'eva, Tetrahedron Lett., 1975, 16, 2451–2454 CrossRef.
- (a) R. Livingstone, Chapter 1-Compounds with three- and four-membered heterocyclic rings, in Rodd's chemistry of carbon compounds, 2nd edn, 1964, vol. IV, pp. 1–82 Search PubMed; (b) J. Parrick and L. K. Mehta, Chapter 4–Four-membered ring systems, in Progress in heterocyclic chemistry, 1995, vol. 7, pp. 64–81 Search PubMed.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed; (b) D. Hazelarda and P. Compain, Org. Biomol. Chem., 2017, 15, 3806–3827 RSC.
- G. Goswami, N. Chauhan, A. Mal, S. Das, M. Das and M. K. Ghorai, ACS Omega, 2018, 3, 17562–17572 CrossRef CAS.
- D. N. Zakusilo, D. S. Ryabukhin, I. A. Boyarskaya, O. S. Yuzikhin and A. V. Vasilyev, Tetrahedron, 2015, 71, 102–108 CrossRef CAS.
- D. Clemett and B. Jarvis, Drugs Aging, 2001, 18, 277–304 CrossRef CAS PubMed.
- K. L. Platt, C. Degenhardt, S. Grupe, H. Frank and A. Seidel, Chem. Res. Toxicol., 2002, 15, 332–342 Search PubMed.
- (a) T. Kuljukka-Rabb, L. Nylund, R. Vaaranrinta, K. Savela, P. Mutanen, T. Veidebaum, M. Sorsa, A. Rannug and K. Peltonen, J. Exposure Sci. Environ. Epidemiol., 2002, 12, 81–91 Search PubMed; (b) M. Sakai, D. Yoshida and S. Mizusaki, Mutat. Res., 1985, 156, 61–67 CrossRef CAS PubMed.
- C. Ran, D. Xu, Q. Dai, T. M. Penning, I. A. Blair and R. G. Harvey, Tetrahedron Lett., 2008, 49, 4531–4533 CrossRef CAS PubMed.
- R. G. Harvey, Q. Dai, C. Ran, K. Lim, I. Blair and T. M. Penning, Polycyclic Aromat. Compd., 2005, 25, 371–391 CrossRef CAS.
Footnote |
| † Dedicated to Professor Parimal K. Bharadwaj on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2020 |