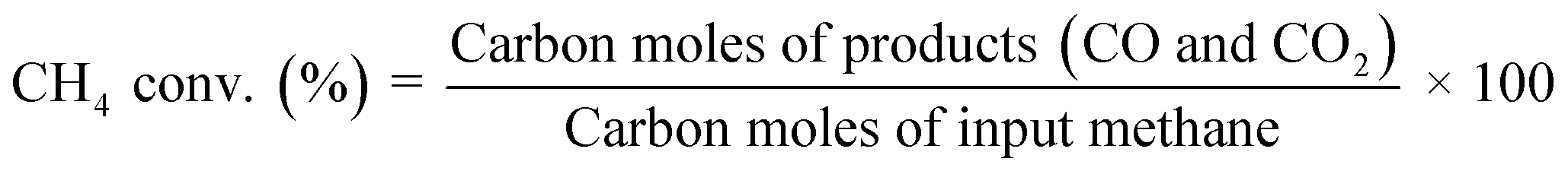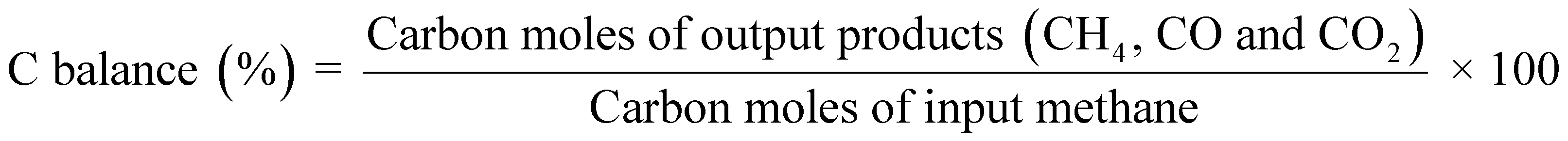DOI:
10.1039/D0RA04717A
(Paper)
RSC Adv., 2020,
10, 26418-26424
Support effects on catalysis of low temperature methane steam reforming†
Received
28th May 2020
, Accepted 8th July 2020
First published on 14th July 2020
Abstract
Low temperature (<500 K) methane steam reforming in an electric field was investigated over various catalysts. To elucidate the factors governing catalytic activity, activity tests and various characterization methods were conducted over various oxides including CeO2, Nb2O5, and Ta2O5 as supports. Activities of Pd catalysts loaded on these oxides showed the order of CeO2 > Nb2O5 > Ta2O5. Surface proton conductivity has a key role for the activation of methane in an electric field. Proton hopping ability on the oxide surface was estimated using electrochemical impedance measurements. Proton transport ability on the oxide surface at 473 K was in the order of CeO2 > Nb2O5 > Ta2O5. The OH group amounts on the oxide surface were evaluated by measuring pyridine adsorption with and without H2O pretreatment. Results indicate that the surface OH group concentrations on the oxide surface were in the order of CeO2 > Nb2O5 > Ta2O5. These results demonstrate that the surface concentrations of OH groups are related to the proton hopping ability on the oxide surface. The concentrations reflect the catalytic activity of low-temperature methane steam reforming in the electric field.
1. Introduction
Recently, H2 demand has been increasing as an alternative energy source. About half of the worldwide demand for hydrogen is met by steam reforming of methane (MSR), described by eqn (1).1–5 The water gas shift reaction (WGS), as shown in eqn (2), proceeds sequentially. Because MSR is a highly endothermic reaction that is controlled by thermodynamic equilibrium, conventional MSR processes require harsh conditions such as 1000–1200 K.1| | |
CH4 + H2O → CO + 3H2, ΔH298 = 206 kJ mol−1
| (1) |
| | |
CO + H2O → CO2 + H2, ΔH298 = −41.2 kJ mol−1
| (2) |
Under such harsh conditions, multiple heat exchangers and a reaction tube with high heat resistance are necessary, leading to higher costs. Furthermore, considering the use of hydrogen as a future energy resource, on-site and on-demand hydrogen production processes are desirable. Therefore, study and development of a process with high activity at lower temperatures are important.
To secure progress in this area, we have proposed a non-conventional low-temperature catalytic system: steam reforming of methane in an electric field (denoted as ER). In this system, applying a few milliamperes of weak direct current to the catalyst bed can achieve high degrees of methane conversion at considerably lower temperatures (below 500 K), at which conventional MSR proceeds only slightly.6–13 To elucidate the mechanism of MSR in an electric field, kinetic analyses were conducted. Manabe et al.7 found that the water pressure dependency on the reaction rate increased from 0.25 to 0.79 and the apparent activation energy decreased from 54.4 kJ mol−1 to 14.3 kJ mol−1 by applying an electric field. Obviously, the different reaction mechanism occurred by applying an electric field. To summarize the additional investigations, including operando-DRIFTS measurements and kinetic analyses using isotope,7,8,11 the reaction mechanism in an electric field was concluded as below. First, the application of an electric field promotes surface proton conduction via adsorbed water on the catalyst surface, known as the Grotthuss mechanism.14–17 The hopping protons collide with physisorbed methane and promote methane activation at the metal-support interface. Consequently, the ER reaction mechanism is completely different from that of MSR. It is necessary to establish a strategy for designing highly active catalyst on ER.
For this purpose, it is important to optimize the catalyst support, which plays an important role for surface proton conduction. Some reports have described the effect of support in an electric field,6,9,18 but no report to date explains a study of catalytic activity with surface proton conductivity, or clarifies catalytic activity or factors of support affecting surface proton conductivity. Thus, the reason of high activity for some catalysts were still unclear. By clarifying the support property that contributes to the high proton conductivity and high ER activity, we would be able to design catalysts with superior performance.
In this study, activity tests and various characterizations including surface proton conductivity were conducted to clarify the relations among physical properties and surface ion conductivity, and to clarify ER catalytic activity effects on low temperature (<500 K) MSR.
2. Experimental
2.1 Catalyst preparation
We employed various oxide supports with loading of 3 wt% Pd as an active metal. The results are presented in Table S1.† Among them, we chose CeO2, Nb2O5 and Ta2O5 for additional investigation. CeO2 (JRC-CEO-1) and Nb2O5 (JRC-NBO-1) were supplied by the Catalysis Society of Japan. They are used after calcination at 773 K for 3 h. A Ta2O5 support with high surface area was prepared using a solvothermal method based on the reported procedure.18 After 2.5 g of TaCl2 was dissolved into ethanol (250 mL) and was stirred for 2 h, it was heated at 463 K for 72 h. Then, after cooling to room temperature, the white precipitate was filtrated and washed with distilled water. It was then dried at 333 K for 12 h, and was calcined at 973 K for 3 h. On these supports, Pd (3 wt%) was loaded using an impregnation method with Pd(OCOCH3)2 as a precursor. As impregnating solvents, we used acetone for CeO2 and Nb2O5, and ethanol for Ta2O5. After impregnation and drying at 393 K, it was calcined at 773 K for 3 h. The metallic surface area and dispersion of loaded Pd among these three catalysts is almost identical.
2.2 Catalytic activity test
Catalytic activity tests were conducted using a fixed-bed flow-type reactor with a quartz tube (6.0 mm i.d. and 8.0 mm o.d.). A schematic image of an apparatus is shown in Fig. S1.† The catalyst was pressed and crushed to obtain particles of 355–500 μm. Then 100 mg of it was used. A thermocouple was set in the bottom of the catalyst bed to measure the catalyst bed temperature. For ER, two stainless steel rods (2 mm o.d.) were inserted into the catalyst bed as electrodes. A direct current electric field (9 mA) was imposed on the catalyst bed using a high voltage power supply. The response voltage wave was observed using an oscilloscope (TDS 2001C with a voltage probe P6015A; Tektronix Inc.). Activity tests were done under a reaction gas flow (CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:Ar = 1:2:7, total flow rate 100 mL min−1) in a kinetic condition (i.e. low conversion, and diffusion is not a rate-determining factor confirmed by our preliminary tests) at different temperatures (423–773 K for ER and 473–773 K for MSR). A GC-FID and a GC-TCD (Shimadzu Corp.) were used to analyze product gases after removal of H2O using a cold-trap. We used eqn (3) to calculate CH4 conversion and eqn (4) to calculate carbon balance.
:H2O:Ar = 1:2:7, total flow rate 100 mL min−1) in a kinetic condition (i.e. low conversion, and diffusion is not a rate-determining factor confirmed by our preliminary tests) at different temperatures (423–773 K for ER and 473–773 K for MSR). A GC-FID and a GC-TCD (Shimadzu Corp.) were used to analyze product gases after removal of H2O using a cold-trap. We used eqn (3) to calculate CH4 conversion and eqn (4) to calculate carbon balance.| |
 | (3) |
| |
 | (4) |
2.3 XPS measurement
Pd electronic states of Pd-loaded catalysts were evaluated using X-ray photoelectron spectroscopy (XPS; Versa Probe II; ULVAC-PHI, Inc., X-ray source was Al Kα). To evaluate the Pd electronic state during ER, activity tests were conducted before the measurement. The activity tests were conducted at 473 K for 30 min while imposing 9 mA of electrical current. After that, the catalysts were purged with Ar, set on the sample table using a transfer vessel in a gas barrier bag with degassing. Orbitals 3d5/2 and 3d3/2 of Pd were measured and the binding energies were calibrated with the C1s peak 284.8 eV.
2.4 Electrochemical impedance spectroscopy (EIS)
2.4.1 Preparation of pellets for EIS. To prepare sample pellets for EIS measurements, Nb2O5 or Ta2O5 powder (Kanto Chemical Co. Inc.) was crushed into fine particles using a planetary ball mill (Pulverisette 6; Fritsch GmbH). After the obtained powder was pressed to make a pellet (20 mm ϕ), it was calcined at high temperatures. For Nb2O5 pellets, 1.0 g of powder was pressed at 110 kN for 30 min and was sintered at 1473 K for 12 h. For Ta2O5 pellets, 1.1 g of powder was pressed at 110 kN for 30 min while degasified by an aspirator. It was then sintered at 1473 K for 12 h. The relative density of pellets was calculated as 61.1% for Nb2O5 and 61.6% for Ta2O5. For CeO2, data from our earlier work were used.19 As the electrode, Pt ink (Pt ink number 356010; the Nilaco Corp.) was painted on both sides of each pellet and was annealed at 1173 K for 1 h.
2.4.2 EIS measurements. EIS measurements were taken using a two-electrode four-wire cell (ProboStat; NorECs AS), which was connected to an impedance spectrometer (Novocontrol alpha-A) with a ZG4 interface. The frequency range was 106 to 10−3 Hz. The amplitude was 0.1 V RMS. The feed gas was Ar only for a dry condition and Ar + H2O (PH2O = 0.026 atm) for a wet condition. The temperature range was 373–673 K for the dry condition and 323–673 K for the wet condition. The obtained data were analyzed using equivalent circuit fitting software (ZView ver. 3.5a; Scribner Associates Inc.). The conductivity was calculated using eqn (5). Here, σ represents electrical conductivity, l denotes the pellet thickness, S stands for the surface area of the painted electrode, and R expresses the resistance. The activation energy (Ea) of the electrical conductivity was calculated using the Arrhenius expression presented as eqn (6), where A represents the pre-exponential factor and kB is Boltzmann's constant.| | |
σT = Aexp(−Ea/kBT)
| (6) |
2.5 Transmission FT-IR measurements
To evaluate the Lewis acidity of supports, Fourier transform infrared (FT-IR) spectroscopy measurements were conducted using pyridine as a basic probe molecule. An FT-IR spectrometer (FT/IR-6200; Jasco Corp.) with an MCT detector and CaF2 window was used. All spectra were recorded using a transmittance mode with 2 cm−1 resolution and 100 scans. The sample (20–30 mg) was shaped to a very thin disk (10 mm ϕ). Measurement flows of two types, with and without H2O treatment, are presented in Fig. S2.† First, the sample was outgassed in vacuo (p < 5 Pa) and heated at 673 K for 1 h. For H2O pre-treatment, the sample was cooled to 473 K. Then H2O was introduced for 30 min and subsequently outgassed in vacuo. The sample was then cooled to 323 K and background spectra were recorded. Then, pyridine was introduced for 10 min and outgassed in vacuo (p < 5 torr). Spectra of the adsorbed pyridine on the support were recorded.
2.6 Other characterizations
The crystalline structure of catalysts and prepared pellets for EIS were characterized using powder X-ray diffraction (XRD; SmartLab III; Rigaku Corp.) at 40 kV and 40 mA with Cu-Kα radiation (Fig. S3, and S4†). Field emission-scanning electron microscope (FE-SEM; S400S; Hitachi Ltd.) images were taken to observe the morphology of the prepared pellets (Fig. S5†). The specific surface area of each support was measured by N2 adsorption using Brunauer–Emmett–Teller (BET) method with an automated specific surface area analyzer (Gemini VII; Micromeritics Instrument Corp.).
3. Results
3.1 Evaluating catalytic activity over three catalysts
To evaluate the effects of metal oxide supports on the catalytic activity for MSR with and without the electric field, catalytic activity tests were conducted using Pd catalysts supported on various metal oxide supports. We selected CeO2, Nb2O5, and Ta2O5 supports for comparison because the Pd catalysts loaded on these three supports only showed both MSR and ER activities, and other Pd-supported catalysts showed no ER or MSR activities or electric field was unstable because of their insulation characteristic (details are presented in ESI Table S1†).
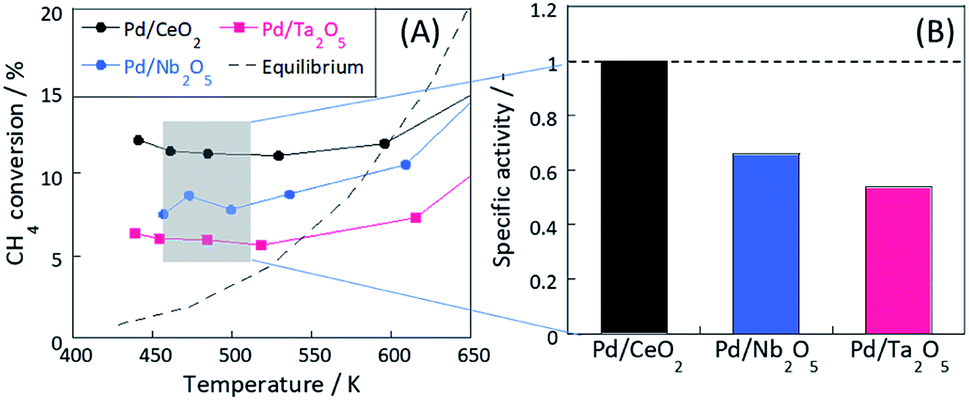
Fig. 1(A) and S6(A)† present results of activity tests over Pd-loaded catalysts. In these experiments, coke formation on these catalysts was negligible for both ER and SR, because carbon balance were almost 100%. These three catalysts showed higher activity at lower temperatures in the electric field (i.e. ER), exceeding the thermodynamic equilibrium. From Fig. S6(b),† apparent activation energies for these catalysts on MSR (i.e. heated catalyst) were almost identical. However, the trend is completely different on ER from MSR, as shown in Fig. S6(C).† In the electric field, CH4 is known to be activated via a three-atom intermediate [CH3–H–H]+ by the collision of hopping H+ and CH4 at the metal-support interface (CH4 + H+ → CH3+ + H2).7,8 In this step, the activation energy of its reverse reaction of CH3+ + H2 → CH4 + H+ is very high, making the step almost irreversible and exceeding the thermodynamic equilibrium.8
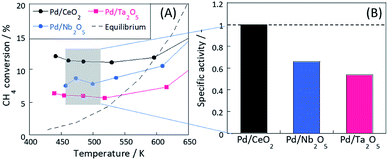 |
| | Fig. 1 (A) Temperature dependence of catalytic MSR activity in the electric field and (B) specific activity at 473 K. Reaction conditions: pre-set temperature, 323–773 K (ER); catalyst weight, 100 mg; flow, CH4:H2O:Ar = 1:2:7, total 100 SCCM; current, 9 mA. | |
The specific activity based on the CH4 conversion of Pd/CeO2 around 473 K was calculated (Fig. 1(B), and Table S2†). At 473 K, the activity without the electric field over these catalysts was sufficiently small to be ignored (0.074% for Pd/CeO2, 0.023% for Pd/Nb2O5, and 0.023% for Pd/Ta2O5). The electric field promoted the activity drastically even at such low temperatures at around 473 K. The effects of Joule heating by the electric field on the reaction rate are negligibly small in this case and no plasma has caused. The specific activities of Pd/Nb2O5 and Pd/Ta2O5 normalized by activity of Pd/CeO2 were, respectively, 0.66 and 0.53. Although these three catalysts showed almost equal furnace temperature and response voltage (i.e. the same input power), conversions of these catalysts in the electric field were clearly different.
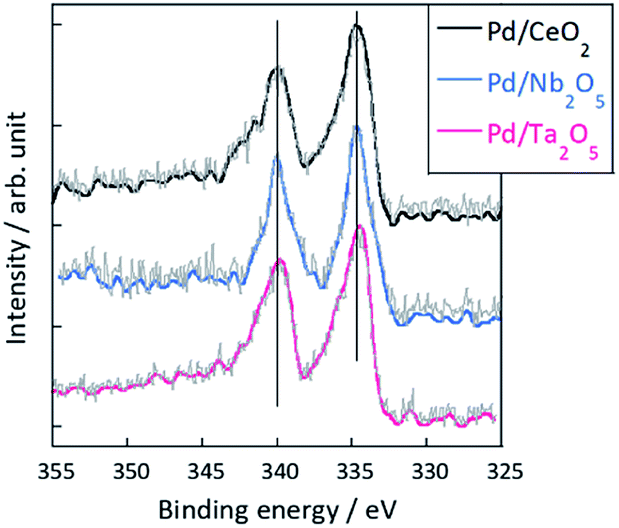
Furthermore, XPS measurement of these three catalysts were conducted to evaluate Pd electronic states during ER. In the case of MSR, the catalytic activity depends on the electronic state of active metal, and it is considered that ER activity is affected by the metal state. Thus, before considering the effect of support for ER activity, it should be confirmed that the electronic state of Pd during ER is the same or not. Fig. 2 shows results of XPS measurement for Pd 3d5/2 and 3d3/2 for Pd/CeO2, Pd/Nb2O5, and Pd/Ta2O5 after the ER activity tests. In this experiment, we used the gas barrier bag, not to be exposed to air. As shown in Fig. 2, the binding energy of Pd 3d5/3 and 3d3/2 of these three catalysts are almost identical. Thus, the difference among the ER activity at lower temperature region (Fig. 1) were not caused by the difference of Pd electronic state. Considering that collision with hopping protons in the electric field activates methane molecules, the differences of activities over these catalysts might derive from the proton conduction ability on the catalyst support surface.
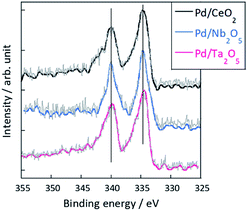 |
| | Fig. 2 XPS results for Pd 3d region after the ER activity tests of Pd/CeO2, Pd/Nb2O5, and Pd/Ta2O5. | |
3.2 Evaluating surface proton hopping capability using electrochemical impedance spectroscopy (EIS) measurement
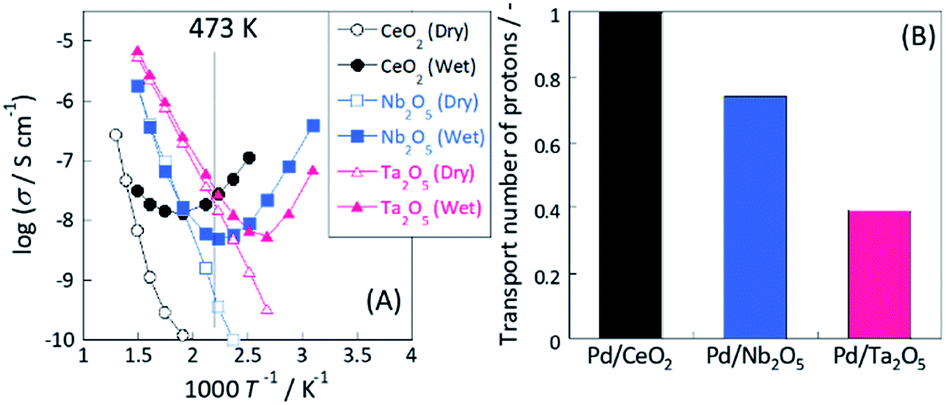
To evaluate the proton conductivity on the oxide support, EIS measurements were taken under dry and wet conditions. It has been widely reported that a porous pellet with lower relative density (R. D. = 50–60%) can be used feasibly for extracting surface ion conductivity because such samples have plenty of vacancy sites for H2O molecules to adsorb on the oxide surface.19–26 Fig. 3(A) presents the temperature dependences of conductivity among three samples (CeO2, Nb2O5 and Ta2O5) under dry and wet conditions. Under a dry condition, all samples showed typical Arrhenius behaviour: conductivity decreased concomitantly with decreasing temperature. In this temperature range (373 K < T < 673 K), the main conduction pathway might be attributed to the electron conduction in bulk.19,21 The calculated apparent activation energy was 1.27 eV (300 °C < T < 500 °C) for CeO2, 0.89 eV for Nb2O5 and 0.71 eV for Ta2O5, respectively. However, all samples showed anti-Arrhenius behaviour27 at lower temperature regions under wet conditions. Such conductive increases at low temperatures under a wet atmosphere are attributable to the increase in the water adsorption amount or relative humidity with decreasing temperature, resulting in enhancement of surface proton conduction via the Grotthuss mechanism.19–30
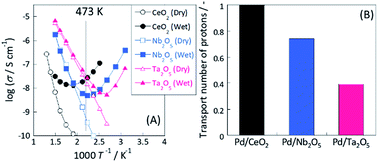 |
| | Fig. 3 (A) Comparison of apparent electrical conductivity under dry or wet condition by electrochemical impedance spectroscopy measurement and (B) calculated transport number of protons at 473 K. Measurement conditions: temperature, 323–773 K; partial pressure of H2O, PH2O = 0.026. | |
Consequently, the measured conductivity under dry conditions includes the electron conductivity in bulk and that under wet conditions includes not only the electron conductivity in bulk but also the proton conductivity on the oxide surface. Assuming that the contributions of electron conduction under both dry and wet conditions are approximately equal (because no effect of gas phase exists on electron conductivity in bulk), the contribution of proton conduction under wet conditions can be estimated as eqn (7). Furthermore, the proton transport number (TH+) is calculable as eqn (8). As shown in Fig. 3(B), the calculated proton transport numbers at 473 K were 1.0 (CeO2), 0.74 (Nb2O5), and 0.34 (Ta2O5).
| | |
TH+ = σH+/(σH+ + σe−) = (σwet − σdry)/σwet
| (8) |
Therefore, from the EIS measurement results, we inferred that these three oxides showed different proton transport numbers at the same temperature because of the surface proton transport ability.
3.3 Evaluating the amount of surface OH groups using pyridine IR
We sought to clarify the oxide support property which affects surface proton conductivity. The Grotthuss mechanism is known as sequential proton hopping via the H-bonds of water molecules.14–17 Consequently, water adsorption on the oxide surface might have a strong relation with the proton conduction. Generally, for H2O adsorption on metal oxides, the metal cation and oxygen species act respectively as Lewis acid and base sites, forming a hydroxyl group on the oxide surface (eqn (9) and (10)).23,31 Such mechanisms are also well discussed by ab initio molecular dynamics (AIMD) simulation.32–34| | |
Msurf + H2O → Msurf–OH2
| (9) |
| | |
Msurf–OH2 + Osurf → Msurf–OH + Osurf–H
| (10) |
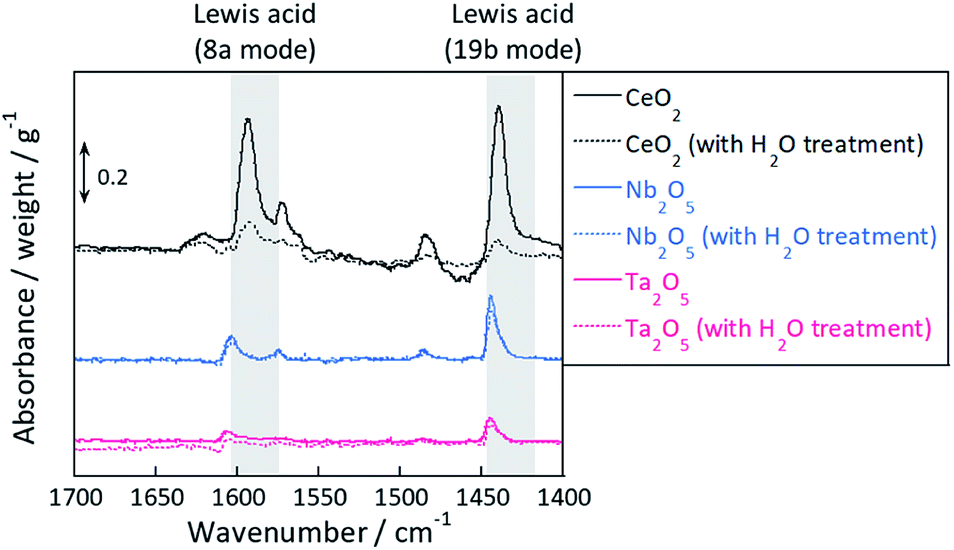
To evaluate the Lewis acidity of CeO2, Nb2O5, and Ta2O5, the adsorbed pyridine species on three oxides were measured using transmission IR. Considering that Lewis acid sites (metal cation) were able to serve as adsorption sites for hydroxyl group, the exposed Lewis acid amount would decrease after pretreatment with H2O. Therefore, we conducted pretreatments of two types, with and without H2O.
Fig. 4 presents transmission FT-IR spectra of supports after exposure to pyridine with or without H2O pretreatment. In this figure, the solid line shows spectra without H2O treatment and the dotted line shows the spectra with H2O treatment. All samples show peaks at 1455–1438 cm−1,35–37 which is assigned to the 19b mode of pyridine, and 1632–1580 cm−1, which is assigned to the 8a mode of pyridine bonded on the Lewis acid site. However, pyridine adsorbed onto Brønsted acid, of which peaks appear around 1540 cm−1, was not observed for any sample. The peak area was quantified using the peak at 1455–1438 cm−1, normalized by the weight or surface area of catalyst and presented in Table 1. As shown in Fig. 4 and Table 1, the exposed Lewis acid amount decreased because of the H2O treatment. The value [Larea (without H2O) − Larea (with H2O)], which was calculated as presented in Table 1, represents the decrement of Lewis acidity and formation of OH groups per unit area by H2O treatment. The order of surface OH group concentration is in the order of CeO2 > Nb2O5 >Ta2O5. This order is identical to that of the proton transport numbers shown in Fig. 3(B). Considering the proton hopping via H-bond of water molecule, the OH group distance is important: if the OH group density is low, then protons have difficulty hopping to the neighbouring site.38 Consequently, the OH group concentration has a strong relation with the proton conduction ability.
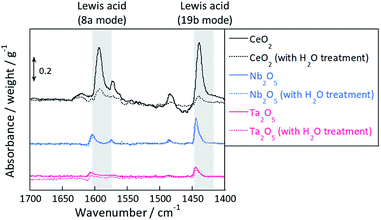 |
| | Fig. 4 Transmission FT-IR spectra of supports after exposure to pyridine and in vacuo with or without H2O treatment. | |
Table 1 Comparison of Lewis acid amount among CeO2, Nb2O5, Ta2O5a
| Sample |
Condition |
W/mg |
SSA (specific surface area)/m2 g−1 |
L/a.u. |
Lweight/a.u. |
Larea/a.u. |
Larea (without H2O) − Larea (with H2O)/a.u. |
| W: sample weight [mg], L: area value of Lewis acid peak, Lweight: L per unit weight, Larea: L per unit area. |
| CeO2 |
Without H2O |
19.9 |
122.9 |
5.77 |
290 |
2.36 |
2.02 |
| With H2O |
20.9 |
0.88 |
42.0 |
0.34 |
| Nb2O5 |
Without H2O |
20.0 |
71.29 |
1.58 |
79.2 |
1.11 |
0.25 |
| With H2O |
20.0 |
1.23 |
61.4 |
0.86 |
| Ta2O5 |
Without H2O |
27.8 |
36.32 |
0.71 |
25.4 |
0.70 |
0.21 |
| With H2O |
27.2 |
0.48 |
17.7 |
0.49 |
4. Discussion
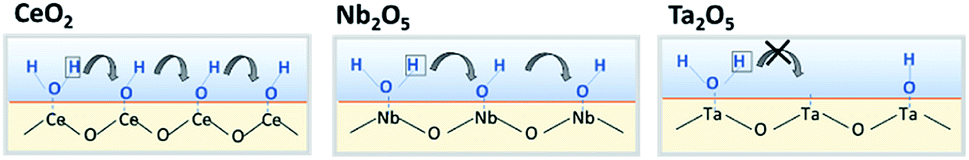
Results of activity tests demonstrated that the activities in the electric field for Pd/CeO2, Pd/Nb2O5, and Pd/Ta2O5 were clearly different. The order was Pd/CeO2 > Pd/Nb2O5 > Pd/Ta2O5. These three catalysts showed almost identical activity by heat, as presented in Fig. S6(A) and S6(b),† but exhibited completely different trends in the electric field, as shown in Fig. S6(C).† XPS revealed that Pd electronic states after ER activity tests were almost identical for the three catalysts, and the difference of ER activity were not caused by the difference of Pd loading state. In the electric field, methane is activated by the collision of hopping protons from the oxide surface.7,8 Therefore, the difference in activity is regarded as the proton conductivity on the oxide surface. The EIS measurements for CeO2, Nb2O5, and Ta2O5 under dry and wet conditions showed that the oxide conductivity increased at low temperatures with decreasing temperature under wet conditions, indicating an increase of proton conductivity via the Grotthuss mechanism. Furthermore, the calculated transport numbers of protons at 473 K were 1.0 for CeO2, 0.74 for Nb2O5 and 0.34 for Ta2O5. The order of transport number (CeO2 > Nb2O5 > Ta2O5) was identical to that of the ER activity. The anti-Arrhenius trend is also the same, as reflected in a comparison of Fig. S6(C)† and 3. Consequently, results showed that proton conductivity on the surface strongly affects the ER activity. The transport numbers of protons differ among the oxides even if the same H2O amount is introduced. Therefore, the proton hopping ability among these oxides is probably different. For proton conduction by the Grotthuss mechanism, the proton hops to the neighbour site via H-bond of adsorbed water (Fig. 5). The density of OH groups might be important: if the interval of OH groups is long, then it is difficult for a proton to hop to the neighbouring site. The amount of formed OH groups on Lewis acid was evaluated by estimating the Lewis acid amount (metal cation) on the oxide surface with or without H2O pretreatment. Furthermore, the order of formed OH groups per unit area was CeO2 > Nb2O5 > Ta2O5, reflecting the same trend at that of catalytic activity in the electric field and of proton hopping ability by EIS measurements.
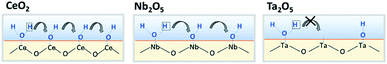 |
| | Fig. 5 Schematic image of relationship between formed OH group and proton conduction on surface. | |
By combining the results of activity tests in the electric field with the EIS and IR results, the factors governing catalytic activity can be considered. For CeO2, great amounts of water dissociated on its surface. Moreover, the density of OH groups was high, thereby producing the high proton-hopping ability. However, the H2O only slightly dissociates on the Ta2O5 surface. The proton conduction barrier becomes much higher; also, the proton hopping ability is low on Ta2O5. Results show that Nb2O5 has intermediate properties between those of CeO2 and Ta2O5. In the electric field, methane is activated by hopping protons. Thereby, oxides with high proton hopping ability on the surface were able to achieve high activity.
5. Conclusions
Methane steam reforming was conducted with and without an electric field over Pd catalysts loaded on various oxides including CeO2, Nb2O5, and Ta2O5 to elucidate the factors controlling activity of the catalyst support. These catalysts showed almost identical and low activity by heated catalysis. Therefore, the structure of supported metal has almost identical structure (i.e. particle diameter, dispersion). Results of activity tests conducted with the electric field demonstrated that all catalysts showed activity at low temperatures exceeding the thermal equilibrium. The order of activity was Pd/CeO2 > Pd/Nb2O5 > Pd/Ta2O5. Electrochemical impedance spectroscopy (EIS) measurements under dry and wet conditions were conducted to evaluate the surface proton conduction. The order of proton transport ability was CeO2 > Nb2O5 > Ta2O5, indicating that H2O adsorption and activation properties over these oxide supports differ. Finally, transmittance FT-IR measurements of the adsorbed pyridine species on these oxide supports were measured to evaluate the Lewis acid amount. By introducing H2O before measurements, the Lewis acid amount decreased, indicating that OH groups formed on a Lewis acid (metal cation). The order of the amount of formed OH groups was CeO2 > Nb2O5 > Ta2O5. The results obtained for EIS and IR revealed that the dissociative adsorption property of H2O and the amount of formed OH groups are related strongly to the proton hopping ability. To summarize this work, as the adsorbed and activated amounts of H2O become larger, the proton conductivity becomes higher, then the catalyst was able to achieve high activity in the electric field for low-temperature MSR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST CREST JPMJCR1423, Japan.
Notes and references
- A. Iulianelli, S. Liguori, J. Wilcox and A. Basile, Catal. Rev.: Sci. Eng., 2016, 58, 1–35 CrossRef CAS.
- J. N. Armor, Appl. Catal., A, 1999, 176, 159–176 CrossRef CAS.
- M. A. Peña, J. P. Gómez and J. L. G. Fierro, Appl. Catal., A, 1996, 144, 7–57 CrossRef.
- K. Delgado, L. Maier, S. Tischer, A. Zellner, H. Stotz and O. Deutschmann, Catalysts, 2015, 5, 871–904 CrossRef CAS.
- J. G. Xu and G. F. Froment, AIChE J., 1989, 35, 88–96 CrossRef CAS.
- K. Oshima, T. Shinagawa and Y. Sekine, J. Jpn. Pet. Inst., 2013, 56, 11–21 CrossRef CAS.
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 3–4 CrossRef PubMed.
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS.
- Y. Sekine, M. Haraguchi, M. Matsukata and E. Kikuchi, Catal. Today, 2011, 171, 116–125 CrossRef CAS.
- S. Ogo and Y. Sekine, Chem. Rec., 2017, 17, 726–738 CrossRef CAS PubMed.
- M. Torimoto, K. Murakami and Y. Sekine, Bull. Chem. Soc. Jpn., 2019, 92, 1785–1792 CrossRef CAS.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J. G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
- Y. Sekine, M. Haraguchi, M. Tomioka, M. Matsukata and E. Kikuchi, J. Phys. Chem. A, 2010, 114, 3824–3833 CrossRef CAS PubMed.
- T. Kumagai, A. Shiotari, H. Okuyama, S. Hatta, T. Aruga, I. Hamada, T. Frederiksen and H. Ueba, Nat. Mater., 2012, 11, 167–172 CrossRef CAS PubMed.
- T. Norby, MRS Bull., 2009, 34, 923–928 CrossRef CAS.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- Z. Zuo, Y. Fu and A. Manthiram, Polymers, 2012, 4, 1627–1644 CrossRef CAS.
- K. N. Manukumar, B. Kishore, K. Manjunath and G. Nagaraju, Int. J. Hydrogen Energy, 2018, 43, 18125–18135 CrossRef CAS.
- R. Manabe, S. Ø. Stub, T. Norby and Y. Sekine, Solid State Commun., 2018, 270, 45–49 CrossRef CAS.
- S. Ø. Stub, K. Thorshaug, P. M. Rørvik, T. Norby and E. Vøllestad, Phys. Chem. Chem. Phys., 2018, 20, 15653–15660 RSC.
- S. Ø. Stub, E. Vøllestad and T. Norby, J. Phys. Chem. C, 2017, 121, 12817–12825 CrossRef CAS.
- B. Scherrer, M. V. F. Schlupp, D. Stender, J. Martynczuk, J. G. Grolig, H. Ma, P. Kocher, T. Lippert, M. Prestat and L. J. Gauckler, Adv. Funct. Mater., 2013, 23, 1957–1964 CrossRef CAS.
- S. Miyoshi, Y. Akao, N. Kuwata, J. Kawamura, Y. Oyama, T. Yagi and S. Yamaguchi, Chem. Mater., 2014, 26, 5194–5200 CrossRef CAS.
- I. G. Tredici, F. Maglia, C. Ferrara, P. Mustarelli and U. Anselmi-Tamburini, Adv. Funct. Mater., 2014, 24, 5137–5146 CrossRef CAS.
- S. Raz, K. Sasaki, J. Maier and I. Riess, Solid State Ionics, 2001, 143, 181–204 CrossRef CAS.
- Y. Hisai, K. Murakami, Y. Kamite, Q. Ma, E. Vøllestad, R. Manabe, T. Matsuda, S. Ogo, T. Norby and Y. Sekine, Chem. Commun., 2020, 56, 2699–2702 RSC.
- K. Murakami, Y. Tanaka, R. Sakai, Y. Hisai, S. Hayashi, Y. Mizutani, T. Higo, S. Ogo, J.-G. Seo, H. Tsuneki and Y. Sekine, Chem. Commun., 2020, 56, 3365–3368 RSC.
- F. Maglia, I. G. Tredici, G. Spinolo and U. Anselmi-Tamburini, J. Mater. Res., 2012, 27, 1975–1981 CrossRef CAS.
- G. Gregori, M. Shirpour and J. Maier, Adv. Funct. Mater., 2013, 23, 5861–5867 CrossRef CAS.
- M. Takayanagi, T. Tsuchiya, K. Kawamura, M. Minohara, K. Horiba, H. Kumigashira and T. Higuchi, Solid State Ionics, 2017, 311, 46–51 CrossRef CAS.
- D. Fernández-Torre, K. Kośmider, J. Carrasco, M. V. Ganduglia-Pirovano and R. Pérez, J. Phys. Chem. C, 2012, 116, 13584–13593 CrossRef.
- R. Sato, S. Ohkuma, Y. Shibuta, F. Shimojo and S. Yamaguchi, J. Phys. Chem. C, 2015, 119, 28925–28933 CrossRef CAS.
- M. Farnesi Camellone, F. Negreiros Ribeiro, L. Szabová, Y. Tateyama and S. Fabris, J. Am. Chem. Soc., 2016, 138, 11560–11567 CrossRef CAS PubMed.
- G. Tocci and A. Michaelides, J. Phys. Chem. Lett., 2014, 5, 474–480 CrossRef CAS PubMed.
- D. T. Lundie, A. R. McInroy, R. Marshall, J. M. Winfield, P. Jones, C. C. Dudman, S. F. Parker, C. Mitchell and D. Lennon, J. Phys. Chem. B, 2005, 109, 11592–11601 CrossRef CAS PubMed.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- A. Takahashi, R. Inagaki, M. Torimoto, Y. Hisai, T. Matsuda, Q. Ma, J.-G. Seo, T. Higo, H. Tsuneki, S. Ogo, T. Norby and Y. Sekine, RSC Adv., 2020, 10, 14487–14492 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04717a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yudai Hisaia,
Naoya Nakanoa,
Ayako Takahashia,
Quanbao Mab,
Jeong Gil Seo
a,
Yudai Hisaia,
Naoya Nakanoa,
Ayako Takahashia,
Quanbao Mab,
Jeong Gil Seo