
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly imprinted polymeric coatings for sensitive and selective gravimetric detection of artemether†
Usman Arshada,
Adnan Mujahid *a,
Peter Lieberzeitb,
Adeel Afzal*c,
Sadia Zafar Bajwad,
Naseer Iqbalc and
Sumaira Roshana
*a,
Peter Lieberzeitb,
Adeel Afzal*c,
Sadia Zafar Bajwad,
Naseer Iqbalc and
Sumaira Roshana
aInstitute of Chemistry, University of the Punjab, Lahore-54590, Pakistan. E-mail: adnanmujahid.chem@pu.edu.pk
bDepartment of Physical Chemistry, University of Vienna, Waehringer Strasse 42, Vienna, A-1090, Austria
cDepartment of Chemistry, College of Science, University of Hafr Al Batin, PO Box 1803, Hafr Al Batin, 39524, Saudi Arabia. E-mail: aa@aafzal.com
dNational Institute for Biotechnology and Genetic Engineering, Jhang Road, Faisalabad, Pakistan
First published on 16th September 2020
Abstract
Monitoring antimalarial drugs is necessary for clinical assays, human health, and routine quality control practices in pharmaceutical industries. Herein, we present the development of sensor coatings based on molecularly imprinted polymers (MIPs) combined with quartz crystal microbalance (QCM) for sensitive and selective gravimetric detection of an antimalarial drug: artemether. The MIP coatings are synthesized by using artemether as the template in a poly(methacrylic acid-co-ethylene glycol dimethacrylate) matrix. Artemether-MIP and the non-imprinted polymer (NIP) control or reference layers are deposited on 10 MHz dual-electrode QCM by spin coating (187 ± 9 nm layer thickness after optimization). The coatings are characterized by FTIR spectroscopy and atomic force microscopy that reveal marked differences among the MIP and NIP. The MIP-QCM sensor exhibits high sensitivity (0.51 Hz ppm−1) with sub-10 ppm detection and quantification limits. The MIP-QCM sensor also exhibits a 6-fold higher sensitivity compared to the NIP-QCM, and a dynamic working range of 30–100 ppm. The response time of MIP-QCM devices for a single cycle of analyte adsorption, signal saturation, and MIP regeneration is less than 2.5 min. The sensor also demonstrates selectivity factors of artemether-MIP of 2.2 and 4.1 compared to artemisinin and lumefantrine, respectively. Reversibility tests reveal less than 5% variation in sensor responses over three cycles of measurements at each tested concentration. The MIP-QCM showed lower detection limits than conventional HPLC-UV, and faster response time compared to HPLC-UV and liquid chromatography-mass spectrometry (LC-MS).
1. Introduction
Malaria affects almost 214 million people and causes over 435![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths around the world every year.1 It is a serious parasitic infection that poses a developmental challenge to third-world countries. The drugs used for treating malaria vary significantly in their chemical nature and mode of action. For instance, the major classes of antimalarial drugs include 8-aminoquinolines, 4-aminoquinolines, artemisinins, arylamino-alcohols, and antifolates.2 Certain antibiotics have also been used in treating malarial patients. Malaria parasites have developed resistance to traditional antimalarial drugs. Therefore, different combinations of antimalarial drugs belonging to different classes are commonly prescribed instead of a single drug. The World Health Organization (WHO) suggests the artemisinin-based combination therapy (ACT) for uncomplicated malaria caused by Plasmodium falciparum.3 This therapy uses an artemisinin derivative that significantly reduces the gametocyte and biomass of the parasite,4 along with another drug with a different mechanism of action that uproots the remaining parasites.5
000 deaths around the world every year.1 It is a serious parasitic infection that poses a developmental challenge to third-world countries. The drugs used for treating malaria vary significantly in their chemical nature and mode of action. For instance, the major classes of antimalarial drugs include 8-aminoquinolines, 4-aminoquinolines, artemisinins, arylamino-alcohols, and antifolates.2 Certain antibiotics have also been used in treating malarial patients. Malaria parasites have developed resistance to traditional antimalarial drugs. Therefore, different combinations of antimalarial drugs belonging to different classes are commonly prescribed instead of a single drug. The World Health Organization (WHO) suggests the artemisinin-based combination therapy (ACT) for uncomplicated malaria caused by Plasmodium falciparum.3 This therapy uses an artemisinin derivative that significantly reduces the gametocyte and biomass of the parasite,4 along with another drug with a different mechanism of action that uproots the remaining parasites.5
One of the most effective drug combinations used in the ACT is a mixture of artemether and lumefantrine. The usual fixed-dose combination containing 20 mg artemether and 120 mg of lumefantrine per tablet is manufactured by Novartis Pharmaceuticals and sold under the brand name Coartem.6 It is recommended for treating Plasmodium falciparum induced malaria. Lumefantrine has extensive conjugation and high molar absorptivity. Therefore, it is relatively easy to detect and quantify lumefantrine using UV-visible spectrophotometry.7 Artemether, on the other hand, has ether functionality (as shown in Fig. 1), and has an extremely low absorption coefficient in the UV-visible region. It is also present in lesser quantity in the tablet, i.e. artemether and lumefantrine come with a 1:6 ratio in combined formulations. It does not contain any prominent UV-active functional group. Therefore, it is challenging to detect artemether at low concentrations directly via standard optical methods.
 | ||
| Fig. 1 Chemical structures of the antimalarial drugs: artemisinin, artemether (a methyl ether derivative of artemisinin), and lumefantrine. | ||
Several methods have been developed for the determination of artemether in complex mixtures including biological samples. These include high-performance liquid chromatography (HPLC) coupled with a UV detector or mass spectrometer.8,9 For UV detection, one either adds a standard to compensate for the low molar absorbance of the drug10 or derivatizes it. Another approach comprises electrochemical detection combined with HPLC; it addresses the peroxide linkage present in artemether.11 Each of these methods has its limitations, e.g. large instrumentation, tedious sample preparation, and lengthy protocols, which may include derivatization or extraction steps before liquid chromatography. Electrochemical detection requires an inert environment. Quantitative detection of artemether without derivatization or any labeling indicator in drug formulations as well as in complex biological samples is greatly desirable for quality control assays in the pharmaceutical industry and clinical analysis. HPLC has been frequently used for routine quality control analysis of artemether in pharmaceuticals;12,13 however, a sensitive and selective method is required that should be fast, robust, cost-effective, and is capable of label-free sensing of artemether in the presence of potential interferents offering detection limits comparable to conventional methods.
Chemical sensors comprising tailored chemical recognition interface could be a smart choice for label-free detection of a diverse range of analytes.14,15 In this regard, molecular imprinting16,17 represents a well-known method for generating synthetic receptor materials offering adequate affinity for target analytes.18 This technique uses the target analyte as a template structure to generate highly tailored cavities in the polymeric receptors that are the replica of the template and that are capable of selectively binding the target molecules through reversible non-covalent interactions. Molecularly imprinted polymers (MIPs) possess specific rebinding sites that offer suitable geometrical fitting to target analyte.19 Furthermore, the functional groups of these sites offer adequate chemical adaptation. This makes MIPs highly competitive20 for selectively recognizing the target analyte in the presence of structurally similar molecules. Moreover, MIP nanostructures21–23 offer the advantages of higher selectivity and sensitivity due to the presence of a larger number of tailored interaction sites and therefore, considered as efficient sensor interfacial coatings for a wide range of analytes.24–26 Moreover, nanosized MIPs exhibited shorter response time due to faster mass transfer rates.
Quartz crystal microbalances (QCMs) are well known acoustic or mass-sensitive transducers that are widely used for gravimetric sensing of various analytes. They allow for label-free sensing since mass is a fundamental property of any target analyte. Thus, one can use them to sense molecules lacking optically or electrochemically active functionalities quite easily. Furthermore, high sensitivity, miniaturized design, rapid response, and low cost are other benefits of these devices. Combining the selective properties of MIPs with QCM devices27–29 is a promising strategy for highly sensitive label-free sensing. Hence, we herein utilized the approach for sensitive and selective detection of artemether.
2. Experimental
2.1. Materials
The chemicals used for the synthesis of different molecularly imprinted polymers (MIPs) include methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA), and 2,2-azobisisobutyronitrile (AIBN) that were purchased from Merck Chemicals, Germany, whereas methyl methacrylate (MMA), styrene and divinylbenzene (DVB) were obtained from Sigma-Aldrich. Dimethyl sulphoxide (DMSO), dimethylformamide (DMF), tetrahydrofuran (THF), and absolute ethanol were purchased from VWR Chemicals. Artemether, artemisinin, and lumefantrine were provided by Shazoo Pharmaceuticals, Pakistan.2.2. Synthesis of molecularly imprinted polymers (MIPs)
For synthesizing optimized MIP, we used different combinations of monomers and crosslinkers, and different solvents for polymerization. Table S1† summarizes the respective polymerization recipes. Among these combinations, MIPs synthesized by the first method (recipe 1) demonstrated the highest sensor response, therefore, this MIP combination was selected to evaluate the sensor characteristics for molecular recognition of artemether.The optimized procedure was as follows: 15 μL of MAA and 5 mg of artemether were mixed with 600 μL DMSO in an Eppendorf vial and sonicated for 10 minutes. This led to the formation of a pre-formed complex between monomers and template molecules. After sonication, 30 μL of EGDMA and 5 mg AIBN were added to this mixture and subjected to vigorous magnetic stirring. The reaction mixture was heated to 60 °C in a water-bath until a change in viscosity of the pre-polymer was noticed. At this point, we stopped heating and stored the obtained viscous MIP for characterization and sensor fabrication. Synthesis of non-imprinted polymer (NIP) followed the same steps without adding the template (artemether).
2.3. Characterization
FTIR spectroscopy served to follow polymerization and to characterize the respective materials and evaluate the presence of artemether in the MIP. FTIR spectra were recorded on PerkinElmer Spectrum 100 spectrometer in the range of 4000–600 cm−1 using attenuated total reflectance (ATR) mode. A drop of the sample was placed on ATR crystal and was covered from the top to avoid any interference. The surface morphology of MIP and NIP coatings is studied with Shimadzu WET-SPM 9600 atomic force microscopy (AFM) to characterize differences in surface topography and roughness.2.4. Fabrication of sensing devices
MIP and NIP spin-coated onto AT-cut quartz crystal microbalance (QCM) with a dual-electrode geometry having fundamental resonance frequency of 10 MHz. We fabricated QCM by screen printing30 gold paste onto commercially available quartz blanks (14 mm diameter, 168 μm thickness, purchased from Roditi Inc). Different quantities of oligomer solutions were spin-coated for varied revolution speeds and time to form homogenous sensor coatings with controlled thickness. In an optimized procedure, 4 μL of the respective pre-polymer solution was spin-coated on the gold electrode at 2000 rpm for 5 seconds to result in a layer thickness of approximately 200 nm. At least six QCM devices were fabricated; average layer thickness and standard deviation served to validate the reproducibility of fabrication. The MIP coated electrode acts as the working electrode, while the NIP coated electrode acts as a reference electrode or control to quantify non-specific polymer–analyte interactions. During the spin coating process, the alternate electrode was covered with a thin polythene foil to avoid any interference. After fabrication, the MIP-QCM devices were hardened in an oven at 80 °C overnight.2.5. Layer thickness measurements
The precise layer thickness of spin-coated MIP was measured by comparing the frequency of the QCM electrode before and after coating. For this purpose, the damping spectra of the QCM electrodes before and after layer deposition were recorded with a network analyzer. After coating, the resonance frequency of the QCM electrode decreases indicating the deposition of the MIP layer of a certain thickness. The MIP layer coated QCM was washed with deionized water on a magnetic plate for two hours to remove the analyte from the polymer layer. The removal of template molecules from the MIP layer was also monitored by the network analyzer, where an increase in the resonance frequency of QCM upon washing indicates the removal of analyte molecules.2.6. Preparation of analyte solutions
Artemether is hardly soluble in water, only up to 133 ppm. To prepare a stock solution we added 10 mg artemether to 100 mL water and sonicated for 30 minutes to obtain a clear solution. This stock was subsequently used to prepare the standard solutions with 30–100 ppm artemether. For selectivity studies, ethanol was used as solvent because lumefantrine and artemisinin are practically insoluble in water.2.7. Sensor measurements
The MIP- and NIP-coated QCM was placed in a customized flow cell connected to an HP 53131A frequency counter via an oscillator. Firstly, 200 μL distilled water was passed through the cell to get a stable frequency baseline at room temperature. Then, 200 μL of artemether solution of 100 ppm was injected in the cell with the help of a micropipette and the resulting frequency drop was monitored. When the frequency reached a stable value or established equilibrium, 200 μL of distilled water was flushed thrice to completely remove artemether molecules from the MIP surface. As a result, the frequency increased due to the removal of analyte molecules and reached its initial baseline frequency. Similarly, the frequency dropped when the next concentrations of artemether were measured under similar conditions. Due to the dual-electrode design, the response of the NIP coated QCM electrode was simultaneously monitored for different concentrations of artemether.To assess selectivity, we exposed QCM to other related antimalarial drug solutions, such as artemisinin and lumefantrine, respectively, at the same concentrations, i.e. 100–70 ppm. Reproducibility of sensor responses and reusability of the sensor coatings were measured by exposing them to artemether solutions in the concentration range of 100–40 ppm; each measurement was carried out in triplicate.
3. Results and discussion
FT-IR spectra served to access the conversion of monomer mixture into viscous MIP oligomer: Fig. 2 shows the fingerprint region of the FTIR spectra of artemether (template), monomer mixture, and pre-polymer MIP and NIP stocks. They reveal that the monomer mixture contains a band at around 1630 cm−1 (ref. 31) which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C region and thus to the polymerizable double bonds in both monomer (MMA) and crosslinker (EGDMA). After polymerization, this peak disappears in both MIP and NIP batches indicating that the system has indeed (partly) polymerized. As expected, the FTIR spectra of both MIP and NIP batches look similar. However, the characteristic absorptions of artemether at 1035 cm−1 and 1103 cm−1 (corresponding to C–O–O and C–O functional groups, respectively) are only visible in the MIP. This indicates the presence of the template in MIP.
C region and thus to the polymerizable double bonds in both monomer (MMA) and crosslinker (EGDMA). After polymerization, this peak disappears in both MIP and NIP batches indicating that the system has indeed (partly) polymerized. As expected, the FTIR spectra of both MIP and NIP batches look similar. However, the characteristic absorptions of artemether at 1035 cm−1 and 1103 cm−1 (corresponding to C–O–O and C–O functional groups, respectively) are only visible in the MIP. This indicates the presence of the template in MIP.
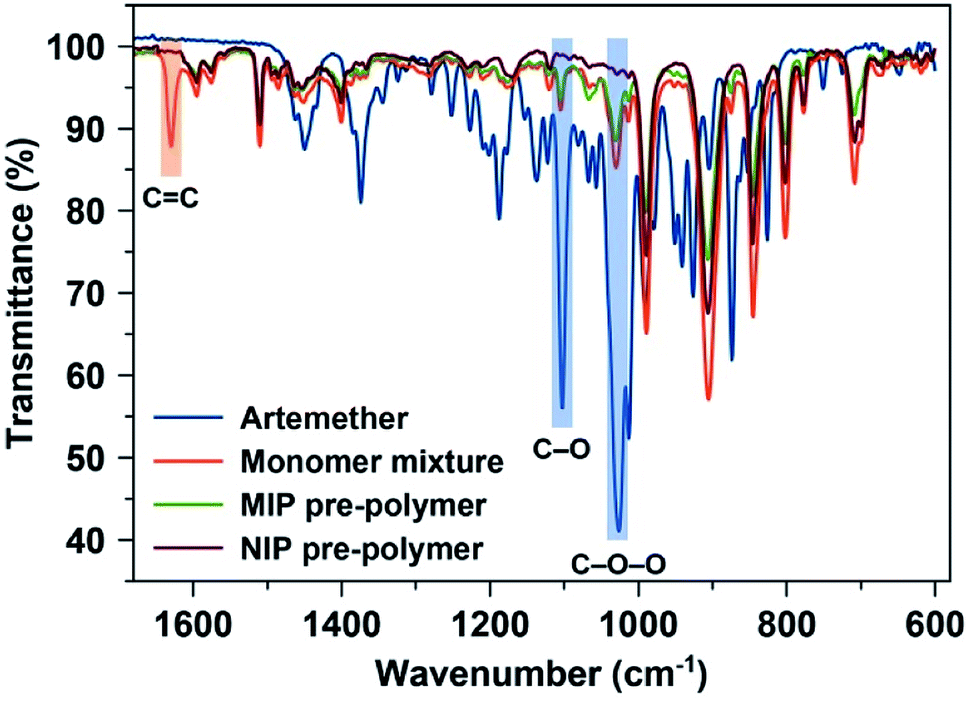 | ||
| Fig. 2 FTIR spectra of artemether (template), monomer mixture before starting polymerization, and pre-polymer MIP and NIP stocks. | ||
Fig. 3 shows the variations in polymer layer thickness as a function of spinning speed (rpm) varying between 1000–2000 rpm. Furthermore, we tested spin coating times from 5–30 seconds. It is evident that spinning speed is the main influence factor for layer thickness: Increasing this parameter decreases polymer layer thickness, while the spin coating time has little to no effect. Based on previous experience with MIP for small molecules, optimal layer heights are in the range of 200 nm. The following spin coating conditions allowed for reaching them: 2000 rpm for 5 seconds led to average layer thicknesses of 187 ± 9 nm.
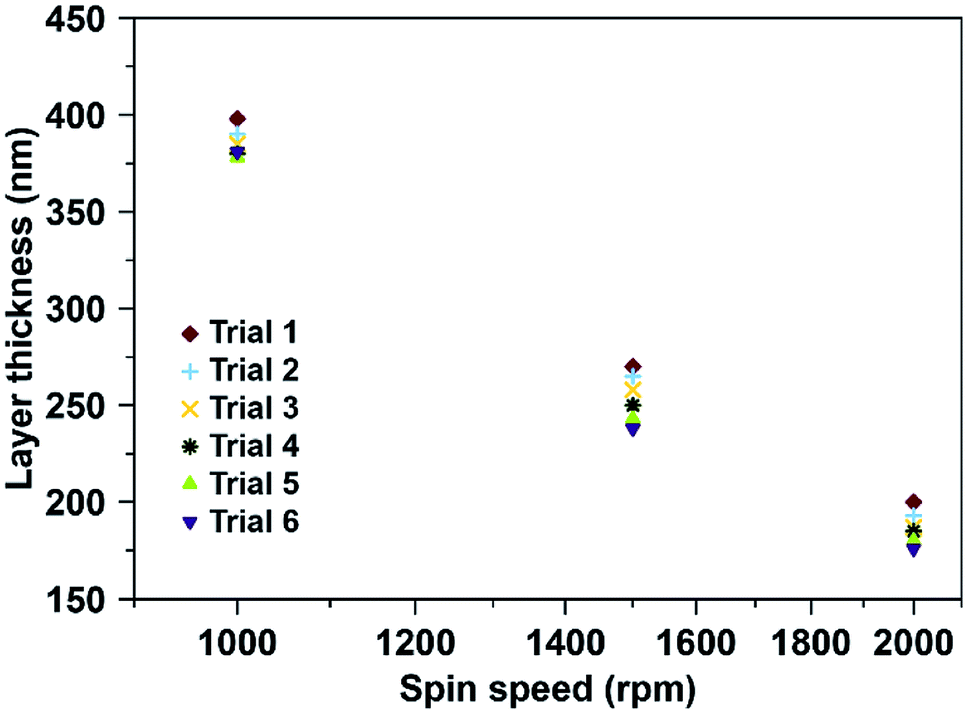 | ||
| Fig. 3 Fabrication of sensor coatings: layer thickness plotted as a function of spin speed in revolutions per minute (rpm); 4 μL of MIP or NIP oligomer solutions per QCM electrode; 5 s at room temperature. | ||
In the next step, it is necessary to demonstrate that one can remove the template from the MIP. Fig. 4 shows this: it contains typical damping spectra of QCM electrodes before and after coating with MIP thin film, and after removing/washing out template molecules from the matrix. Coating the QCM with the polymer decreases the frequency by 4.5 kHz and slightly increases damping (from −4.2 dB to −5.5 dB). After washing with water, the frequency again increases by 0.75 kHz. As previously shown, the relation between frequency shift and layer height is 1 kHz ≅ 40 nm.32 Hence, the layer in Fig. 4 is about 150 nm thick. The damping values and the shape of damping spectra suggest that the layers do not substantially impede QCM resonance. It also indicates that the MIP coating is rigidly bound to QCM, which ensures stable and repeatable sensor measurements as well as reduced noise and improved detection limits.
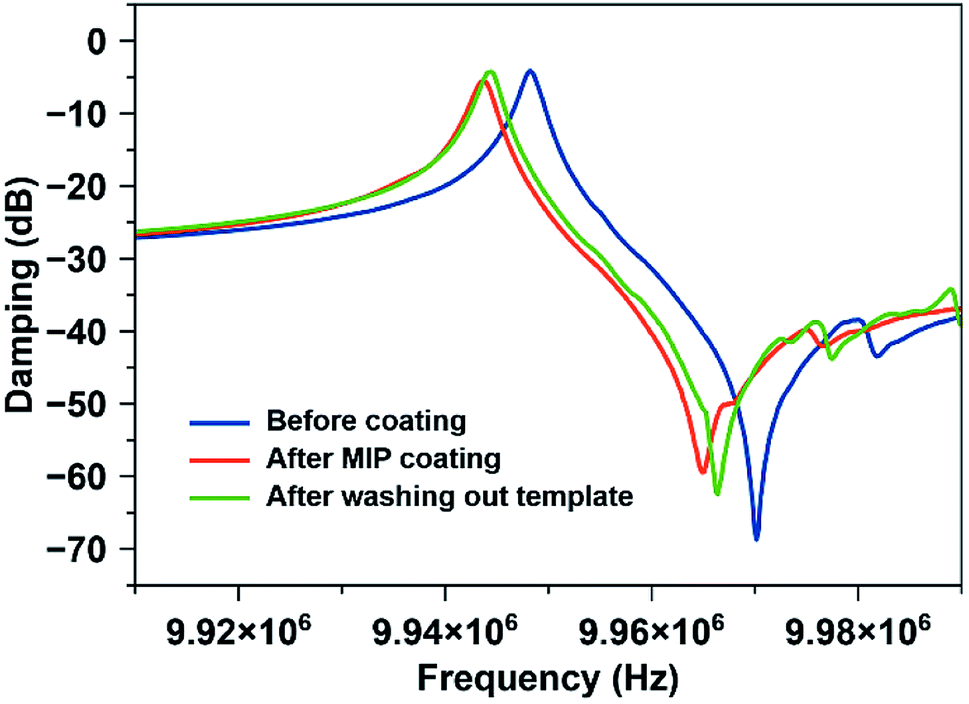 | ||
| Fig. 4 Damping spectra of the QCM electrode before and after coating with MIP, respectively, and after removing the template. | ||
Fig. 5 exhibits surface morphology, topography, and depth profiles for AFM images of MIP and NIP coatings, respectively. Both coatings exhibit homogenous surfaces. The MIP surface shows the root mean square roughness (Rrms) of 1.202 nm and an average surface height (hav) of 8.865 nm. In contrast, the NIP surface has significantly higher Rrms and hav that is 2.492 nm and 45.798 nm, respectively. The low Rrms of MIP than NIP33 could be attributed to the denser MIP structures developed due to the interactions between monomer and template molecules. The homogeneity of both surfaces (as indicated by the surface profiles) signifies the formation of uniform coatings on the QCM electrodes.
 | ||
| Fig. 5 (top) 3-Dimensional AFM surface topography, the x-axis and y-axis scales for both MIP and NIP are 10 × 10 μm whereas the z-axis values for MIP and NIP are 15 nm and 70 nm respectively. (middle) 2-Dimensional AFM images, and (bottom) surface profiles of MIP and NIP coatings fabricated on the surface of the QCM electrodes. | ||
Fig. 6(a) shows the shift in frequency of MIP and NIP sensors on exposure to different artemether solutions (c = 30–100 ppm) as a function of time. On exposing 100 ppm solution, the drop in the frequency of the MIP-QCM electrode is about 53 Hz, whereas at the same time NIP-QCM electrode shows a frequency shift of about 18 Hz. The higher frequency shift of the MIP-QCM sensor indicates the presence of tailored interaction sites for artemether recognition. It is proposed that during molecular imprinting, artemether interacts with MAA (the monomer) through non-covalent interactions (hydrogen bonds) and develops a pre-polymer complex. MAA is a typical H-bond donor and therefore, the crosslinked polymer network can provide hydrogen atoms to the template molecule, i.e. artemether, which has multiple electron-pair donating oxygen atoms, i.e. peroxide and ether groups in artemether have the probability to act as H-bond acceptors34 and thus, they could develop hydrogen bonding interactions15 with the MIP, as shown in Fig. S1.† Whereas the NIP-QCM sensor lacks such interaction centers and thus, exhibits only non-specific bindings and lower sensor response. After washing, the sensor signals recover to the baseline value, which demonstrates the reversible (non-covalent) nature of sensor–analyte interactions. One can observe a similar trend for all other artemether solutions.
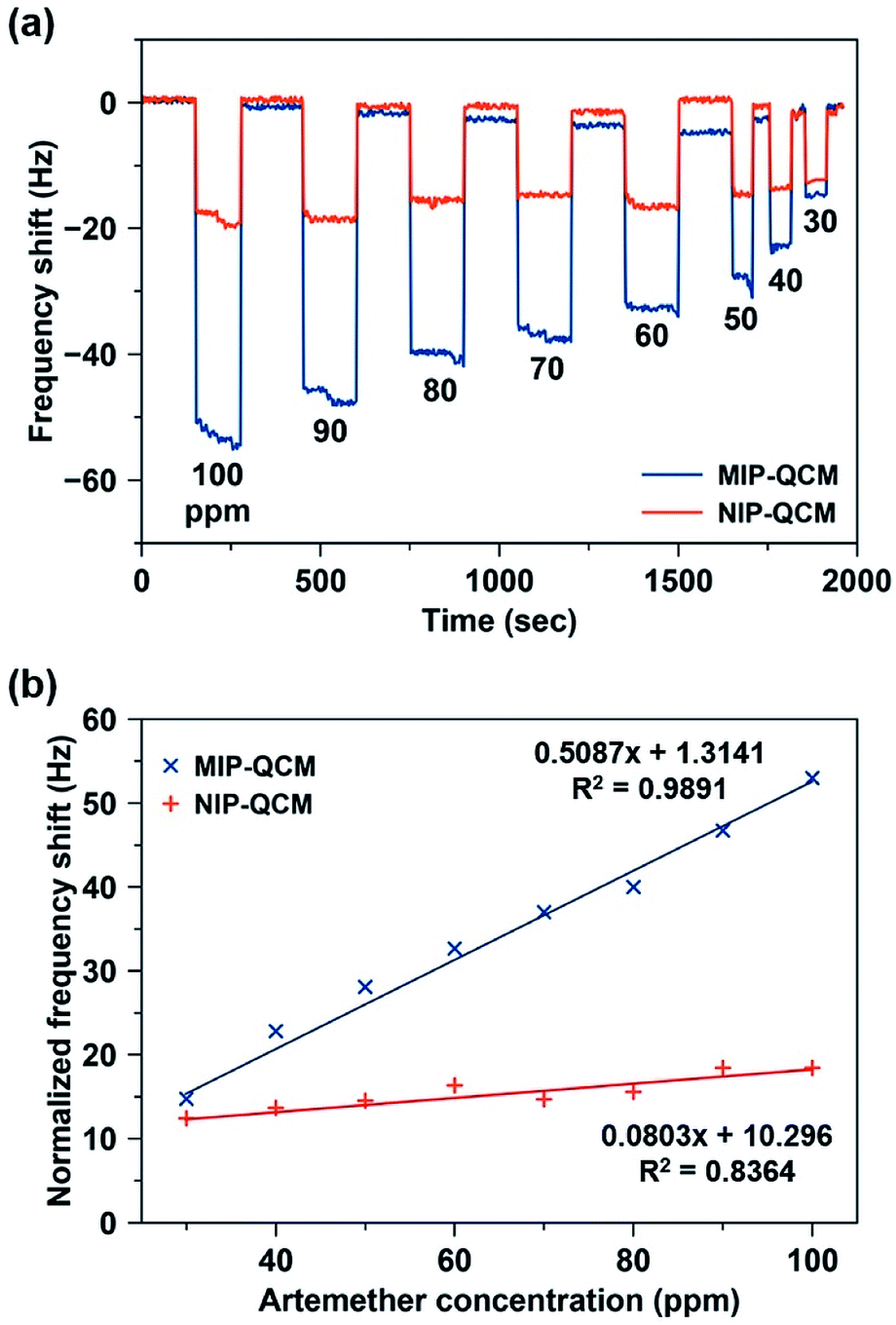 | ||
| Fig. 6 (a) Sensor response of MIP and NIP coatings on exposure to standard artemether solutions in the concentration range of 30–100 ppm. (b) Sensor characteristics of MIP and NIP sensors, respectively, where slopes represent their respective sensitivities toward artemether (in Hz ppm−1). | ||
Fig. 6(b) shows the sensor characteristics for the average responses of MIP- and NIP-coated QCM sensors, respectively, in the concentration range of 30–100 ppm. The MIP-QCM sensor exhibits a linear response in the concentration range 30–100 ppm with a slope of 0.51 Hz ppm−1, which is substantially higher than the NIP-QCM sensor response with a sensitivity of 0.08 Hz ppm−1. Thus, the MIP-QCM sensor exhibits approximately 6.3-fold higher sensitivity for artemether compared to the NIP-QCM. This remarkable sensitivity of the MIP-QCM sensor is due to imprinting effects whereas the NIP-QCM sensor lacks such affinity.
The limit of detection (LOD) was calculated by the linear regression method,35 i.e. LOD = 3.3 × (standard deviation)/slope. The standard deviation of the MIP-QCM sensor is 0.2567, whereas the slope of the MIP calibration curve was 0.51 Hz ppm−1. The calculated LOD of the MIP-QCM sensor is 1.67 ppm, which is considerably low. The limit of quantification (LOQ) was also calculated by a similar method,35 i.e. LOQ = 10 × (standard deviation)/slope, which comes out to be 5.05 ppm. These values suggest that the MIP-QCM sensor can be applied for the detection of low (sub-10 ppm level) concentrations of artemether.
Since artemisinin has close structural resemblance with artemether (as shown in Fig. 1), it is an interesting candidate for determining the selectivity of the MIP sensors. On the other hand, artemether and lumefantrine are used in binary pharmaceutical combinations to treat malaria. Therefore, it is important to assess the sensor responses for lumefantrine as well. Fig. 7(a) shows the response of the MIP-QCM and NIP-QCM sensors toward artemether, artemisinin, and lumefantrine, respectively. At 100 ppm, the MIP-QCM sensor shows a frequency shift of ∼60 Hz for artemether, whereas the same concentration of artemisinin results in 30 Hz only that is about half of the response toward artemether. Furthermore, lumefantrine at 100 ppm yields even lower frequency response, i.e. 15 Hz, that is about four times less. The higher response of artemisinin than lumefantrine is due to the close resemblance of artemisinin structure with artemether. The selectivity measurements suggest that the MIP-QCM sensor possesses highly tailored recognition cavities generated by template (artemether) imprinting, which can better accommodate artemether than artemisinin and lumefantrine. For other tested concentrations of artemether, artemisinin, and lumefantrine, a similar trend was observed. It is worth mentioning that the NIP-QCM responses for artemether and artemisinin are very similar, which in a way reflects their similar structures. Furthermore, it makes the imprinting effect even more remarkable. On the other hand, the lumefantrine signals are lower for both MIP- and NIP-QCM that proves the extent to which these coatings are optimized. In general, the response of the NIP-QCM sensor was only due to non-specific binding interactions and therefore, it was much less than the MIP-QCM sensor. It is due to non-specific binding at the NIP surface that the NIP-QCM sensor exhibits similar frequency shifts for artemether and artemisinin.
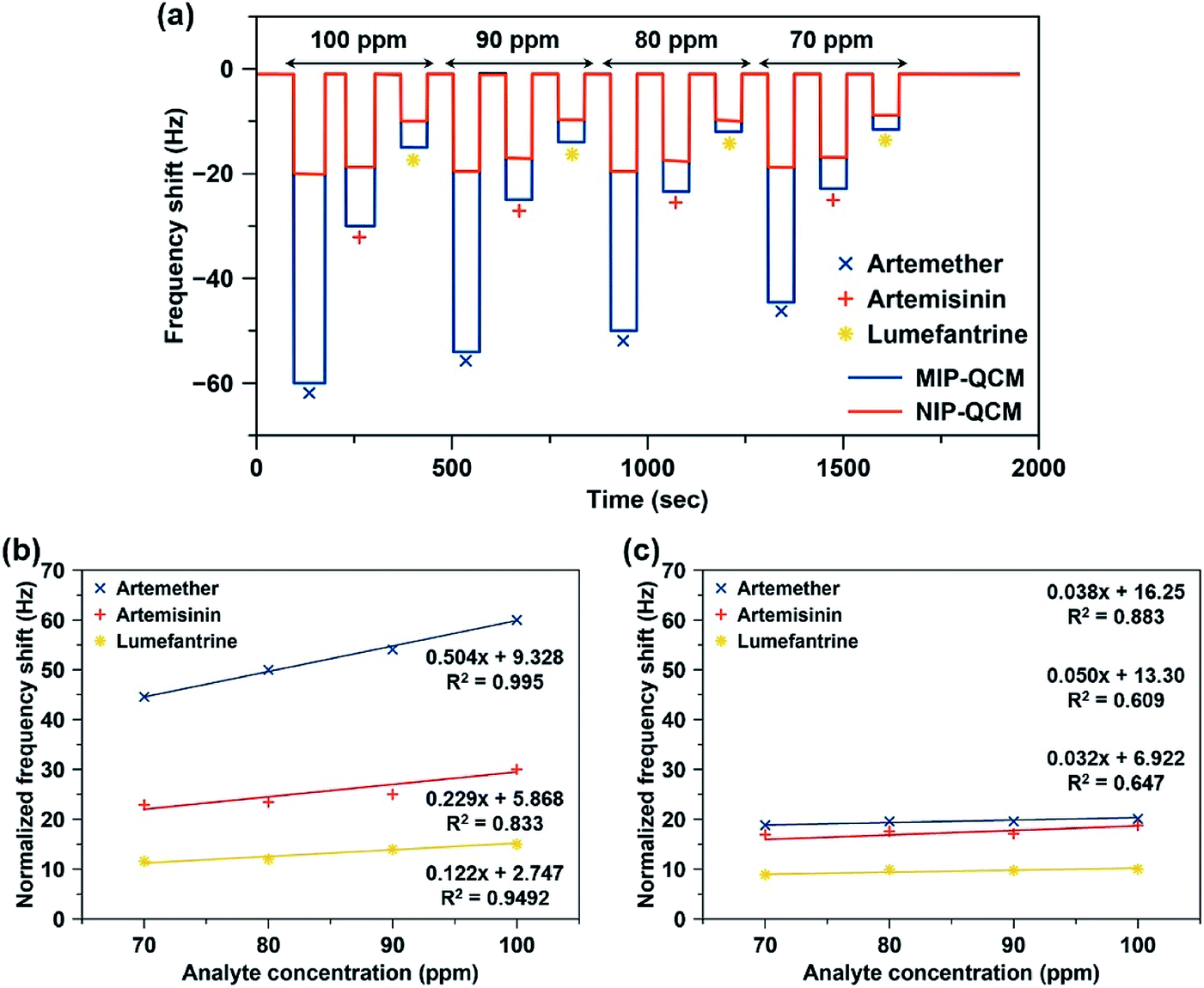 | ||
| Fig. 7 (a) Frequency shifts of the MIP-QCM and NIP-QCM sensors toward artemether, artemisinin, and lumefantrine in the concentration range of 70–100 ppm. (b and c) The calibration curves showing a comparison of relative slopes (sensitivities) and coefficient of correlation (R2) of (b) MIP-QCM, and (c) NIP-QCM sensors toward artemether, artemisinin, and lumefantrine. | ||
Fig. 7(b and c) shows the sensor characteristics resulting from selectivity measurements, i.e. relative slopes and coefficients of correlation (R2). Evidently, MIP-QCM sensors respond selectively to artemether (0.504 Hz ppm−1) compared to artemisinin (0.229 Hz ppm−1) and lumefantrine (0.122 Hz ppm−1). Moreover, their respective R2 values for artemether, artemisinin, and lumefantrine are 0.995, 0.833, and 0.949, respectively. QCM-MIP sensor hence demonstrates 2.2-fold and 4.1-fold higher sensitivities toward artemether compared to artemisinin and lumefantrine, respectively. In contrast, NIP-QCM sensors show much smaller sensitivities toward these analytes, as shown in Fig. 7(c). The NIP-QCM sensor reveals similar sensitivity in the range of 0.032–0.050 Hz ppm−1 for all three drugs indicating the lack of selectivity. This demonstrates that molecular imprinting leads to practicable, selective sensor matrices for artemether.
Fig. 8 shows the frequency shifts of the MIP-QCM and NIP-QCM sensors on multiple exposures to different concentrations of artemether. One can see that the frequency shifts of the MIP-QCM sensor are highly consistent and reproducible: variations between successive measurements are less than 5% at each concentration. The sensor response of the MIP-QCM is much higher than NIP-QCM at all concentrations due to the molecular imprinting effect, as already explained. Furthermore, non-covalent interactions between the MIP-QCM sensor and analyte ensure the complete reversibility of the sensor signal. The response of the NIP-QCM sensor is almost the same at all concentrations of artemether indicating that NIP coating is an effective control for subtracting the non-specific interactions. The high reproducibility on successive measurements of the MIP-QCM sensor suggests that the developed sensor coatings remain stable for repeated cycles of analyte adsorption, signal saturation, and sensor regeneration and demonstrate reliable sensor performance at low concentrations of artemether. Moreover, the inter-day stability of the designed QCM sensor is also tested after a one-week time. The sensor response of MIP and NIP coated QCM is measured against 100 ppm artemether solution and compared to one-week-old QCM sensor measurements made under exactly similar conditions. The results, as shown in Fig. S2,† suggest that sensor response by both MIP and NIP layer remains almost the same with variations less than 5% after a one-week time which signifies the inter-day stability of sensor coatings.
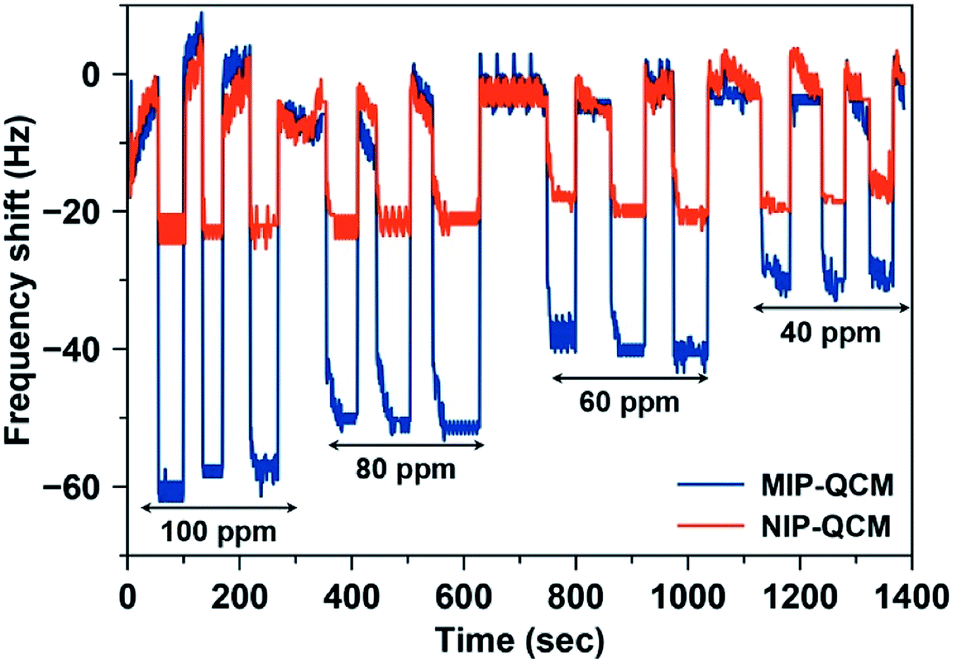 | ||
| Fig. 8 Reproducibility of the MIP-QCM and NIP-QCM sensors on exposure to the standard artemether solutions with 40–100 ppm concentration. | ||
There are several studies related to detecting artemether in complex mixtures, such as high-performance liquid chromatography (HPLC) coupled with ultraviolet (UV) and mass spectrometry (MS) detectors. However, there are no reports on the chemical sensing of artemether. Hence, Table 1 compares the sensing characteristics of MIP-QCM with well-established analytical methods. It demonstrates that the MIP-QCM sensor allows for short analysis time and leads to sensitivity in the range of HPLC-UV. However, LC-MS methods offer a wider dynamic range and lower detection limit for artemether. Nevertheless, the MIP-QCM sensor is an easy-to-use, miniaturized device that offers high sensitivity, selectivity, and low-cost detection of antimalarial drugs. It also reveals rapid response and short analysis time and therefore is inherently suitable for routine quality control, clinical assays, and other applications.
| Technique | Analysis time (min) | Concentration range (ppm) | Regression coefficient, R2 | LOD (ppm) | LOQ (ppm) | Ref. |
|---|---|---|---|---|---|---|
| a HPLC: high-performance liquid chromatography; UV: ultraviolet; ESI: electrospray ionization; LC-MS: liquid chromatography-mass spectrometry; LPME: liquid-phase microextraction. | ||||||
| HPLC/UV | 5 (run time) | 375–425 | 0.9984 | 5 | 15 | 7 |
| Derivative spectroscopy/UV | 30 (reaction time) | 5–30 | 0.9990 | 0.349 | 1.057 | 36 |
| HPLC/UV | 15 (run time) | 20–200 | 0.994 | 7.202 | 21.831 | 8 |
| HPLC/ESI-MS | 9 (run time) | 0.01–1 | 0.998 | — | 0.01 | 9 |
| HPLC/UV | 7 (run time) | 0.4–2.8 | 0.998 | 0.1 | 0.6 | 37 |
| HPLC/UV | 8 (run time) | 32–192 | 0.999 | 0.6 | 1.95 | 38 |
| Microemulsion electrokinetic chromatography | 6 (run time) | 600–1400 | 0.998 | 164 | 548 | 39 |
| LC/MS | 7.5 (run time) | 0.002–0.5 | — | — | 0.002 | 40 |
| LPME/LC-MS | 5 (run time) | 0.005–1 | 0.9931 | — | 0.005 | 41 |
| MIP-QCM sensor | 2.5 (one cycle: adsorption–saturation–regeneration) | 30–100 | 0.9891 | 1.6 | 5.04 | This work |
4. Conclusions
In this study, a facile molecular imprinting approach is used to develop a miniaturized, dual-electrode QCM sensor setup for rapid and highly sensitive label-free detection of artemether. The procedures for the synthesis of coating materials (MIP) and fabrication of (MIP-QCM) sensors are optimized to control and tailor microstructure, thickness, and sensor response leading to enhanced molecular recognition of artemether. Due to the imprinting effect, the MIP-QCM sensor shows high sensitivity and selectivity for artemether compared to other structurally similar and interfering analytes, e.g. artemisinin and lumefantrine. Furthermore, the imprinted sensor demonstrates appreciable reversibility, reproducibility, and repeatability of the sensor response, which is favorable for several sensor measurements without losing recognition features. Compared to advanced analytical techniques such as HPLC and LC-MS, the MIP-QCM sensor is a cost-effective and easy-to-use device with shorter response time. These properties show its potential for rapid, sensitive, and selective analysis of antimalarial drugs in complex mixtures with adequate precision. Thus, the drug-MIP-QCM approach can be extended to other drugs and to monitor the concentration of various drugs in biofluids.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. A., A. M. and N. I. extend their appreciation to the Deanship of Scientific Research, University of Hafr Al Batin for funding this work through the research group project number G-107-2020. U. A. thanks the Higher Education Commission (Pakistan) for funding his stay at the University of Vienna (Austria) under the International Research Support Initiative Program scholarship. U. A. and A. M. also thank Prof. Peter A. Lieberzeit for providing the lab space, resources, and guidance to carry out this work.References
- W. H. Organization, Guidelines for the treatment of malaria, World Health Organization, 2015 Search PubMed.
- M. Schlitzer, Arch. Pharm., 2008, 341, 149–163 CrossRef CAS.
- G. G. Rivelli, L. B. M. Ricoy, I. C. César, C. Fernandes and G. A. Pianetti, J. Pharm. Biomed. Anal., 2018, 155, 262–269 CrossRef CAS.
- J. Golenser, J. H. Waknine, M. Krugliak, N. H. Hunt and G. E. Grau, Int. J. Parasitol., 2006, 36, 1427–1441 CrossRef CAS.
- M. Delves, D. Plouffe, C. Scheurer, S. Meister, S. Wittlin, E. A. Winzeler, R. E. Sinden and D. Leroy, PLoS Med., 2012, 9(2), e1001169 CrossRef.
- L. A. Resende, P. H. R. da Silva and C. Fernandes, J. Pharm. Biomed. Anal., 2019, 165, 304–314 CrossRef CAS.
- I. da Costa César, F. H. A. Nogueira and G. A. Pianetti, J. Pharm. Biomed. Anal., 2008, 48, 951–954 CrossRef.
- V. Y. Jadhav and K. Singh, J. Pharmacogn. Phytochem., 2018, 7(6), 75–78 CrossRef CAS.
- I. C. César, J. A. de Aquino Ribeiro, L. de Souza Teixeira, K. B. Bellorio, F. C. de Abreu, J. M. Moreira, P. R. Chellini and G. A. Pianetti, J. Pharm. Biomed. Anal., 2011, 54, 114–120 CrossRef.
- P. Debrah, H. Nettey, K. K. Miltersen, P. Ayeh-Kumi, B. Brock, J. A. Sarkodie, I. Akwo-Kretchy, P. Owusu-Danso, S. Adjei and E. Petersen, Am. J. Trop. Med. Hyg., 2016, 95, 158–163 CrossRef CAS.
- S. Mohamed, S. Khalid, S. Ward, T. Wan, H. Tang, M. Zheng, R. Haynes and G. Edwards, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 731, 251–260 CrossRef CAS.
- I. d. C. César and G. A. Pianetti, Braz. J. Pharm. Sci., 2009, 45, 737–742 CrossRef.
- J. Prah, E. O. Ameyaw, R. Afoakwah, P. Fiawoyife, E. Oppong-Danquah and J. N. Boampong, J. Trop. Med., 2016, 2016, 8602619 CrossRef.
- W. Zhao, B. Li, S. Xu, X. Huang, J. Luo, Y. Zhu and X. Liu, J. Mater. Chem. B, 2019, 7, 2311–2319 RSC.
- H. Bai, C. Wang, J. Chen, J. Peng and Q. Cao, Biosens. Bioelectron., 2015, 64, 352–358 CrossRef CAS.
- W. J. Cheong, F. Ali, J. H. Choi, J. O. Lee and K. Y. Sung, Talanta, 2013, 106, 45–59 CrossRef CAS.
- L. Figueiredo, G. Erny, L. Santos and A. Alves, Talanta, 2016, 146, 754–765 CrossRef CAS.
- M. R. Younis, S. Z. Bajwa, P. A. Lieberzeit, W. S. Khan, A. Mujahid, A. Ihsan and A. Rehman, J. Sep. Sci., 2016, 39, 793–798 CrossRef CAS.
- A. Afzal, S. Feroz, N. Iqbal, A. Mujahid and A. Rehman, Sens. Actuators, B, 2016, 231, 431–439 CrossRef CAS.
- A. Mujahid, P. A. Lieberzeit and F. L. Dickert, Materials, 2010, 3, 2196–2217 CrossRef CAS.
- H. R. Rajabi, A. Zarezadeh and G. Karimipour, RSC Adv., 2017, 7, 14923–14931 RSC.
- H. R. Rajabi and A. Zarezadeh, J. Mater. Sci.: Mater. Electron., 2016, 27, 10911–10920 CrossRef CAS.
- A. Zarezadeh, H. R. Rajabi, O. Sheydaei and H. Khajehsharifi, Mater. Sci. Eng., C, 2019, 94, 879–885 CrossRef CAS.
- H. R. Rajabi, M. Roushani and M. Shamsipur, J. Electroanal. Chem., 2013, 693, 16–22 CrossRef CAS.
- O. Sheydaei, H. Khajehsharifi and H. R. Rajabi, Sens. Actuators, B, 2020, 309, 127559 CrossRef CAS.
- M. B. Gholivand, M. Shamsipur, S. Dehdashtian and H. R. Rajabi, Mater. Sci. Eng., C, 2014, 36, 102–107 CrossRef CAS.
- K. Kotova, M. Hussain, G. Mustafa and P. A. Lieberzeit, Sens. Actuators, B, 2013, 189, 199–202 CrossRef CAS.
- M. Zeilinger, H. Sussitz, W. Cuypers, C. Jungmann and P. Lieberzeit, Sensors, 2019, 19, 2366 CrossRef CAS.
- R. Suedee, W. Intakong and F. L. Dickert, Talanta, 2006, 70, 194–201 CrossRef CAS.
- U. Latif, S. Can, O. Hayden, P. Grillberger and F. L. Dickert, Sens. Actuators, B, 2013, 176, 825–830 CrossRef CAS.
- N. V. Gupta and H. Shivakumar, Iran. J. Pharm. Res., 2012, 11, 481 CAS.
- P. A. Lieberzeit, A. Afzal, A. Rehman and F. L. Dickert, Sens. Actuators, B, 2007, 127, 132–136 CrossRef CAS.
- C.-Y. Huang, T.-C. Tsai, J. L. Thomas, M.-H. Lee, B.-D. Liu and H.-Y. Lin, Biosens. Bioelectron., 2009, 24, 2611–2617 CrossRef CAS.
- X.-Y. Gong and X.-J. Cao, J. Biotechnol., 2011, 153, 8–14 CrossRef CAS.
- A. Shrivastava and V. B. Gupta, Chron. Young Sci., 2011, 2, 21 CrossRef.
- J. Christian, P. Shah, M. Patel, K. Patel and T. Gandhi, Journal of Taibah University for Science, 2017, 11, 729–740 CrossRef.
- S. Sandhya, P. Shijikumar and S. Meena, Indo Am. J. Pharm. Res., 2015, 5, 720–726 CAS.
- R. Arun and A. A. Smith, Int. J. Pharm. Biomed. Res., 2011, 2, 201–205 CAS.
- N. C. C. Amin, H. Fabre, M.-D. Blanchin, J. Montels and M. Aké, Malar. J., 2013, 12, 202 CrossRef CAS.
- L. Wiesner, K. Govender, S. A. Meredith, J. Norman and P. J. Smith, J. Pharm. Biomed. Anal., 2011, 55, 373–378 CrossRef CAS.
- I. R. Magalhães, V. A. Jabor, A. M. Faria, C. H. Collins, I. C. Jardim and P. S. Bonato, Talanta, 2010, 81, 941–947 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04785f |
| This journal is © The Royal Society of Chemistry 2020 |