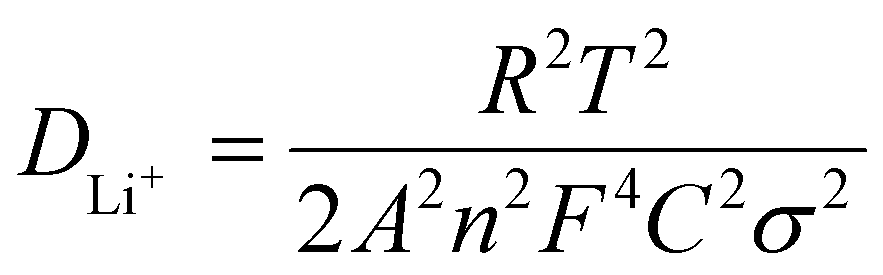DOI:
10.1039/D0RA04754F
(Paper)
RSC Adv., 2020,
10, 30406-30414
Electrochemical performance of nano-sized LiFePO4-embedded 3D-cubic ordered mesoporous carbon and nitrogenous carbon composites†‡
Received
29th May 2020
, Accepted 2nd August 2020
First published on 17th August 2020
Abstract
Herein, we report a single-step synthesis, characterization, and electrochemical performance of nano-sized LiFePO4 (LFP)-embedded 3D-cubic mesoporous carbon (CSI-809) and nitrogenous carbon (MNC-859) composites. Furthermore, in order to investigate the effects of both CSI-809 and MNC-859 on the electrochemical characteristics of LFP, a systematic study was performed on the morphology and microstructure of the composites, viz., LFP/CSI-809 and LFP/MNC-859, using XRD, FE-SEM, FT-Raman, and BET surface area analyses. Among these composites, LFP/MNC-859 exhibited better electrochemical performance with higher specific capacity and rate capability as compared to those of LFP/CSI-809. In addition, even after 100 cycles, LFP/MNC-859 retained 97% of its initial discharge capacity at 1C rate. The enhanced electrochemical performance of the nano-sized LFP-embedded MNC-859 can be attributed to the conductive nitrogenous carbon and mesoporosity, which facilitate electrolyte diffusion, and improved conductivity of the advanced LFP-nitrogenous porous carbon matrix.
Introduction
The exploration of promising cathode materials is vital for the development of Li-ion-based energy storage systems. Olivine-structured LiFePO4 is generally regarded as one of the most versatile cathode materials for lithium batteries due to the relative low cost of its raw materials, environmental benignity, non-toxicity, high chemical stability, and safety.1 Over the past two decades, many attempts have been made to ameliorate the low intrinsic electronic conductivity and sluggish mass and charge transport issues.2 Accordingly, decreasing the particle size, doping with foreign atoms and mixing with conductive carbon materials or conducting polymers have been used to overcome the abovementioned limitations.3 However, surface carbon coating is a straightforward technique to improve the poor electronic conductivity of materials.4
Nano-sized LiFePO4 cathode materials have attracted much attention because diminishing the particle size to the nano scale dimension has substantially shortened the time required for the diffusion of Li-ions into the crystal lattice.5–7 Nevertheless, these nano-sized particles are difficult to handle, especially while mixing them with carbon black during electrode fabrication. Moreover, coating LiFePO4 nanoparticles uniformly with conductive carbon is always a big challenge.8 The extensive liquid electrolyte/electrode interface emerging from their nano-scale size contributes amply to unwanted side reactions. All these factors are responsible for the poor cycling performance of nano-sized LiFePO4 particles.9
In the case of synthesizing mesoporous LiFePO4, ordered mesoporous carbon or silica cannot be directly used as a hard template to prepare an inverse replica of the original template. LiFePO4 is soluble in both strong acids (HF and HCl) and strong bases (NaOH) and hence it is not possible to directly utilize the nano-casting strategy (hard-template route). Therefore, an optimised approach has been applied such that the nano-sized LiFePO4 nanoparticles can be successfully embedded inside the pores of a mesoporous carbon matrix.10 Herein, raw materials were selected such that a LiFePO4 solution could be formed at room temperature and it was facilely infiltrated into the pores of mesoporous carbon using the wet impregnation strategy.
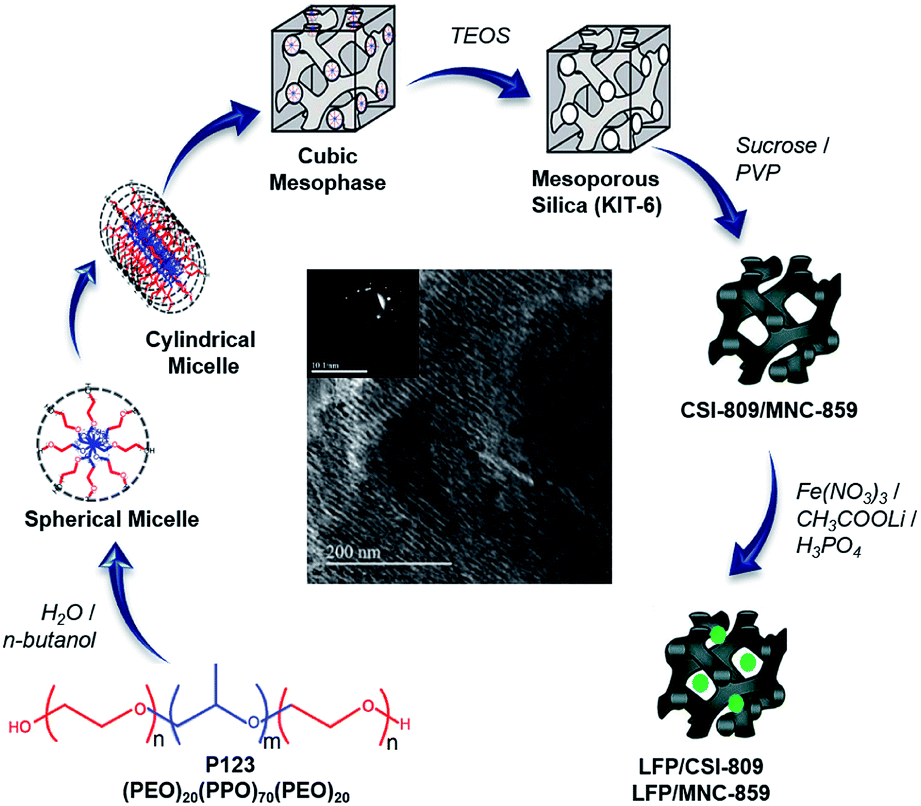
To achieve porous cathode materials for lithium-ion battery applications,11 it is crucial to investigate novel material architectures with hierarchically organized interconnected pores decorated with conductive carbon.12,13 In continuation of our recently published paper14 we show, for the first time, that there exists a dependency regarding the effect of pore dimensionality of mesoporous carbon coating on the electrochemical performance of LFP/carbon composite. In this study, we have made an attempt to simultaneously consider two things, viz., the nano-size of LiFePO4 and a 3D-coating with conductive mesoporous nitrogenous carbon, during the synthesis (Scheme 1) of robust cathode materials for Li-ion batteries. Finally, the development of mesoporous nitrogenous carbon-coated LiFePO4 has been discussed, highlighting key points on how the presence of nitrogen can significantly improve the electronic conductivity as well as excellent lithium storage properties of the prepared cathode material.15,16 Furthermore, we indicated that 3D cubic ordered mesoporous carbon is a competent carbon matrix for the fabrication of mesostructured LiFePO4-based cathode materials for the first time.
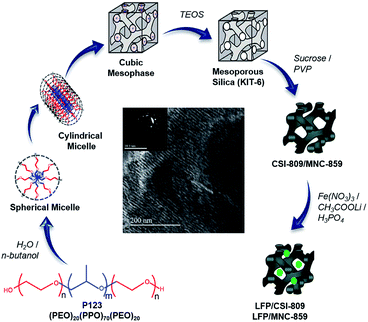 |
| | Scheme 1 Synthesis strategy of ordered mesoporous carbon/nitrogenous carbon-coated LFP composites. | |
Experimental
Starting materials
All the chemicals employed in this study were analytical-grade reagents and used without any further purification. An ordered mesoporous silica (OMS) hard template, viz., KIT-6, was hydrothermally synthesized as per the literature procedure.17,18 Then, the resulting sample was used as a starting material for the preparation of ordered mesoporous carbon (denoted by the code CSI) and ordered mesoporous nitrogenous carbon (denoted by the code MNC).
Synthesis of CSI-809
3D cubic-ordered mesoporous carbon, designated as CSI-809, was prepared by a previously reported nano-casting method17 using sucrose as a carbon precursor. Typically, 1.0 g of KIT-6 was impregnated with an acidified sucrose solution; the mixture was dried at 100 °C for 6 h followed by heating at 160 °C for 6 h in a hot-air oven. Furthermore, the impregnation step was performed twice for complete infiltration, and subsequently polymerized in the mesopores. The resulting black silica-polymer composite was carbonized at 900 °C under an argon atmosphere for 6 h. The required carbon sample was recovered after dissolving the silica framework in 15 wt% hydrofluoric acid followed by filtration, washing the material multiple times with deionized water, and drying at 100 °C in air. The obtained template-free ordered mesostructured carbon material synthesized from the SBA-15 template was labeled as CSI-809.
Synthesis of MNC-859
3D cubic-ordered mesoporous nitrogenous carbon, designated as MNC-809, was prepared by a similar procedure as used for CSI-809. In a typical synthesis, 1.0 g of KIT-6 was added to a solution of 5 g of polyvinylpyrrolidone (PVP) in 20 mL of dichloromethane followed by stirring for 6 h at room temperature. The mixture was placed in a drying oven at 70 °C for 6 h. Carbonization was completed by pyrolysis at 900 °C for 6 h at a heating rate of 5 °C min−1 under inert argon gas flow. The resulting carbon/silica composite was dissolved in 15 wt% HF at room temperature to remove the silica template. The template-free carbon product thus obtained was washed with ethanol, dried at 100 °C in air, and denoted as MNC-859.
Preparation of LFP/CSI-809
A facile synthesis of LFP/CSI-809 was carried out using ordered mesoporous carbon (CSI-809) as a hard template agent. For the synthesis of the mesoporous nanocomposite, the CSI-809 template powder (0.250 g) was dispersed in 5.0 M HNO3 followed by stirring for 0.5 h at 60 °C to induce hydrophilicity for facile wet impregnation. The template was filtered, washed with water, and dried at 90 °C. In the second step, Fe(NO3)3·9H2O in a stoichiometric amount was dissolved in 10 mL deionized water, and then, ascorbic acid, CH3COOLi (5 mmol), and phosphoric acid in requisite quantities were added followed by stirring for 2 h to ensure homogeneity. Subsequently, functionalized CSI-809 (0.25 g) was added to the abovementioned solution followed by stirring overnight. The mixture was dried and ground. The powder was finally sintered at 650 °C for 6 h under a 5% H2/Ar atmosphere. The material prepared was designated as LFP/CSI-809.
Preparation of LFP/MNC-859
For the facile synthesis of the nitrogen-containing ordered mesoporous composite, 5 mmol of Fe(NO3)3·9H20 and ascorbic acid in requisite amount were dissolved in a water–ethanol (50% v/v) mixture. CH3COOLi (5 mmol) and phosphoric acid (H3PO4) in stoichiometric amounts were added followed by stirring to ensure homogeneity. Then, MNC-859 (0.250 g) was added to the resulting solution followed by stirring overnight. The mixture was dried at 90 °C and ground. The powder was calcined at 650 °C for 6 h under a 5% H2/Ar atmosphere. The material prepared was designated as LFP/MNC-859.
Characterization
The samples LFP/CSI-809 and LFP/MNC-859 including CSI-809 and MNC-859, respectively, were systematically characterized by various analytical, spectroscopic, and imaging techniques. X-ray diffraction (XRD) patterns were recorded using a Bruker D8 Focus Diffractometer. It is furnished with a detector (Lynx Eye) and a Ni-filtered Cu Kα radiation source (λ = 1.5406 Å) operating at 40 kV and 40 mA in the 2θ range from 1.0 to 8.0° with a 2θ step size of 0.01°. Rietveld refinement of the XRD data was carried out using a general structure analysis system (GSAS) to determine structural parameters in a precise manner.
Nitrogen adsorption measurements were conducted using a Micrometrics ASAP 2020 instrument. Before analysis, the samples were degassed at 200 °C for 10 h, and the sorption measurements were carried out at −196 °C. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) equation. The total pore volume was calculated from the total nitrogen uptake under a relative pressure of P/P0 = 0.99. The pore size distribution was computed using the BJH method. In addition, elemental analysis was performed using a Perkin Elmer 2400 Series II CHNS/O elemental analyser to examine the carbon and nitrogen contents in the calcined samples.
SEM images were acquired using a FEI Quanta FEG 200 scanning electron microscope (HR-SEM). Transition electron microscopy (TEM) images were obtained using a JEOL JEM 2010 microscope, operated at 200 keV. The samples were ultrasonicated in ethanol, and then, a few drops of the dispersed materials were placed on a copper grid (carbon-coated). The grid was then dried before analysis. Raman measurements were performed using a Bruker FT-MultiRAM Raman spectrometer equipped with a 1064 nm laser excitation source and a LnGe detector in the spectral range from 3600 cm−1 to 50 cm−1. The measurements were carried out using a laser power of 20 mW, resolution of 4 cm−1, aperture of 5 nm, and 1024 scans.
Electrode fabrication
The fabrication of two-electrode Swagelok-type cells for electrochemical measurements was performed in an argon filled glove box (MBraun). The concentrations of H2O and O2 were maintained below 0.1 and 0.6 ppm, respectively. For cathode fabrication, the active material (LFP/carbon), Super P carbon black, and polyvinylidene difluoride (PVDF) were taken in a weight ratio 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10, respectively. The slurry was prepared by homogeneously grinding the abovementioned mixture using the N-methyl-2-pyrrolidinone (NMP) solvent, and it was coated on an aluminum foil (φ = 12 mm) followed by drying in an air oven at 90 °C for 12 h. The foil was cut into circular discs with an area of 1.13 cm2, and the active material loading on the aluminium foil was ∼2 mg cm−2. In order to synthesise the LFP + CSI-809 and LFP + MNC-859 cathodes, the active material, mesoporous carbon (CSI-809 or MNC-859), Super P carbon-black, and PVDF were taken in a weight ratio of 60:20:10:10, respectively, and the abovementioned procedure was used. For comparison, we fabricated cells using nano-sized LFP prepared by the procedure reported in a previous study.14 A Li metal foil (99.9%, Alfa Aesar) was used as both the reference and counter electrode. Whatman glass microfiber (GF/D) was used as a separator. Moreover, 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 vol%, Sigma-Aldrich) was used as a Li+ ion conducting electrolyte. After fabrication, the coin cells were maintained at room temperature for 24 h for the effective percolation of electrolyte into the electrode active materials. After this, the cells were galvanostatically charged and discharged in the voltage window of 2.5–4.2 V versus Li+/Li at room temperature using an Arbin battery testing system (Model BT2000, USA). Cyclic voltammograms (CV) were obtained using an Autolab instrument in the potential range of 2.5–4.2 V vs. Li+/Li at various scan rates (from 0.1 to 0.6 mV s−1). Electrochemical impedance spectroscopy (EIS) measurements were carried out using a Bio-logic VSP instrument.
:10:10, respectively. The slurry was prepared by homogeneously grinding the abovementioned mixture using the N-methyl-2-pyrrolidinone (NMP) solvent, and it was coated on an aluminum foil (φ = 12 mm) followed by drying in an air oven at 90 °C for 12 h. The foil was cut into circular discs with an area of 1.13 cm2, and the active material loading on the aluminium foil was ∼2 mg cm−2. In order to synthesise the LFP + CSI-809 and LFP + MNC-859 cathodes, the active material, mesoporous carbon (CSI-809 or MNC-859), Super P carbon-black, and PVDF were taken in a weight ratio of 60:20:10:10, respectively, and the abovementioned procedure was used. For comparison, we fabricated cells using nano-sized LFP prepared by the procedure reported in a previous study.14 A Li metal foil (99.9%, Alfa Aesar) was used as both the reference and counter electrode. Whatman glass microfiber (GF/D) was used as a separator. Moreover, 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 vol%, Sigma-Aldrich) was used as a Li+ ion conducting electrolyte. After fabrication, the coin cells were maintained at room temperature for 24 h for the effective percolation of electrolyte into the electrode active materials. After this, the cells were galvanostatically charged and discharged in the voltage window of 2.5–4.2 V versus Li+/Li at room temperature using an Arbin battery testing system (Model BT2000, USA). Cyclic voltammograms (CV) were obtained using an Autolab instrument in the potential range of 2.5–4.2 V vs. Li+/Li at various scan rates (from 0.1 to 0.6 mV s−1). Electrochemical impedance spectroscopy (EIS) measurements were carried out using a Bio-logic VSP instrument.
Results and discussion
Structure and morphology
Fig. 1 depicts the wide-angle XRD patterns of the mesoporous LFP/CSI-809 and LFP/MNC-859 composites. All the reflections of both samples could be indexed to those of olivine-structured LiFePO4 (JCPDS file number 89-6192) with the space group Pnma. Moreover, no impurity phases of iron phosphides or LiPO4 could be detected; this indicated the formation of well-crystallized single-phase electrode materials free from anti-site defects.19 This was also affirmed by Rietveld refinement profiles (ESI Fig. S1‡),20,21 and the structural parameters including the acceptable ‘R’ values obtained from the refinement of the LiFePO4/carbon composites are listed in Table 1. Furthermore, the low-angle diffraction pattern (Fig. 2B) of the calcined LFP/MNC-859 composite showed a sharp peak around 2θ = 1.02°, suggesting the presence of a long-range ordered 3D-cubic mesostructure. There is a clear shift in the position of the 211 reflection peak for the LFP/MNC-859 composite as compared to that of the MNC-859 template, indicating the filling of mesopores during the impregnation process. A similar shifting of the peak to lower angles was also noticed in the case of the LFP/CSI-809 composite (see Fig. 2A).
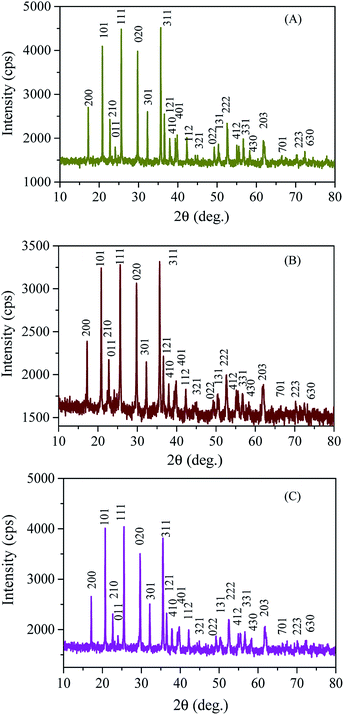 |
| | Fig. 1 Wide-angle XRD patterns of (A) LFP/CSI-809, (B) LFP/MNC-859, and (C) LFP. | |
Table 1 Structural parameters of LFP, LFP/carbon, and LFP/nitrogenous carbon composites
| Material |
a (Å) |
b (Å) |
c (Å) |
V (Å)3 |
RP (%) |
RWP (%) |
χ2 |
| LFP |
10.322(4) |
6.001(3) |
4.693(2) |
290.733(5) |
1.73 |
2.16 |
0.79 |
| LFP/CSI-809 |
10.329(2) |
6.008(2) |
4.696(2) |
291.418(8) |
1.81 |
2.33 |
0.83 |
| LFP/MNC-859 |
10.329(8) |
6.007(4) |
4.698(3) |
291.554(2) |
1.84 |
2.35 |
0.81 |
 |
| | Fig. 2 Low-angle XRD patterns of mesoporous pristine carbon (A) and nitrogenous carbon (B): (a) CSI-809, (b) LFP/CSI-809, (c) MNC-859, and (d) LFP/MNC-859. | |
Fig. 3 shows the typical N2 sorption isotherms and the corresponding BJH pore size distribution curves of the mesoporous LiFePO4/C composite. Textural properties of the LFP/C composites are provided in Table 1. The adsorption isotherm of the LFP/CSI-809 composite is a typical type-IV isotherm with an H1-type hysteresis loop, which is a prominent feature of mesoporous materials. The BET surface areas computed from the nitrogen desorption isotherms of the LFP/CSI-809 and LFP/MNC-859 composites are 127 and 77 m2 g−1, respectively. The larger specific surface area of the LFP/carbon composite can be attributed to the mesoporous carbon support left in the sample after calcination under an inert atmosphere.22 Therefore, the 3D cubic structure of the mesoporous carbon composite would promote the facile penetration of electrolyte and hence lead to high electrolyte–electrode interface area.23
 |
| | Fig. 3 N2 adsorption–desorption isotherms of pristine OMC (A) and nitrogenous carbon (B): (a) CSI-809, (b) LFP/CSI-809, (c) MNC-859, and (d) LFP/MNC-859. | |
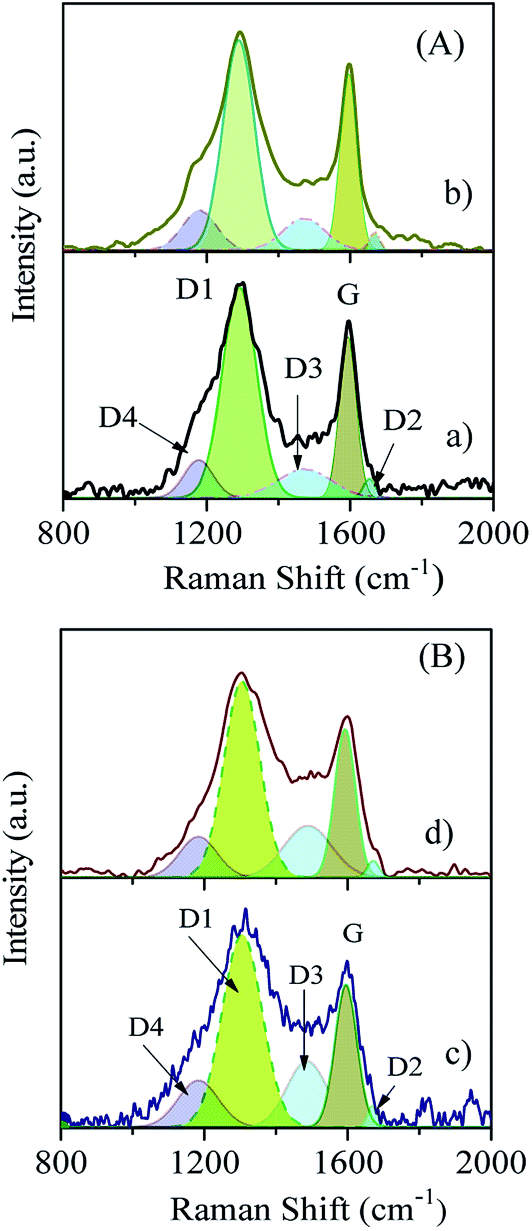
The typical Raman spectra of LFP/carbon composites are shown in Fig. 4. The two intense Raman bands (D-band and G-band) can be deconvoluted into four Lorentzian bands (D1, D2, D4, and G) and a Gaussian band (D3). The G-band at around 1595 cm−1 is generally related to the optically allowed E2g mode of crystalline graphite, and the D1-band (symmetry: A1g) is usually associated with the disordered graphitic lattice vibration mode.24,25 There is a distinct shift in the position of the D1 band (1307 cm−1 and 1296 cm−1 in the cases of the LFP/MNC-859 and LFP/CSI-809 composites, respectively); this clearly indicates that nitrogen is doped into the mesoporous carbon matrix in the case of the LFP/MNC-859 composite. However, the D2 band, which emerged as a shoulder on the G-band for both the composites, represented a highly defective graphitic lattice mode (E2g symmetry). The wide D3 band (∼1490 cm−1) mainly arises from the amorphous carbon fraction primarily contributed by the pore walls of mesoporous CSI-809 or MNC-859 carbon present in the LFP/carbon composite. The D4 band for both mesoporous CSI-809 and MNC-859 carbon can be attributed to diamond-like carbon (DLC) with short-range vibrations resulting from the substantial fraction of sp3 carbon (Table 2).26
 |
| | Fig. 4 Raman spectra of pristine OMC (A) and nitrogenous carbon (B): (a) CSI-809, (b) LFP/CSI-809, (c) MNC-859, and (d) LFP/MNC-859. | |
Table 2 Textural and spectral properties of mesoporous pristine and nitrogenous carbon, LEP, and LFP/carbon and LFP/nitrogenous carbon composites
| Material |
BET |
FT-Raman |
| SBET (m2 g−1) |
DBJH (nm) |
VP (cm3 g−1) |
G (cm−1) |
D (cm−1) |
D1/G |
| CSI-809 |
1168 |
3.80 |
1.22 |
1594 |
1296 |
3.05 |
| MNC-859 |
512 |
3.88 |
1.24 |
1597 |
1308 |
2.42 |
| LEP |
9 |
— |
— |
— |
— |
— |
| LFP/CSI-809 |
127 |
3.77 |
0.09 |
1596 |
1296 |
2.91 |
| LFP/MNC-859 |
77 |
3.81 |
0.11 |
1595 |
1307 |
2.31 |
Further, it can be seen from this table that the ratio of the integrated area of the D1-band and G-band is lower for LFP/MNC-859 indicating a uniform coating of large domains of graphene on the surface. The porous morphology of the LFP/CSI-809 and LFP/MNC-859 samples could be easily visualized from the SEM monographs (see Fig. 5A and B). To further analyse the fine microstructure of the mesoporous composites, high-resolution TEM analysis was employed. It can be clearly observed that the LiFePO4 nanoparticles (∼4 nm) are embedded in the channels of the mesoporous 3D nitrogenous carbon matrix (see Fig. 5F). The selected-area electron diffraction (SAED) patterns of the LFP/CSI-809 and LFP/MNC-859 samples (inset of Fig. 5E and F) show a typical bright spot mainly attributed to crystalline LiFePO4.27 It was anticipated that the 3D cubic mesoporous framework of the LFP/carbon composite could substantially allow the electrolyte to penetrate into the embedded LiFePO4 nanoparticles (see Scheme 1).
 |
| | Fig. 5 FE-SEM images of LFP/CSI-809 (A) and LFP/MNC-859 (B) and HR-TEM images of CSI-809 (C), MNC-859 (D), LFP/CSI-809 (E), and LFP/MNC-859 (F). Insets: SAED patterns. | |
Electrochemical testing
To evaluate the primary electrochemical behaviours of porous LFP/CSI-809 and LFP/MNC-859 electrodes, we obtained the corresponding cyclic voltammograms in the potential range of 2.5–4.2 V vs. Li+/Li using the Swagelok cells (Fig. 6). The CV curves of both electrodes show only one pair of cathodic/anodic peak at 3.3 and 3.6 V vs. Li+/Li, which was attributed to the intercalation/deintercalation of Li-ions in the LFP-based cathodes. The higher peak current in the case of the LFP/MNC-859 composite clearly depicts enhancement in the electronic conductivity caused by the nitrogenous carbon matrix. In the case of the LFP/CSI-809 composite (Fig. 6A), the anodic peak currents are relatively higher than the corresponding cathodic peak currents at higher scan rates; this clearly indicates that the Li-ion deintercalation process is facile as compared to the intercalation process. Fig. 6B shows a shift in the peak position towards a higher potential with the increasing scan rate for the LFP/MNC-859 composite. At a high scan rate of 0.6 mV s−1, the sharp redox peaks were still maintained during the entire deintercalation/intercalation process for both the electrodes. As the entire Li+ intercalation/deintercalation process was found to proceed through diffusion, the Li-ion diffusion paths in the electrodes were presumed to be semi-infinite diffusion paths.28 For comparison, the cyclic voltammograms of physical mixtures, viz., LFP + CMK-8 and LFP + MNC-81, at different scan rates are presented in the ESI Fig. S2.‡
 |
| | Fig. 6 Cyclic voltammograms of (A) LFP/CSI-809 and (B) LFP/MNC-859 at different scan rates. Insets: relationship between the peak currents and ν1/2 at various scan rates. | |
The Li-ion diffusion coefficient (DLi+) was evaluated according to the following classical Randles–Sevcik equation:
| | |
ip = 2.69 × 105n3/2AC0D01/2ν1/2
| (1) |
where
n is the number of electrons transferred in the redox reaction (
n = 1 for LiFePO
4),
A is the electrode surface area equivalent to one-third of the BET surface area (cm
2),
C0 is the concentration of the active material (mol cm
−3),
D0 is the diffusion coefficient (cm
2 s
−1), and
ν is the potential scan rate (V s
−1). The computed cathodic and anodic Li-ion diffusion coefficients of the LFP/MNC-859 composite are 1.4 × 10
−11 and 2.1 × 10
−12 cm
2 s
−1, respectively. In case of the LFP/CSI-809 composite, the corresponding values are 4.6 × 10
−13 and 1.4 × 10
−12 cm
2 s
−1 (see
Table 3). These values are in good agreement with the values previously reported in the literature.
29 Note that the Li-ion diffusion coefficient of pure LFP is around 1.8 × 10
−18 cm
2 s
−1. Hence, the
DLi+ of the LFP/MNC-859 composite is around six orders of magnitude greater than that of pure LFP.
Table 3 Electrochemical parameters of the LEP and LFP/carbon and LFP/nitrogenous carbon composites
| Material |
RCT (Ω) |
DLi+ (cm2 s−1) |
| Cathodica |
Anodica |
Impedance |
| From cyclic voltammogram studies. Values taken from ref. 14 for comparison purpose. |
| LFPb |
1089 |
— |
— |
6.9 × 10−18 |
| LFP/CSI-809 |
232 |
4.6 × 10−13 |
1.4 × 10−12 |
9.6 × 10−15 |
| LFP/MNC-859 |
211 |
1.4 × 10−11 |
2.1 × 10−12 |
4.3 × 10−14 |
| LFP + CSI-809 |
653 |
9.5 × 10−17 |
1.6 × 10−16 |
7.2 × 10−16 |
| LFP + MNC-859 |
512 |
7.9 × 10−17 |
5.8 × 10−16 |
1.1 × 10−15 |
In order to study the specific capacity of the LFP/C composite cathodes, we carried out galvanostatic charging and discharging experiments of the cells at 0.1C rate in the potential window of 2.7–4.0 V vs. Li+/Li, as shown in Fig. 7. Both the LFP/MNC-859 and LFP/CSI-809 electrodes showed flat voltage plateaus in the 3.4–3.5 V range, indicating a two-phase reaction, and delivered the first discharge capacities of 132.3 mA h g−1 and 117.7 mA h g−1 at 0.1C, respectively. Furthermore, the observed polarization difference between the charge and the discharge plateaus for both the cathode materials is highly consistent with the cyclic voltammogram results. The LFP/CSI-809 cathode material has high polarization difference than that of the LFP/MNC-859 cathode material because of the low electronic conductivity of CSI-809 as compared to that of the MNC-859 carbon matrix. As depicted in Fig. 7, the reversible discharge capacities of 106.5 mA h g−1 and 110.8 mA h g−1 were delivered by the LFP + CMK-8 and LFP + MNC-81 electrodes, respectively.
 |
| | Fig. 7 Galvanostatic charge–discharge profiles obtained at 0.1C rate for mesoporous LFP/MNC-859, LFP/CSI-809, LFP + MNC-859, LFP + CSI-809, and LFP. | |
For the LFP/MNC-859 composite, the obtained discharge capacities were 132.3, 124.7, 119.7, 111.1, 95.8, and 82.3 mA h g−1 at 0.1, 0.5, 1, 2, 3, and 5C, respectively (Fig. 8). The discharge capacities for the LFP/CSI-809 composites were 117.7, 105.8, 92.3, 80.3, 73.1, and 66.7 mA h g−1 at 0.1, 0.5, 1, 2, 3, and 5C, respectively. Moreover, when a lower C-rate (0.1C) was applied, the LFP/MNC-859 electrode could retrieve 98% of its initial discharge capacity. This clearly indicates that the carbonaceous 3D network of the LFP/MNC-859 composite can handle the interfacial volume strain associated with the Li-ion de-intercalation/intercalation process during charge–discharge.
 |
| | Fig. 8 Rate performance of LFP/carbon. Inset: cycling stability studies (discharge capacity) of the LFP/CSI-809 and LFP/MNC-859 electrodes at the current rate of 0.1C. | |
In addition, we fabricated the LFP/MNC-859 composite without Super P carbon black and performed galvanostatic charging and discharging experiments at 0.1C rate, as shown in the ESI Fig. S3.‡ Moreover, the observed polarization difference between the charge and the discharge plateaus for the cathode material was large when the conductive additive (Super P carbon black) was not added during fabrication. These results also indicate that the addition of Super P plays an important and positive role in ameliorating the electrochemical performance by enhancing the electronic conductivity of the LFP/MNC-859 composite. The enhanced capacity of the LFP/MNC-859 composite fabricated using Super-P carbon black is mainly ascribed to the reversible redox reaction between the Li-ions of the electrolyte and the conductive electrode–electrolyte interface formed by the combination of Super P and the residual nitrogenous mesoporous carbon.
To examine the electrochemical cycling stability of the electrodes, we selected two nominal C-rates, viz., 0.1C and 1C. A reversible capacity of 129.1 mA h g−1 was achieved (see inset of Fig. 8) at 0.1C rate even after 50 cycles, implying good cycling stability and reversibility of the LFP/MNC-859 electrode. The LFP/MNC-859 cathode exhibited a discharge capacity of 116.2 mA h g−1 at 1C after 100 cycles with an excellent capacity retention of 97.1% and high coulombic efficiency, as depicted in Fig. 9. For the LFP/CSI-809 cathode, a discharge capacity of 88.5 mA h g−1 was obtained after 100 cycles at 1C rate, and it still had a nominal (4.1%) capacity fading. It is evident that the LFP/MNC-859 cathode was stable during cycling performance tests at various C-rates, whereas the LFP/CSI-809 electrode exhibited gradual capacity fading. The improved cycling performance of the LIP-MNC-859 cathode is because the mesoporous structure originated from the nitrogenous carbon matrix of MNC-859 furnished enough 3D channels for facile Li+ ion transport.
 |
| | Fig. 9 Cycling stability studies of the LFP/CSI-809 and LFP/MNC-859 electrodes at the current rate of 1C. | |
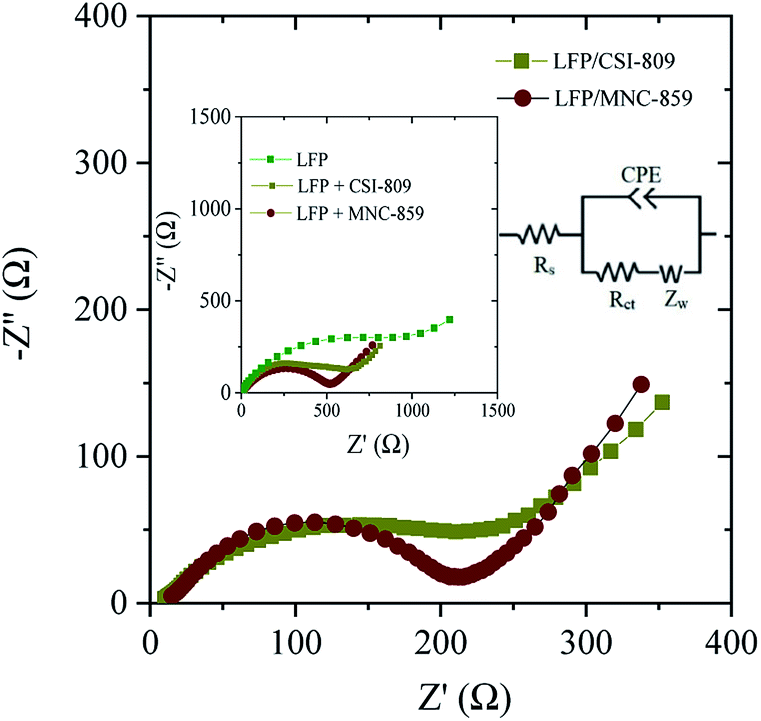
Electrochemical impedance spectroscopy (EIS) experiments were carried out to exemplify the improved electrochemical performance of the LFP/MNC-859 cathode material. Fig. 10 shows the Nyquist plots for both composites, and the data were investigated based on the specified equivalent circuit. Re denotes mainly the ohmic resistances due to electrolyte, current collector including active materials particle to particle contact resistances. The Rct (charge-transfer resistance) is generally associated with electrode surface reactions at the electrode/electrolyte interface. The obtained plot consists of a depressed semicircle in the intermediate and high frequency ranges, representing the charge transfer process. The sloping straight line in the very low frequency range represents the Warburg impedance (ZW) related to the solid-state diffusion of Li-ions into both the composites.30,31 The Nyquist plot also indicated that the charge transfer resistances (intermediate frequency) of the LFP/CSI-809 and LFP/MNC-859 composites were 231.6 Ω and 194.2 Ω, respectively; this implied that the nitrogen-containing mesoporous carbon (MNC-859) matrix significantly enhanced the electronic conductivity of the composite. More specifically, the surface nitrogen present in the MNC-859 matrix could construct a conductive network over the nano-sized LFP particles embedded in the LFP/MNC-859 composite. Moreover, the obtained Rct values could be well correlated to the electrochemical performance test results. The Li-ion diffusion coefficient (DLi+) was computed from the Warburg impedance (low frequency) for both the composites according to the following equation:
| |
 | (2) |
where
R is the gas constant,
T is the absolute operating temperature,
A is the surface area of the electrode,
n is the number of electrons per molecule during the oxidation process,
F is the Faraday's constant,
C is the molar concentration of Li-ions (7.69 × 10
−3 mol cm
−3), and
σ is the Warburg factor obtained from the slope of the plot of
Zreal versus the reciprocal square root of angular frequencies (
ω−1/2). The calculated
DLi+ values were 9.6 × 10
−15 and 4.3 × 10
−14 cm
2 s
−1 for LFP/CSI-809 and LFP/CSI-859, respectively (
Table 3). The
DLi+ value of the LFP/MNC-859 composite is about one order of magnitude higher than that of the LFP/CSI-809 composite, which clearly manifests the enhanced mobility of Li-ions in the case of the LFP/MNC-859 composite and the superior cycling performance of this composite. The estimated Li-ion diffusion coefficients are consistent with the calculated values reported by various research groups.
29,32
 |
| | Fig. 10 Electrochemical impedance spectra (Nyquist plot) of the LFP/CSI-809 and LFP/MNC-859 electrodes. Inset: Nyquist plots of LFP, LFP + CSI-809, and LFP + MNC-859. | |
Conclusions
Herein, we developed a 3D-cubic mesocomposite where the LiFePO4 nanoparticles were embedded on the mesoporous nitrogenous carbon matrix by a hard template approach, and the roles of nitrogen doping and pore dimensionality of the mesoporous structure were vividly assessed. This cubic mesoporous nitrogenous carbon, i.e., MNC-859, matrix acts as an electrolyte reservoir, ameliorating the electronic conductivity of the carbon matrix and subsequently cutting down the diffusion pathway for the lithium ions. This straightforward one-pot synthesis method can be employed to obtain various mesostructured cathode materials for fabricating Li-ion-based efficient energy storage devices. The favourable impacts of using cubic ordered mesoporous nitrogenous carbon materials along with unique in situ carbon coating strategies were unified for the development of highly electroactive 3D cubic mesostructured LiFePO4/carbon composite cathode materials for Li-ion batteries.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by Australia-India Strategic Research Fund by DST under grant No. INT/AUS/P-53/2012(G), and MNRE under project no. 103/140/2008-NT. The authors would like to thank Department of Science and Technology, New Delhi, for funding NCCR, IIT-Madras under SERB (Project No. IR/S1/CU-01/2002). The authors thank Professor B. Viswanathan and Professor S. Hayami for encouragement and support; Professor U. V. Varadaraju for fruitful discussion. The initiative and support of Dr G. S. K. Kannangara and Dr A. Milev, in early stages of the project, is gratefully acknowledged. Thanks are also due to CSIR for SRF (TVRM) and IITM for IPDF (RPR).
References
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- H. L. Zhang, H. B. Zhao, M. A. Khan, W. W. Zou, J. Q. Xu, L. Zhang and J. J. Zhang, J. Mater. Chem. A, 2018, 6, 20564–20620 RSC.
- M. Gaberscek, R. Dominko, M. Bele, M. Remskar, D. Hanzel and J. Jamnik, Solid State Ionics, 2005, 176, 1801–1805 CrossRef CAS.
- X.-L. Wu, L.-Y. Jiang, F.-F. Cao, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS.
- A. Milev, L. George, S. Khan, P. Selvam and G. S. K. Kannangara, Electrochim. Acta, 2016, 209, 565–573 CrossRef CAS.
- C. J. Yang, D. J. Lee, H. Kim, K. Kim, J. Joo, W. B. Kim, Y. B. Song, Y. S. Jung and J. Park, RSC Adv., 2019, 9, 13714–13721 RSC.
- H. Yang, X. L. Wu, M. H. Cao and Y. G. Guo, J. Phys. Chem. C, 2009, 113, 3345–3351 CrossRef CAS.
- R. R. Kapaev, S. A. Novikova, A. A. Chekannikov, D. Y. Gryzlov, T. L. Kulova, A. M. Skundin and A. B. Yaroslavtsev, Rev. Adv. Mater. Sci., 2018, 57, 183–192 CAS.
- N. Mohamed and N. K. Allam, RSC Adv., 2020, 10, 21662–21685 RSC.
- M. Yang and Q. Gao, J. Alloys Compd., 2011, 509, 3690–3698 CrossRef CAS.
- C. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132–2135 CrossRef CAS PubMed.
- R. Dominko, M. Bele, J.-M. Goupil, M. Gaberscek, D. Hanzel, I. Arcon and J. Jamnik, Chem. Mater., 2007, 19, 2960–2969 CrossRef CAS.
- Y. Zhang, F. S. Qi and Y. J. Liu, RSC Adv., 2020, 10, 11210–11218 RSC.
- S. Khan, R. P. Raj, T. V. R. Mohan, S. Bhuvaneswari, U. V. Varadaraju and P. Selvam, J. Electroanal. Chem., 2019, 848, 113242 CrossRef CAS.
- S. Yoon, C. Liao, X. G. Sun, C. A. Bridges, R. R. Unocic, J. Nanda, S. Dai and M. P. Paranthaman, J. Mater. Chem., 2012, 22, 4611–4614 RSC.
- Q. Q. Xiong, J. J. Lou, X. J. Teng, X. X. Lu, S. Y. Liu, H. Z. Chi and Z. G. Ji, J. Alloys Compd., 2018, 743, 377–382 CrossRef CAS.
- K. P. Gierszal, M. Jaroniec, T. W. Kim, J. Kim and R. Ryoo, New J. Chem., 2008, 32, 981–993 RSC.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- J. Wu, G. K. P. Dathar, C. Sun, M. G. Theivanayagam, D. Applestone, A. G. Dylla, A. Manthiram, G. Henkelman, J. B. Goodenough and K. J. Stevenson, Nanotechnology, 2013, 24, 424009 CrossRef PubMed.
- J. Wang, J. Yang, Y. Zhang, Y. Li, Y. Tang, M. N. Banis, X. Li, G. Liang, R. Li and X. Sun, Adv. Funct. Mater., 2013, 23, 806–814 CrossRef CAS.
- J. Wang, Y. Tang, J. Yang, R. Li, G. Liang and X. Sun, J. Power Sources, 2013, 238, 454–463 CrossRef CAS.
- N. N. Sinha, C. Shivakumara and N. Munichandraiah, ACS Appl. Mater. Interfaces, 2010, 2, 2031–2038 CrossRef CAS.
- T. Kozawa, N. Kataoka, A. Kondo, E. Nakamura, H. Abe and M. Naito, Mater. Chem. Phys., 2015, 155, 246–251 CrossRef CAS.
- R. Baddour-Hadjean and J. P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278–1319 CrossRef CAS PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- Q. Zhang, S. Z. Huang, J. Jin, J. Liu, Y. Li, H. E. Wang, L. H. Chen, B. J. Wang and B. L. Su, Sci. Rep., 2016, 6, 25942 CrossRef CAS PubMed.
- Y. C. Li, Y. P. Zhang, J. J. Ma, L. Yang, X. B. Li, E. Q. Zhao, S. M. Fan, G. R. Xu, S. T. Yang and C. C. Yang, J. Electrochem. Soc., 2019, 166, A410–A415 CrossRef CAS.
- D. Y. W. Yu, C. Fietzek, W. Weydanz, K. Donoue, T. Inoue, H. Kurokawa and S. Fujitani, J. Electrochem. Soc., 2007, 154, A253 CrossRef CAS.
- D. Qu, G. Wang, J. Kafle, J. Harris, L. Crain, Z. Jin and D. Zheng, Small Methods, 2018, 2, 1700342 CrossRef.
- D. Zane, M. Carewska, S. Scaccia, F. Cardellini and P. P. Prosini, Electrochim. Acta, 2004, 49, 4259–4271 CrossRef CAS.
- X. Tu, Y. Zhou, X. Tian, Y. Song, C. Deng and H. Zhu, Electrochim. Acta, 2016, 222, 64–73 CrossRef CAS.
Footnotes |
| † Dedicated to Professor U. V. Varadaraju on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04754f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Rayappan Pavul Raj
a,
Rayappan Pavul Raj