
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of mercaptopropanamide-substituted aryl tetrazoles as new broad-spectrum metallo-β-lactamase inhibitors†
Yu-Hang Yan,
Jian Chen,
Zhen Zhan,
Zhu-Jun Yu,
Gen Li,
Li Guo,
Guo-Bo Li ,
Yong Wu and
Yongxiang Zheng*
,
Yong Wu and
Yongxiang Zheng*
Key Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, Sichuan Engineering Laboratory for Plant-Sourced Drug, Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Sichuan 610041, China. E-mail: yongxiangzheng@scu.edu.cn; Tel: +86 199 3880 2868
First published on 25th August 2020
Abstract
β-Lactam antibiotic resistance mediated by metallo-β-lactamases (MBL) has threatened global public health. There are currently no available inhibitors of MBLs for clinical use. We previously reported the ruthenium-catalyzed meta-selective C–H nitration synthesis method, leading to some meta-mercaptopropanamide substituted aryl tetrazoles as new potent MBL inhibitors. Here, we described the structure–activity relationship of meta- and ortho-mercaptopropanamide substituted aryl tetrazoles with clinically relevant MBLs. The resulting most potent compound 13a showed IC50 values of 0.044 μM, 0.396 μM and 0.71 μM against VIM-2, NDM-1 and IMP-1 MBL, respectively. Crystallographic analysis revealed that 13a chelated to active site zinc ions via the thiol group and interacted with the catalytically important residues Asn233 and Tyr67, providing further structural information for the development of thiol based MBL inhibitors.
Introduction
β-Lactam antibiotics remain the most important class of antibiotics due to their safety, high efficacy and broad-spectrum antibacterial activity.1 The emergence and rapid worldwide distribution of β-lactam-resistant bacteria pathogens poses an increasing clinical threat to human health and life.2–4 β-Lactam hydrolysis mediated by β-lactamase is a major contributor to the resistance in bacteria.5,6 Based on different catalytic mechanisms, β-lactamases are categorized into two groups: serine β-lactamases (SBLs) and metallo-β-lactamases (MBLs).5 SBLs employ an active-site nucleophilic serine to hydrolyze the β-lactam ring, while MBLs utilize a Zn(II)-bound hydroxyl group to achieve nucleophilic attack on the β-lactam carbonyl, ultimately leading to the cleavage of the β-lactam ring.7,8 Several SBL inhibitors, such as clavulanic acid, tazobactam, sulbactam, avibactam, and vaborbactam, have been successfully applied in combination with beta-lactam antibiotics to treat infections caused by SBL-producing bacteria in clinics.9,10 However, there are no FDA-approved MBL inhibitors.11,12On the basis of sequence and structural similarities, MBL enzymes can be divided into three subclasses (B1, B2, B3).7 Subclass B1 MBLs contain the most clinically significant types, such as the New Delhi MBL (NDMs), imipenemase (IMPs), and Verona integron-encoded MBL (VIMs), representing the major targets for developing inhibitors with therapeutic value.13 So far, various types of MBL inhibitors have been reported, such as thiol-containing compounds, cyclic boronates, succinic acids, carboxylates and others.14–20 Most MBL inhibitors work via active-site metal chelation manner, and more importantly, the broad-spectrum MBL inhibitors have metal chelation and anchor residue binding features.14 Despite progress has been made, there is still a need to develop new broad-spectrum MBL inhibitors to provide candidates for the development of clinically useful MBL inhibitors.
In our previous study, we described a new synthesis method, namely ruthenium-catalyzed meta-selective C–H nitration, which led to new potent MBL inhibitors; these inhibitors contain an aryl tetrazole fragment and a mercaptopropanamide moiety as the metal binding pharmacophore.21 This work presents structure–activity relationship (SAR) studies of mercaptopropanamide substituted aryl tetrazoles with clinically relevant MBLs and crystallographic analyses of the mechanism-of-inhibition of this chemotype inhibitors. We synthesized a series of ortho- and meta-mercaptopropanamide substituted aryl tetrazole derivatives through C–H activation methods, and tested for their inhibitory activity against three MBL enzymes, including VIM-2, NDM-1 and IMP-1. Among all the synthesized target compounds, meta-mercapto-2-methylpropanamide substituted aryl tetrazole 13a had the strongest inhibitory activity against clinically relevant VIM-2, NDM-1 and IMP-1 with IC50 values of 0.044 μM, 0.396 μM and 0.71 μM, respectively. Crystallographic analyses showed that 13a chelated to both active site zinc ions via thiol group and interacted with catalytically important residues on the L1 and L3 loop.
Results and discussion
Chemistry
Schemes 1 and 2 show the synthetic routes and total yields of all target compounds. Compounds 6a, 7a–7i and 8 were synthesized using the reaction sequence shown in Scheme 1. Compound 2 was obtained by using benzonitrile to react with NaN3 in the presence of ZnBr2 and H2O stirring at 100 °C for 24 h.22 Compounds 3a–3h were synthesized by the reactions of compound 2 and different iodoalkanes.23 Then, compounds 3a–3h were reacted with 3-phenyloxazolidinone to afford the corresponding amides 4a–4h via the C–H activation strategy as described previously, followed by reacting with sodium hydroxide to produce 2-(2H-tetrazol-5-yl)anilines (5a–5h).21 Subsequently, Compounds 5a–5h were treated with different carboxylic acids, giving the thioesters (6a–6i) in high yields. Finally, the desired compounds 7a–7i were furnished through the hydrolysis reactions of 6a–6i in the presence of NH3·H2O under argon atmosphere. Compound 5a was subjected to the condensation reaction by isobutyryl chloride to give the desired compound 8.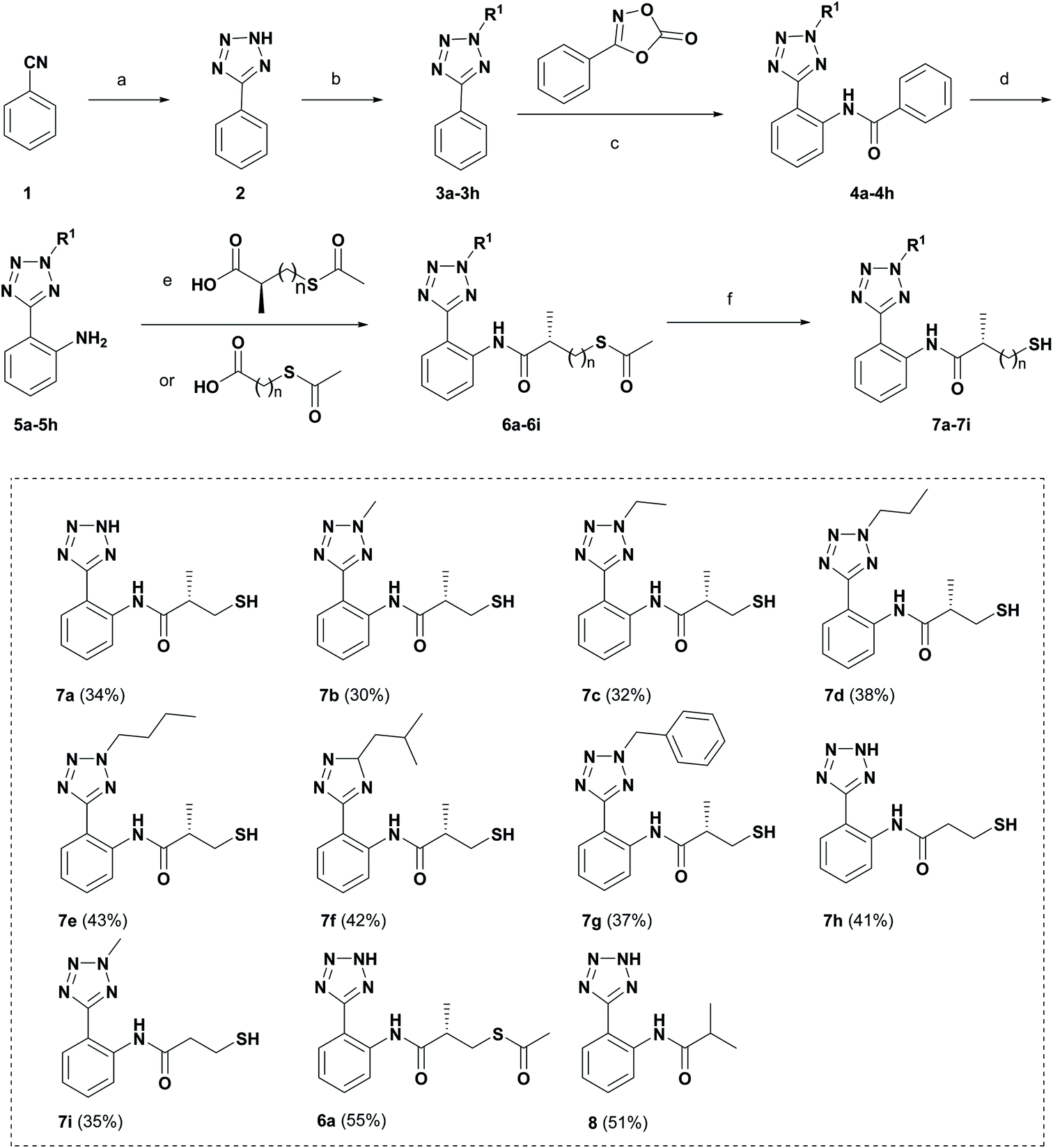 | ||
| Scheme 1 Preparation of target compounds 7a–7i, 6a and 8. Reagents and conditions: (a) NaN3, ZnBr2, H2O, reflux, 24 h; (b) K2CO3, rt, 5 min; R–I, 85 °C, 4 h; (c) [Cp*RhCl2]2, Ag2SO4, DCE, 80 °C, 24 h; (d) NaOH, EtOH, reflux, 1 h; (e) IBCF, NMM, dry DCM, rt, 24 h; (f) NH3·H2O, Ar, rt, 2 h. The overall yield for each target compound is shown in brackets behind the compound number. | ||
Scheme 2 shows the synthetic route for target compounds 12a, 13a–13k and 14. Compounds 9a–9j were reacted with Cu(NO3)2·3H2O to afford the meta-nitrated products 10a–10j via the C–H activation strategy, followed by the reduction reactions to give the compounds 11a–11j.24 Then, the thioesters 12a–12k were readily synthesized by the condensation reactions of 11a–11j with different carboxylic acids. Subsequently, the hydrolysis reactions of 12a–12k produced the target compounds 13a–13k. Compound 14 was obtained by the condensation reaction of compound 9a and isobutyryl chloride.
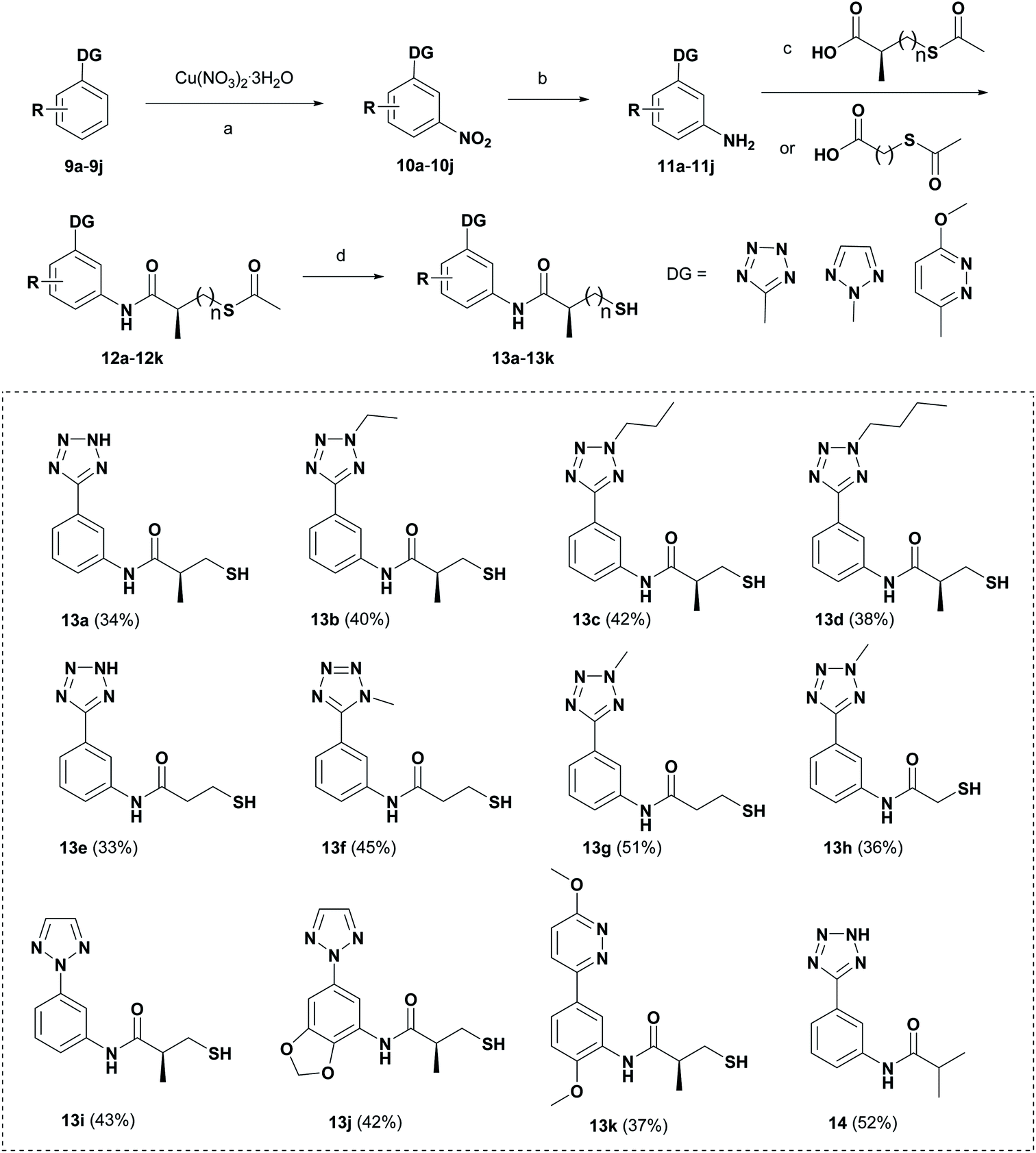 | ||
| Scheme 2 Preparation of target compounds 13a–13k and 14. Reagents and conditions: (a) Ru3(CO)12, PPh3, PhI(TFA)2, HFIP, 100 °C, 24 h; (b) H2, 10% Pd/C, MeOH; (c) IBCF, NMM, dry DCM, rt, 24 h; (d) NH3·H2O, Ar, rt, 2 h. | ||
SAR of mercaptopropanamide substituted aryl tetrazole derivatives with MBLs
We evaluated the inhibitory activity of all the targeted compounds against clinically important VIM-2, NDM-1 and IMP-1 by using a fluorescence-based assay (Fig. S1†).25 As shown in Table 1, 7a–7g contain mercapto-2-methylpropanamide substituents on the ortho position of the phenyl ring. 7a displayed potent inhibition on VIM-2 with IC50 value of 0.035 μM and moderate inhibition on NDM-1 and IMP-1. Addition of alkyl or benzyl substituents to the N-2 of tetrazole of 7a slightly decreased the inhibitory activity of VIM-2, but increased the inhibitory activity on NDM-1 and IMP-1, except for 7b with methyl substitution on the tetrazole, which showed less potent inhibition against all the tested enzymes, especially for NDM-1. The ortho-mercaptopropionamide-substituted derivatives 7h and 7i showed comparable inhibition with 7a. Comparison of 7a, 7h, and 7i indicated that the effect of methyl substitution on the tetrazole of 7h on the activity of NDM-1 was not as great as that of 7a, which was reduced by about 5 times. Two examples showed that the thiol group was important for interacting with the active site: (1) the product 6a with acetylation modification on the thiol group exhibited significantly reduced inhibitory activity; (2) no inhibition against tested MBL enzymes was observed for 8 without the thiol group.| Cpds | Chemical structure | IC50 value (μM)/pIC50/s.e. pIC50 | ||
|---|---|---|---|---|
| VIM-2 | NDM-1 | IMP-1 | ||
| 7a | 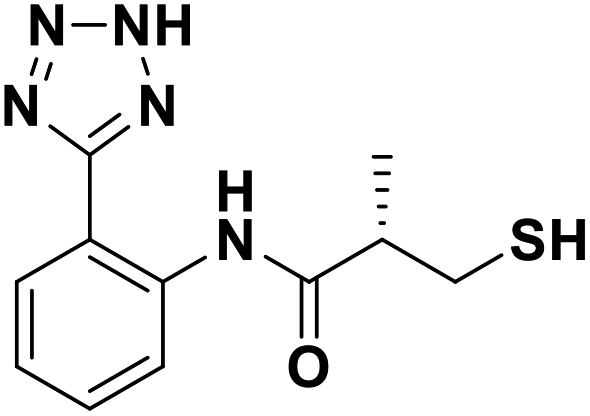 |
0.035/7.45/0.039 | 10.75/4.97/0.046 | 9.87/5.01/0.118 |
| 7b | 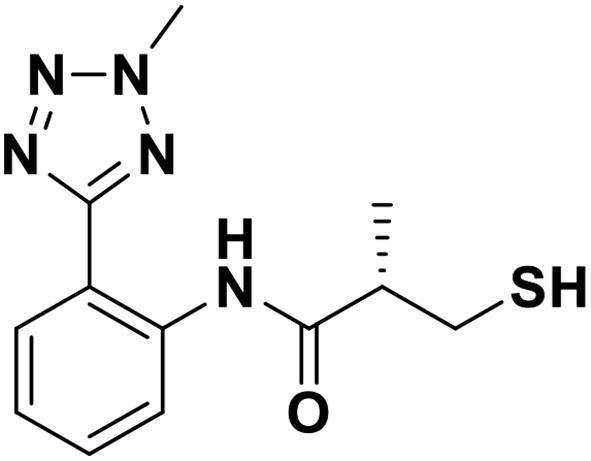 |
1.82/5.74/0.050 | >100/—/— | 12.26/4.90/0.127 |
| 7c | 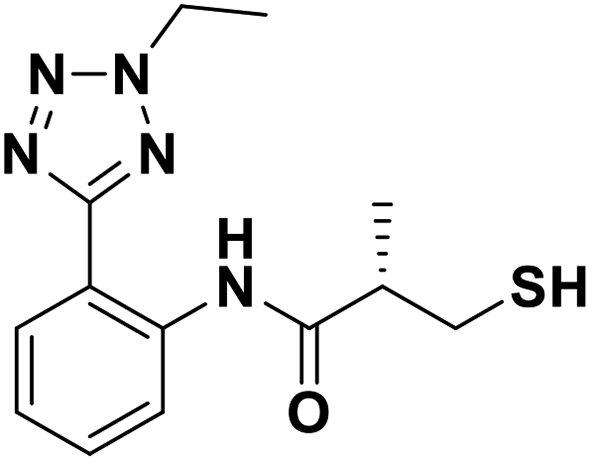 |
0.06/7.22/0.050 | 1.84/5.74/0.048 | 0.13/6.89/0.107 |
| 7d | 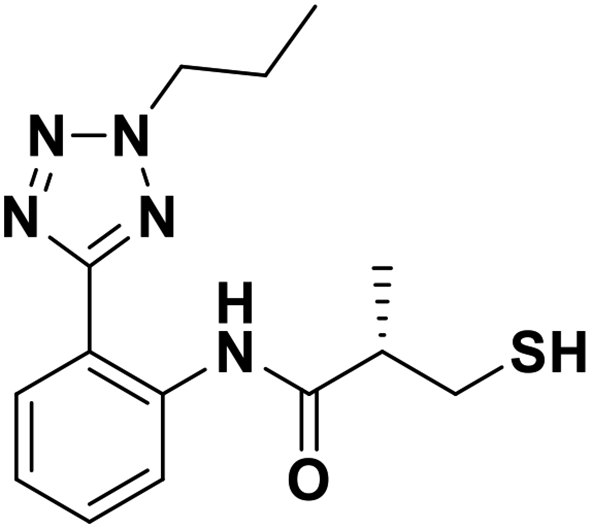 |
0.49/6.37/0.024 | 0.83/6.08/0.138 | 0.88/6.056/0.057 |
| 7e | 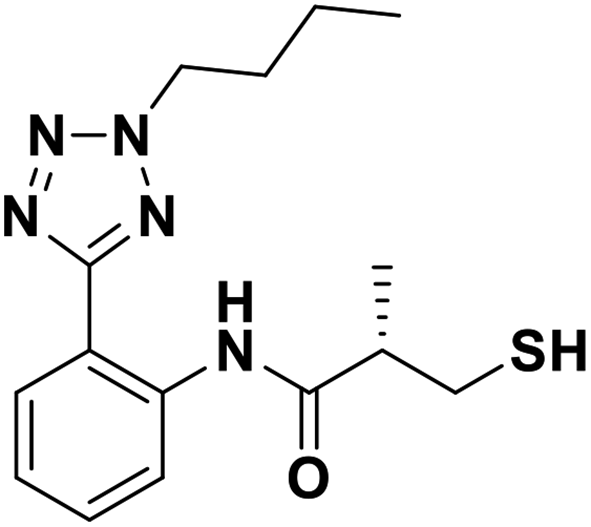 |
0.16/6.81/0.062 | 2.0/5.69/0.056 | 1.49/5.83/0.084 |
| 7f | 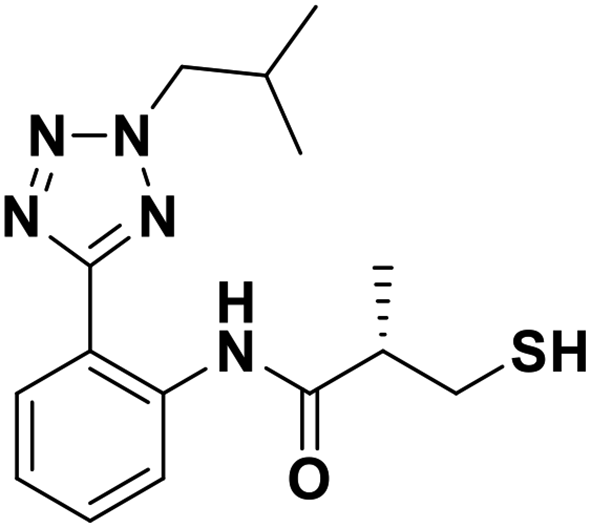 |
1.70/5.77/0.039 | 2.59/5.59/0.076 | 1.34/5.87/0.099 |
| 7g | 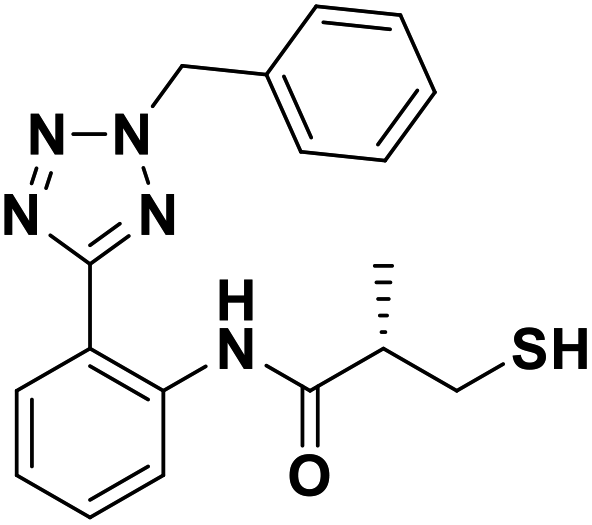 |
0.16/6.81/0.047 | 1.46/5.84/0.036 | 1.36/5.87/0.043 |
| 7h | 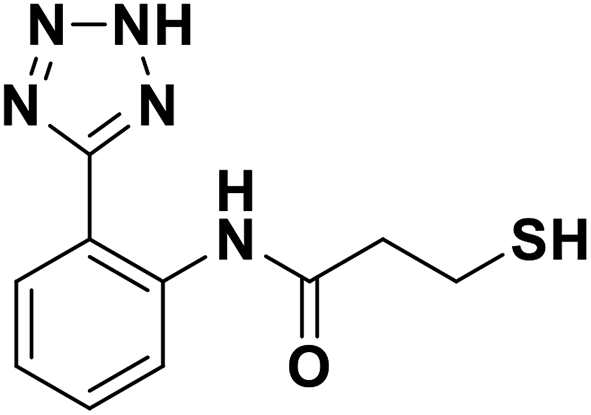 |
0.028/7.55/0.044 | 3.16/5.50/0.041 | 10.17/4.99/0.076 |
| 7i | 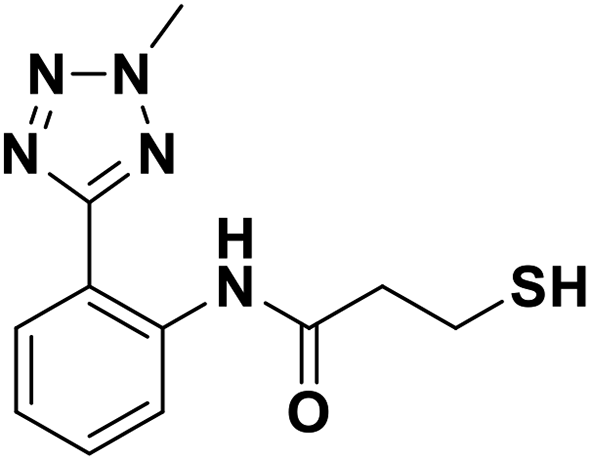 |
0.026/7.59/0.033 | 19.52/4.70/0.094 | 4.68/5.33/0.166 |
| 6a | 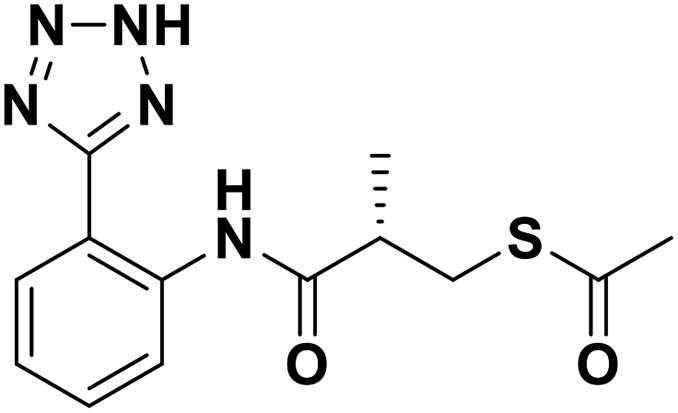 |
0.63/6.20/0.049 | 30.16/4.52/0.077 | 30.41/4.52/0.088 |
| 8 | 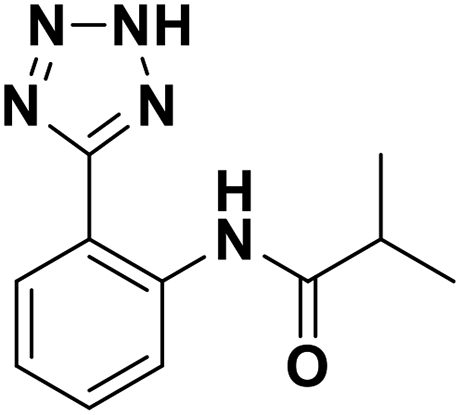 |
>100/—/— | >100/—/— | >100/—/— |
Then, we investigated the activity of compounds with mercaptopropionamide substitutions on the meta position of the phenyl ring (Table 2, Fig. S1†). Among all the synthesized mercaptopropanamide derivatives, 13a manifested the most potent inhibition, displaying IC50 values of 0.044 μM, 0.396 μM and 0.71 μM against VIM-2, NDM-1 and IMP-1, respectively. Introduction of alkane chain on the 2-N of tetrazole resulted in 13b–13d. Compared to 13a, 13b–13d exhibited compared inhibition on VIM-2 and decreased inhibition on NDM-1 and IMP-1. Mercaptopropionamide substituted compound 13e showed IC50 values of 0.021 μM, 6.10 μM and 0.44 μM against VIM-2, NDM-1 and IMP-1, respectively, indicating that the methyl substitution on the α-C of the carbonyl group had a great influence on the activity of NDM-1. For the methyl substituted on N-1 or N-2 of 13e derivatives 13f and 13g, no significant impact of the substitution position on NDM-1 and IMP-1 inhibition was found, but methyl substitution on N-1 was unfavorable for VIM-2 activity. In addition, the shortening of the alkyl chain was also adverse to the inhibition on VIM-2, as exemplified by 13h. Furthermore, we also explored the effect of different heterocycles-replacing biphenyltetrazole (13i–13k) on MBL inhibitory activity. Obviously, substitution of aromatic ring had the greatest effect on NDM-1 and IMP-1 inhibition, which led to at least 3-fold drop in activity. The activity of 13i–13k on VIM-2 was comparable to or slightly weaker than that of 13a. Similar to what was observed before, 14 without thiol group had very low inhibitory activity on tested MBLs.
| Cpds | Chemical structure | IC50 value (μM)/pIC50/s.e. pIC50 | ||
|---|---|---|---|---|
| VIM-2 | NDM-1 | IMP-1 | ||
| 13a | 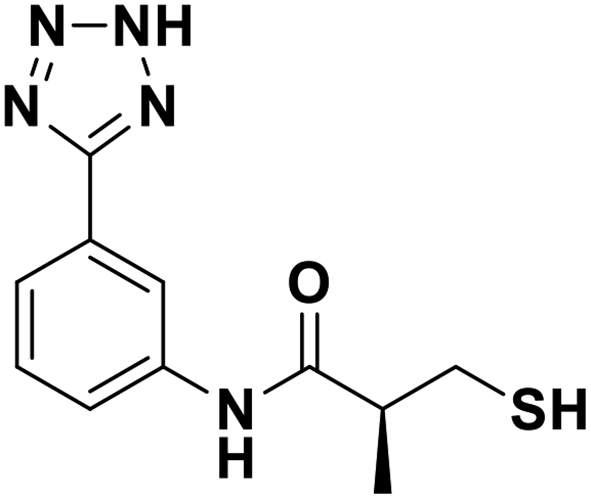 |
0.044/7.35/0.022 | 0.396/6.40/0.036 | 0.71/6.15/0.089 |
| 13b | 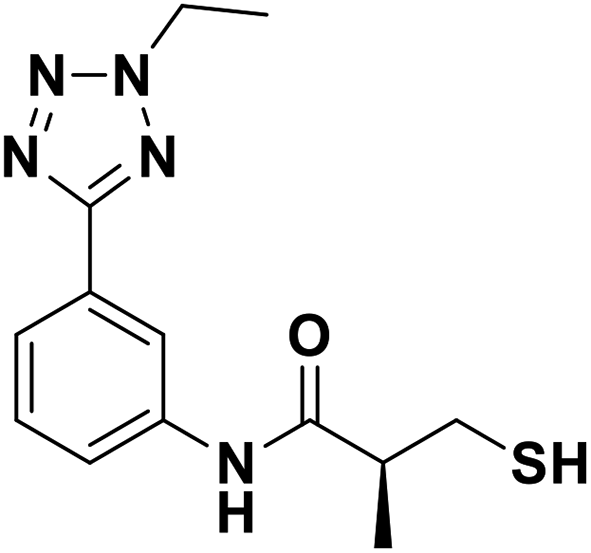 |
0.082/7.08/0.026 | 3.02/5.52/0.032 | 6.23/5.21/0.153 |
| 13c | 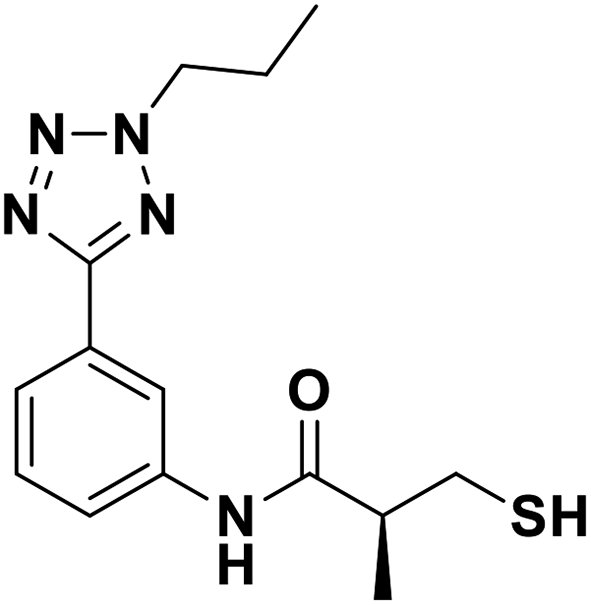 |
0.018/7.74/0.039 | 2.37/5.63/0.040 | 1.23/5.91/0.050 |
| 13d | 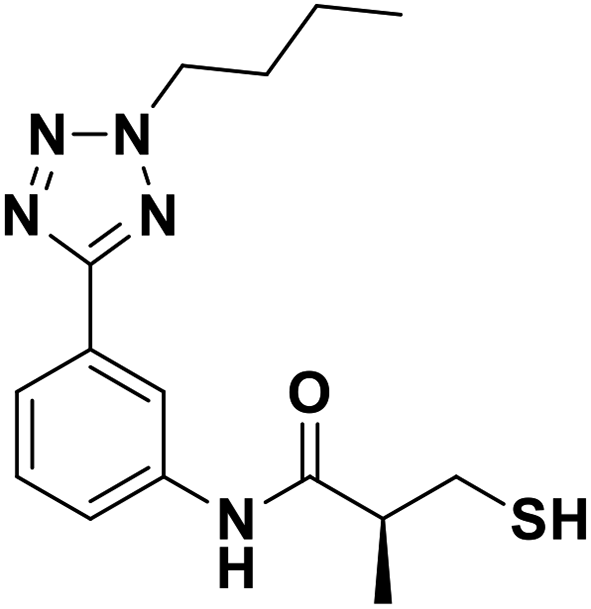 |
0.025/7.60/0.066 | 2.65/5.58/0.030 | 4.97/5.30/0.384 |
| 13e | 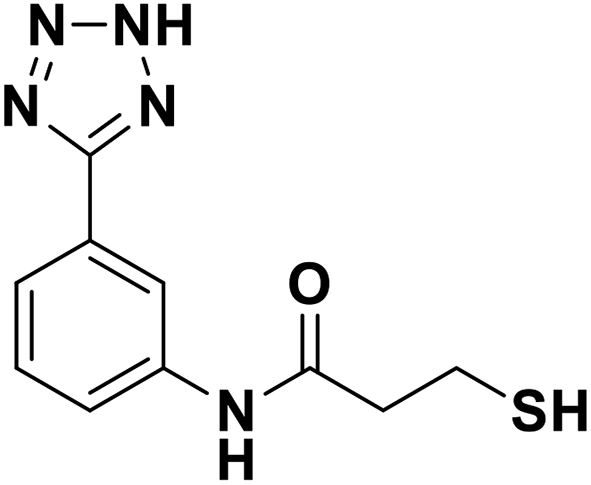 |
0.021/7.67/0.038 | 6.10/5.21/0.028 | 0.44/6.30/0.192 |
| 13f | 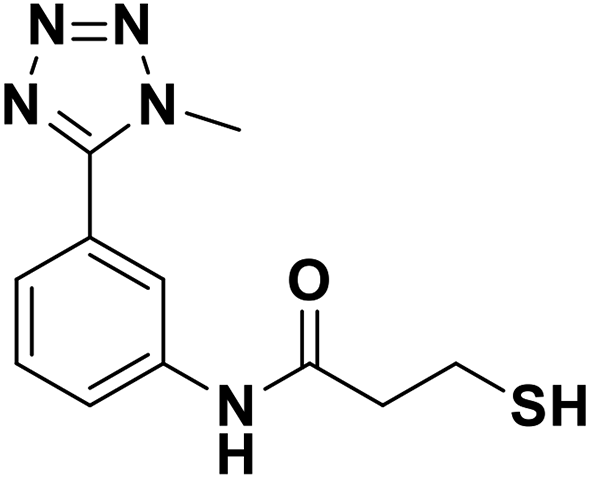 |
0.48/6.32/0.056 | 3.23/5.49/0.023 | 1.54/5.81/0.106 |
| 13g | 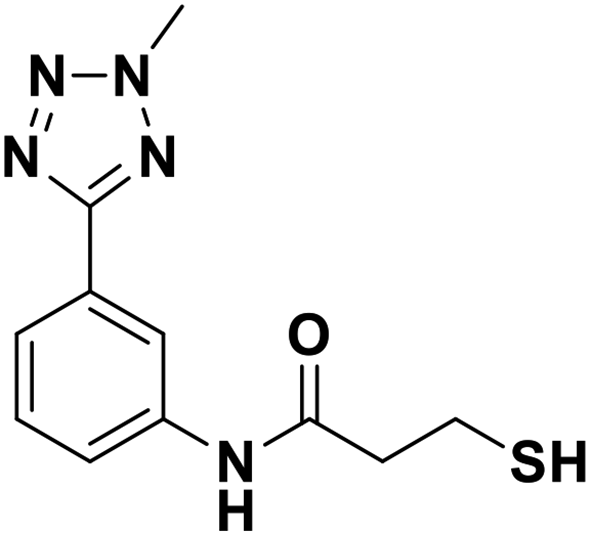 |
0.079/7.10/0.040 | 4.19/5.38/0.065 | 1.18/5.93/0.067 |
| 13h | 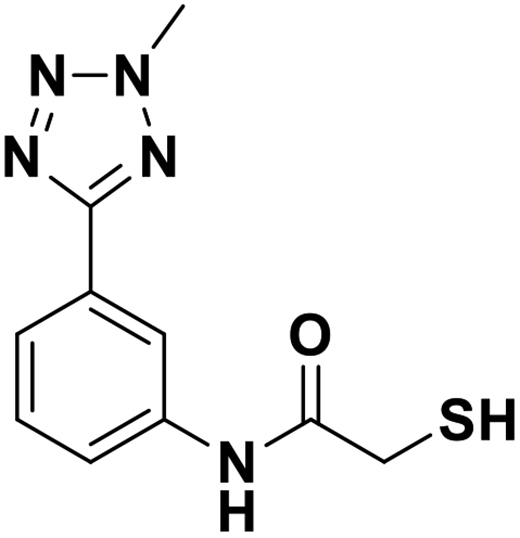 |
1.38/5.86/0.091 | 3.22/5.49/0.024 | 0.94/6.03/0.053 |
| 13i | 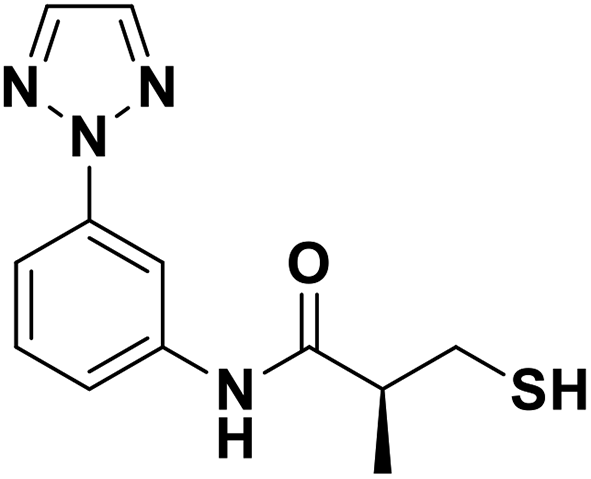 |
0.11/6.97/0.036 | 2.10/5.68/0.048 | 3.70/5.43/0.062 |
| 13j | 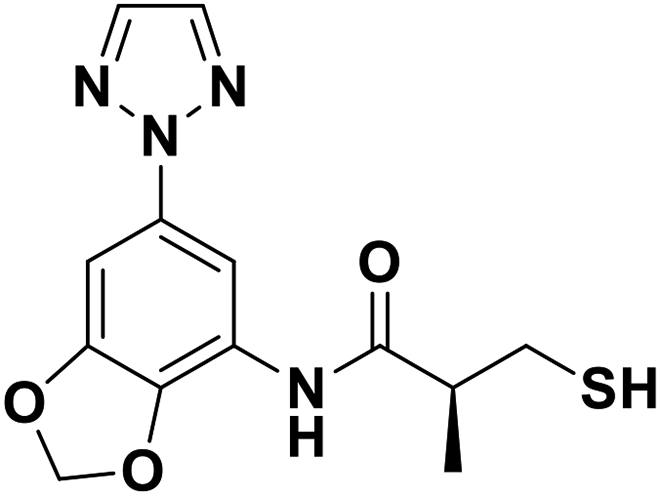 |
0.025/7.60/0.034 | 1.27/5.90/0.058 | 4.19/5.38/0.399 |
| 13k | 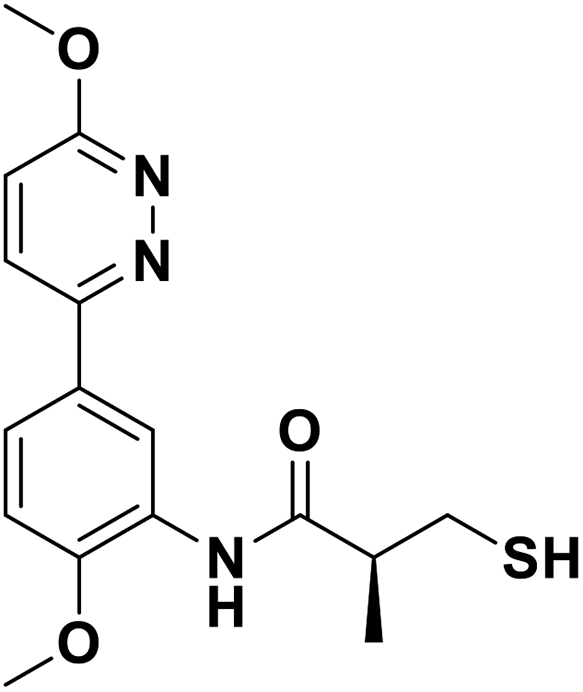 |
0.056/7.25/0.041 | 1.27/5.90/0.067 | 1.56/5.81/0.199 |
| 14 | 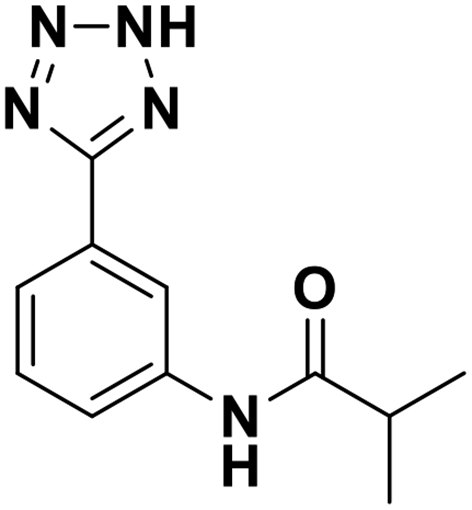 |
>100/—/— | >100/—/— | >100/—/— |
Moreover, we assessed the effect of the most potent inhibitor 13a on a cellular level through minimum inhibitory concentrations (MIC) assay (Table S2†). The results indicated that 13a potentiated the efficacy of meropenem at 100 μM against laboratory-constructed VIM-2 expressing E. coli strain with MIC value of 4 μg mL−1.
Crystallographic analysis
In order to investigate the binding mode of the most potent compound 13a, we obtained the X-ray structure of VIM-2 in complex with 13a by co-crystal experiments. The VIM-2: 13a (PDB ID 7CJL) complex structure was solved to 1.79 Å resolution (Table S1†). The VIM-2:13a complex crystallized in a P 212121 space group with two molecules in the asymmetric unit (ASU) and was refined to Rwork/Rfree value of 20.53/24.64% (Table S1†). The crystal structure revealed that 13a was positioned to bridge two active site zinc ions replacing the nucleophilic water the thiol group, which was common for the binding of thiol-containing MBL inhibitors with VIM-2 (Fig. 1).15,17,19 And the carbonyl group makes hydrogen-bonding interactions with catalytically important residue Asn233 on the L3 loop. In addition, the phenyl group forms face-to-face π–π interactions with Tyr67 on the L1 loop. Although 13a did not have negatively charged group to interact with the positively charged residue Arg228, it manifested potent MBL inhibition via specific binding with the flexible L1 loop, which provide new insights into inhibitor design of MBL inhibitors.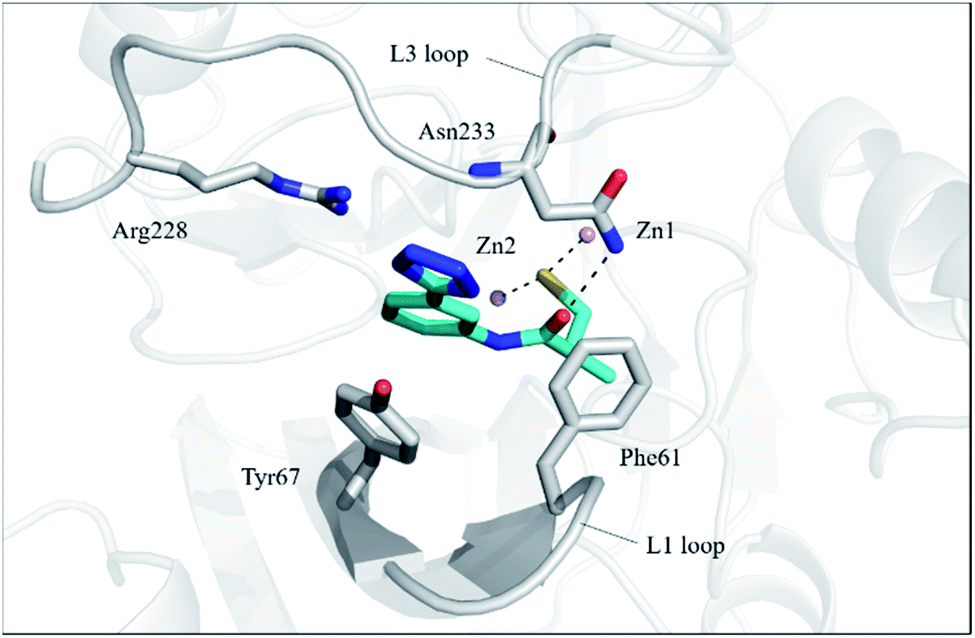 | ||
| Fig. 1 Crystallographic analyses reveal the binding modes of 13a to VIM-2. The active-site zinc ions are shown as pink spheres. The key residues and 13a are shown as sticks. View from crystal structures of VIM-2 in complex with 13a (PDB ID 7CJL) reveal that the inhibitor binds to chelate with active site zinc ions and form interactions with residues on the L1 and L3 loops. | ||
Conclusions
In conclusion, a series of mercaptopropionamide-substituted aryl tetrazole derivatives were synthesized via the C–H activation strategy, which will provide new strategies to readily derive new meta- and ortho- substituted aryl tetrazoles or other related derivatives. The SAR studies of these synthesized derivatives with three representative MBL enzymes led to the identification of 13a as a potent, broad-spectrum MBL inhibitor with IC50 values of 0.044 μM, 0.396 μM and 0.71 μM against VIM-2, NDM-1 and IMP-1, respectively. The crystal structure of VIM-2 in complex with 13a revealed that 13a was positioned to bridge the active site zinc ions via the thiol group and interacted with catalytically important residues on the L1 and L3 loop. Notably, 13a was observed to fit well with the flexible L1 loop, providing new insights for further development of potent MBL inhibitors of clinical relevance.Experimental
Protein expression and purification
VIM-2 (27–266 aa), NDM-1 (1–270 aa) and IMP-1 (19–246 aa) were expressed and purified as previously published procedures.15Inhibition assays
The experiments were carried out by using Tecan microplate reader and flat-bottom 96-well black plates at room temperature. Compounds were dissolved into 100 mM with DMSO. All the MBL enzymes were dissolved in the assay buffer: 20 mM Tris–HCl (pH 7.5), 200 mM NaCl. The compounds (with 10 different concentrations in 3-fold dilution) pe-incubated with appropriate amount of MBL enzymes for 10 min. The reactions were initiated by adding of the substrate FC5. The IC50 values for the compounds were determined by monitoring the fluorescence at λex of 380 nm and λem of 460 nm. All determinations were tested in triplicate. The IC50 values were fitted by using GraphPad Prism5 software.Microdilution broth minimum inhibitory concentrations (MICs) assay
Strains of E. coli DH5a containing plasmids pET28-lacUV5-VIM-2 (E. coli-VIM-2) and pET28-lacUV5 (as control) were used to assess the inhibitors.17 Single colonies of the E. coli-VIM-2 strain on MH agar plates were transferred to 5 mL of MH liquid medium, and grown at 37 °C to an OD630 of ∼0.5. The concentrations of E. coli-VIM-2 and E. coli strains were diluted to ∼106 colony forming units (CFU) per mL in the MH medium, then transferred to the microtiter plates, and treated with meropenem (final concentrations of 128 mg mL−1 to 0.25 mg mL−1 in 2-fold dilution) or/and the inhibitors 13a (final concentrations of 25–200 μM, 2-fold concentration gradient). The microtiter plates were incubated for 16–20 h at 37 °C and visually evaluated for bacterial growth. Each determinate was performed in duplicate.Crystallization and data collection
X-ray structure of VIM-2 in complex with inhibitors 13a was obtained as previously described. Briefly, the mixture of 10 mg mL−1 VIM-2 protein and 5 mM inhibitor was incubated in crystallization buffer (20 mM Tris–HCl, pH 7.5, 200 mM NaCl, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)) for 60 min at 4 °C. The crystallization drops constituted of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of protein-inhibitor mixture:reservoir solutions (25–32% PEG 3350, 0.2 M magnesium formate). The crystals were cryo-protected using a solution of 30% (v/v) glycerol, and flash-cooled in liquid nitrogen. Data were collected at the Shanghai Synchrotron Radiation Facility, and processed using HKL2000 (Otinowski and Minor, 1997). The molecular replacement and refinement were carried out by using Phenix and Wincoot.15
:1 ratio of protein-inhibitor mixture:reservoir solutions (25–32% PEG 3350, 0.2 M magnesium formate). The crystals were cryo-protected using a solution of 30% (v/v) glycerol, and flash-cooled in liquid nitrogen. Data were collected at the Shanghai Synchrotron Radiation Facility, and processed using HKL2000 (Otinowski and Minor, 1997). The molecular replacement and refinement were carried out by using Phenix and Wincoot.15
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation (81874291), Sichuan Science and Technology Program (2018HH0100), and Outstanding Interdiscipline Project of West China Hospital of Sichuan University (grant no: ZYJC18024).Notes and references
- T. P. Van Boeckel, S. Gandra, A. Ashok, Q. Caudron, B. T. Grenfell, S. A. Levin and R. Laxminarayan, Lancet Infect. Dis., 2014, 14, 742–750 CrossRef PubMed.
- K. Garber, Nat. Rev. Drug Discovery, 2015, 14, 445–447 CrossRef CAS PubMed.
- D. Brown, Nat. Rev. Drug Discovery, 2015, 14, 821–832 CrossRef CAS PubMed.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein, P. Huovinen, G. A. Jacoby, R. Kishony, B. N. Kreiswirth, E. Kutter, S. A. Lerner, S. Levy, K. Lewis, O. Lomovskaya, J. H. Miller, S. Mobashery, L. J. V. Piddock, S. Projan, C. M. Thomas, A. Tomasz, P. M. Tulkens, T. R. Walsh, J. D. Watson, J. Witkowski, W. Witte, G. Wright, P. Yeh and H. I. Zgurskaya, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS PubMed.
- K. Bush and G. A. Jacoby, Antimicrob. Agents Chemother., 2010, 54, 969–976 CrossRef CAS PubMed.
- M. W. Crowder, J. Spencer and A. J. Vila, Acc. Chem. Res., 2006, 39, 721–728 CrossRef CAS PubMed.
- M.-N. Lisa, A. R. Palacios, M. Aitha, M. M. González, D. M. Moreno, M. W. Crowder, R. A. Bonomo, J. Spencer, D. L. Tierney, L. I. Llarrull and A. J. Vila, Nat. Commun., 2017, 8, 538 CrossRef PubMed.
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J.-C. Jiménez-Castellanos, M. B. Avison, J. Spencer, C. W. G. Fishwick and C. J. Schofield, Nat. Commun., 2016, 7, 12406–12413 CrossRef CAS PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- K. A. Toussaint and J. C. Gallagher, Ann. Pharmacother., 2015, 49, 86–98 CrossRef PubMed.
- K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS PubMed.
- A. Krajnc, J. Brem, P. Hinchliffe, K. Calvopina, T. D. Panduwawala, P. A. Lang, J. Kamps, J. M. Tyrrell, E. Widlake, B. G. Saward, T. R. Walsh, J. Spencer and C. J. Schofield, J. Med. Chem., 2019, 62, 8544–8556 CrossRef CAS PubMed.
- R. R. Watkins and R. A. Bonomo, Expert Rev. Anti-Infect. Ther., 2013, 11, 543–545 CrossRef CAS PubMed.
- Y. H. Yan, G. Li and G. B. Li, Med. Res. Rev., 2020, 40(5), 1558–1592 CrossRef CAS PubMed.
- Y. L. Wang, S. Liu, Z. J. Yu, Y. Lei, M. Y. Huang, Y. H. Yan, Q. Ma, Y. Zheng, H. Deng, Y. Sun, C. Wu, Y. Yu, Q. Chen, Z. Wang, Y. Wu and G. B. Li, J. Med. Chem., 2019, 62, 7160–7184 CrossRef CAS PubMed.
- Z. J. Yu, S. Liu, S. Zhou, H. Li, F. Yang, L. L. Yang, Y. Wu, L. Guo and G. B. Li, Bioorg. Med. Chem. Lett., 2018, 28, 1037–1042 CrossRef CAS PubMed.
- S. Liu, L. Jing, Z. J. Yu, C. Wu, Y. Zheng, E. Zhang, Q. Chen, Y. Yu, L. Guo, Y. Wu and G. B. Li, Eur. J. Med. Chem., 2018, 145, 649–660 CrossRef CAS PubMed.
- C. Zhang, Y.-c. Pu, Z.-J. Yu, C.-y. Wu, J. Brem, M. A. McDonough, C. J. Schofield, G.-B. Li and Y. Wu, Org. Chem. Front., 2018, 5, 1288–1292 RSC.
- G. B. Li, J. Brem, R. Lesniak, M. I. Abboud, C. T. Lohans, I. J. Clifton, S. Y. Yang, J. C. Jimenez-Castellanos, M. B. Avison, J. Spencer, M. A. McDonough and C. J. Schofield, Chem. Commun., 2017, 53, 5806–5809 RSC.
- G.-B. Li, M. I. Abboud, J. Brem, H. Someya, C. T. Lohans, S.-Y. Yang, J. Spencer, D. W. Wareham, M. A. McDonough and C. J. Schofield, Chem. Sci., 2017, 8, 928–937 RSC.
- J. Chen, T. Huang, X. Gong, Z. J. Yu, Y. Shi, Y. H. Yan, Y. Zheng, X. Liu, G. B. Li and Y. Wu, Adv. Synth. Catal., 2020, 362(14), 2984–2989 CrossRef CAS.
- L. Wang, W. Wu, Q. Chen and M. He, Org. Biomol. Chem., 2014, 12, 7923–7926 RSC.
- C. S. Chang, Y. T. Lin, S. R. Shih, C. C. Lee, Y. C. Lee, C. L. Tai, S. N. Tseng and J. H. Chern, J. Med. Chem., 2005, 48, 3522–3535 CrossRef CAS PubMed.
- S. C. Kohler, S. Vahdati, M. S. Scholz and M. Wiese, Eur. J. Med. Chem., 2018, 146, 483–500 CrossRef PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06405j |
| This journal is © The Royal Society of Chemistry 2020 |