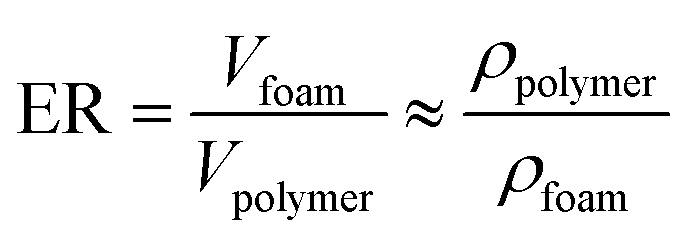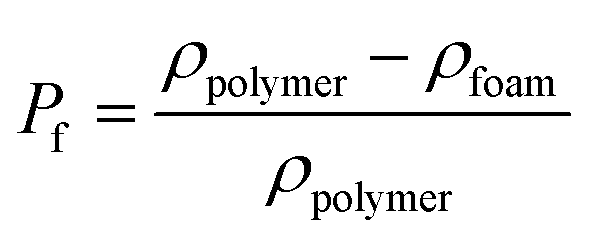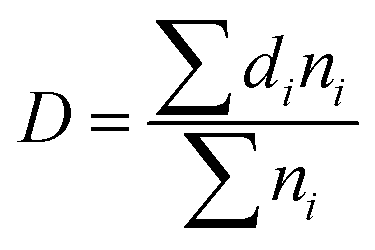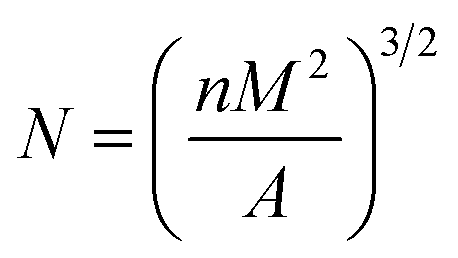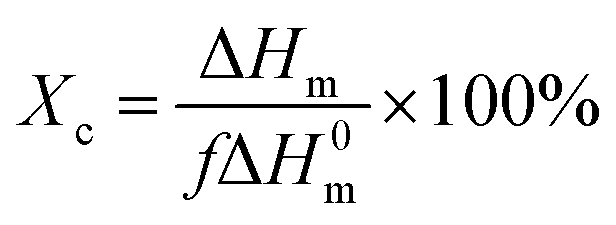DOI:
10.1039/D0RA04486E
(Paper)
RSC Adv., 2020,
10, 31355-31362
The effects of rubber nanoparticles with different polarities on thermal properties and foaming performance of polypropylene blends
Received
20th May 2020
, Accepted 6th August 2020
First published on 25th August 2020
Abstract
Polypropylene blends with both polybutadiene rubber (PB) and polycarboxylbuturonile rubber (xNBR) and the required amount of acrylamide (AM) was prepared by blending with water, and the crystallinity, rheological behaviour and thermal performance were analysed and compared. The results of DSC and XRD characterization showed an obvious enhancement in the crystallization of the PP matrix in PP/xNBR/AM blends compared to PP/PB/AM blends, due to the strong incompatibility between xNBR nanoparticles and the PP polymer matrix leading to the inhibition of segmental mobility and induced formation of heterogeneous nuclei. Rheological analysis showed that the dynamical mobility of polymer chains was retarded while the AM monomer was incorporated, due to strengthening interfacial interactions by grafts through hydrogen bonding. The foaming performance was clearly improved, as reflected in the uniform cell morphology and higher cell density, and the expansion ratio achieved was 13-fold. In addition, the decomposition temperature increased from 403 °C to 465 °C by nearly 62 °C as compared with neat PP, which is ascribed to the inhibition of segmental mobility due to the cyclization reaction of nitriles. The increase in the surface energy was about 2.2-fold, which resulted in a decrease of the water contact angle from 105.3° to 83.7°, attributed due to AM addition to the composition.
Introduction
The mechanical properties and ease of processability are important for the commercial plastic like linear isotactic polypropylene, which has attracted much attention of researchers worldwide.1–6 The incorporation of rubber is a common technique used to endow blends with toughening properties, but it also has obvious disadvantages either in poor processing or in lower modulus due to the enhanced viscosity of blends.7–11 Inhibiting the chain movement can enhance the melt strength and is achieved via long chain entanglement or strong polar interaction between groups by co-polymerization (known as the in-reactor method) or modification (known as the past-reactor method). This can greatly improve thermoformability,12,13 but balancing the isotacticity and selectivity in the confined geometric catalyst technique limits its wide application; the β-degradation of the linear PP in the modification of melt-blending leads to serious deterioration of mechanical properties. Therefore, incorporating rubber latex nanoparticles into PP using unique methods can restrict rubber particle aggregation and improve the dispersion of the rubber particles in the PP matrix, which might improve its mechanical performance and thermoformability simultaneously; thus, this approach has great significance both in academia and industry.5,14–18
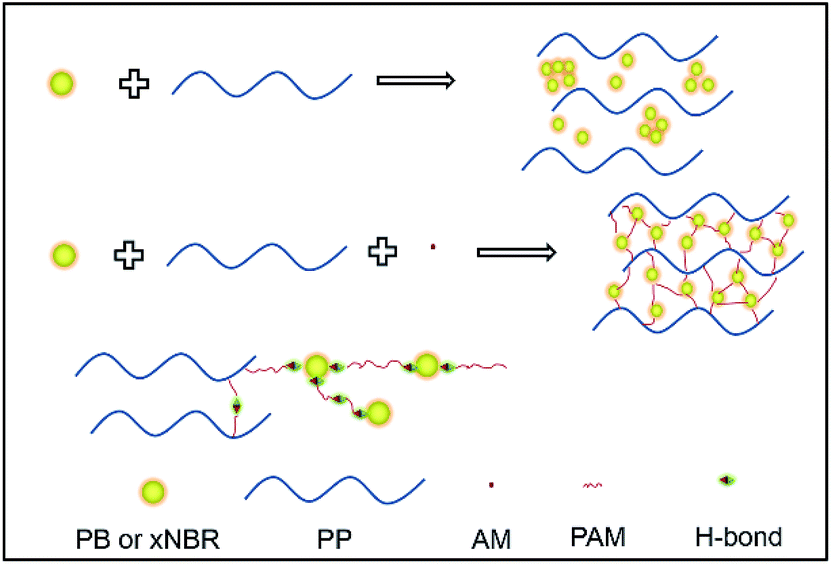
The invention of fully vulcanized nanoparticle rubber has opened a new era for the post-reactor modification of plastic materials.11,19,20 However, there are significant challenges to be addressed for their wide practical application, such as the aggregation and uniform nanoparticle rubber powder dispersion in polymer matrices. For instance, it is difficult to both inhibit the aggregation of the nanoparticle rubber powder and realize an even dispersion in the polyolefin matrix after spray-drying from their latex form, which greatly limits the application of the nanoparticle rubber powders to modify plastic materials.21–23 A feasible strategy is proposed to address this problem: (I) the rubber latex is pre-irradiated to enhance the cross-linking density and inhibit the fusion of latex particles, so as to fix the native size of latex particles effectively; (II) blending24 assisted by water is used to ensure that latex particles are premixed in their emulsion state (primary entity particle size), which can effectively restrict the aggregation of nanoparticles during demulsification and dehydration process; (III) the grafting reaction in situ during blending imparts grafts both on nanoparticles and the PP matrix while incorporating water-soluble monomers, which can inhibit the aggregation of nanoparticles and strengthen interfacial interactions between the rubber powder and PP matrix, promoting more uniform dispersion in the melt.
Both PP/(xNBR)/AM and PP/PB/AM blends were prepared via water-assisted reactive blending, and more uniform nanoparticle dispersions in the PP matrix were achieved. The effect of rubber nanoparticles with different polarities on the crystalline behaviour, rheological response, foaming performance and thermal properties was compared and analysed in this work (Scheme 1).
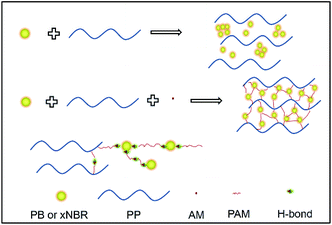 |
| | Scheme 1 Schematic diagram of the melting reaction of the blend. | |
Materials and methods
Materials
PP T30S with a melt flow rate of 2.8 g/10 min and a melting temperature of 162 °C was obtained from PetroChina Daqing Refinery and Chemical Plant (Daqing, China). Polybutadiene latex (PBL), 42 wt% solids, was obtained from China Daqing Petrochemical Plant (Daqing, China). Carboxyl butyronitrile latex (xNBRL), brand FM301, solid content 41 wt%, nitrile content ≥30, was obtained from Feima Chemical Co., Ltd. (Xinxiang, China). AM, content (C3H5NO) ≥98 wt%, pH 5.5–7.5, was obtained from Kermel Chemical Reagent Co., Ltd. (Tianjin, China). Antioxidant Irganox 1076 was obtained from Milan Chemical Co., Ltd. (Nanjing, China). Carbon dioxide with a purity of 99.95% was used as the blowing agent.
Pre-irradiation
60Co γ source with a capacity of 3.7 × 1015 Bq was applied as the radiation source. PP powder was packed into a polyethylene bag and irradiated with a dose of 0.2 kGy and a dose rate of 50 Gy h−1, which was labeled as rPP. PBL was packed into a PET bottle and irradiated with a dose of 250 kGy and a dose rate of 20 kGy h−1, labeled as rPBL. xNBRL was packed into a PET bottle and irradiated with a dose of 250 kGy, labeled as rxNBRL.
Preparation of PP and its blends
PP powder was added to the torque rheometer (RM200C, Harbin, China), the temperature was set at 30 °C, the speed was set at 60 rad min−1, 5 wt% rPBL (or rxNBRL), 5 wt% AM, and the mixture was blended in a rheometer for about 10 min. At a heating rate of 15 °C min−1, from 30 to 180 °C, 1 wt% antioxidant 1076 and rPP were added until a constant torque was achieved. The schematic reaction is shown in Fig. 1. All samples were pressed by a plate vulcanizer (KY-3201A-30T, Dongguan, China); the pressure was 10 MPa, and the samples were pressed to 150 × 150 × 1 mm. The experimental formulations are listed in Table 1.
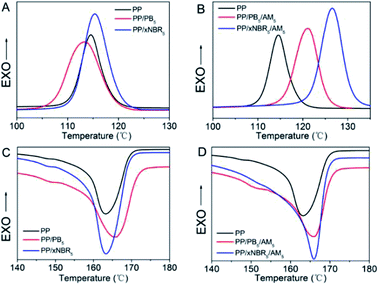 |
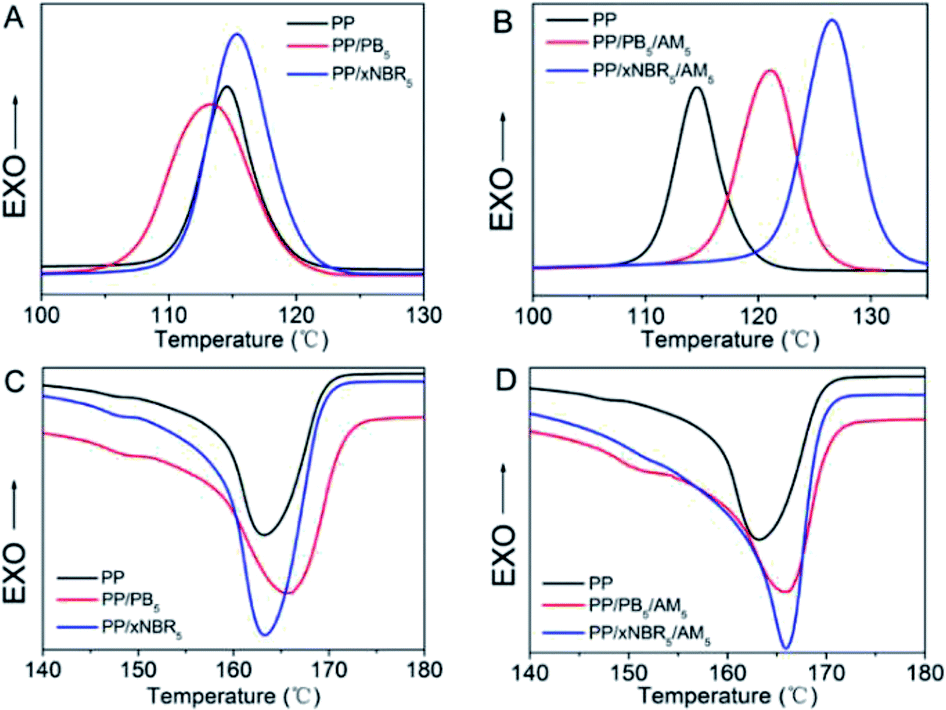
| | Fig. 1 Crystallization of PP blends (A and B); melting curves (C and D). | |
Table 1 PP blends component mass percentage
| Sample |
PP (wt%) |
PBL (wt%) |
xNBRL (wt%) |
AM (wt%) |
rPP (wt%) |
Antioxidant 1076 (wt%) |
| PP |
99 |
0 |
0 |
0 |
0 |
1 |
| PP/PB5 |
89 |
5 |
0 |
0 |
5 |
1 |
| PP/xNBR5 |
89 |
0 |
5 |
0 |
5 |
1 |
| PP/PB5/AM5 |
84 |
5 |
0 |
5 |
5 |
1 |
| PP/xNBR5/AM5 |
84 |
0 |
5 |
5 |
5 |
1 |
Foaming test
About 1.6 g of sample was put into the tube, which was placed in the lab-scale foaming equipment (homemade), and supercritical CO2 was used as the foaming gas. The purge time was 10 min. As mentioned above, the PP, PP/PB5, PP/xNBR5, PP/PB5/AM5, and PP/xNBR5/AM5 were foamed at temperatures of 155 °C, 158 °C, 157 °C, 160 °C, and 162 °C, respectively, at a pressure of 15–20 MPa, for 2–4 h. The autoclave was decompressed within 2 s, and the tube with sample was placed into ice water to cool down to room temperature. The expansion ratio (ER) was determined according to Archimedes' principle (drainage method) ASTM a792-00, and the formula for the foaming test is as follows:| |
 | (1) |
where Vfoam and Vpolymer are denoted as the volume of sample after foaming and unfoamed, respectively, and ρpolymer and ρfoam are denoted as the densities of sample before and after foaming.
The percentage of bubbles in sample after foaming is denoted as Pf:
| |
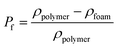 | (2) |
Image Pro Plus software was used to analyse the SEM images of the foaming materials. The average diameter D of bubble hole is shown in the following formula:
| |
 | (3) |
where,
n is the number of bubble holes,
d is the perimeter-equivalent diameter of bubble holes, and
i is greater than 200.
Bubble density (N) is the number of bubble holes per unit volume, as shown in formula (4):
| |
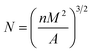 | (4) |
where,
n is the number of bubbles in the photomicrograph,
M is the magnification factor of the photomicrograph, and
A is the area of the photomicrograph (cm
2).
Characterization
The crystalline behaviour and crystallinity of PP/PB/AM and PP/xNBR/AM blends were studied using a differential scanning calorimeter (DSC), DSC2500 (US waters). The samples were scanned at a heating rate of 10 °C min−1 under nitrogen protection. The melting enthalpy was calculated according to the area of the melting peak, and the crystallinity (Xc) was calculated according to the following formula:| |
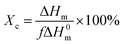 | (5) |
where ΔH0m is the melting enthalpy of 100% crystalline PP, while for isotactic PP, ΔH0m is 209 J g−1;25 ΔHm is the melting enthalpy of blends and f is the volume fraction of PP in the blend. The samples were melted at 190 °C for 5 min, then quenched to 135 °C under 10 MPa for 2 h, and observed by a polarized optical microscope (PLM), DM2500P (Germany Leica). The rheological performance was analysed by using a rotary rheometer, Physica MCR102 (Austria antongpa co. LTD), in dynamically scanning mode, frequency (ω) 100–0.05 rad s−1, strain (γ) of 1%, at a temperature of 200 °C, to ensure all the samples were in the linear viscoelastic region. The foaming sample was dipped into liquid nitrogen for 5 min and then quenched. The morphology of the foaming sample was observed with a scanning electron microscope (SEM), JSM-IT300LV (JEOL corporation of Japan). The thermal properties of the samples were measured by using a thermogravimetric analyser (TGA), TGA5500 (USA, Waters), at a temperature of 25–800 °C under nitrogen protection.26 The surface energy was tested by using a Data physics OCA 15EC Goniometer (Filderstadt, Germany), and pure water and ethylene glycol were used as the liquid. The contact angle was measured at the same time interval (30 s), and each sample was measured at different positions at least 5 times. The surface energy was calculated from the contact angle of water and ethylene glycol using the Owens two-night method; the formula is as follows:27| |
rl(1 + cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) = 2(rPl × rPs)1/2 + (rDl × rDs)1/2 θ) = 2(rPl × rPs)1/2 + (rDl × rDs)1/2
| (6) |
In eqn (6)–(8), rl is the surface energy of the liquid; rPl and rDl are the polar component and the dispersive component of the liquid surface energy, respectively; rs is the surface energy of a solid; rPs and rDs are the polar component and the dispersion component of the solid surface energy, respectively. The surface energies and components of pure water and ethylene glycol are shown in Table 2.
Table 2 Surface energy parameters of liquid
| Liquid |
rl |
rPl |
rDl |
| Water |
72.8 |
51.0 |
21.8 |
| Ethylene glycol |
48.0 |
19.0 |
29.0 |
Results and discussion
Crystallization behaviour
The two kinds of rubber have different effects on the crystalline behaviour of the PP matrix, especially with incorporation of the AM monomer. The results are shown in Fig. 1 and Table 3. As shown in Fig. 1A and C, there were distinct changes in the peak area and peak width in both PP/xNBR5 and PP/PB5 blends compared to that of the neat PP, which is related to the crystallinity and crystalline integrity. The most obvious influence on the crystallization of the PP matrix was observed in the PP/xNBR5 blend compared to that of the PP/PB5 blend, which was ascribed to the heterogeneous nucleation of the xNBR nanoparticles.
Table 3 Thermal properties of PP and various PP blends
| Sample |
Tc (°C) |
Tm (°C) |
ΔHm (J) |
Ti (°C) |
Xc (%) |
| PP |
114.58 |
163.15 |
56.47 |
118.44 |
0.27 |
| PP/PB5 |
113.26 |
165.60 |
80.36 |
119.26 |
0.40 |
| PP/xNBR5 |
115.34 |
163.25 |
89.82 |
120.27 |
0.45 |
| PP/PB5/AM5 |
121.12 |
165.77 |
77.84 |
125.41 |
0.41 |
| PP/xNBR5/AM5 |
126.54 |
165.96 |
94.29 |
130.85 |
0.50 |
The differences in the polarity resulted in significant incompatibility with the PP matrix, which was probably heavily impacted by the chains folding in an orderly and segmental alignment in the vicinity, leading to increase of both the initial crystallization temperature (Ti) and peak crystallization temperature (Tc), and enhanced crystallinity (χc); however, the reduced crystalline integrity was ascribed to inadequate relaxation due to the hindrance of nanoparticles, resulted in the corresponding widening of the peaks. The deviation of the melting temperature (Tm) is related to the reduced crystalline integrity, and the PP/xNBR blend demonstrated much more influence on the crystallization compared to the PP/PB blend. The crystalline morphology observed also certified the reduction of the spherulite size with increasing crystallinity when nanoparticles were incorporated, and the PP/xNBR blend exhibited much less crystallinity compared to the neat PP, as shown in Fig. 2A–C. Moreover, the significantly heterogeneous nucleation was found in the PP/xNBR/AM blends compared to the PP/PB/AM blends. The enhanced interfacial interaction between the xNBR and PP matrix restricted aggregation and enabled a more uniform dispersion of nanoparticles that provided more sites for nucleation. The Ti increased to 130.85 °C and 125.41 °C, Tc increased to 126.64 °C and 121.12 °C, and Tm increased to 165.96 °C and 165.77 °C for the PP/xNBR/AM and PP/PB/AM blends, respectively. Both increases in the crystallinity and viscosity due to increasing polarity resulted in larger melting peak areas, as shown in Fig. 1B and D. The POM morphology showed that the spherulite size decreased significantly both in the PP/xNBR and PP/PB blends after incorporation of the AM, which was ascribed to the more heterogeneous nucleation, as shown in Fig. 2C and D.
 |
| | Fig. 2 PLM of the neat PP and various blends: (A) neat PP; (B) PP/PB5; (C) PP/xNBR5; (D) PP/PB5/AM5; and (E) PP/xNBR5/AM5. | |
Generally speaking, the PP spherulites were large and the grains grew and developed completely, as shown in Fig. 2A–C. However, in the PP/xNBR/AM and PP/PB/AM blends, the PP matrix crystallization mode changed, showing that the size of spherulites became much smaller, the grains increased, and the integrity decreased, which is attributed to the changes in the crystallization process. As can be seen from Fig. 2D and E, the size of spherulites obviously shrank but increased in number, which indicates that the development of the PP spherulite was hindered, but the nucleation rate and crystallinity were promoted due to more heterogeneous nucleation. The observation is consistent with the results given in Fig. 1 and Table 3.
Rheological analysis
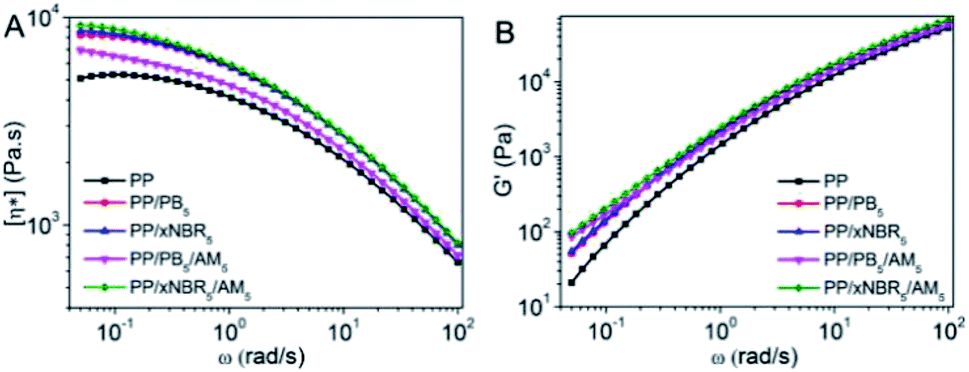
The complex viscosity (η*) vs. frequency (ω) for the neat PP and PP blends with different compositions are shown in Fig. 4A. In Fig. 4A, PP shows typical Newtonian fluid rheological behaviour. A Newtonian plateau appears in the low frequency region, and η* decreases as ω increases, which is attributed to the disentanglement and alignment of the polymer chains and segments under shearing, which resulted in decreased viscosity. However, for the PP/PB5 and PP/xNBR5 blends, with inclusion of the PB and xNBR latex to the composition, they exhibited pseudoplastic rheological characteristics like on-Newtonian fluids, showing increases of η* in the low-frequency region. The hydrodynamic effect is considerably stronger in PP/xNBR5 compared to the PP/PB5 blend, due to higher aggregation and segregation of the xNBR nanoparticles. The much larger sizes of aggregates blocked and restricted the dynamics of the PP matrix heavily, resulting in the η* of PP/xNBR5 that was slightly higher than that of PP/PB5. Increases in the viscosity and storage modulus after incorporating rubber particles in the blend also occurred, as shown in Fig. 3A and B. The interaction between grafts both on the particles and PP matrix endowed the melt with the characteristics of a heterogeneous structure, exhibiting a shearing thinning of η* and decreasing slope of G′ at low ω after inclusion of AM in the blend. In addition, this phenomenon was more obvious in PP/xNBR5/AM5 compared to the PP/PB5/AM5 blend, which was attributed to xNBR-g-PAM and PP-g-PAM that formed in situ during blending to first serve as a compatibilizer to promote the uniform dispersion of nanoparticles in the PP matrix, then, act as a physical crosslinking site between grafts to inhibit the chain mobility and increase the viscosity. Moreover, the nitrile groups of xNBR were also involved in interactions, which additionally contributed to PP/xNBR5/AM5 to enhance the viscosity and storage modulus when compared to the PP/PB5/AM5 blend, as shown in Fig. 3.
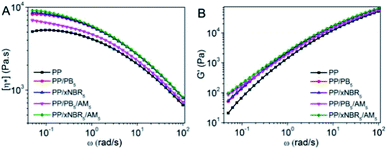 |
| | Fig. 3 Relationship between PP blends: (A) (η*)–ω and (B) (G′)–ω. | |
The sensitivity of the structural change to the relaxation time can be expressed by the storage modulus (G′) at low frequencies.22–25 Fig. 3B shows the changes of G′ vs. ω for the neat PP and PP blends. Compared with that of the neat PP, the G′ of PP/xNBR5 increased and the slope decreased at the low frequency, which indicated that the aggregation of xNBR nanoparticles restricted the segmental mobility of the PP matrix and retarded the relaxation of polymer chains, resulting a prolonged relaxation time and enhanced G′. Moreover, the restriction become even stronger after inclusion of AM in the blend, G′ further increased and the terminal slope became much smaller, especially in the PP/xNBR5/AM5 blend compared to the PP/PB5/AM5 blend, indicating that the interaction between PAM grafts on both particle and PP matrix inhibited the aggregation of xNBR5 particles and promoted the uniform dispersion in the PP matrix, leading to increases in G′ and a smaller slope at low ω.
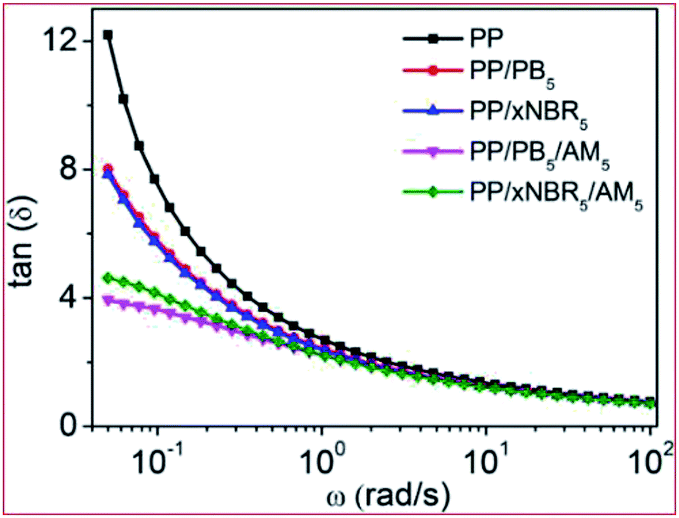
The plot of loss tangent (tanδ) vs. ω of the neat PP and PP blends28–33 is shown in Fig. 4. Compared with the neat PP, there were decreases in tanδ for both the PP/xNBR5 and PP/PB5 blends, showing that the elasticity increased due to the hydrodynamic effect by incorporation of nanoparticles in the blend. In addition, the contribution is slightly less for PP/xNBR5 than the PP/PB5 blend, probably because of the poor dispersion of xNBR in the PP matrix. However, the elastic contribution is much larger for PP/xNBR5/AM5 than PP/PB5/AM5, due to the enhanced interfacial interactions that greatly improved its dispersion.
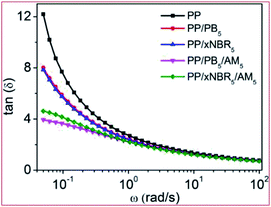 |
| | Fig. 4 Loss angle tangent–ω relation curve of PP blends. | |
SEM analysis
The morphology of samples foamed by supercritical CO2 was analysed by SEM; the results are shown in Fig. 5. The uneven cells with thick walls were observed in the neat PP (see Fig. 5A) since it is easy for the cell to collapse and coalescence due to the inability to withstand the stretch during foaming. This is a consequence of the lower melt strength and lower elasticity in the linear PP. In addition, as shown in Fig. 5B and C, the increased viscosity due to the hydrodynamic effect did not improve the melt strength. Increased cell sizes and decreased cell density in both PP/PB and PP/xNBR blends deteriorated the melt continuity, as caused by incompatibility between the particles and PP matrix. However, the cell morphologies were obviously improved after incorporating AM, as shown in both PP/xNBR5/AM5 and PP/PB5/AM5. The dispersion of particles in the PP matrix was greatly ameliorated by in situ compatibilization, which also served as physical cross-linking sites to restrict the mobility of polymer chains, leading to a simultaneous enhancement of the melt elasticity. Compared with PP/PB5/AM5, PP/xNBR5/AM5 exhibited more uniform cell sizes and higher cell density. The nitrile group serving as the nucleation site of foaming might be a reason for the improvement in cell morphology. Furthermore, the polygonal cell morphology observed in the PP/xNBR5/AM5 blend, as shown in Fig. 5E, was ascribed to the uneven distribution of stress caused by the heterogeneous structure of the melt, which could endow the sample with better mechanical properties compared to the circular cell morphology.34
 |
| | Fig. 5 SEM of PP blends: (A) PP; (B) PP/PB5; (C) PP/xNBR5; (D) PP/PB5/AM5; (E) PP/xNBR5/AM5. | |
The results of the cell morphology analysis are shown in Table 4, and as mentioned above, the slight increase of cell size in the binary blend of PP/PB and PP/xNBR is attributed to the collapse and coalescence of cells that were incompatible in the blends, which resulted in decreased cell density compared to the neat PP. Meanwhile, the higher bubble and expansion ratio also resulted from the less continuous melt. However, the decreased cell size in the ternary blend of PP/xNBR5/AM5 and PP/PB5/AM5 is attributed to the enhanced elasticity of the melt, which suppresses the collapse and coalescence of cells, leading to increased cell density. So, only in this situation, the increased bubble ratio and expansion ratio have practical significance.
Table 4 Density, bubble ratio, and expansion ratio of PP blends before and after foaming
| Sample |
Density (cells per cm3) × 109 |
Diameter of cell (μm) |
Bubble ratio (%) |
Expansion ratio |
| PP |
81 ± 3 |
17 ± 2 |
77.68 |
4.48 |
| PP/PB5 |
63 ± 2 |
18 ± 1 |
88.38 |
8.70 |
| PP/xNBR5 |
52 ± 1 |
20 ± 3 |
85.98 |
7.13 |
| PP/PB5/AM5 |
89 ± 1 |
16 ± 2 |
91.66 |
11.99 |
| PP/xNBR5/AM5 |
100 ± 2 |
14 ± 1 |
92.38 |
13.12 |
TGA analysis
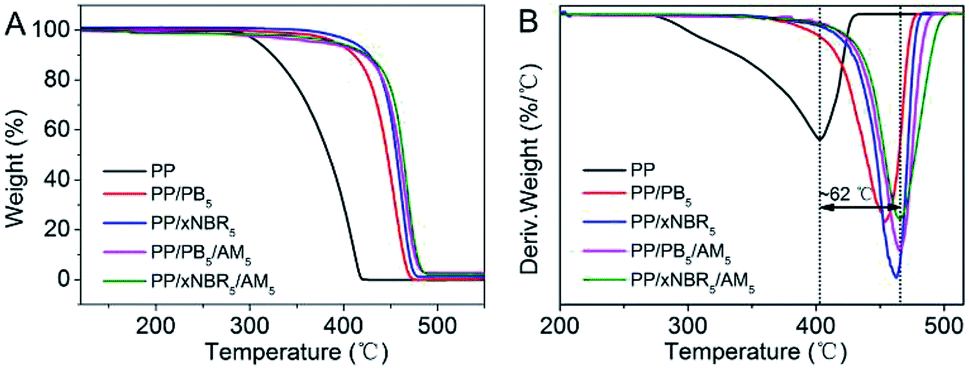
The thermal analysis of the neat PP and PP blends is shown in Fig. 6A and B. Compared with the neat PP, the initial decomposition temperature and peak temperature at the maximum degradation rate for the binary and ternary blends increased significantly, and that of the ternary blend was even more than the binary blend. In addition, PP/xNBR was superior to PP/PB as presented in the TGA and DTA analysis shown in Fig. 6A and B. The mechanism of thermal degradation of the PP matrix is induced by radicals. First, the increased thermal stability for the binary blends is attributed to the presence of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– in both PB and xNBR rubber particles, which can capture and stabilize the radicals. Besides, the cyclization reaction of the nitrile groups in xNBR improved the thermal stability considerably compared to PB.35–37 Second, the thermal resistance of the ternary blends improved even further, which was attributed to the enhanced interfacial interaction between the particles and PP matrix that inhibited the mobility of PP chains heavily, leading to an increase in the peak temperature at the maximum degradation rate of nearly 62 °C higher than that of the neat PP.
C– in both PB and xNBR rubber particles, which can capture and stabilize the radicals. Besides, the cyclization reaction of the nitrile groups in xNBR improved the thermal stability considerably compared to PB.35–37 Second, the thermal resistance of the ternary blends improved even further, which was attributed to the enhanced interfacial interaction between the particles and PP matrix that inhibited the mobility of PP chains heavily, leading to an increase in the peak temperature at the maximum degradation rate of nearly 62 °C higher than that of the neat PP.
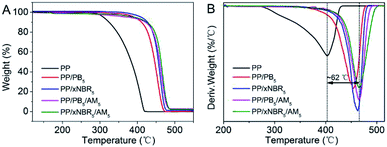 |
| | Fig. 6 TGA (A) and DTG (B) of PP blends. | |
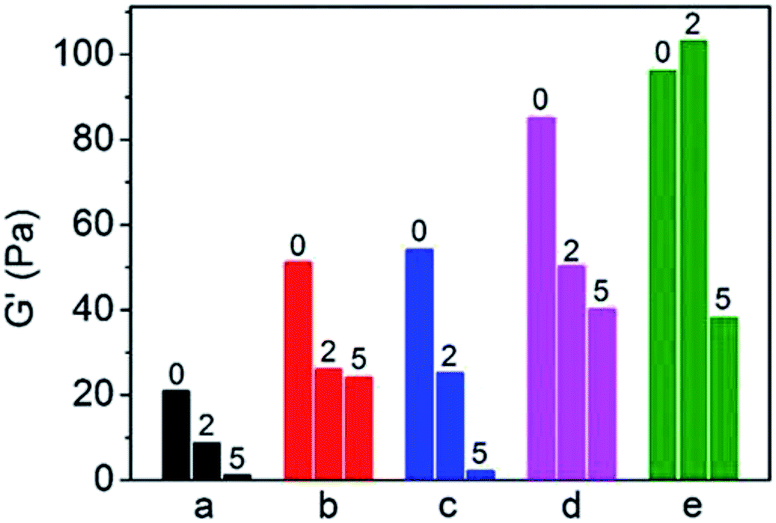
γ-Ray irradiation provides an extreme environment to test the aging performance of the PP blends.38 The neat PP and PP blends were irradiated at doses of 2 kGy and 5 kGy, respectively, and the acceleration of aging performance of the blends were compared based on the terminal storage module of the samples before and after irradiation. The results are shown in Fig. 7. As can be seen in Fig. 7, for the neat PP, the storage modulus decreases gradually with an increasing irradiation dose from 2 kGy to 5 kGy. The dramatic degradation induced by irradiation resulted in loss of the mechanical properties even at the lower dose of 2 kGy. However, the storage modulus increased for the binary blend of PP/PB and PP/xNBR compared to the neat PP, and the residual storage modulus under a dose of 2 kGy was almost identical to the un-irradiated PP, probably because the function of radical capture and stabilization by –CC– groups inhibited the degradation efficiently. In particular, the PP/PB blend had a much higher residual modulus even at a dose of 5 kGy, as shown in Fig. 6. Furthermore, like the binary blend, the ternary blend showed a much higher modulus at first, and the residual modulus under a dose of 2 kGy almost achieved that of the non-irradiated binary blend, which is attributed to improved dispersion of the nanoparticles in the PP matrix, especially in the PP/xNBR/AM blend, so the resistance of aging is improved at the lower dose of irradiation.
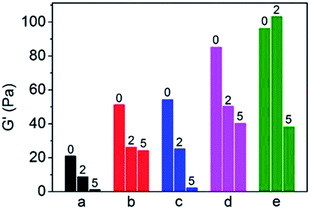 |
| | Fig. 7 Storage modulus of PP blends with different radiation doses G′: (a) PP; (b) PP/PB5; (c) PP/xNBR5; (d) PP/PB5/AM5; (e) PP/xNBR5/AM5. All samples are measured at 200 °C. | |
Surface energy analysis
The contact angle and surface energy of the neat PP and PP blends were measured with water and glycol, and the results are shown in Table 5. Compared with the neat PP, the contact angles of the two liquids for both the binary and ternary blends decreased while the surface energy respectively increased. For the binary blends, the rough surface as a consequence of incompatibility between the particles and PP matrix might lead to decreased contact angles, besides, the increased polarity in the PP/xNBR blend resulted in a decreased contact angle. However, for the ternary blends, the deceased contact angle is attributed to the enhanced polarity, which also endowed the blends with uniform nanoparticle dispersions.
Table 5 Contact angle and surface energy parameters of neat PP and PP blend measured with water and glycol
| Sample |
Contact angle/° |
Surface free energy/mJ m−2 |
| Water |
Ethyl glycol |
| PP |
105.20 ± 0.52 |
88.44 ± 0.89 |
13.32 |
| PP/PB5 |
92.02 ± 5.11 |
69.38 ± 3.20 |
23.17 |
| PP/xNBR5 |
90.88 ± 5.00 |
69.00 ± 0.78 |
23.20 |
| PP/PB5/AM5 |
86.28 ± 4.53 |
65.08 ± 1.21 |
24.46 |
| PP/xNBR5/AM5 |
83.70 ± 2.27 |
64.60 ± 0.66 |
29.59 |
Conclusions
The polypropylene blends incorporated with two kinds of nanoparticle rubber with different polarities were prepared through water-assisted reactive blending. The aggregation of the nanoparticles was inhibited by the enhanced interfacial interactions between the nanoparticles and PP matrix through in situ compatibilization after incorporation of AM monomer. The crystalline behaviour showed increases in the initial crystallization and peak temperature due to the heterogeneous nucleation effect of the nanoparticles in the binary blend, which was enhanced further in the ternary blends due to the strengthened interfacial interactions and amelioration of the dispersion of the nanoparticles in the PP matrix. The foaming performance was also improved for the uniform dispersion and continuity of the melt, and the foaming ratio increased 13-fold in the PP/xNBR/AM blend. In addition, the enhanced thermal properties and aging performance are attributed to the radical capture and stabilization of the –CC– groups in the component, which inhibited the degradation efficiently, and exhibited an increase in the peak decomposition temperature by nearly 62 °C. The surface properties were especially improved in the ternary blends, such as a lower contact angle and higher surface energy, which is mainly ascribed to the enhancement of the polarity. This work might provide a method for preparing polyolefin blends modified with other rubber nanoparticles.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Natural Science Foundation for Youths of Heilongjiang Province of China (QC2018070), the Heilongjiang Academy of Sciences Fund for Distinguished Young Scholars (CXJQ2019WL01), and the Heilongjiang Academy of Sciences Fund for Pilot and Promoting Project (ZSSH2019WL01).
References
- E. Manias, A. Touny, L. Wu, K. E. Strawhecker, B. Lu and T. C. Chung, Chem. Mater., 2001, 13, 3516–3523 CrossRef CAS.
- P. Wu, Q. Yang, Z. Zhao, T. Zhang, Y. Huang and X. Liao, Polymer, 2019, 161, 109–121 CrossRef CAS.
- M. M. Haque, K. Goda, H. Ito, S. Ogoe, M. Okamot, T. Ema, K. Kagawa and H. Nogami, Adv. Ind. Eng. Polym. Res., 2019, 2, 42–50 Search PubMed.
- K. Zhao, S. Li, M. Huang, X. Shi, G. Zheng, C. Liu, K. Dai, C. Shen, R. Yin and J. Z. Guo, Chem. Eng. J., 2019, 358, 924–935 CrossRef CAS.
- R. Wiwattananukul, B. Fan and M. Yamaguchi, Compos. Sci. Technol., 2017, 141, 106–112 CrossRef CAS.
- A. R. Albooyeh, Polym. Test., 2019, 74, 86–98 CrossRef CAS.
- Z. Martin, I. Jimenez, M. A. Gomezfatou, M. M. West and A. P. Hitchcock, Macromolecules, 2011, 44, 2179–2189 CrossRef CAS.
- X. D. Qi, D. X. Sun, C. J. Yang, W. Y. Wang and Y. Wang, Polym. Test., 2019, 75, 185–191 CrossRef CAS.
- E. Jia, S. Zhao, Y. Shangguan and Q. Zheng, Polymer, 2019, 178, 121602 CrossRef CAS.
- F. S. Li, Y. B. Gao and W. Jiang, Polymer, 2019, 170, 101–106 CrossRef CAS.
- W. Dong, Y. Liu, X. Zhang, J. Gao, F. Huang, Z. Song and J. Qiao, Macromolecules, 2005, 38, 4551–4553 CrossRef CAS.
- T. Shiono, S. M. Azad and T. Ikeda, Macromolecules, 1999, 32, 5723–5727 CrossRef CAS.
- A. Lugao, B. W. H. Artel, A. Yoshiga, L. F. C. P. Lima, D. Parra, J. Bueno, S. Liberman, M. Farrah, W. R. Terçariol and H. Otaguro, Radiat. Phys. Chem., 2007, 76, 1691–1695 CrossRef CAS.
- M. Hasanpour, M. Mehrabi mazidi and M. Aghjeh, Polym. Test., 2019, 79, 106018 CrossRef.
- Y. Chen, Z. Wu, Q. Fan, S. Yang, E. Song and Q. Zhang, Compos. Sci. Technol., 2018, 167, 277–284 CrossRef CAS.
- W. Yuan, F. Wang, Z. Chen, C. Gao, P. Liu, Y. Ding, S. Zhang and M. Yang, Polymer, 2018, 151, 242–249 CrossRef CAS.
- J. Xu, J. M. Eagan, S.-S. Kim, S. Pan, B. Lee, K. Klimovica, K. Jin, T. W. Lin, M. J. Howard, C. J. Ellison, A. M. LaPointe, G. W. Coates and F. S. Bates, Macromolecules, 2018, 51, 8585–8596 CrossRef CAS.
- L. Liu, Y. Xu, S. Li, M. Xu, Y. He, Z. Shi and B. Li, Composites, Part B, 2019, 176, 107218 CrossRef CAS.
- Y. Q. Liu, X. H. Zhang, J. M. Gao, F. Huang, B. H. Tan, G. S. Wei and J. L. Qiao, Polymer, 2004, 45, 275–286 CrossRef CAS.
- W. Si, W. Yuan, Y. Li, Y. Chen and J. Zeng, Polym. Test., 2017, 65, 249–255 CrossRef.
- F. Tang, P. Bao, A. Roy, Y. Wang and Z. Su, Polymer, 2018, 142, 155–163 CrossRef CAS.
- F. Tang, P. Bao and Z. Su, Anal. Chem., 2016, 88, 4926–4930 CrossRef CAS PubMed.
- M. Rezaei and A. Jalali, Thermochim. Acta, 2015, 617, 120–128 CrossRef.
- K. Suzuki, A. Sato, H. Okumura, T. Hashimoto, A. Nakagaito and H. Yano, Cellulose, 2014, 21, 507–518 CrossRef CAS.
- Q. Guo, X. Li, W. Li and Z. Yao, Ind. Eng. Chem. Res., 2018, 57, 6696–6703 CrossRef CAS.
- L. Liu, Y. Xu, M. Xu, Z. Li, Y. Hu and B. Li, Composites, Part B, 2019, 167, 422–433 CrossRef CAS.
- S. Mei, L. Yang, Y. Pan, D. Wang, X. Wang, T. Tang and J. Wei, Colloids Surf., B, 2019, 174, 207–215 CrossRef CAS PubMed.
- F. Su, J. H. Yan and H. X. Huang, J. Appl. Polym. Sci., 2011, 119, 1230–1238 CrossRef CAS.
- E. Ueda, X. Liang, M. Ito and K. Nakajima, Macromolecules, 2019, 52, 311–319 CrossRef CAS.
- M. Hussain, S. Yasin, M. Adnan Akram, H. Xu, Y. Song and Q. Zheng, Ind. Eng. Chem. Res., 2019, 58, 18205–18212 CrossRef CAS.
- M. A. Malik and C. S. Saini, Food Hydrocolloids, 2018, 81, 229–241 CrossRef CAS.
- W. Sui, X. Xie, R. Liu, T. Wu and M. Zhang, Food Hydrocolloids, 2018, 84, 571–580 CrossRef CAS.
- G. Romeo, G. Filippone, A. Fernandez-Nieves, P. Russo and D. Acierno, Rheol. Acta, 2008, 47, 989–997 CrossRef CAS.
- C. G. Yang, Z. Xing, M. H. Wang, Q. Zhao, M. L. Wang, M. J. Zhang and G. Z. Wu, Ind. Eng. Chem. Res., 2018, 57, 15916–15923 CrossRef CAS.
- X. Liu, J. Zhao, R. Yang, R. Iervolino and S. Barbera, Polym. Degrad. Stab., 2018, 151, 136–143 CrossRef CAS.
- Z. Q. Fan, X. L. Tan, J. W. Chen, Z. L. Liu, J. F. Kuang, W. J. Lu, W. Shan and J. Y. Chen, J. Agric. Food Chem., 2018, 66, 9399–9408 CrossRef CAS PubMed.
- S. Bhattacharjee, A. K. Bhowmick and B. N. Avasthi, Polym. Degrad. Stab., 1991, 31, 71–87 CrossRef CAS.
- R. Perera, C. Albano, J. González, P. Silva and M. Ichazo, Polym. Degrad. Stab., 2004, 85, 741–750 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Zhigang Lib,
Jinfeng Lib,
Gang Yaob,
Wei Dong*c,
Yuguang Liu*b and
Mingwei Di*a
ab,
Zhigang Lib,
Jinfeng Lib,
Gang Yaob,
Wei Dong*c,
Yuguang Liu*b and
Mingwei Di*a