
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly synthesis of fused pyrano[2,3-b]pyrans via ammonium acetate-mediated formal oxa-[3 + 3]cycloaddition of 4H-chromene-3-carbaldehydes and cyclic 1,3-dicarbonyl compounds†
Vitaly A. Osyanin*a,
Dmitry V. Osipov a,
Irina A. Semenovaa,
Kirill S. Korzhenkoa,
A. V. Lukashenkoa,
Oleg P. Demidovb and
Yuri N. Klimochkina
a,
Irina A. Semenovaa,
Kirill S. Korzhenkoa,
A. V. Lukashenkoa,
Oleg P. Demidovb and
Yuri N. Klimochkina
aDepartment of Organic Chemistry, Chemical Technological Faculty, Samara State Technical University, 244 Molodogvardeyskaya St., Samara 443100, Russia. E-mail: VOsyanin@mail.ru
bDepartment of Chemistry, North Caucasus Federal University, 1 Pushkin St., Stavropol 355009, Russia
First published on 16th September 2020
Abstract
Various substituted polycyclic pyrano[2,3-b]pyrans were synthesized via the condensation of 4H-chromene-3-carbaldehydes and their areno-condensed analogues with hetero- and carbocyclic 1,3-dicarbonyl compounds in acetic acid. Ammonium acetate was used as a green catalyst for the reaction. The process also involves the subsequent Knoevenagel condensation and 6π-electrocyclization of the 1-oxatriene intermediates formed. Fused pyridines were isolated as the products of the conjugated addition of ammonia to 1-oxatriene intermediates while using carbocyclic 1,3-dicarbonyl compounds and increasing the reaction time, indicating the reversibility of the electrocyclization stage. The calculated values of the Gibbs free energies and reaction rate constants for the 1-oxatriene – 2H-pyran equilibrium also testified to the irreversibility of pyrano[2,3-b]pyran formation in the case of using of heterocyclic 1,3-dicarbonyl compounds.
Introduction
Among the effective approaches to the synthesis of six-membered oxygen- and nitrogen-containing heterocycles are the reactions of formal [3 + 3]-cycloaddition, including the Knoevenagel condensation followed by 6π-electrocyclization.1 The net result is the formation of a new stereocenter adjacent to the pyran oxygen atom, two σ-bonds and ring (Scheme 1). | ||
| Scheme 1 The formal [3 + 3]-cycloaddition approach leading to the fused 2H-pyran motifs. | ||
Oxa-[3 + 3]-annulation is a powerful synthetic strategy in the preparation of natural compounds and represents a complementary approach to oxa-[4 + 2]-cycloaddition. This method has already been widely used in the synthesis of various terpenes and alkaloids.1a–c At the same time, the set of possible substrates is limited almost exclusively to acyclic α,β-unsaturated aldehydes. The involvement of other types of conjugated aldehydes, in particular 4H-chromen-3-carbaldehydes, in this type of transformation stimulates significant interest because it will provide access to the synthesis of a large number of new polycondensed heterocycles.
Despite the obvious advantages of formal oxa-[3 + 3]-cycloaddition, the use of this methodology has long been limited due to the presence of a large number of competitive processes that reduce the yield of the target product. These include competition between the 1,2- and 1,4-addition to the α,β-unsaturated aldehyde, while the ambident nature of the nucleophile can lead to the initial O- or C-attack of the conjugated system (Fig. 1). At the same time, acyclic α,β-unsaturated enones and enals usually give 1,4-addition products (Michael adducts) as the predominant or sole products.2
 | ||
| Fig. 1 Possible reaction pathways of 1,3-dicarbonyl compounds with α,β-unsaturated aldehydes. | ||
2H,5H-Pyrano[4,3-b]pyran-5-one and 2,6-dihydro-5H-pyrano[3,2-c]pyridin-5-one are important scaffolds that are present in many natural products with potent biological activities and structural diversity. The pyrano[3,2-c]quinolone moiety is a structural motif present in several pyranoquinolone alkaloids including haplophytin A,3a oricine3b and simulenoline,3c which is a potent inhibitor of platelet aggregation. Among the derivatives of 2H-pyran-2-one and its benzo-condensed analogues are triptiliospinocoumarin,3d vismiaguian A,3e pyripyropene T,3f an antithrombinic sesquiterpenoid pyranocoumarin ferprenin3g and some others.3h Besides, the polycyclic acetal-fused pyrano[2,3-b]pyran skeleton is present in a variety of biologically active natural products, for example, welwitschin H,3i oleracone G3j and some alkaloids3k,l (Fig. 2).
 | ||
| Fig. 2 Natural compounds containing fused 2H,5H-pyrano[4,3-b]pyran-5-one, 2,6-dihydro-5H-pyrano[3,2-c]pyridin-5-one and pyrano[2,3-b]pyran scaffolds. | ||
Fused 2H,5H-pyrano[4,3-b]pyran-5-ones and 2,6-dihydro-5H-pyrano[3,2-c]pyridin-5-ones are important structural motifs in bioactive synthetic products. For instance, compound A shows phytotoxic activity,4a pyrones B–E strongly inhibit acetylcholinesterase activity, DNA synthesis, and leukemic cell growth.4b Pyrano[4,3-b]pyranone F is cytotoxic to neuroblastoma and melanoma cells.4c The tricyclic 2H-pyrans G–I may serve as lead compounds for the discovery of new drugs for the prevention and treatment of neurodegenerative diseases (for example, Alzheimer's disease).4d,e Pyrano[3,2-c]quinolone and pyrano[3,2-c]pyranone scaffolds J are present in a number of antiproliferative agents (against breast and liver cancer cell) (Fig. 3).4f–h
 | ||
| Fig. 3 Biologically active compounds containing fused 2H,5H-pyrano[4,3-b]pyran-5-one and 2,6-dihydro-5H-pyrano[3,2-c]pyridin-5-one scaffolds. | ||
Results and discussion
3-Formyl-4H-chromenes and their benzo-condensed derivatives are promising starting compounds for the synthesis of a wide variety of heterocyclic systems due to the presence of two nonequivalent electrophilic centers (C-2 and carbonyl carbons). We have previously shown that the three-component condensation of 3-formyl-4H-chromenes, ammonium acetate and 1,3-dicarbonyl compounds leads to β-(2-hydroxybenzyl)-substituted pyridine derivatives.5a However, we have found that the use of the 1,3-dicarbonyl compounds of the heterocyclic series in this reaction leads to pyrano[2,3-b]pyrans as formal [3 + 3]-cycloaddition products. In continuation of our interest in chromene chemistry,5 here we report an effective eco-friendly synthesis of polycyclic pyrano[2,3-b]pyrans from β-formyl-substituted 4H-chromenes, ammonium acetate and cyclic β-dicarbonyl compounds.As a model reaction, we chose the reaction between the aldehyde 1a and 4-hydroxycoumarin 2a. It is known that formal [3 + 3]-cycloaddition is catalyzed by both Lewis and Brønsted acids and bases.6 The annulation reaction of 1a and 2a was initially performed under refluxing conditions in AcOH with piperazine as a catalyst. As depicted in Table 1 (entry 1), 3aa was obtained in 34% yield along with the recovery of the starting materials in 2 min in the presence of 0.2 equiv. of piperidine. Increasing the used catalyst did not affect the yield to a large extent (entry 2). However, increasing the reaction time to 30 min allowed us to isolate 3aa in 71% yield (entry 3). A further increase in the reaction time up to 2 hours did not affect the reaction yield (entry 4). To improve the yields and reaction rates, several experimental conditions were evaluated. The use of CH3CN, 1,2-dichloroethane, dioxane, or DMF resulted in lower yields and longer reaction times (entries 5–8). As AcOH was found to be the best solvent, we screened different catalysts. In the case of TMG, Et3N, DBU, N-methylimidazole and even pyridine, we did not observe any significant differences in the yields and reaction rates (entries 9, 10, 12, 14, 16). The best results were obtained in AcOH as a solvent and in the presence of 1.0 equiv. of ammonium acetate. Under these conditions, the reaction was complete in 1 h, and the yield was 95% (entry 18). Further increase in the quantity of ammonium acetate did not affect the yield. It is important to note that the use of even 0.2 equiv. of ammonium acetate led to 3aa in comparable yield (entry 19). Ethanol was also used in the ammonium acetate catalyzed reaction as a solvent, but the yield of 3aa was lower (entry 20). When we carried out the same transformation in the presence of 1.0 equiv. of p-TSA in AcOH, a complex mixture of unidentified products was obtained along with the starting aldehyde 1a (entry 21). To determine the role of the catalyst, the reaction was carried out in the absence of ammonium acetate in AcOH or ethanol and 3aa was isolated from the reaction mixture in 43–46% yield (entries 22–24). This indicates that a secondary amine, or ammonium acetate, is not necessary for this transformation, but they accelerate the reaction and increase the yields. The synthesis of compound 3aa was repeated under optimized conditions on several different scales (up to 20 mmol), all with comparable yields.
|
|||
|---|---|---|---|
| Entry | Catalyst (x equiv.) | Time | Yieldb (%) |
| a All reactions were carried out with 1a (1 mmol), 2a (1 mmol) and catalyst in 3 mL of AcOH under reflux (unless otherwise indicated) and an air atmosphere.b Isolated yields.c CH3CN.d 1,2-Dichloroethane.e 1,4-Dioxane.f DMF at 100 °C.g 1,1,3,3-Tetramethylguanidine.h EtOH. | |||
| 1 | Piperidine (0.2) | 2 min | 34 |
| 2 | Piperidine (1.0) | 2 min | 39 |
| 3 | Piperidine (0.2) | 30 min | 71 |
| 4 | Piperidine (0.2) | 2 h | 73 |
| 5c | Piperidine (0.2) | 5 h | 40 |
| 6d | Piperidine (0.2) | 14 h | 49 |
| 7e | Piperidine (0.2) | 14 h | 46 |
| 8f | Piperidine (0.2) | 30 min | 31 |
| 9 | Et3N (0.2) | 2 h | 78 |
| 10 | DBU (0.2) | 2 h | 79 |
| 11 | N-Methylimidazole (0.2) | 2 min | 13 |
| 12 | N-Methylimidazole (0.2) | 2 h | 74 |
| 13 | TMGg (0.2) | 2 min | 13 |
| 14 | TMG (1.0) | 1 h | 80 |
| 15 | Py (0.2) | 2 min | 37 |
| 16 | Py (1.0) | 1 h | 76 |
| 17 | AcONH4 (1.0) | 20 min | 89 |
| 18 | AcONH4 (1.0) | 1 h | 95 |
| 19 | AcONH4 (0.2) | 2 h | 93 |
| 20h | AcONH4 (0.2) | 2 h | 52 |
| 21 | p-TSA (1.0) | 2 h | — |
| 22 | None | 8 h | 43 |
| 23h | None | 1 h | 46 |
| 24h | None | 2 h | 44 |

With the optimized reaction parameters, we extended this methodology to various formylchromones 1a–f and cyclic 1,3-dicarbonyl compounds 2a–g such as 4-hydroxy-6-methyl-2H-pyran-2-one, 4-hydroxy-6-methylpyridin-2(1H)-ones and their benzocondensed analogues. As shown in Table 2, polycyclic acetals 3 were prepared in 64–85% isolated yields. All reactions were carried out using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of chromenecarbaldehyde 1 and 1,3-dicarbonyl compound 2. Ammonium acetate is a very cheap and nontoxic catalyst, therefore, this procedure presents a green method for the diversity-oriented synthesis of biologically active compounds.
:1 molar ratio of chromenecarbaldehyde 1 and 1,3-dicarbonyl compound 2. Ammonium acetate is a very cheap and nontoxic catalyst, therefore, this procedure presents a green method for the diversity-oriented synthesis of biologically active compounds.

The structures of all products were determined on the basis of their analytical data. The cyclic nature of products 3 was confirmed by the absence of phenolic OH-stretching frequencies in the IR spectra and phenolic proton signals in the 1H NMR spectra. A characteristic feature of the 1H NMR spectra is the presence of two one-proton singlet signals in the region of 6.62–7.03 ppm, corresponding to the acetal and olefinic protons. Methylene protons of 3 were observed, as a rule, as two separate doublets at 3.54–5.30 ppm (JAB = 15.8–19.2 Hz) due to the chiral acetal center in these molecules. NH protons in the 1H NMR spectra of the N-unsubstituted derivatives of pyridinone and quinolinone were seen as singlets at 10.93–12.20 ppm. In the 13C NMR spectra of 3, a signal in the region of 95.8–98.2 ppm was assigned to the acetal carbon atom. Methylene carbon atoms appeared at 29.8–37.2 ppm. The DEPT spectra showed that the number of protons directly attached to the carbon atoms corresponds to the assigned structures. The structure of 3cc was also confirmed by single-crystal X-ray diffraction analysis.
We also studied the reaction of 1H-benzo[f]chromene-2-carbaldehyde 1a with 2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione 2h in the presence of ammonium acetate. In this reaction, the formation of two electrocyclic products was potentially possible (Scheme 2). According to NMR spectroscopy, only one cyclic regioisomer was obtained. In the 13C NMR spectrum, the carbonyl carbon atom appeared at 161.1 ppm. A comparison of the chemical shifts of the carbonyl carbon atoms in the product and in model compounds7 K–N was in favor of the linear condensed structure 3ah. Thus, the carbonyl carbon atom bonded to nitrogen at the double bond was more unshielded and appeared at 168.3–170.3 ppm (pyrimidin-4(1H)-one K and 2H-pyrido[1,2-a]pyrimidin-2-ones L1, L2), while carbonyl carbons in pyrimidin-4(3H)-one M and 4H-pyrido[1,2-a]pyrimidin-4-one N appeared at 161.3 and 162.0 ppm respectively. This does not attribute structure 3ah′ to the formed isomer.
 | ||
| Scheme 2 Reaction of 1H-benzo[f]chromene-2-carbaldehyde 1a with 2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione 2h. | ||
Scheme 3 outlines a proposed mechanism for the formation of fused 2H-pyrans 3. The Knoevenagel condensation between the enol derivatives of the β-ketolactones or β-ketolactams 2 and the electrophilic formyl carbon atom of the chromenecarbaldehydes 1 furnishes the conjugated 1-oxatriene intermediate, which undergoes a 6π-electrocyclization to form the heterocyclic compounds. Ammonium acetate might play the role of a Lewis base (to activate the enol form of the cyclic 1,3-dicarbonyl compounds) and the role of a Brønsted acid (to activate the aldehydes via hydrogen bonding or immonium salt formation). It should be noted that the studied 1,3-dicarbonyl compounds are strong OH-acids. For example, the pKa of 4-hydroxycoumarin 2a is 4.1.8 On the other hand, the vinyliminium ion arising from the formyl group and ammonia in situ is a better electrophile than 1 and undergoes selective C-1,2-addition with the 1,3-dicarbonyl compound. Thus, the activation of both partners of this reaction by the same catalyst might explain the highest yields in the case of using ammonium acetate. At the same time, the formation of the iminium salt is not obligatory. It was shown that the reaction was catalyzed not only by piperidine and ammonium acetate but also pyridine, Et3N and N-methylimidazole (Table 1). Therefore, we consider that the formation of the enolate anion from 2 is more important. Both chromenecarbaldehydes 1 and cyclic 1,3-dicarbonyl compounds 2 are polydentate reagents, and several reaction modes are possible by the combination of 1,4- vs. 1,2-addition. Nevertheless, the reaction is regioselective and the path II including the initial carbo-Michael addition was not realized. Electrocyclization involving the carbonyl group of the lactone or lactam moiety was also not observed (path III).
 | ||
| Scheme 3 Putative formal [3 + 3]-cycloaddition pathway. | ||
In order to broaden the scope of the present method, we attempted this protocol using dimedone 2i and syncarpic acid 2j as cyclic 1,3-dicarbonyl compounds. The products of electrocyclization, 3ai, 3aj, were isolated in low yields when the reaction was carried out for 1 min in boiling acetic acid. However, the boiling of an equimolar mixture of reagents for 20 min resulted exclusively in 7,8-dihydroquinolin-5(6H)-one derivatives 4ai, 4aj (Scheme 4) as described in literature.5a Compounds 4ai, 4aj arose from 3ai, 3aj via an oxa-6π-electrocyclic ring-opening of 3, a 1,6-addition of ammonia, dehydration and closure of the dihydropyran fragment. This assumption was confirmed by the fact that the isolated compound 3aj was converted to 4aj in 83% yield under the mentioned conditions. Since both the Knoevenagel condensation and electrocyclic ring-closure are reversible,9 such preference could simply be a reflection of the stability of the respective products being in favour of 4.
 | ||
| Scheme 4 Reaction of 1a with enolizable cyclic β-diketones (dimedone and syncarpic acid). | ||
We also observed an intriguing contrast between 1,3-indandione 2k and oxindole 2l (Scheme 5). In the case of 1,3-indandione, the 1-oxatriene 5ak was isolated in 21% yield after boiling for 5 min. Prolonged heating (5 h) of an equimolar mixture of reagents in AcOH in the presence of ammonium acetate gave the 1,6-addition product 6ak of ammonia generated from ammonium acetate. No desired electrocyclization product was obtained. At the same time, the reaction of 1a with oxindole was terminated at the stage of the Knoevenagel adduct 5al. The last was isolated in 78% yield as a mixture of E- and Z-isomers in an approximately 3.5:1 molar ratio, judging from the 1H NMR spectrum of the crude product. Pure E-indolinone was prepared by multiple recrystallizations from DMF, and its structure was confirmed by X-ray analysis. The Knoevenagel condensation products 5ak and both Z- and E-isomers of 5al that can undergo electrocyclization only after isomerization to the Z-form failed to give the fused pyrano[2,3-b]pyran derivatives. In contrast to the ketone carbonyls of dimedone, syncarpic acid and 1,3-indandione, it appears that the amide carbonyl of oxindole are not sufficiently electrophilic to react with ammonia. Thus, for reactions of 1,3-indandione and oxindole with 1a, the electrocyclic ring-closure step is either very slow or is arrested and, in the case of 1,3-indandione, the 1,6-addition of ammonia either occurs faster or is more favored.
 | ||
| Scheme 5 Reaction of 1a with 1,3-indandione and oxindole. aX-ray with 50% probability displacement. | ||
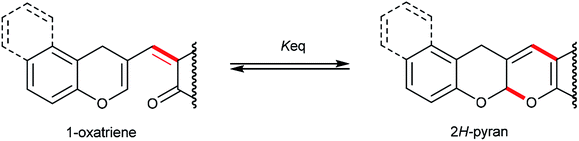
Based on the literature,4g,9 we can presume that compounds 3 equilibrate with 1-oxatriene intermediates. The energy calculation was done to determine the energy differences between some fused pyrano[2,3-b]pyrans 3 and their 1-oxatriene valence tautomers. It turned out that the energy differences between 2H-pyran and 1-oxatriene forms of 5ak and 5al were significantly larger as compared to other 1,3-dicarbonyl compounds (Table 3).
|
||
|---|---|---|
| Compound | ΔG, kJ mol−1 | Keq |
| a Calculated at 298.15 K and 1 atm using the IEFPCM solvent model (acetic acid). | ||
| 3ab | −14.05 | 290.1 |
| 3ad | −20.73 | 4303.5 |
| 3ca | −9.71 | 50.4 |
| 3ai | −5.48 | 9.1 |
| 3aj | −1.65 | 1.9 |
| 5ak | 49.02 | 2.6 × 10−9 |
| Z-5al | 57.63 | 7.9 × 10−11 |
We proposed that the reaction of 1a with 3-(benzylamino)-5,5-dimethylcyclohex-2-enone 2m by analogy with 1,3-dicarbonyl compounds would result in the formation of a formal aza- or oxa-[3 + 3]-cycloaddition product as a result of the Knoevenagel condensation and 6π-electrocyclization (Scheme 6). The use of such cyclic vinylogous amide 2m in a formal [3 + 3]-cycloaddition, including the 1-azatriene intermediate, has already been described in the literature.10 However, the treatment of 1a with enaminoketone 2m in the presence of catalytic amounts of ammonium acetate/piperidine in acetic acid or p-TSA in toluene under reflux resulted in the recovery of the starting material. In the presence of an excess of ammonium acetate, 7,8-dihydroquinolin-5(6H)-one 4ai was isolated in 70% yield. The benzylamino fragment in enaminone 2m was first replaced by an amino group with the formation of 3-amino-5,5-dimethylcyclohex-2-en-1-one 2n. Then, enaminone 2n reacted with chromene 1a to form the Michael adduct. The subsequent opening of the dihydropyran fragment and intramolecular nucleophilic addition led to the formation of a pyridine ring.
 | ||
| Scheme 6 Reaction of 1a with enaminone 2m. | ||
Conclusion
We have developed the first ammonium acetate-catalyzed formal oxa-[3 + 3]-annulation allowing the synthesis of a variety of fused pyrano[2,3-b]pyrans from cyclic β-dicarbonyl compounds and 4H-chromene-3-carbaldehydes. In some cases, 1-oxatriene compounds were proposed as key intermediates, and fused pyridines were isolated as 1,6-addition products of ammonia. The calculated values of the Gibbs free energies and reaction rate constants for 1-oxatriene – 2H-pyran equilibrium and experimental data indicated the reversibility of the electrocyclization stage, while the direction of the equilibrium shift was determined by the nature of the 1,3-dicarbonyl compound. Several new polycondensed heterocyclic systems were obtained in good yields in the green one-pot reaction. This method does not require strong bases for successful annulation, perhaps owing to the high acidity of 1,3-dicarbonyl compounds. Besides, the advantages of this eco-friendly procedure include the use of available and inexpensive reagents, atom- and step-economy, simple reaction conditions, broad substrate scope, no metal catalysis, scalability, and easy isolation by a chromatography-free protocol.Experimental
General information
Melting points were determined by the capillary method on an SRS OptiMelt MPA100 apparatus and were uncorrected. FTIR-spectra were obtained on a Shimadzu IR Affinity-1 spectrophotometer with the Specac Diamond ATR GS 10800-B attachment and are reported in cm−1. 1H and 13C NMR spectra (including DEPT-135, HMBC, HETCOR, COSY, and NOESY experiments) were recorded on a JEOL JNM-ECX 400 spectrometer (400 and 100 MHz, respectively) in DMSO-d6 solutions at ambient temperature unless otherwise noted, relative to the residual solvent signal [DMSO-d6 δ = 2.50 ppm (1H), δ = 39.5 ppm (13C)]. Chemical shifts and coupling constants were recorded in units of parts per million and hertz, respectively. 13C NMR spectroscopy of 3ac, 3af and 3fc was hindered due to its poor solubility in common solvents. High-resolution mass spectra (HRMS) were recorded on an Agilent 6230 TOF instrument using an electrospray (ESI) ionization source. The reaction progress was controlled by TLC on aluminum foil-backed silica gel plates (Merck, Kiesgel 60 F254); visualization was conducted under UV light and in iodine vapor. X-ray diffraction data were collected by using STOE STADI VARI PILATUS-100K (for 3cc), or SuperNova, Dual, Cu at zero, AtlasS2 (for 5al) diffractometers. All commercial solvents and reagents were used without additional purification. Computational calculations were carried out using the hybrid functionals of Becke11 and Lee, Yang and Parr.12 The solvent effect was computed by carrying out the single-point energy calculations of the gas-phase optimized geometries using the polarized continuum model (PCM).13 The Gaussian 03 program package14 was used for all calculations. The reported 4H-chromene-3-carbaldehydes and 1H-benzo[f]chromene-2-carbaldehydes were prepared according to literature procedures.15General procedure
A mixture of 3-formylchromone 1a–f (1 mmol), 1,3-dicarbonyl compound 2a–g (1 mmol) and ammonium acetate (15 mg, 0.2 mmol or more) in acetic acid (3 mL, unless otherwise noted) was stirred under reflux for 1 h. The progress of the reaction was monitored by TLC. After completion, the mixture was cooled to room temperature, and the formed precipitate was collected. The crude product was purified by recrystallization.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). HRMS (ESI, m/z) calcd for C23H15O4 [M + H]+: 355.0970, found: 355.0967.O). HRMS (ESI, m/z) calcd for C20H16NO3 [M + H]+: 318.1130, found: 318.1137.:5). IR νmax: 2961, 2889, 1661, 1620, 1595, 1510, 1464, 1425, 1406, 1393, 1333, 1227, 1206, 1188, 1175, 1144, 1067, 1036, 974, 955, 912, 814, 772, 752, 741. 1H NMR (400 MHz, DMSO-d6) δ: 1.00 (s, 3H), 1.03 (s, 3H), 2.20 (d, J = 15.8 Hz, 1H), 2.29 (d, J = 15.8 Hz, 1H), 2.48 (d, J = 18.1 Hz, 1H), 2.56 (d, J = 18.1 Hz, 1H), 3.94 (s, 2H), 6.64 (s, 1H), 6.68 (s, 1H), 7.05 (d, J = 8.9 Hz, 1H), 7.39 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.53 (ddd, J = 8.5, 7.1, 1.4 Hz, 1H), 7.72 (d, J = 8.9 Hz, 1H), 7.79–7.84 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ: 27.8 (CH3), 28.9 (CH3), 29.8 (CH2), 32.6 (C), 40.9 (CH2), 50.3 (CH2), 96.5 (CH), 108.4 (C), 112.5 (CH), 115.8 (C), 119.0 (CH), 122.3 (C), 122.9 (CH), 124.6 (CH), 127.4 (CH), 128.9 (2CH), 129.5 (C), 132.2 (C), 150.3 (C), 166.4 (C), 194.5 (C). HRMS (ESI, m/z) calcd for C22H21O3 [M + H]+: 333.1491, found: 333.1486.
O). HRMS (ESI, m/z) calcd for C23H15O4 [M + H]+: 355.0970, found: 355.0967.O). HRMS (ESI, m/z) calcd for C20H16NO3 [M + H]+: 318.1130, found: 318.1137.:5). IR νmax: 2961, 2889, 1661, 1620, 1595, 1510, 1464, 1425, 1406, 1393, 1333, 1227, 1206, 1188, 1175, 1144, 1067, 1036, 974, 955, 912, 814, 772, 752, 741. 1H NMR (400 MHz, DMSO-d6) δ: 1.00 (s, 3H), 1.03 (s, 3H), 2.20 (d, J = 15.8 Hz, 1H), 2.29 (d, J = 15.8 Hz, 1H), 2.48 (d, J = 18.1 Hz, 1H), 2.56 (d, J = 18.1 Hz, 1H), 3.94 (s, 2H), 6.64 (s, 1H), 6.68 (s, 1H), 7.05 (d, J = 8.9 Hz, 1H), 7.39 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.53 (ddd, J = 8.5, 7.1, 1.4 Hz, 1H), 7.72 (d, J = 8.9 Hz, 1H), 7.79–7.84 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ: 27.8 (CH3), 28.9 (CH3), 29.8 (CH2), 32.6 (C), 40.9 (CH2), 50.3 (CH2), 96.5 (CH), 108.4 (C), 112.5 (CH), 115.8 (C), 119.0 (CH), 122.3 (C), 122.9 (CH), 124.6 (CH), 127.4 (CH), 128.9 (2CH), 129.5 (C), 132.2 (C), 150.3 (C), 166.4 (C), 194.5 (C). HRMS (ESI, m/z) calcd for C22H21O3 [M + H]+: 333.1491, found: 333.1486.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3–C-11), 24.60 (H3–C-11), 25.21 (H3–C-9), 25.23 (H3–C-9), 29.7 (CH2-14), 47.2 (C-9), 55.4 (C-11), 96.7 (CH-7a), 106.3 (C-12a), 113.0 (CH-13), 115.7 (C-14a), 119.0 (CH-6), 122.9 (CH-1), 123.7 (C-13a), 124.7 (CH-3), 127.5 (CH-2), 128.89 (CH-5), 128.92 (CH-4), 129.5 (C-4a), 132.2 (C-14b), 150.2 (C-6a), 167.4 (C-8a), 195.0 (C-12), 211.6 (C-10). HRMS (ESI, m/z) calcd for C24H23O4 [M + H]+: 375.1596, found: 375.1602.
H3–C-11), 24.60 (H3–C-11), 25.21 (H3–C-9), 25.23 (H3–C-9), 29.7 (CH2-14), 47.2 (C-9), 55.4 (C-11), 96.7 (CH-7a), 106.3 (C-12a), 113.0 (CH-13), 115.7 (C-14a), 119.0 (CH-6), 122.9 (CH-1), 123.7 (C-13a), 124.7 (CH-3), 127.5 (CH-2), 128.89 (CH-5), 128.92 (CH-4), 129.5 (C-4a), 132.2 (C-14b), 150.2 (C-6a), 167.4 (C-8a), 195.0 (C-12), 211.6 (C-10). HRMS (ESI, m/z) calcd for C24H23O4 [M + H]+: 375.1596, found: 375.1602.Compound 4ai was also prepared from 1H-benzo[f]chromene-2-carbaldehyde 1a (210 mg, 1 mmol), 3-(benzylamino)-5,5-dimethylcyclohex-2-enone 2m and ammonium acetate (385 mg, 5 mmol) in acetic acid (4 mL) after refluxing for 10 min in 70% yield (232 mg).
:1 in 78% yield (507 mg). After three-fold recrystallization from DMF, the pure E-isomer was isolated in 38% yield (247 mg). Bright-yellow solid, mp 264–265 °C. IR νmax: 3400–2700, 1705, 1639, 1574, 1462, 1437, 1400, 1331, 1308, 1298, 1231, 1215, 1204, 1132, 1086, 874, 854, 808, 781. 1H NMR (400 MHz, DMSO-d6) δ: 4.17 (s, 2H), 6.85 (d, J = 7.6 Hz, 1H), 7.00 (t, J = 7.4 Hz, 1H), 7.16–7.25 (m, 3H), 7.50 (t, J = 7.3 Hz, 1H), 7.61–7.66 (m, 2H), 7.80 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.92 (d, J = 7.6 Hz, 2H), 10.50 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.7 (CH2), 110.5 (CH), 112.1 (C), 117.8 (CH), 121.87 (CH), 121.92 (C), 123.3 (CH), 124.3 (CH), 124.9 (C), 125.5 (CH), 127.8 (CH), 128.9 (CH), 129.2 (CH, C), 129.6 (CH), 131.0 (C), 131.9 (C), 135.4 (CH), 143.0 (C), 147.1 (C), 148.1 (CH), 169.6 (C). HRMS (ESI, m/z) calcd for C22H16NO2 [M + H]+: 326.1181, found: 326.1175.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was supported by the Russian Science Foundation (Grant No. 19-13-00421, development of method for pyrano[2,3-b]pyrans synthesis) and by the Russian Foundation for Basic Research (Grant No. 18-33-20249, computational study of the reaction mechanism). The X-ray diffractometer used in this research was purchased under the Moscow State University Program of Development. We thank Dr V. B. Rybakov (MSU) for the X-ray diffraction analysis.Notes and references
- For reviews on formal [3 + 3]-cycloaddition, see: (a) R. P. Hsung, A. V. Kurdyumov and N. Sydorenko, Eur. J. Org. Chem., 2005, 23 CrossRef CAS; (b) J. P. A. Harrity and O. Provoost, Org. Biomol. Chem., 2005, 3, 1349 CAS; (c) G. S. Buchanan, J. B. Feltenberger and R. P. Hsung, Curr. Org. Synth., 2010, 7, 363 CrossRef CAS; (d) D. Tejedor, S. Delgado-Hernández, R. Diana-Rivero, A. Díaz-Díaz and F. García-Tellado, Molecules, 2019, 24, 2904 CrossRef.
- D. H. Hua, Y. Chen, H.-S. Sin, M. J. Maroto, P. D. Robinson, S. W. Newell, E. M. Perchellet, J. B. Ladesich, J. A. Freeman, J.-P. Perchellet and P. K. Chiang, J. Org. Chem., 1997, 62, 6888 CrossRef CAS and references therein.
- (a) M. S. Ali, M. K. Pervez, M. Saleem and R. B. Tareen, Phytochemistry, 2001, 57, 1277 CrossRef CAS; (b) S. A. Khalid and P. G. Waterman, Phytochemistry, 1981, 20, 2761 CrossRef CAS; (c) I.-S. Chen, I.-W. Tsai, C.-M. Teng, J.-J. Chen, Y.-L. Chang, F. N. Ko, M. C. Lu and J. M. Pezzuto, Phytochemistry, 1997, 46, 525 CrossRef CAS; (d) M. Bittner, J. Jakupovic, F. Bohlmann, M. Grenz and M. Silva, Phytochemistry, 1988, 27, 3263 CrossRef CAS; (e) E.-K. Seo, M. C. Wani, M. E. Wall, H. Navarro, R. Mukherjee, N. R. Farnsworth and A. D. Kinghorn, Phytochemistry, 2000, 55, 35 CrossRef CAS; (f) C. Prompanya, C. Fernandes, S. Cravo, M. M. M. Pinto, T. Dethoup, A. M. S. Silva and A. Kijjoa, Mar. Drugs, 2015, 13, 1432 CrossRef CAS; (g) G. Appendino, S. Tagliapietra, P. Gariboldi, G. M. Nano and V. Picci, Phytochemistry, 1988, 27, 3619 CrossRef CAS; (h) M. Jonassohn, H. Anke, O. Sterner and C. Svensson, Tetrahedron Lett., 1994, 35, 1593 CrossRef CAS; (i) M. Moriyasu, N. Nakatani, M. Ichimaru, Y. Nishiyama, A. Kato, S. G. Mathenge, F. D. Juma and P. B. C. Mutiso, J. Nat. Med., 2011, 65, 313 CrossRef CAS; (j) Y. Xixiang, Z. Chengcheng, Z. Wenjie and M. Yufei, CN109824685A, 2019; (k) J. S.-Y. Yeap, S. Navanesan, K.-S. Sim, K.-T. Yong, S. Gurusamy, S.-H. Lim, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2018, 81, 1266 CrossRef CAS; (l) K. Gherida, M. Zeches-Hanrot, B. Richard, G. Massiot, L. Le Men-Olivier, T. Sevenet and S. H. Goh, Phytochemistry, 1988, 27, 3955 CrossRef.
- (a) M. Valente, L. C. A. Barbosa, A. H. Rathi, T. J. Donohoe and A. L. Thompson, Molecules, 2009, 14, 4973 CrossRef; (b) D. H. Hua, Y. Chen, H.-S. Sin, M. J. Maroto, P. D. Robinson, S. W. Newell, E. M. Perchellet, J. B. Ladesich, J. A. Freeman, J.-P. Perchellet and P. K. Chiang, J. Org. Chem., 1997, 62, 6888 CrossRef CAS; (c) H. Leutbecher, L. A. D. Williams, H. Rösner and U. Beifuss, Bioorg. Med. Chem. Lett., 2007, 17, 978 CrossRef CAS; (d) L. Pokhrel, I. Maezawa, T. D. T. Nguyen, K.-O. Chang, L.-W. Jin and D. H. Hua, J. Med. Chem., 2012, 55, 8969 CrossRef CAS; (e) S. Rana, H.-S. Hong, L. Barrigan, L.-W. Jin and D. H. Hua, Bioorg. Med. Chem. Lett., 2009, 19, 670 CrossRef CAS; (f) P. Kumari, C. Narayana, S. Dubey, A. Gupta and R. Sagar, Org. Biomol. Chem., 2018, 16, 2049 RSC; (g) P. Kumari, S. Gupta, C. Narayana, S. Ahmad, S. Singh and R. Sagar, New J. Chem., 2018, 42, 13985 RSC; (h) R. Sagar, J. Park, M. Koh and S. B. Park, J. Org. Chem., 2009, 74, 2171 CrossRef CAS.
- For chemistry of push-pull chromenes, see: (a) D. V. Osipov, V. A. Osyanin and Yu. N. Klimochkin, Chem. Heterocycl. Compd., 2018, 54, 1121 CrossRef CAS; (b) D. V. Osipov, V. A. Osyanin and Y. N. Klimochkin, Targets Heterocycl. Syst., 2018, 22, 436 CAS and references therein; (c) D. V. Osipov, A. A. Artyomenko, V. A. Osyanin and Yu. N. Klimochkin, Chem. Heterocycl. Compd., 2019, 55, 261 CrossRef CAS; (d) V. A. Osyanin, D. V. Osipov, I. V. Melnikova, K. S. Korzhenko, I. A. Semenova and Yu. N. Klimochkin, Synthesis, 2020, 52 DOI:10.1055/s-0040-1707209; for articles covered 2-aryl-4H-chromenes, see: (e) M. R. Demidov, M. Yu. Lapshina, D. V. Osipov, V. A. Osyanin and Yu. N. Klimochkin, Chem. Heterocycl. Compd., 2017, 53, 1053 CrossRef CAS; (f) A. A. Spasov, D. A. Babkov, D. V. Osipov, V. G. Klochkov, D. R. Prilepskaya, M. R. Demidov, V. A. Osyanin and Yu. N. Klimochkin, Bioorg. Med. Chem. Lett., 2019, 29, 119 CrossRef CAS.
- (a) E. J. Jung, Y. R. Lee and H.-J. Lee, Bull. Korean Chem. Soc., 2009, 30, 2833 CrossRef CAS; (b) A. D. Fotiadou and A. L. Zografos, Org. Lett., 2012, 14, 5664 CrossRef CAS; (c) Y. R. Lee, D. H. Kim, J.-J. Shim, S. K. Kim, J. H. Park, J. S. Cha and C.-S. Lee, Bull. Korean Chem. Soc., 2002, 23, 998 CrossRef CAS; (d) H. Leutbecher, J. Conrad, I. Klaiber and U. Beifuss, QSAR Comb. Sci., 2004, 23, 895 CrossRef CAS; (e) V. R. Narayana, Z. Pudukulathan and R. Varala, Org. Commun., 2013, 6, 110 CAS; (f) C. Hubert, J. Moreau, J. Batany, A. Duboc, J.-P. Hurvois and J.-L. Renaud, Adv. Synth. Catal., 2008, 350, 40 CrossRef CAS; (g) S. Fernandes, R. Rajakannu and S. V. Bhat, RSC Adv., 2015, 5, 67706 RSC; (h) M. J. Riveira and M. P. Mischne, Synth. Commun., 2013, 43, 208 CrossRef CAS.
- (a) H. Bibas, D. W. J. Moloney, R. Neumann, M. Shtaiwi, P. V. Bernhardt and C. Wentrup, J. Org. Chem., 2002, 67, 2619 CrossRef CAS; (b) G. M. Kheifets and V. A. Gindin, Russ. J. Org. Chem., 2004, 40, 560 CrossRef CAS; (c) T. A. Alanine, W. R. J. D. Galloway, S. Bartlett, J. J. Ciardiello, T. M. McGuire and D. R. Spring, Org. Biomol. Chem., 2016, 14, 1031 RSC.
- P. M. Nowak, F. Sagan and M. P. Mitoraj, J. Phys. Chem. B, 2017, 121, 4554 CrossRef CAS.
- (a) H. C. Shen, J. Wang, K. P. Cole, M. J. McLaughlin, C. D. Morgan, C. J. Douglas, R. P. Hsung, H. A. Coverdale, A. I. Gerasyuto, J. M. Hahn, J. Liu, H. M. Sklenicka, L. L. Wei, L. R. Zehnder and C. A. Zificsak, J. Org. Chem., 2003, 68, 1729 CrossRef CAS; (b) Z. A. Krasnaya, Chem. Heterocycl. Compd., 1999, 35, 1255 CrossRef CAS.
- (a) Q. H. To, Y. R. Lee and S. H. Kim, Monatsh. Chem., 2012, 143, 1421 CrossRef CAS; (b) J. Moreau, C. Hubert, J. Batany, L. Toupet, T. Roisnel, J.-P. Hurvois and J.-L. Renaud, J. Org. Chem., 2009, 74, 8963 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- J. Tomasi, B. Mennucci and E. Cances, J. Mol. Struct.: THEOCHEM, 1999, 464, 211 CrossRef CAS.
- Gaussian 09, Revision B.05, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. V. Lukashenko, D. V. Osipov, V. A. Osyanin and Yu. N. Klimochkin, Russ. J. Org. Chem., 2016, 52, 1817 CrossRef CAS; (b) A. V. Lukashenko, V. A. Osyanin, D. V. Osipov and Yu. N. Klimochkin, Chem. Heterocycl. Compd., 2016, 52, 711 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of products and copies of 1H and 13C NMR spectra. CCDC 1939651 and 1943197. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06450e |
| This journal is © The Royal Society of Chemistry 2020 |