
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali modified P25 with enhanced CO2 adsorption for CO2 photoreduction
Jeannie Z. Y. Tan *a,
Stelios Gavrielidesa,
Hao R. Xub,
Warren A. Thompsona and
M. Mercedes Maroto-Valera
*a,
Stelios Gavrielidesa,
Hao R. Xub,
Warren A. Thompsona and
M. Mercedes Maroto-Valera
aResearch Centre for Carbon Solutions (RCCS), Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: j.tan@hw.ac.uk
bDepartment of Chemical Engineering, Loughborough University, Loughborough, UK
First published on 27th July 2020
Abstract
To improve the CO2 adsorption on the photocatalyst, which is an essential step for CO2 photoreduction, solid solutions were fabricated using a facile calcination treatment at 900 °C. Using various alkalis, namely NaOH, Na2CO3, KOH, K2CO3, the resulted samples presented a much higher CO2 adsorption capacity, which was measured with the pulse injection of CO2 on the temperature programmed desorption workstation, compared to the pristine Evonik P25. As a result, all of the fabricated solid solutions produced higer yield of CO under UV light irradiation due to the increased basicity of the solid solutions even though they possessed only the rutile polymorph of TiO2. The highest CO2 adsorption capacity under UV irradiation was observed in the sample treated with NaOH, which contained the highest amount of isolated hydroxyls, as shown in the FTIR studies.
Introduction
The utilisation of fossil fuels and associated greenhouse effect have raised lots of concerns. Solar energy has the potential to meet our energy demands if it can be efficiently harvested and transformed into fuels. Photocatalytic reduction of CO2 into valuable hydrocarbons, such as CO and CH4, is promising to reduce CO2 emissions by offering a renewable energy alternative.1Photocatalytic reduction of CO2 involves multiple reaction steps before producing the hydrocarbon products.2,3 However, as the most abundant and stable oxidised form of carbon, CO2 is difficult to activate.4 Surface chemistry studies have demonstrated that adsorption of CO2 molecules on the surface of metal oxides is always accompanied by an activation step.5 CO2 in the chemisorption state (mainly carbonate and/or CO2− anion) has a bent O–C–O bond angle and a decreased LUMO, which will favour the charge transfer from photo-excited semiconductors to the surface adsorbed CO2 molecules.6,7
Adsorption of CO2 on the surface of the photocatalyst is known as the first essential step in a CO2 photoreduction reaction. However, the competition between CO2 and H2O (i.e., reducing agent) for the adsorption sites on the surface of the photocatalyst could significantly affect the CO2 photoreduction efficiency. To understand the effects of the strength and state of CO2 adsorption on photocatalytic CO2 reduction, various methods have been proposed to enhance the adsorption of CO2 as well as to enhance the chemical interaction of CO2 with the photocatalyst. One simple way is through the addition of alkalis and alkaline earth metals, such as NaOH and Na2CO3,8 in order to enhance the adsorption capacity9 and activation of CO2 (ref. 5 and 10) without the use of noble metals, such as Pt.5 Nie et al. proposed that the modification of P25 with NaOH to introduce more surface hydroxyl groups that would establish strong hydrogen bonding on TiO2, and subsequently promote the electron transfer between the reactant and TiO2.9 As a result, the photocatalytic conversion of the NaOH modified P25 titania was improved approximately 12 times when compared to the pristine P25 photocatalyst.
A better understanding of the surface hydroxyls of TiO2, especially their role in the photocatalytic process, may provide insight into the photocatalytic mechanism. A previous study proposed that the type of hydroxyl group within TiO2 could significantly influence the photocatalytic reaction due to the difference in acidic–basic strength of the hydroxyl group.11 Liu et al. recently proposed that isolated hydroxyls could act as an effective adsorption site for CO2 as well as enhanced the selectivity for the production of hydrocarbon fuels from CO2 and H2O.12 Using infrared spectroscopy, the bands observed between 3715–3630 and 3675 cm−1 were assigned to the stretching modes of isolated –OH, whereas bands at lower frequencies of 1640–1625 cm−1 were assigned to the bending modes of adsorbed water.13–15 The isolated –OH was categorised into terminal and bridged –OH groups. Some studies also indicated that the preparation method, which produced different size and morphology of particles, had a great effect on the –OH frequencies.16,17 Moreover, UV irradiation would lead to the changes of the concentration and structure of the hydroxyl groups on TiO2 surface.18,19 The behaviour of the surface –OH as source of hydroxyl radicals as well as the adsorption sites for the reactant in the photocatalytic processes has been described.13 However, the relationship between the surface –OH and the capability of CO2 adsorption under light irradiation is not fully understood.
Surface modification with alkalis has shown advantages for CO2 photocatalytic reduction. However, the influence of anionic and cationic moieties of the alkalis on CO2 adsorption capacity and the CO2 photoreduction activity has not been systematically studied. Therefore, this study aims to understand the influence of commonly used alkalis, namely NaOH, Na2CO3, KOH, K2CO3, in modifying commercial TiO2, P25 photocatalyst. The effect of anions and cations on the CO2 adsorption capability and CO2 photoreduction under UV is investigated here using temperature programmed desorption analysis.
Experimental
Materials
Aeroxide® P25 (≥99.5%), referred here to as P25 in short, NaOH (BioXtra, ≥98.0%), Na2CO3 (ACS reagent, ≥99.5%), KOH (BioXtra, ≥85.0%) and K2CO3 (BioXtra, ≥99.0%) were purchased from Sigma Aldrich. All reagents were used without further purification. Milli-Q water was collected from a Millipore academic purification system with resistivity 18.2 MΩ cm.Methods
P25 (0.1 g) was mixed and ground thoroughly with 5 wt% of NaOH, Na2CO3, KOH or K2CO3. The samples were annealed at 900 °C (ramping rate: 10 °C min−1) for 4 h and they were denoted as P25-X, in which X = NO, NC, KO and KC represents the NaOH, Na2CO3, KOH and K2CO3, respectively. For comparison, P25 was also calcined under the same conditions and this sample was denoted as P25-C.Characterisation
Crystallinity and phase identification of the synthesized samples were conducted using powder X-ray diffraction XRD (Bruker D8 Advanced Diffractormeter) equipped with Cu Kα radiation (λ = 1.5418 Å) and compared with the ICDD–JCPDS Powder Diffraction File database. Transient photocurrent response was measured using Autolab PGSTAT 302N electrochemical workstation with a standard three-electrode system, in which fabricated thin film acted as the working electrode (2.5 × 2.5 cm2), Pt wire as counter electrode and Ag/AgCl (KCl 1 M) as reference electrode, and 0.1 M Na2CO3 aqueous solution was used as the electrolyte. Zeta potential was measured using a Zetasizer Nano Z from Malvern Panalytical. Each sample was measured with 4 replicas. The measurement was conducted with the fabricated samples diluted in Milli-Q water (0.167 mg mL−1) at 22 °C. Fourier transform infrared (FTIR) spectroscopy (Frontier from PerkinElmer) was used to obtain the fingerprint of the powder samples. Surface area was measured using Gemini VII Surface Area Analyser (Micromeritics). Temperature Programmed Desorption (TPD, ChemBET Pulsar TPD from Quantachrome Instruments) studies were performed using the pulse injection method to measure the volume of CO2 adsorbed under UV irradiation (OmniCure S2000, 75 mW cm−1, 365 nm) at 25 °C. Prior to the measurement, 0.01 g of sample was filled into a U-shape quartz tube and degassed at 150 °C for 4 h. After cooling down to 25 °C, the quartz tube was filled with CO2 (99.99%) and the measurement was proceeded for 5500 s under UV irradiation. Then, the quartz tube was purged with CO2 for 24 h before the next measurement for another 4500 s. To estimate the amount of CO2 adsorbed, the area under the curve obtained in each injection was compared with the area under the curve obtained when an empty (no sample loaded in the quartz tube) was used in the injection of CO2.Photocatalytic testing
CO2 photoreduction testing of the produced samples was conducted in a customised stainless steel photoreactor with a quartz window.20 0.01 g of sample was distributed as powder on the bottom of the photoreactor. To purge and equilibrate the system, a flow rate of 0.42 mL min−1 CO2 was passed through an aluminium bubbler set at 20 ± 2 °C and charged the photoreactor overnight. The reaction was performed at 24 ± 2 °C. An optical fibre lamp (OmniCure S2000) was used as the light source (75 mW cm−1, 365 nm). The irradiance was measured using a radiometer (OmniCure R2000). The outlet gas was analysed hourly online by a gas chromatography (GC, Agilent, Model 7890 B series) with a Hayesep Q column (1.5 m, 1/16 inch OD, 1 mm ID), molecular sieve 13X (1.2 m, 1/16 inch OD, 1 mm ID), thermal conductivity detector (TCD), nickel catalysed methaniser and flame-ionization detector (FID).The quantum yield (ϕ) was measured under similar photocatalytic reaction conditions using the same UV lamp (75 mW cm−1, 365 nm). The incident flux was determined by a Laboratory Spectroradiometer (Apogee Instruments). The ϕ values of CO evolution for the CO2 photoreduction reaction were calculated according to the following equation:
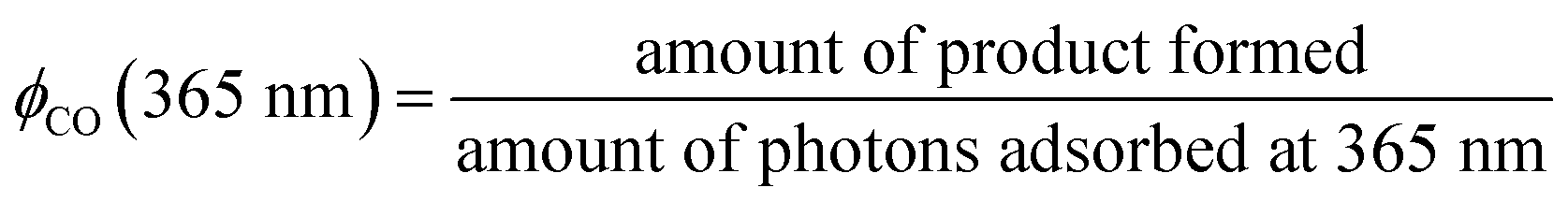
Results and discussion
Four types of alkalis, namely NaOH, Na2CO3, KOH and K2CO3, were used to modify the commercial P25, which has been extensively used as a benchmark photocatalyst.21 To distinguish the effect of high temperature calcination and salt modification, pristine P25 was treated at 900 °C without the addition of salt. The modified P25, and resultant P25-C, P25-NO, P25-NC, P25-KO and P25-KC, revealed 100% rutile crystal phase, whereas pristine P25 had a mixture of anatase and rutile (Fig. 1A). This observation indicated that the phase transformation was primarily due to the calcination treatment.22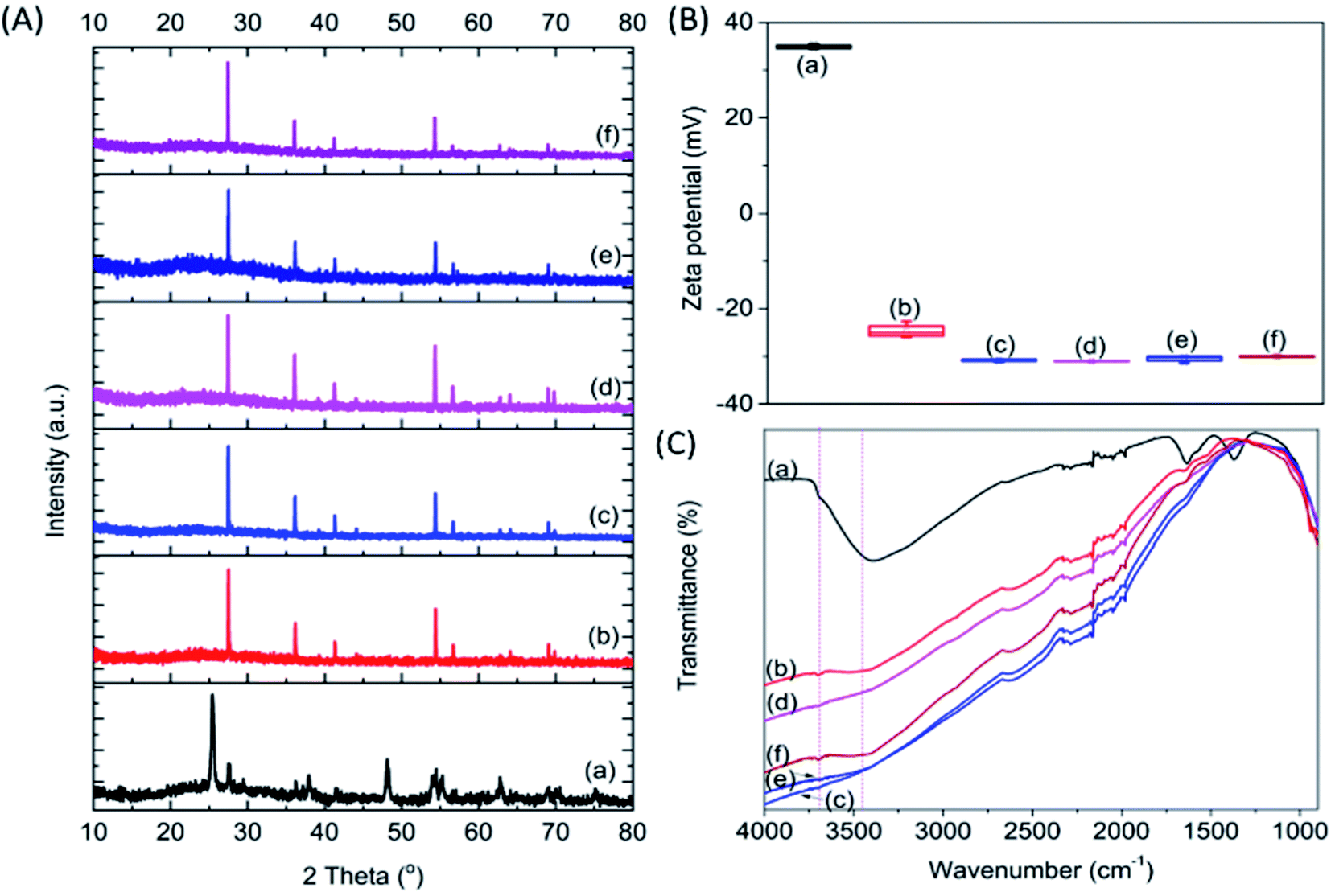 | ||
| Fig. 1 XRD (A), zeta potential (B) and FTIR (C) patterns of P25 (a) P25-C (b) P25-NO (c) P25-NC (d) P25-KO (e) and P25-KC (f). | ||
The surface area of the samples after the calcination treatment with (46.4–48.3 m2 g−1) and without (48.5 m2 g−1) the incorporation of alkali was reduced slightly compared to the pristine P25 (51.7 m2 g−1). However, the calcination treatment reduced the zeta potential value significantly from 35 mV (pristine P25) to −24.5 mV (P25-C). The addition of salts further decreased the zeta potential value to about −32 mV (Fig. 1B). This was due to the increase of the basicity of the materials upon the addition of alkalis. However, the type of salt added did not significantly affect the zeta potential.
Pristine P25 revealed a strong trough centred at 3391 cm−1, which was assigned to the O–H stretching vibration bands, due to water adsorption (Fig. 1C).23 Upon calcination, this trough was weakened and this was also the case of the trough at 1630 cm−1, which was assigned to the bending modes of adsorbed water,24,25 indicating the dehydration of the sample.11 In addition to the decline of troughs, a very broad band emerged between 3600–3800 cm−1 due to the formation of Ti3+–OH and/or Ti4+–OH moieties (i.e., isolated hydroxyl groups) after the addition of alkalis as proposed previously.26
The calcination treatment with or without the incorporation of alkalis with different anions (i.e., –OH− and –CO32−) and cations (i.e., Na+ and K+) changed the basicity, resulting from the formation of different amounts of isolated –OH groups in the modified P25. The amount of isolated –OH formed after calcination within the samples decreased in the order of P25-NO > P25-KO > P25-NC > P25-KC > P25-C (Fig. 1C). Therefore, these observations proposed that hydroxides could induce more isolated –OH groups than carbonates during the alkali treatment.
To investigate the CO2 adsorption capacity of the samples, pulse injection of CO2 under UV light irradiation studies were performed using a TPD instrument (Fig. 2A and B). The pulse injection pattern showed that pristine P25 exhibited the highest signal amongst the samples, indicating the least amount of CO2 injected into the sample. However, all calcined samples exhibited a much lower signal compared to pristine P25, indicating a much higher amount of CO2 adsorbed by those calcined samples. The area under the curve was determined to estimate the volume of CO2 adsorbed (Fig. 2C). The area under the curve reached a plateau after 6 injections, indicating that the samples had been saturated with CO2 after being equilibrated with CO2 for 24 h (purple region in Fig. 2C). Based on the integration of the graphs obtained, the area under the curve increased in the order of P25-NO < P25-KO < P25-NC < P25-KC < P25-C (Fig. 2C). However, the estimated volume of CO2 adsorbed by the samples had an inverse relation to the area under the curve (Table 1). In other words, the capability of CO2 adsorption decreased in the order of P25-NO > P25-KO > P25-NC > P25-KC > P25-C, which revealed similar trend as shown in the FTIR pattern in Fig. 1C. In other words, the amount of isolated –OH present in the samples directly influenced the CO2 adsorption capacity, as previously proposed.26
 | ||
| Fig. 2 Pulse injection of CO2 performed on TPD before (A) and after (B) CO2 equilibrated for 24 h, and the integrated area under the curve (C) for pristine P25 (a) P25-C (b) P25-NO (c) P25-NC (d) P25-KO (e) and P25-KC (f) under UV irradiation. | ||
| Sample | Area under the curve | Estimated CO2 volume adsorbed (mL g−1) |
|---|---|---|
| P25 | 316.4 | 16.3 |
| P25-C | 181.8 | 20.0 |
| P25-NO | 92.1 | 22.4 |
| P25-NC | 106.7 | 21.9 |
| P25-KO | 99.6 | 22.2 |
| P25-KC | 128.9 | 21.4 |
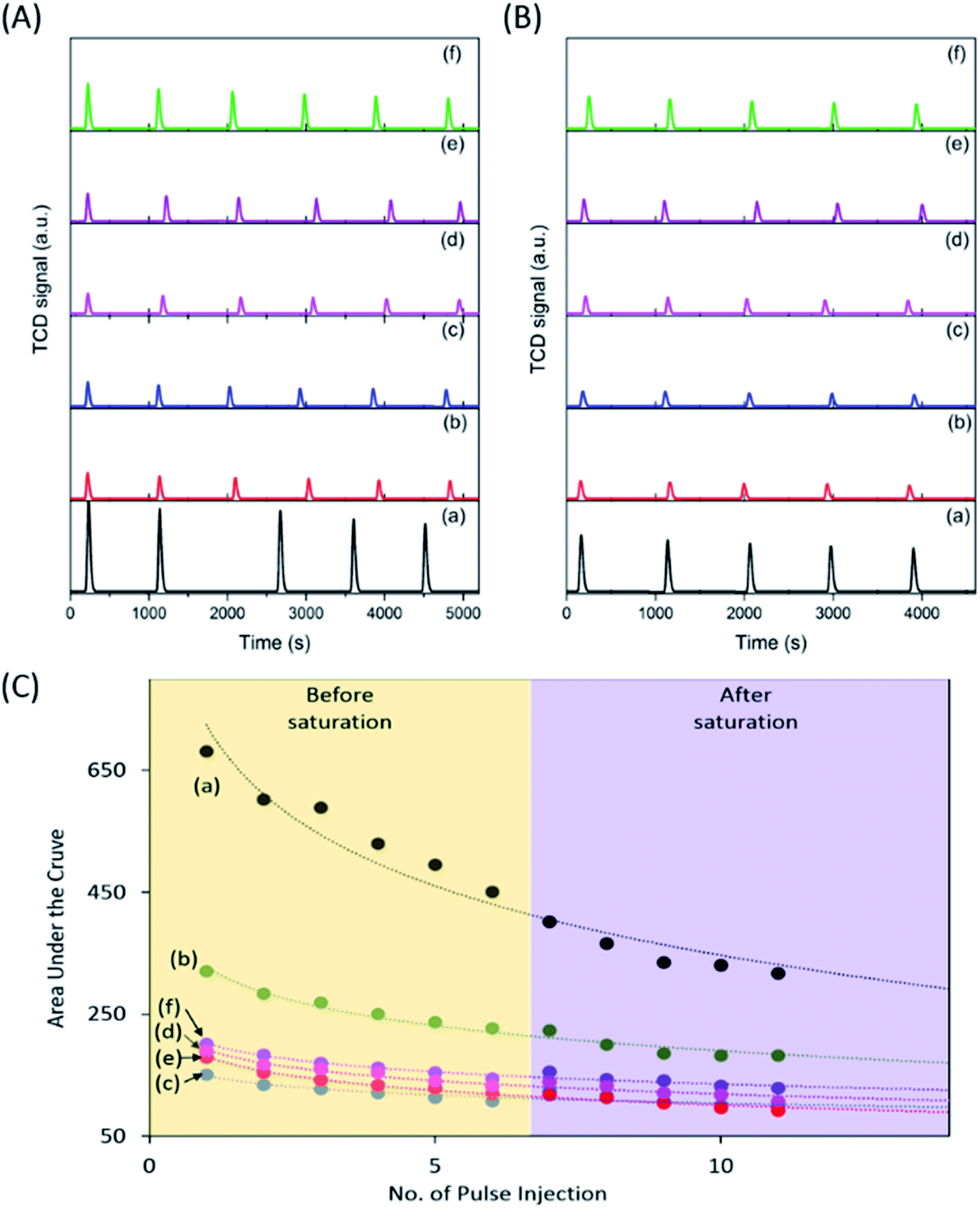
The photocatalytic reduction of CO2 was performed under UV irradiation for 8 h (Fig. 3A). No carbonaceous product was observed when the samples were tested in the dark. The pristine P25 sample did not produce observable products, whereas, the calcined samples produced CO under UV irradiation. The CO production decreased in the order of P25-NO > P25-KO > P25-NC > P25-KC > P25-C. The higher CO production was observed for the P25 treated with hydroxides, namely P25-NO produced 12.3 μmol g−1 h−1 with a ϕCO = 0.0167, followed by P25-KO (10.1 μmol g−1 h−1). Samples treated with carbonates presented lower conversion, e.g. 7.4 and 5.9 μmol g−1 h−1 of CO by P25-NC and P25-KC, respectively.
 | ||
| Fig. 3 CO production (A) and photocurrent (B) obtained from for the pristine P25 (a) P25-C (b) P25-NO (c) P25-NC (d) P25-KO (e) and P25-KC (f) under UV irradiation. | ||
Transient photocurrent response is used to reveal the migration characteristics of photogenerated electrons.27 Overall, the samples treated with alkalis exhibited higher photocurrent than the pristine P25 and P25-C samples (Fig. 3B). Among the modified P25 samples, P25-NO revealed the highest photocurrent, indicating the resulted P25-NO solid solution had enhanced the transportation of the photogenerated electrons, that subsequently improved the photocatalytic conversion of CO2 into CO.27 The superior electronic property of P25-NO was probably due to higher relative amount of Ti3+–OH/Ti4+–OH formation than P25-KO. Hence, P25-NO revealed a much higher photocatalytic activity than P25-KO although they revealed very small difference in their CO2 adsorption capacities (Table 1).
In summary, the sample capable to adsorb the highest amount of CO2 (Fig. 2) exhibited highest isolated –OH concentration, as shown in the FTIR pattern (Fig. 1C). In addition, the modification of P25 with alkalis generally enhanced the transport of photogenerated electrons, as shown in the photocurrent analysis (Fig. 3B). Among the modified samples, P25-NO revealed the highest photocurrent. The synergistic effects of the enhanced CO2 adsorption and the transportation of photogenerated electrons had significantly promoted the photoreduction of CO2 into CO under UV irradiation.
Conclusions
The modification of P25 with different alkalis showed significant CO2 enhancement attributed to the increase of surface basicity as well as isolated hydroxyls. In addition, the fabricated solid solutions could produce more photogenerated electrons and transported effectively to the surface for CO2 photoreduction. As a result, the fabricated solid solutions exhibited much higher CO production from CO2 under UV light irradiation. The sample treated with NaOH exhibited the highest CO2 adsorption capacity (∼22.4 mL g−1) and produced ∼13 μmol gcatalyst−1 h−1 of CO from the CO2 photoreduction reaction with H2O under UV irradiation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support provided by the Engineering and Physical Sciences Research Council (EP/K021796/1) and the Research Centre for Carbon Solutions (RCCS) at Heriot-Watt University.Notes and references
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC
.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 CrossRef PubMed
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef
- J. Guo, K. Wang and X. Wang, Catal. Sci. Tech., 2017, 7, 6013–6025 RSC
- L. Nie, J. Yu, X. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777–2783 CrossRef CAS PubMed
- X. Xiong, X. Zhang and Y. Xu, J. Phys. Chem. C, 2016, 120, 25689–25696 CrossRef CAS
- H. Lin, J. Long, Q. Gu, W. Zhang, R. Ruan, Z. Li and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 9468–9474 RSC
- L. Liu, C. Zhao, J. Xu and Y. Li, Appl. Catal., B, 2015, 179, 489–499 CrossRef CAS
- C. Peng, G. Reid, H. Wang and P. Hu, J. Chem. Phys, 2017, 147, 030901 CrossRef
- L. Liu, C. Zhao and Y. Li, J. Phys. Chem. C, 2012, 116, 7904–7912 CrossRef CAS
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS
- J. Lee, D. C. Sorescu and X. Deng, J. Am. Chem. Soc., 2011, 133, 10066–10069 CrossRef CAS
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS
- H. He, P. Zapol and L. A. Curtiss, J. Phys. Chem. C, 2010, 114, 21474–21481 CrossRef CAS
- W. A. Thompson, C. Perier and M. M. Maroto-Valer, Appl. Catal., B, 2018, 238, 136–146 CrossRef CAS
- B. Ohtani, O. O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS
- G. Ma, Q. Guo, Y. Hou, Z. Huang, X. Han and Y. Fang, J. Nanosci. Nanotechnol., 2014, 14, 7181–7188 CrossRef CAS
- A. León, P. Reuquen, C. Garín, R. Segura, P. Vargas, P. Zapata and P. A. Orihuela, Appl. Sci., 2017, 7, 49 CrossRef
- A. Šurca Vuk, R. Ješe, M. Gaberšček, B. Orel and G. Dražič, Sol. Energy Mater. Sol. Cells, 2006, 90, 452–468 CrossRef
- K. S. Finnie, D. J. Cassidy, J. R. Bartlett and J. L. Woolfrey, Langmuir, 2001, 17, 816 CrossRef CAS
- K. Suriye, R. J. Lobo-Lapidus, G. J. Yeagle, P. Praserthdam, R. D. Britt and B. C. Gates, Chem.–Eur. J., 2008, 14, 1367 CrossRef
- Q. Bi, Y. Gao, C. Dang, Z. Wang and J. Xue, CrystEngComm, 2019, 21, 6744–6757 RSC
| This journal is © The Royal Society of Chemistry 2020 |